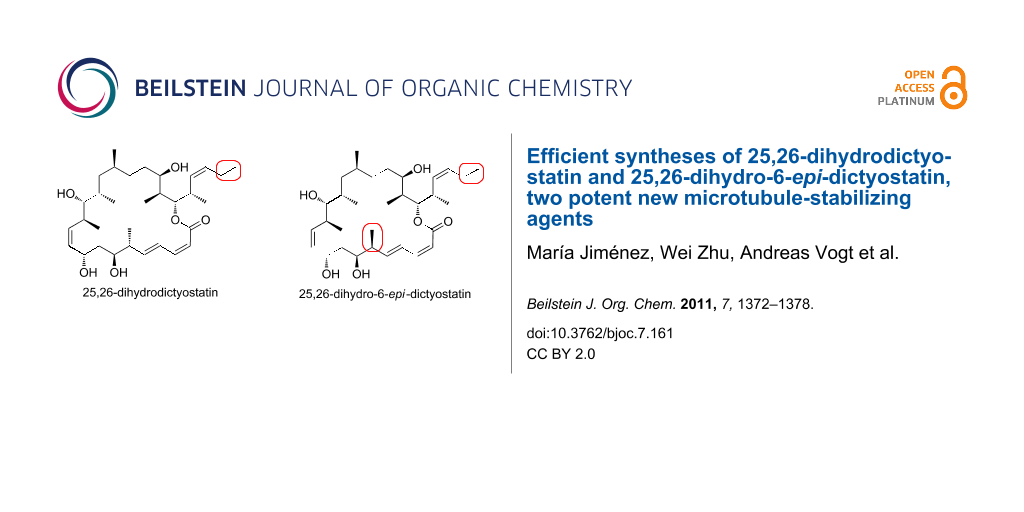
Abstract
The dictyostatins are powerful microtubule-stabilizing agents that have shown antiproliferative activity against a variety of human cancer cell lines. Two highly active analogs of dictyostatin, 25,26-dihydrodictyostatin and 25,26-dihydro-6-epi-dictyostatin, were prepared by a new streamlined total synthesis route. Three complete carbon fragments were prepared to achieve maximum convergency. These were coupled by a Horner–Wadsworth–Emmons reaction sequence and an esterification. A late stage Nozaki–Hiyama–Kishi reaction was then used to form the 22-membered macrolide. The stereoselectivity of this reaction depended on the configurations of the nearby stereocenter at C6.

Graphical Abstract
Introduction
The discovery of compounds that function as anticancer agents by altering the dynamics of microtubules continues to be an important goal in medicinal chemistry. Such agents can force the cell to exit mitosis aberrantly, leading to apoptosis [1,2]. Important classes of microtubule-stabilizing agents include taxanes, epothilones, and discodermolides, among others [3,4]. Dictyostatin (1a) is an exceptionally potent microtubule-stabilizing agent that has shown antiproliferative activity in a variety of human cancer cell lines in the low nanomolar range. Isolated first in 1994 by Pettit and coworkers [5,6], its complete stereostructure was proposed by Paterson and Wright in 2004 [7]. The structure assignment phase was finalized in 2004 when total syntheses by Paterson and our group confirmed the assignment [8,9].
The two synthetic samples of dictyostatin provided exciting biological results [10], which in turn spurred structure–activity studies in both Paterson’s group [11-17] and ours [18-23]. These studies, founded on total synthesis, were largely complementary and together provide a solid if still evolving [24] picture of dictyostatin SAR. Phillips [25], Ramachandran [26] and Gennari [27] have also developed efficient synthetic routes to the natural product or fully functionalized analogs.
