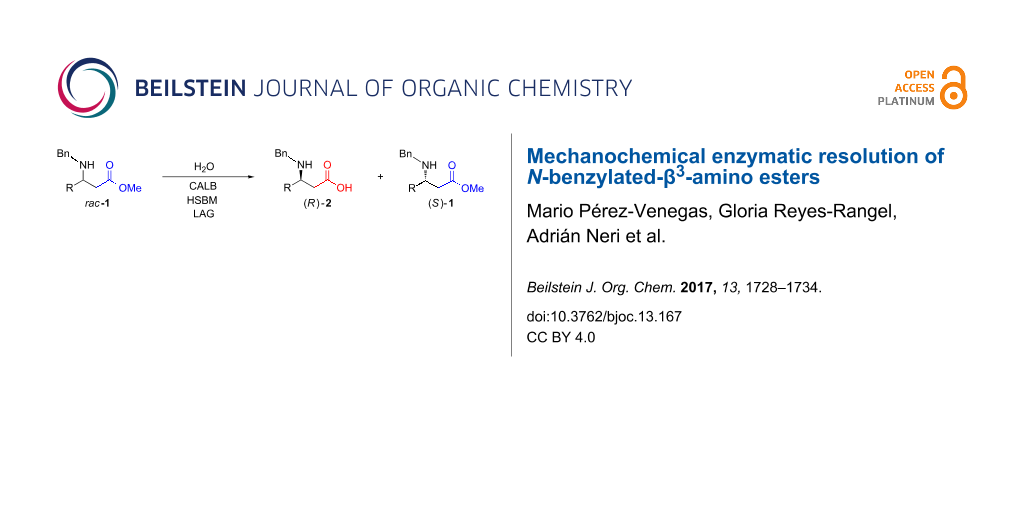
Abstract
The use of mechanochemistry to carry out enantioselective reactions has been explored in the last ten years with excellent results. Several chiral organocatalysts and even enzymes have proved to be resistant to milling conditions, which allows for rather efficient enantioselective transformations under ball-milling conditions. The present article reports the first example of a liquid-assisted grinding (LAG) mechanochemical enzymatic resolution of racemic β3-amino esters employing Candida antarctica lipase B (CALB) to afford highly valuable enantioenriched N-benzylated-β3-amino acids in good yields. Furthermore the present protocol is readily scalable.

Graphical Abstract
Introduction
β-Amino acids are rather interesting molecules that frequently exhibit exceptional biological properties [1-3]; for instance, some of them are efficient inhibitors of several enzymes [4,5]. Furthermore, β-amino acid residues can be used to protect peptides and proteins against the activity of proteolytic enzymes [6,7], or are precursors of numerous active compounds such as β-lactams [8,9]. Finally, β-amino acids are present in numerous natural products [10]. These properties have generated great interest in the development of synthetic methods for the preparation of β-amino acids, especially protocols leading to products with high enantiomeric excess (ee), which are required to test the pharmacological activity of each enantiomer [11-13]. In this regard, several methods for the asymmetric synthesis of β-amino acids have been documented [14-22] including strategies based on organocatalysis [23-26] and kinetic resolution using enzymes such as Candida antarctica lipase B, which was shown to be efficient in the resolution of racemic β-amino acids under various conditions [27-30].
Among recent developments in instrumentation for synthetic chemistry, mechanochemistry has proved a rather attractive and useful technique [31-37]. In particular, it has been demonstrated that mechanochemistry allows for the generation of products through catalysts that can be recovered and reused [38-44], so this converts mechanochemistry into a green technique, whose field of application is still very wide.
In this context, the use of a minimal amount of solvent (LAG) enable the development of convenient ball-milling protocols. In particular, LAG facilitates mechanochemical applications on a large scale [45,46].
Very recently, Hernández, Frings, and Bolm developed a method to carry out the kinetic resolution of secondary alcohols through selective acylation using Candida antarctica lipase B, under solvent-free ball-milling conditions [47,48]. Inspired by this ground-breaking report, which is in line with our continuous interest in developing new sustainable organocatalytic protocols [39,49-51], and taking advantage of previous experience with the enzymatic hydrolysis of a racemic mixture of N-protected-β3-amino acid methyl esters [52], we decided to examine the use of CALB enzyme under high-speed ball-milling (HSBM) conditions as a method to obtain enantiopure N-benzylated-β3-amino acids (Scheme 1).
![[1860-5397-13-167-i1]](/bjoc/content/inline/1860-5397-13-167-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Enantioselective enzymatic hydrolysis of racemic β3-amino ester rac-1a using CALB in solution [52] (top) and under HSBM conditions (button). 2M2B: 2-methyl-2-butanol.
Scheme 1: Enantioselective enzymatic hydrolysis of racemic β3-amino ester rac-1a using CALB in solution [52] (top...
Results and Discussion
A racemic mixture of substrate rac-1a (82 mg, 1 equiv) was milled in an Agate jar (12 mm of diameter, 4.6 mL) with an Agate ball (6 mm of diameter, 480 mg weight) using water (3.6 μL, 0.5 equiv), 0.2 mL of 2-methyl-2-butanol (2M2B) as a LAG additive (η = 1.63) and 40 mg of CALB (Novozym 435, Novozymes, recombinant, expressed in Aspergillus niger, immobilized in acrylic resin, >10000 U/g) at 25 Hz during 30 min. Gratifyingly, 55% conversion to the enantioenriched (R)-N-benzylated-β3-amino acid (R)-2a was observed, recovering 51% of enantioenriched starting material. It could be established by chiral HPLC that the ee of the product amounted 80% (Table 1, entry 1). This assay demonstrated that enzymatic hydrolysis can indeed be carried out under HSBM conditions. A second reaction was carried out under the same conditions but in the absence of the enzyme, which did not proceed and the starting material was recovered in its totality. This result shows that the observed hydrolysis is induced by CALB and not by the milling process per se. Furthermore, it could be established that the CALB enzyme and N-benzylated-β3-amino esters are stable to the mechanical force caused by HSBM. We then focused our attention on the search of the best conditions for this enzymatic mechanochemical resolution.