Based on the biological profile of over 30 analogs of dictyostatin synthesized in Pittsburgh, we selected 6-epi-dictyostatin (1b) for scale-up and in vivo testing [28]. Indeed, 1b proved to be more effective than paclitaxel in treating mice bearing human breast cancer xenografts. Encouraged by these results, we have pursued both new analogs and improved synthetic routes. We recently reported a streamlined synthesis of dictyostatin (1a) and used it to prepare two new analogs: 16-desmethyl-25,26-dihydrodictyostatin (2a) and its C6 epimer 2b (Figure 1) [29]. The terminal C25–C26 alkene of the C23–C26 diene is a synthetic liability, so it was welcome news when the biological data revealed that the analogs lacking this alkene retained significant activity. Prior results suggested that most if not all of the lost activity could be regained by reinstating the missing C16-methyl group [11,19].
![[1860-5397-7-161-1]](/bjoc/content/figures/1860-5397-7-161-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structures of dictyostatin and selected analogs varying at C6, C16, and C25–C26.
Figure 1: Structures of dictyostatin and selected analogs varying at C6, C16, and C25–C26.
We thus set out to synthesize and test two new analogs of dictyostatin: 25,26-dihydrodictyostatin (3a) and 25,26-dihydro-6-epi-dictyostatin (3b) (Figure 1). The later parts of this synthesis have been briefly communicated in a recent paper whose primary focus was biological evaluation [30]. Indeed, 3a and 3b prove to be promising anticancer agents with in vitro and cellular testing data superior to those of the 16-desmethyl analogs 2a and 2b, and roughly comparable to those of dictyostatin (1a) and 6-epi-dictyostatin (1b). Here we report the full details of the synthesis of 3a and 3b.
Results and Discussion
Figure 2 shows key aspects of the retrosynthetic analysis to make analogs 3a and 3b, which follows after the successful streamlined route to make 1a [29]. For high convergence, the analogs were dissected strategically into three complete fragments called top (8, C18–C26), middle (7, C10–C17), and bottom (6a,b, C1–C9), respectively. The top 8 and middle 7 fragments were first combined through an established Horner–Wadsworth–Emmons (HWE) reaction sequence to give 5 [8,9]. The bottom fragment 6a or 6b was then attached to the top/middle fragment 5 through an esterification reaction. Finally, an intramolecular Nozaki–Hiyama–Kishi (NHK) reaction [31,32] of compounds 4a and 4b was used to form the macrolactone at C9–C10.
![[1860-5397-7-161-2]](/bjoc/content/figures/1860-5397-7-161-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Retrosynthetic analysis for 3a and 3b.
Figure 2: Retrosynthetic analysis for 3a and 3b.
We have previously described in detail the synthesis of the top fragment 8, which is shared by both analogs 3a,b [29,33]. As summarized in Scheme 1, this fragment was made on multigram scale in seven steps starting from the well-known intermediate 9.
![[1860-5397-7-161-i1]](/bjoc/content/inline/1860-5397-7-161-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of top fragment 8 (C18–C26).
Scheme 1: Synthesis of top fragment 8 (C18–C26).
The shared middle fragment 7 was conveniently made by the pathway shown in Scheme 2, which is a streamlined version of prior routes in the 16-desmethyl series [14]. (However, some material 7 was also made by an extension of a prior middle fragment route, as described in Supporting Information File 1, Scheme S1). Myers alkylation [17] of commercially available pseudoephedrine amide 10 and alkyl iodide 11 afforded amide 12 in 95% yield. The chiral auxiliary was then removed with BH3·NH3 (92%) [34], then the resulting primary alcohol was oxidized under Swern conditions to provide aldehyde 13 (86%). A Marshall palladium-catalyzed addition reaction [35] between aldehyde 13 and mesylate 14 [36] gave alcohol 15 in 77% yield. Interestingly, the Marshall reaction works well even with mesylates containing a terminal alkyne; reaction of 13 with 16 provided 17 in 72% yield.
![[1860-5397-7-161-i2]](/bjoc/content/inline/1860-5397-7-161-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of middle fragment 7 (C10–C17).
Scheme 2: Synthesis of middle fragment 7 (C10–C17).