Table 1: Search of the best parameters in the enzymatic enantioselective hydrolysis of rac-1a under ball milling.
![[Graphic 1]](/bjoc/content/inline/1860-5397-13-167-i2.svg?max-width=637&scale=1.0)
|
|||||||
| entrya | LAG additiveb | yield (%)c (S)-1a/(R)-2a | time (h) | ee (S)-1a (%)d | ee (R)-2a (%)d | ce (%) | Ef |
| 1g | 2M2B | 51/49 | 0.5 | 99 | 80 | 55 | 46 |
| 2 | 2M2B | 70/30 | 0.5 | 89 | 77 | 54 | 23 |
| 3 | 2M2B | 51/49 | 1 | 99 | 95 | 51 | >200 |
| 4 | AcOEt | 86/13 | 1 | 69 | 95 | 42 | 81 |
| 5 | IPA | 82/21 | 1 | 48 | 95 | 34 | 63 |
| 6 | CH3CN | 65/29 | 1 | 65 | 95 | 41 | 77 |
| 7 | hexane | 40/60 | 1 | 97 | 86 | 53 | 55 |
| 8 | – | 58/41 | 1 | 95 | 92 | 51 | 89 |
| 9g | – | 58/42 | 1 | 93 | 86 | 52 | 45 |
| 10h | – | 68/31 | 1 | 74 | 80 | 48 | 20 |
aReactions were carried out with 0.5 equivalents of water and 15 Hz of frequency. b0.2 mL of LAG additive was used. cDetermined after purification by flash chromatography. dDetermined by HPLC with chiral stationary phase. eCalculated from c = ees/(ees + eep). fE = ln[1 − c(1 + eep)]/ln[1 − c(1 − eep)]. g25 Hz of frequency was used. h0.25 equivalents of water were used.
First of all, we examined the effect of the milling frequency, 15 Hz (Table 1, entry 2). Both yield and ee decreased substantially in comparison with the initial approach carried out at 25 Hz (Table 1, entry 1). Nevertheless, when the reaction time was increased from 30 min to 1 h at 15 Hz (Table 1, entry 3) the yield of the N-benzylated-β3-amino acid reached 49%, and presented high ee (95%, E > 200). These data represent an improvement both in ee and yield compared with the data recorded in solution [52]. Motivated by this result, we investigated the effect of other LAG additives in the reaction (see Supporting Information File 1, Table S1, entries 4–10). When 2M2B was replaced with other LAG additives a lower yield was observed (Table 1, entries 4–6). Nevertheless, the enantioselectivity of the process is maintained (95% ee), except when hexane was used (Table 1, entry 7), where a higher yield was observed (60%) although with a lower enantiomeric excess (86% ee). In the absence of a LAG additive and using 0.25 equivalents of water (Table 1, entries 8–10) both yield and ee were lower.
Water plays an important role in the reaction controlling the activity of the enzyme; for example, the use of 0.5 equivalents of water yielded 49% of product 2a (Table 1, entry 3). However, when 1 equivalent of water was employed the yield of the product increased to 92%. By contrast, when the reaction was carried out in the absence of water only traces of product were detected (see Supporting Information File 1 Table S1).
To determine the substrate scope, the conditions that led to the best results in the enzymatic resolution of substrate rac-1a (Table 1, entry 3) were employed with other racemic N-benzylated-β3-amino esters as substrates (Table 2). It can be appreciated that reaction yields decrease when longer aliphatic chains are present in the substrate (Table 2, entries 1–5), although the ee in products 2b–f remained rather high (>90%). Notably, this aliphatic chain-length effect has been studied in other systems with similar results [53].
Table 2: Substrate scope for the enzymatic resolution of N-benzylated-β3-amino esters.
![[Graphic 2]](/bjoc/content/inline/1860-5397-13-167-i3.svg?max-width=637&scale=1.0)
|
||||||||||
| entrya | rac | R | yield (%)b (S)-1/(R)-2 | eec (S)-1 (%) |
![[Graphic 3]](/bjoc/content/inline/1860-5397-13-167-i4.svg?max-width=637&scale=1.0)
|
eec (R)-2 (%) |
![[Graphic 4]](/bjoc/content/inline/1860-5397-13-167-i5.svg?max-width=637&scale=1.0)
|
cf (%) | Eg | absolute configurationh |
| 1 | 1b | CH3-(CH2)- | 51/49 | 91 | 4.5 | 97 | −36.5 | 48 | >200 | R |
| 2 | 1c | CH3-(CH2)2- | 53/43 | 84 | 2.1 | 98 | −45.2 | 46 | >200 | R |
| 3 | 1d | CH3-(CH2)3- | 68/29 | 23 | 2.0 | 94 | −35.3 | 20 | 40 | R |
| 4 | 1e | CH3-(CH2)4- | 74/24 | 57 | 0.2 | 94 | −40.0 | 15 | 38 | R |
| 5 | 1f | CH3-(CH2)5- | 79/18 | 13 | 0.8 | 91 | −39.7 | 13 | 24 | R |
| 6i | 1g | Ph | 92/10 | 18 | 3.4 | 83 | −35.0 | 18 | 13 | S |
| 7i | 1h | 4-MeO-Ph | 89/10 | 1 | −0.5 | 80 | −31.7 | 1 | 9 | S |
| 8 | 1i | t-Bu | 89/4 | 4 | −0.6 | 94 | 12.8 | 4 | 34 | S |
aReactions were carried out with 0.5 equivalents of water and 0.2 mL of 2M2B at 15 Hz during 1 h. bDetermined after purification by flash chromatography. cDetermined by HPLC with chiral stationary phase. dc = 0.33 in CH3Cl. ec = 0.33 in MeOH. fCalculated from c = ees/(ees + eep). gE = ln[1 − c(1 + eep)]/ln[1 − c(1 − eep)]. hAssigned by chemical correlation and by HPLC with chiral stationary phase. i0.75 equivalents of water were used.
The introduction of an aromatic ring (either unsubstituted or para-substituted) in the substrate resulted in diminished yields (Table 2, entries 6 and 7) but good ee (≥80%). With bulky groups, such as tert-butyl, the experimentally observed low yield was accompanied nevertheless by high ee (Table 2, entry 8). Other reaction conditions were tested aiming of increasing both yield and ee (see Supporting Information File 1); however, the best results continued to be obtained by using the conditions indicated in Table 1, entry 3.
To establish the absolute configuration of product 2a, a sample was crystallized to give a suitable single-crystal for X-ray diffraction analysis. The resulting structure showed the R configuration (Flack parameter = 0.154) in the stereocenter delimited by the atoms marked as C1, N1 and C3 (Figure 1). The R configuration in hydrolyzed product 2a was confirmed by comparison with literature data [52]. The configuration of products 2b, 2g to 2i was also assigned by comparison with literature data [52,54,55]. Finally, in the case of products 2c–f, comparison of the elution order for both enantiomers with the tendency found in 2a and 2b suggested that the configuration is the same in all of them (see Supporting Information File 1) [56,57].
![[1860-5397-13-167-1]](/bjoc/content/figures/1860-5397-13-167-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: X-ray crystallographic structure of product (R)-2a (50% of probability ellipsoids). CCDC registry number 1552645.
Figure 1: X-ray crystallographic structure of product (R)-2a (50% of probability ellipsoids). CCDC registry n...
The enzyme employed in these experiments was recovered by centrifugation of the reaction crude followed by drying under vacuum (90% of recovered enzyme; we will call it rCALB). This recovered material was reused to evaluate the enzyme recyclability after the mechanochemical protocol. When the reaction was carried out using the recovered enzyme the yield was not as good as the obtained with fresh catalyst (compare entries 1 and 2 in Table 3). This might suggest that the enzyme undergoes partial denaturation and/or partial destruction of the support, within each cycle (Table 3, entry 3). Interestingly, however, ee values of the isolated β-amino acid still resulted quite acceptable. On the other hand, no product was detected after the third cycle. To evaluate the denaturalization of the enzyme provoked by the milling process, a sample of fresh catalyst was milled for 1 h at 15 Hz under solvent-free conditions and in the presence of a LAG additive, finding that both reaction yield (38%) and ee (>90%) are higher (see Supporting Information File 1, Table S3, entries 5 and 6), compared with results from the hydrolysis using the catalyst recovered after the first cycle (Table 4, entry 2). The milling process carried out using the catalyst milled with 2M2B presents a slight decrease in ee compared with the resolution reaction using the milled enzyme under solvent-free conditions. This observation suggests that the LAG additive increases to some extent the degree of denaturation of the enzyme, reducing the enantiodiscrimination (ee = 91%) although maintaining significant catalytic activity (yield = 38%).
Table 3: Recycling capacity of immobilized CALB under HSBM conditions.
![[Graphic 5]](/bjoc/content/inline/1860-5397-13-167-i6.svg?max-width=637&scale=1.0)
|
||||||
| entrya | recycling cycle | yield (%)b (S)-1a/(R)-2a | eec (S)-1a (%) | eec (R)-2a (%) | cd (%) | Ee |
|---|---|---|---|---|---|---|
| 1 | – | 51/49 | 49 | 95 | 51 | >200 |
| 2 | 1 | 65/37 | 35 | 88 | 59 | 22 |
| 3 | 2 | 80/20 | 6 | 80 | 51 | 10 |
| 4 | 3 | 100/0 | 0 | – | – | – |
aReactions were carried out with 0.5 equivalents of water and 0.2 mL of 2M2B at 15 Hz during 1 h. bDetermined after purification by flash chromatography. cDetermined by HPLC with chiral stationary phase. dCalculated from c = ees/(ees + eep). eE = ln[1 − c(1 + eep)]/ln[1 − c(1 − eep)].
Finally, to test the scalability of the process, a set of reactions was carried out increasing the amount of substrate rac-1a under the optimized reaction parameters. (Table 4).
Table 4: Scaling-up of the enzymatic hydrolysis reaction under ball-milling using substrate rac-1a.
![[Graphic 6]](/bjoc/content/inline/1860-5397-13-167-i7.svg?max-width=637&scale=1.0)
|
||||||
| entrya | catalyst/substrate (equiv) b | yield (%)c (S)-1a/(R)-2a | eed (S)-1a (%) | eed (R)-2a (%) | ce (%) | Ef |
|---|---|---|---|---|---|---|
| 1g | 1/1 | 51/49 | >99 | 95 | 51 | >200 |
| 2 | 1/3 | 52/48 | 62 | 93 | 40 | 52 |
| 3 | 1/6 | 61/42 | 53 | 93 | 36 | 47 |
| 4 | 1/9 | 59/40 | 49 | 94 | 34 | 53 |
aReactions were carried out with 0.5 equivalents of water at 15 Hz during 1 h. b1 equivalent of enzyme = 40 mg; 1 equivalent of susbtrate = 82 mg. cDetermined after purification by flash chromatography. dDetermined by HPLC with chiral stationary phase. eCalculated from c = ees/(ees + eep). fE = ln[1 − c(1 + eep)]/ln[1 − c(1 − eep)]. g0.2 mL of LAG additive were used.