The TIPS and primary TBS groups of 15 were simultaneously cleaved under basic conditions with TBAF [37] (88%), followed by protection of both primary and secondary hydroxy groups with TBSOTf to afford 18 in 96% yield. The terminal alkyne of 18 was then converted to the iodoalkyne with BuLi and I2 (92%), followed by diimide reduction with o-nitrobenzene sulfonylhydrazide (NBSH) [38] to the cis-vinyl iodide 19 in 94% yield. The primary TBS group of 19 was selectively removed with HF·pyridine in pyridine/THF to afford a primary alcohol (97%), which was then oxidized to aldehyde 7 with the aid of the Dess–Martin reagent (85%). Overall, the synthesis of the middle fragment was accomplished in 10 steps in over 40% yield starting from commercially available pseudoephedrine amide 10.
The syntheses of the bottom fragments 6a and 6b were achieved by applying the previously reported cross-metathesis reactions of readily available 20a,b with (2Z,4E)-methyl hexa-2,4-dienoate followed by silylation to provide 21a,b [39]. These dienoates were converted to the carboxylic acids by using TMSOK [40], followed by transformation into the acid chloride 6a and 6b with the Ghosez reagent [41] (Scheme 3). This acid chloride was then used directly in the subsequent esterification reaction.
![[1860-5397-7-161-i3]](/bjoc/content/inline/1860-5397-7-161-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of bottom fragments 6a,b (C1–C9).
Scheme 3: Synthesis of bottom fragments 6a,b (C1–C9).
With the top, middle and bottom fragments in hand, we turned to the initial fragment couplings as summarized in Scheme 4. Fragments 7 and 8 were first combined by a proven sequence starting with an HWE reaction mediated by Ba(OH)2 [42] to provide enone 22 in good yield (80%). This enone was then treated with Stryker’s reagent [43] to selectively reduce the conjugated alkene, followed directly by removal of the PMB group with DDQ. This sequence afforded alcohol 23 in 76% yield over two steps. A stereoselective 1,3-syn reduction was then performed under Prasad conditions [44] to give the target diol (90%) as a single isomer. The stereochemistry at C19 was confirmed by NMR analysis of the corresponding acetonide [45] (Supporting Information File 1). This diol was then selectively protected at the less hindered hydroxy group (C19), with TBSOTf at −78 °C [46], to provide 5 in 86% yield.
![[1860-5397-7-161-i4]](/bjoc/content/inline/1860-5397-7-161-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Coupling of the top and middle fragments.
Scheme 4: Coupling of the top and middle fragments.
The synthetic route to 3a and 3b continues in Scheme 5 with the coupling of alcohol 5 with the bottom fragments 6a and 6b. The hydroxy group in 5 was deprotonated with NaHMDS followed by addition of the crude reaction mixture containing acid chloride 6a or 6b. The coupled products 24a and 24b proved difficult to purify by flash chromatography. In the C6 (S)-series, 24b was successfully isolated in 57% yield (82% BRSM). In the C6 (R)-series, the crude coupled product 24a could not be separated from 5, and was immediately subjected to HF·pyridine deprotection. The resulting primary alcohol 25a was isolated in 71% yield (90% BRSM) after careful flash chromatography. Selective deprotection of purified 24b with HF·pyridine afforded the respective primary alcohol 25b in 84% yield.
![[1860-5397-7-161-i5]](/bjoc/content/inline/1860-5397-7-161-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Coupling with the bottom fragment and end game.
Scheme 5: Coupling with the bottom fragment and end game.
Next, treatment of each primary alcohol 25a,b with Dess–Martin reagent provided aldehydes 4a and 4b in comparable yields (95% and 94%). Initial NHK reactions were performed in THF with 15 equiv of CrCl2, 0.2 equiv of NiCl2(dppf) and 15 equiv of 4,4'-di-tert-butyl-2,2'-dipyridine [47]. The target product 26b for the C6 (S)-series was obtained in respectable yield (42%). Only the target C9-β epimer was detected in the 1H NMR spectrum of the crude product, though we cannot rule out the presence of small amounts of the α-epimer. However, in the C6 (R)-series, the C9 β/α ratio was only about 75/25 and the isolated yield of pure β-epimer 26a was only 22%. After surveying several other conditions and solvents, we found that treatment of 4a with 15 equiv of CrCl2 and 0.2 equiv of NiCl2 in DMF/THF [48] improved the β/α ratio to about 85/15 and improved the isolated yield of pure 26a to 48%. Again, formation of acetonides confirmed the stereochemical assignment (Supporting Information File 1) [45]. Finally, treatment of 26a and 26b with HF·pyridine afforded the desired analogs 3a and 3b in good yields (86% and 82%, respectively). These compounds were fully characterized by the usual spectroscopic means.
The target compounds 25,26-dihydrodictyostatin (3a) and its C6-epimer 3b were tested in comparison to dictyostatin (1a) and 6-epi-dictyostatin (1b), epothilone B, and paclitaxel, and these data have recently been reported in detail [30]. Briefly, both compounds were potent microtubule-perturbing agents that induced mitotic arrest and microtubule assembly in vitro and in intact cells. Each displaced [3H]paclitaxel and [14C]epothilone B from microtubules with potencies comparable to (−)-dictyostatin and discodermolide. Each compound also inhibited the growth of cell lines resistant to paclitaxel and epothilone B at low nanomolar concentrations, synergized with paclitaxel in MDA-MB-231 human breast cancer cells, and had anti-angiogenic activity in transgenic zebra fish larvae.
Conclusion
We have successfully used a streamlined synthesis to access two new analogs, 3a and 3b, in the dictyostatin class of natural products. This synthesis based on three large fragments is highly convergent, requiring minimal functional group transformations once the coupling events take place. The synthesis of each fragment is amenable to scale-up and takes ten steps or less. Ten more steps are needed from the start of fragment coupling to the end of the synthesis, providing the target compounds in about 7–8% overall yield.
The intramolecular NHK reaction was successful for the formation of the macrolactone. As with the prior 16-desmethyl series compounds, the stereoselectivity of this reaction depended on the configuration of the nearby stereocenter at C6 with the (6S,7S)-epimer giving almost exclusively the target isomer at C9, whereas the (6R,7S)-epimer gave about 25% of a minor isomer along with about 75% of the target isomer. The subsequent testing data identify 25,26-dihydrodictyostatin and 25,26-dihydro-6-epi-dictyostatin as candidates for scale-up synthesis and further preclinical development.
Supporting Information
| Supporting Information File 1: Experimental details, characterization data and copies of NMR spectra of all new compounds. | ||
| Format: PDF | Size: 2.1 MB | Download |
References
-
Zhou, J.; Giannakakou, P. Curr. Med. Chem. - Anti-Cancer Agents 2005, 5, 65–71. doi:10.2174/1568011053352569
Return to citation in text: [1] -
Myles, D. C. Emerging microtubule stabilizing agents for cancer chemotherapy. In Annual Reports in Medicinal Chemistry; Doherty, A. M., Ed.; Academic Press: San Diego, CA, 2002; Vol. 37, pp 125–132.
Return to citation in text: [1] -
Kingston, D. G. I. J. Nat. Prod. 2009, 72, 507–515. doi:10.1021/np800568j
Return to citation in text: [1] -
Dalby, S. M.; Paterson, I. Curr. Opin. Drug Discovery Dev. 2010, 13, 777–794.