Relative to the results obtained with 1 equivalent of rac-1a in the presence of LAG additive (Table 4, entry 1) a slight decrease in yield was observed when 3 equivalents of substrate (and no LAG additive) were used to carry out the reaction (Table 4, entry 2). Nevertheless, the hydrolysis still proceeds with excellent ee (93%). This result confirms that under solvent-free conditions a particular amount of enzyme can catalyze a larger amount of substrate, even up to nine equivalents, without loss of enantiodiscrimination (Table 4, entry 4). It appears that this high efficiency is a consequence of the highly-concentrated medium that is generated under solvent-free mechanochemical conditions, an effect that is not possible to reach in solution [52]. This effect also allows for an increase in the amount of product per cycle of the enzymatic reaction.
Conclusion
The capacity of immobilized CALB to carry out the enzymatic hydrolytic resolution of N-benzylated-β3-amino esters under mechanochemical conditions was demonstrated. The reaction proceeds with an excellent yield (up to 49% of the theoretical 50% maximum) and high enantioselectivity (up to 98% ee). The method proved to be efficient in the resolution of racemic mixtures of β3-amino esters with aliphatic chains of different lengths, affording high ees of the resulting β-amino acids in spite of a decrease in yield in the case of long aliphatic chains. This efficiency of the enzymatic process is also observed in substrates with bulky aromatic groups (ee ≥ 80%). The reaction is best carried out in the presence of the LAG additive 2-methyl-2-butanol when the concentration of the substrate is low. The enzymatic process could be scaled-up to 9-fold affording the hydrolyzed product with high ee (≥93%) and an excellent yield (40% out of a 50% theoretical maximum). Finally, the enzyme catalyst could be recovered and reused several times affording the desired amino acids with good ee although with a decrease in conversion due to a partial denaturation process and partial destruction of the enzyme support.
Supporting Information
| Supporting Information File 1: Experimental section, NMR spectra, chromatograms and X-ray diffraction data. | ||
| Format: PDF | Size: 6.1 MB | Download |
References
-
Seebach, D.; Beck, A. K.; Capone, S.; Deniau, G.; Grošelj, U.; Zass, E. Synthesis 2009, 1–32. doi:10.1055/s-0028-1087490
Return to citation in text: [1] -
Steer, D. L.; Lew, R. A.; Perlmutter, P.; Smith, A. I.; Aguilar, M.-I. Curr. Med. Chem. 2002, 9, 811–822. doi:10.2174/0929867024606759
Return to citation in text: [1] -
Ton, J.; Mauch-Mani, B. Plant J. 2004, 38, 119–130. doi:10.1111/j.1365-313X.2004.02028.x
Return to citation in text: [1] -
Farmer, L. J.; Clark, M. P.; Boyd, M. J.; Perola, E.; Jones, S. M.; Tsai, A.; Jacobs, M. D.; Bandarage, U. K.; Ledeboer, M. W.; Wang, T.; Deng, H.; Ledford, B.; Gu, W.; Duffy, J. P.; Bethiel, R. S.; Shannon, D.; Byrn, R. A.; Leeman, J. R.; Rijnbrand, R.; Bennett, H. B.; O’Brien, C.; Memmott, C.; Nti-Addae, K.; Bennani, Y. L.; Charifson, P. S. ACS Med. Chem. Lett. 2017, 8, 256–260. doi:10.1021/acsmedchemlett.6b00486
Return to citation in text: [1] -
Seebach, D.; Beck, A. K.; Bierbaum, D. J. Chem. Biodiversity 2004, 1, 1111–1239. doi:10.1002/cbdv.200490087
Return to citation in text: [1] -
Gentilucci, L.; De Marco, R.; Cerisoli, L. Curr. Pharm. Des. 2010, 16, 3185–3203. doi:10.2174/138161210793292555
Return to citation in text: [1] -
Zubrzak, P.; Williams, H.; Coast, G. M.; Issac, R. E.; Reyes-Rangel, G.; Juaristi, E.; Zabrocki, J.; Nachman, R. J. Pept. Sci. 2007, 88, 76–82. doi:10.1002/bip.20638
Return to citation in text: [1] -
Magriotis, P. A. Angew. Chem., Int. Ed. 2001, 40, 4377–4379. doi:10.1002/1521-3773(20011203)40:23<4377::AID-ANIE4377>3.0.CO;2-J
Return to citation in text: [1] -
Escalante, J.; González-Tototzin, M. A.; Aviña, J.; Muñoz-Muñiz, O.; Juaristi, E. Tetrahedron 2001, 57, 1883–1890. doi:10.1016/S0040-4020(00)01169-8
Return to citation in text: [1] -
Spiteller, P.; von Nussbaum, F. β-Amino Acids in Natural Products. In Enantioselective Synthesis of β-Amino acids, 2nd ed.; Juaristi, E.; Soloshonok, V. A., Eds.; Wiley-VCH: New York, 2005; pp 19–91. doi:10.1002/0471698482.ch2
Return to citation in text: [1] -
Hoekstra,, W. J.; Maryanoff, B. E.; Damiano, B. P.; Andrade-Gordon, P.; Cohen, J. H.; Costanzo, M. J.; Haertlein, B. J.; Hecker, L. R.; Hulshizer, B. L.; Kauffman, J. A.; Keane, P.; McComsey, D. F.; Mitchell, J. A.; Scott, L.; Shah, R. D.; Yabut, S. C. J. Med. Chem. 1999, 42, 5254–5265. doi:10.1021/jm990418b
Return to citation in text: [1] -
Synthesis of Non-Natural Amino Acids. In Handbook of Chiral Chemicals, 2nd ed.; Ager, D. J., Ed.; DSM Pharma Chemicals: Raleigh, NC, 2006.
Return to citation in text: [1] -
Bandala, Y.; Juaristi, E. Recent Developments in the Synthesis of b-Amino Acids. In Amino Acids, Peptides and Proteins in Organic Chemistry; Hughes, A. B., Ed.; Wiley-VCH: Weinheim, 2009; pp 291–365. doi:10.1002/9783527631766.ch7
Return to citation in text: [1] -
Juaristi, E.; Soloshonok, V. A., Eds. Enantioselective Synthesis of β-Amino Acids, 2nd ed.; John Wiley and Sons: Hoboken, NJ, 2005.
Return to citation in text: [1] -
Weiner, B.; Szymański, W.; Janssen, D. B.; Minnaard, A. J.; Feringa, B. L. Chem. Soc. Rev. 2010, 39, 1656–1691. doi:10.1039/b919599h
Return to citation in text: [1] -
Gedey, S.; Liljeblad, A.; Lázár, L.; Fülöp, F.; Kanerva, L. T. Tetrahedron: Asymmetry 2001, 12, 105–110. doi:10.1016/S0957-4166(01)00002-7
Return to citation in text: [1] -
Juaristi, E.; Quintana, D.; Escalante, J. Aldrichimica Acta 1994, 27, 3–11.
Return to citation in text: [1] -
Juaristi, E.; López-Ruiz, H. Curr. Med. Chem. 1999, 6, 983–1004.
Return to citation in text: [1] -
Abele, S.; Seebach, D. Eur. J. Org. Chem. 2000, 1–15. doi:10.1002/(SICI)1099-0690(200001)2000:1<1::AID-EJOC1>3.0.CO;2-6
Return to citation in text: [1] -
Fülöp, F. Chem. Rev. 2001, 101, 2181–2204. doi:10.1021/cr000456z
Return to citation in text: [1] -
Liu, M.; Sibi, M. P. Tetrahedron 2002, 58, 7991–8035. doi:10.1016/S0040-4020(02)00991-2
Return to citation in text: [1] -
Ma, J.-A. Angew. Chem., Int. Ed. 2003, 42, 4290–4299. doi:10.1002/anie.200301600
Return to citation in text: [1] -
Wenzel, A. G.; Jacobsen, E. N. J. Am. Chem. Soc. 2002, 124, 12964–12965. doi:10.1021/ja028353g
Return to citation in text: [1] -
Ting, A.; Schaus, S. E. Eur. J. Org. Chem. 2007, 35, 5797–5815. doi:10.1002/ejoc.200700409
Return to citation in text: [1] -
Wilson, J. E.; Casarez, A. D.; MacMillan, D. W. C. J. Am. Chem. Soc. 2009, 131, 11332–11334. doi:10.1021/ja904504j
Return to citation in text: [1] -
Meyer, D.; Marti, R.; Seebach, D. Eur. J. Org. Chem. 2015, 2015, 4883–4891. doi:10.1002/ejoc.201500636
Return to citation in text: [1] -
Forró, E.; Fülöp, F. Chem. – Eur. J. 2007, 13, 6397–6401. doi:10.1002/chem.200700257
Return to citation in text: [1] -
Fitz, M.; Forró, E.; Vigóczki, E.; Lázár, L.; Fülöp, F. Tetrahedron: Asymmetry 2008, 19, 1114–1119. doi:10.1016/j.tetasy.2008.04.002
Return to citation in text: [1] -
Heck, T.; Seebach, D.; Steffen, O.; ter Wiel, M. K. J.; Kohler, H.-P. E.; Geueke, B. ChemBioChem 2009, 10, 1558–1561. doi:10.1002/cbic.200900184
Return to citation in text: [1] -
Weise, N. J.; Ahmed, S. T.; Parmeggiani, F.; Turner, N. J. Adv. Synth. Catal. 2017, 359, 1570–1576. doi:10.1002/adsc.201600894
Return to citation in text: [1] -
Hernández, J. G.; Bolm, C. J. Org. Chem. 2017, 82, 4007–4019. doi:10.1021/acs.joc.6b02887
Return to citation in text: [1] -
Rodríguez, B.; Rantanen, T.; Bolm, C. Angew. Chem., Int. Ed. 2006, 45, 6924–6926. doi:10.1002/anie.200602820
Return to citation in text: [1] -
Declerck, V.; Nun, P.; Martinez, J.; Lamaty, F. Angew. Chem., Int. Ed. 2009, 48, 9318–9321. doi:10.1002/anie.200903510
Return to citation in text: [1] -
Bonnamour, J.; Métro, T.-X.; Martinez, J.; Lamaty, F. Green Chem. 2013, 15, 1116–1120. doi:10.1039/c3gc40302e
Return to citation in text: [1] -
Baig, R. B. N.; Varma, R. S. Chem. Soc. Rev. 2012, 41, 1559–1584. doi:10.1039/C1CS15204A
Return to citation in text: [1] -
Jones, W.; Eddleston, M. D. Faraday Discuss. 2014, 170, 9–34. doi:10.1039/C4FD00162A
Return to citation in text: [1] -
Hernández, J. G.; Friščić, T. Tetrahedron Lett. 2015, 56, 4253–4265. doi:10.1016/j.tetlet.2015.03.135
Return to citation in text: [1] -
Lawrenson, S. B.; Arav, R.; North, M. Green Chem. 2017, 19, 1685–1691. doi:10.1039/C7GC00247E
Return to citation in text: [1] -
Hernández, J. G.; Juaristi, E. Chem. Commun. 2012, 48, 5396–5409. doi:10.1039/c2cc30951c
Return to citation in text: [1] [2] -
Schmidt, R.; Stolle, A.; Ondruschka, B. Green Chem. 2012, 14, 1673–1679. doi:10.1039/c2gc16508b
Return to citation in text: [1] -
Hernández, J. G.; Macdonald, N. A. J.; Mottillo, C.; Butler, I. S.; Friščić, T. Green Chem. 2014, 16, 1087–1092. doi:10.1039/C3GC42104J
Return to citation in text: [1] -
Machuca, E.; Juaristi, E. Asymmetric Organocatalytic Reactions Under Ball Milling. In Ball Milling Towards Green Synthesis: Applications, Projects, Challenges; Ranu, B.; Stolle, A., Eds.; Royal Society of Chemistry: Cambridge, UK, 2015; pp 81–95.