Return to citation in text: [1] -
Pettit, G. R.; Cichacz, Z. A.; Gao, F.; Boyd, M. R.; Schmidt, J. M. J. Chem. Soc., Chem. Commun. 1994, 1111–1112. doi:10.1039/C39940001111
Return to citation in text: [1] -
Isbrucker, R. A.; Cummins, J.; Pomponi, S. A.; Longley, R. E.; Wright, A. E. Biochem. Pharmacol. 2003, 66, 75–82. doi:10.1016/S0006-2952(03)00192-8
Return to citation in text: [1] -
Paterson, I.; Britton, R.; Delgado, O.; Wright, A. E. Chem. Commun. 2004, 632–633. doi:10.1039/b316390c
Return to citation in text: [1] -
Paterson, I.; Britton, R.; Delgado, O.; Meyer, A.; Poullennec, K. G. Angew. Chem., Int. Ed. 2004, 43, 4629–4633. doi:10.1002/anie.200460589
Return to citation in text: [1] [2] -
Shin, Y.; Fournier, J.-H.; Fukui, Y.; Brückner, A. M.; Curran, D. P. Angew. Chem., Int. Ed. 2004, 43, 4634–4637. doi:10.1002/anie.200460593
Return to citation in text: [1] [2] -
Madiraju, C.; Edler, M. C.; Hamel, E.; Raccor, B. S.; Balachandran, R.; Zhu, G.; Giuliano, K. A.; Vogt, A.; Shin, Y.; Fournier, J.-H.; Fukui, Y.; Brückner, A. M.; Curran, D. P.; Day, B. W. Biochemistry 2005, 44, 15053–15063. doi:10.1021/bi050685l
Return to citation in text: [1] -
Paterson, I.; Gardner, N. M.; Poullenneca, K. G.; Wright, A. E. Bioorg. Med. Chem. Lett. 2007, 17, 2443–2447. doi:10.1016/j.bmcl.2007.02.031
Return to citation in text: [1] [2] -
Paterson, I.; Gardner, N. M.; Guzmán, E.; Wright, A. E. Bioorg. Med. Chem. Lett. 2008, 18, 6268–6272. doi:10.1016/j.bmcl.2008.09.109
Return to citation in text: [1] -
Paterson, I.; Gardner, N. M.; Poullennec, K. G.; Wright, A. E. J. Nat. Prod. 2008, 71, 364–369. doi:10.1021/np070547s
Return to citation in text: [1] -
Paterson, I.; Naylor, G. J.; Wright, A. E. Chem. Commun. 2008, 4628–4630. doi:10.1039/b811575c
Return to citation in text: [1] [2] -
Paterson, I.; Gardner, N. M.; Guzmán, E.; Wright, A. E. Bioorg. Med. Chem. 2009, 17, 2282–2289. doi:10.1016/j.bmc.2008.10.084
Return to citation in text: [1] -
Paterson, I.; Naylor, G. J.; Fujita, T.; Guzmán, E.; Wright, A. E. Chem. Commun. 2010, 46, 261–263. doi:10.1039/b921237j
Return to citation in text: [1] -
Paterson, I.; Naylor, G. J.; Gardner, N. M.; Guzmán, E.; Wright, A. E. Chem.–Asian J. 2011, 6, 459–473. doi:10.1002/asia.201000541
Return to citation in text: [1] [2] -
Shin, Y.; Choy, N.; Turner, T. R.; Balachandran, R.; Madiraju, C.; Day, B. W.; Curran, D. P. Org. Lett. 2002, 4, 4443–4446. doi:10.1021/ol026942l
Return to citation in text: [1] -
Shin, Y.; Fournier, J.-H.; Balachandran, R.; Madiraju, C.; Raccor, B. S.; Zhu, G.; Edler, M. C.; Hamel, E.; Day, B. W.; Curran, D. P. Org. Lett. 2005, 7, 2873–2876. doi:10.1021/ol050808u
Return to citation in text: [1] [2] -