Return to citation in text: [1] -
McKissic, K. S.; Caruso, J. T.; Blair, R. G.; Mack, J. Green Chem. 2014, 16, 1628–1632. doi:10.1039/c3gc41496e
Return to citation in text: [1] -
Schmidt, R.; Burmeister, C. F.; Baláž, M.; Kwade, A.; Stolle, A. Org. Process Res. Dev. 2015, 19, 427–436. doi:10.1021/op5003787
Return to citation in text: [1] -
Friščić, T.; Trask, A. V.; Jones, W.; Motherwell, W. D. S. Angew. Chem., Int. Ed. 2006, 45, 7546–7550. doi:10.1002/anie.200603235
Return to citation in text: [1] -
Friščić, T.; Jones, W. Cryst. Growth Des. 2009, 9, 1621–1637. doi:10.1021/cg800764n
Return to citation in text: [1] -
Hernández, J. G.; Frings, M.; Bolm, C. ChemCatChem 2016, 8, 1769–1772. doi:10.1002/cctc.201600455
Return to citation in text: [1] -
Hernández, J. G.; Ardila-Fierro, K. J.; Crawford, D.; James, S. L.; Bolm, C. Green Chem. 2017, 19, 2620–2625. doi:10.1039/C7GC00615B
Return to citation in text: [1] -
Hernández, J. G.; Juaristi, E. J. Org. Chem. 2010, 75, 7107–7111. doi:10.1021/jo101159a
Return to citation in text: [1] -
Polindara-García, L. A.; Juaristi, E. Eur. J. Org. Chem. 2016, 1095–1102. doi:10.1002/ejoc.201501371
Return to citation in text: [1] -
Landeros, J. M.; Juaristi, E. Eur. J. Org. Chem. 2017, 687–694. doi:10.1002/ejoc.201601276
Return to citation in text: [1] -
Rangel, H.; Carrillo-Morales, M.; Galindo, J. M.; Castillo, E.; Obregón-Zúñiga, A.; Juaristi, E.; Escalante, J. Tetrahedron: Asymmetry 2015, 26, 325–332. doi:10.1016/j.tetasy.2015.02.007
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Vaysse, L.; Ly, A.; Moulin, G.; Dubreucq, E. Enzyme Microb. Technol. 2002, 31, 648–655. doi:10.1016/S0141-0229(02)00166-7
Return to citation in text: [1] -
Guizzetti, S.; Benaglia, M.; Bonsignore, M.; Raimondi, L. Org. Biomol. Chem. 2011, 9, 739–743. doi:10.1039/C0OB00570C
Return to citation in text: [1] -
Bonsignore, M.; Benaglia, M.; Annunziata, R.; Celentano, G. Synlett 2011, 1085–1088. doi:10.1055/s-0030-1259941
Return to citation in text: [1] -
Péter, A.; Lázár, L.; Fülöp, F.; Armstrong, D. W. J. Chromatogr. A 2001, 926, 229–238. doi:10.1016/S0021-9673(01)01078-0
Return to citation in text: [1] -
Enders, D.; Wahl, H.; Bettray, W. Angew. Chem., Int. Ed. Engl. 1995, 34, 455–457. doi:10.1002/anie.199504551
Return to citation in text: [1]
| 52. | Rangel, H.; Carrillo-Morales, M.; Galindo, J. M.; Castillo, E.; Obregón-Zúñiga, A.; Juaristi, E.; Escalante, J. Tetrahedron: Asymmetry 2015, 26, 325–332. doi:10.1016/j.tetasy.2015.02.007 |
| 52. | Rangel, H.; Carrillo-Morales, M.; Galindo, J. M.; Castillo, E.; Obregón-Zúñiga, A.; Juaristi, E.; Escalante, J. Tetrahedron: Asymmetry 2015, 26, 325–332. doi:10.1016/j.tetasy.2015.02.007 |
| 53. | Vaysse, L.; Ly, A.; Moulin, G.; Dubreucq, E. Enzyme Microb. Technol. 2002, 31, 648–655. doi:10.1016/S0141-0229(02)00166-7 |
| 1. | Seebach, D.; Beck, A. K.; Capone, S.; Deniau, G.; Grošelj, U.; Zass, E. Synthesis 2009, 1–32. doi:10.1055/s-0028-1087490 |
| 2. | Steer, D. L.; Lew, R. A.; Perlmutter, P.; Smith, A. I.; Aguilar, M.-I. Curr. Med. Chem. 2002, 9, 811–822. doi:10.2174/0929867024606759 |
| 3. | Ton, J.; Mauch-Mani, B. Plant J. 2004, 38, 119–130. doi:10.1111/j.1365-313X.2004.02028.x |
| 10. | Spiteller, P.; von Nussbaum, F. β-Amino Acids in Natural Products. In Enantioselective Synthesis of β-Amino acids, 2nd ed.; Juaristi, E.; Soloshonok, V. A., Eds.; Wiley-VCH: New York, 2005; pp 19–91. doi:10.1002/0471698482.ch2 |
| 52. | Rangel, H.; Carrillo-Morales, M.; Galindo, J. M.; Castillo, E.; Obregón-Zúñiga, A.; Juaristi, E.; Escalante, J. Tetrahedron: Asymmetry 2015, 26, 325–332. doi:10.1016/j.tetasy.2015.02.007 |
| 8. | Magriotis, P. A. Angew. Chem., Int. Ed. 2001, 40, 4377–4379. doi:10.1002/1521-3773(20011203)40:23<4377::AID-ANIE4377>3.0.CO;2-J |
| 9. | Escalante, J.; González-Tototzin, M. A.; Aviña, J.; Muñoz-Muñiz, O.; Juaristi, E. Tetrahedron 2001, 57, 1883–1890. doi:10.1016/S0040-4020(00)01169-8 |
| 52. | Rangel, H.; Carrillo-Morales, M.; Galindo, J. M.; Castillo, E.; Obregón-Zúñiga, A.; Juaristi, E.; Escalante, J. Tetrahedron: Asymmetry 2015, 26, 325–332. doi:10.1016/j.tetasy.2015.02.007 |
| 6. | Gentilucci, L.; De Marco, R.; Cerisoli, L. Curr. Pharm. Des. 2010, 16, 3185–3203. doi:10.2174/138161210793292555 |
| 7. | Zubrzak, P.; Williams, H.; Coast, G. M.; Issac, R. E.; Reyes-Rangel, G.; Juaristi, E.; Zabrocki, J.; Nachman, R. J. Pept. Sci. 2007, 88, 76–82. doi:10.1002/bip.20638 |
| 47. | Hernández, J. G.; Frings, M.; Bolm, C. ChemCatChem 2016, 8, 1769–1772. doi:10.1002/cctc.201600455 |
| 48. | Hernández, J. G.; Ardila-Fierro, K. J.; Crawford, D.; James, S. L.; Bolm, C. Green Chem. 2017, 19, 2620–2625. doi:10.1039/C7GC00615B |
| 4. | Farmer, L. J.; Clark, M. P.; Boyd, M. J.; Perola, E.; Jones, S. M.; Tsai, A.; Jacobs, M. D.; Bandarage, U. K.; Ledeboer, M. W.; Wang, T.; Deng, H.; Ledford, B.; Gu, W.; Duffy, J. P.; Bethiel, R. S.; Shannon, D.; Byrn, R. A.; Leeman, J. R.; Rijnbrand, R.; Bennett, H. B.; O’Brien, C.; Memmott, C.; Nti-Addae, K.; Bennani, Y. L.; Charifson, P. S. ACS Med. Chem. Lett. 2017, 8, 256–260. doi:10.1021/acsmedchemlett.6b00486 |
| 5. | Seebach, D.; Beck, A. K.; Bierbaum, D. J. Chem. Biodiversity 2004, 1, 1111–1239. doi:10.1002/cbdv.200490087 |
| 39. | Hernández, J. G.; Juaristi, E. Chem. Commun. 2012, 48, 5396–5409. doi:10.1039/c2cc30951c |
| 49. | Hernández, J. G.; Juaristi, E. J. Org. Chem. 2010, 75, 7107–7111. doi:10.1021/jo101159a |
| 50. | Polindara-García, L. A.; Juaristi, E. Eur. J. Org. Chem. 2016, 1095–1102. doi:10.1002/ejoc.201501371 |
| 51. | Landeros, J. M.; Juaristi, E. Eur. J. Org. Chem. 2017, 687–694. doi:10.1002/ejoc.201601276 |
| 27. | Forró, E.; Fülöp, F. Chem. – Eur. J. 2007, 13, 6397–6401. doi:10.1002/chem.200700257 |
| 28. | Fitz, M.; Forró, E.; Vigóczki, E.; Lázár, L.; Fülöp, F. Tetrahedron: Asymmetry 2008, 19, 1114–1119. doi:10.1016/j.tetasy.2008.04.002 |
| 29. | Heck, T.; Seebach, D.; Steffen, O.; ter Wiel, M. K. J.; Kohler, H.-P. E.; Geueke, B. ChemBioChem 2009, 10, 1558–1561. doi:10.1002/cbic.200900184 |
| 30. | Weise, N. J.; Ahmed, S. T.; Parmeggiani, F.; Turner, N. J. Adv. Synth. Catal. 2017, 359, 1570–1576. doi:10.1002/adsc.201600894 |
| 38. | Lawrenson, S. B.; Arav, R.; North, M. Green Chem. 2017, 19, 1685–1691. doi:10.1039/C7GC00247E |
| 39. | Hernández, J. G.; Juaristi, E. Chem. Commun. 2012, 48, 5396–5409. doi:10.1039/c2cc30951c |
| 40. | Schmidt, R.; Stolle, A.; Ondruschka, B. Green Chem. 2012, 14, 1673–1679. doi:10.1039/c2gc16508b |
| 41. | Hernández, J. G.; Macdonald, N. A. J.; Mottillo, C.; Butler, I. S.; Friščić, T. Green Chem. 2014, 16, 1087–1092. doi:10.1039/C3GC42104J |
| 42. | Machuca, E.; Juaristi, E. Asymmetric Organocatalytic Reactions Under Ball Milling. In Ball Milling Towards Green Synthesis: Applications, Projects, Challenges; Ranu, B.; Stolle, A., Eds.; Royal Society of Chemistry: Cambridge, UK, 2015; pp 81–95. |
| 43. | McKissic, K. S.; Caruso, J. T.; Blair, R. G.; Mack, J. Green Chem. 2014, 16, 1628–1632. doi:10.1039/c3gc41496e |
| 44. | Schmidt, R.; Burmeister, C. F.; Baláž, M.; Kwade, A.; Stolle, A. Org. Process Res. Dev. 2015, 19, 427–436. doi:10.1021/op5003787 |
| 52. | Rangel, H.; Carrillo-Morales, M.; Galindo, J. M.; Castillo, E.; Obregón-Zúñiga, A.; Juaristi, E.; Escalante, J. Tetrahedron: Asymmetry 2015, 26, 325–332. doi:10.1016/j.tetasy.2015.02.007 |
| 23. | Wenzel, A. G.; Jacobsen, E. N. J. Am. Chem. Soc. 2002, 124, 12964–12965. doi:10.1021/ja028353g |
| 24. | Ting, A.; Schaus, S. E. Eur. J. Org. Chem. 2007, 35, 5797–5815. doi:10.1002/ejoc.200700409 |
| 25. | Wilson, J. E.; Casarez, A. D.; MacMillan, D. W. C. J. Am. Chem. Soc. 2009, 131, 11332–11334. doi:10.1021/ja904504j |
| 26. | Meyer, D.; Marti, R.; Seebach, D. Eur. J. Org. Chem. 2015, 2015, 4883–4891. doi:10.1002/ejoc.201500636 |
| 45. | Friščić, T.; Trask, A. V.; Jones, W.; Motherwell, W. D. S. Angew. Chem., Int. Ed. 2006, 45, 7546–7550. doi:10.1002/anie.200603235 |
| 46. | Friščić, T.; Jones, W. Cryst. Growth Des. 2009, 9, 1621–1637. doi:10.1021/cg800764n |
| 14. | Juaristi, E.; Soloshonok, V. A., Eds. Enantioselective Synthesis of β-Amino Acids, 2nd ed.; John Wiley and Sons: Hoboken, NJ, 2005. |
| 15. | Weiner, B.; Szymański, W.; Janssen, D. B.; Minnaard, A. J.; Feringa, B. L. Chem. Soc. Rev. 2010, 39, 1656–1691. doi:10.1039/b919599h |
| 16. | Gedey, S.; Liljeblad, A.; Lázár, L.; Fülöp, F.; Kanerva, L. T. Tetrahedron: Asymmetry 2001, 12, 105–110. doi:10.1016/S0957-4166(01)00002-7 |
| 17. | Juaristi, E.; Quintana, D.; Escalante, J. Aldrichimica Acta 1994, 27, 3–11. |
| 18. | Juaristi, E.; López-Ruiz, H. Curr. Med. Chem. 1999, 6, 983–1004. |
| 19. | Abele, S.; Seebach, D. Eur. J. Org. Chem. 2000, 1–15. doi:10.1002/(SICI)1099-0690(200001)2000:1<1::AID-EJOC1>3.0.CO;2-6 |
| 20. | Fülöp, F. Chem. Rev. 2001, 101, 2181–2204. doi:10.1021/cr000456z |
| 21. | Liu, M.; Sibi, M. P. Tetrahedron 2002, 58, 7991–8035. doi:10.1016/S0040-4020(02)00991-2 |
| 22. | Ma, J.-A. Angew. Chem., Int. Ed. 2003, 42, 4290–4299. doi:10.1002/anie.200301600 |
| 52. | Rangel, H.; Carrillo-Morales, M.; Galindo, J. M.; Castillo, E.; Obregón-Zúñiga, A.; Juaristi, E.; Escalante, J. Tetrahedron: Asymmetry 2015, 26, 325–332. doi:10.1016/j.tetasy.2015.02.007 |
| 54. | Guizzetti, S.; Benaglia, M.; Bonsignore, M.; Raimondi, L. Org. Biomol. Chem. 2011, 9, 739–743. doi:10.1039/C0OB00570C |
| 55. | Bonsignore, M.; Benaglia, M.; Annunziata, R.; Celentano, G. Synlett 2011, 1085–1088. doi:10.1055/s-0030-1259941 |
| 11. | Hoekstra,, W. J.; Maryanoff, B. E.; Damiano, B. P.; Andrade-Gordon, P.; Cohen, J. H.; Costanzo, M. J.; Haertlein, B. J.; Hecker, L. R.; Hulshizer, B. L.; Kauffman, J. A.; Keane, P.; McComsey, D. F.; Mitchell, J. A.; Scott, L.; Shah, R. D.; Yabut, S. C. J. Med. Chem. 1999, 42, 5254–5265. doi:10.1021/jm990418b |
| 12. | Synthesis of Non-Natural Amino Acids. In Handbook of Chiral Chemicals, 2nd ed.; Ager, D. J., Ed.; DSM Pharma Chemicals: Raleigh, NC, 2006. |
| 13. | Bandala, Y.; Juaristi, E. Recent Developments in the Synthesis of b-Amino Acids. In Amino Acids, Peptides and Proteins in Organic Chemistry; Hughes, A. B., Ed.; Wiley-VCH: Weinheim, 2009; pp 291–365. doi:10.1002/9783527631766.ch7 |
| 31. | Hernández, J. G.; Bolm, C. J. Org. Chem. 2017, 82, 4007–4019. doi:10.1021/acs.joc.6b02887 |
| 32. | Rodríguez, B.; Rantanen, T.; Bolm, C. Angew. Chem., Int. Ed. 2006, 45, 6924–6926. doi:10.1002/anie.200602820 |
| 33. | Declerck, V.; Nun, P.; Martinez, J.; Lamaty, F. Angew. Chem., Int. Ed. 2009, 48, 9318–9321. doi:10.1002/anie.200903510 |
| 34. | Bonnamour, J.; Métro, T.-X.; Martinez, J.; Lamaty, F. Green Chem. 2013, 15, 1116–1120. doi:10.1039/c3gc40302e |
| 35. | Baig, R. B. N.; Varma, R. S. Chem. Soc. Rev. 2012, 41, 1559–1584. doi:10.1039/C1CS15204A |
| 36. | Jones, W.; Eddleston, M. D. Faraday Discuss. 2014, 170, 9–34. doi:10.1039/C4FD00162A |
| 37. | Hernández, J. G.; Friščić, T. Tetrahedron Lett. 2015, 56, 4253–4265. doi:10.1016/j.tetlet.2015.03.135 |
| 56. | Péter, A.; Lázár, L.; Fülöp, F.; Armstrong, D. W. J. Chromatogr. A 2001, 926, 229–238. doi:10.1016/S0021-9673(01)01078-0 |
| 57. | Enders, D.; Wahl, H.; Bettray, W. Angew. Chem., Int. Ed. Engl. 1995, 34, 455–457. doi:10.1002/anie.199504551 |
© 2017 Pérez-Venegas et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)