Fukui, Y.; Brückner, A. M.; Shin, Y.; Balachandran, R.; Day, B. W.; Curran, D. P. Org. Lett. 2006, 8, 301–304. doi:10.1021/ol0526827
Return to citation in text: [1] -
Jung, W.-H.; Harrison, C.; Shin, Y.; Fournier, J.-H.; Balachandran, R.; Raccor, B. S.; Sikorski, R. P.; Vogt, A.; Curran, D. P.; Day, B. W. J. Med. Chem. 2007, 50, 2951–2966. doi:10.1021/jm061385k
Return to citation in text: [1] -
Shin, Y.; Fournier, J.-H.; Brückner, A.; Madiraju, C.; Balachandran, R.; Raccor, B. S.; Edler, M. C.; Hamel, E.; Sikorski, R. P.; Vogt, A.; Day, B. W.; Curran, D. P. Tetrahedron 2007, 63, 8537–8562. doi:10.1016/j.tet.2007.05.033
Return to citation in text: [1] -
Raccor, B. S.; Vogt, A.; Sikorski, R. P.; Madiraju, C.; Balachandran, R.; Montgomery, K.; Shin, Y.; Fukui, Y.; Jung, W.-H.; Curran, D. P.; Day, B. W. Mol. Pharmacol. 2008, 73, 718–726. doi:10.1124/mol.107.042598
Return to citation in text: [1] -
Jogalekar, A. S.; Damodaran, K.; Kriel, F. H.; Jung, W.-H.; Alcaraz, A. A.; Zhong, S.; Curran, D. P.; Snyder, J. P. J. Am. Chem. Soc. 2011, 133, 2427–2436. doi:10.1021/ja1023817
Return to citation in text: [1] -
O'Neil, G. W.; Phillips, A. J. J. Am. Chem. Soc. 2006, 128, 5340–5341. doi:10.1021/ja0609708
Return to citation in text: [1] -
Ramachandran, P. V.; Srivastava, A.; Hazra, D. Org. Lett. 2007, 9, 157–160. doi:10.1021/ol062737k
Return to citation in text: [1] -
Zanato, C.; Pignataro, L.; Ambrosi, A.; Hao, Z.; Trigili, C.; Díaz, J. F.; Barasoain, I.; Gennari, C. Eur. J. Org. Chem. 2011, 2643–2661. doi:10.1002/ejoc.201100244
Return to citation in text: [1] -
Eiseman, J. L.; Bai, L.; Jung, W.-H.; Moura-Letts, G.; Day, B. W.; Curran, D. P. J. Med. Chem. 2008, 51, 6650–6653. doi:10.1021/jm800979v
Return to citation in text: [1] -
Zhu, W.; Jiménez, M.; Jung, W.-H.; Camarco, D. P.; Balachandran, R.; Vogt, A.; Day, B. W.; Curran, D. P. J. Am. Chem. Soc. 2010, 132, 9175–9187. doi:10.1021/ja103537u
Return to citation in text: [1] [2] [3] -
Vollmer, L. L.; Jiménez, M.; Camarco, D. P.; Zhu, W.; Daghestani, H. N.; Balachandran, R.; Reese, C. E.; Lazo, J. S.; Hukriede, N. A.; Curran, D. P.; Day, B. W.; Vogt, A. Mol. Cancer Ther. 2011, 10, 994–1006. doi:10.1158/1535-7163.MCT-10-1048
Return to citation in text: [1] [2] -
Wessjohann, L. A.; Scheid, G. Synthesis 1999, 1–36. doi:10.1055/s-1999-3672
Return to citation in text: [1] -
Takai, K. Org. React. 2004, 64, 253–612.
Return to citation in text: [1] -
Myers, A. G.; Yang, B. H.; Chen, H.; McKinstry, L.; Kopecky, D. J.; Gleason, J. L. J. Am. Chem. Soc. 1997, 119, 6496–6511. doi:10.1021/ja970402f
Return to citation in text: [1] -
Myers, A. G.; Yang, B. H.; Kopecky, D. J. Tetrahedron Lett. 1996, 37, 3623–3626. doi:10.1016/0040-4039(96)00652-1
Return to citation in text: [1] -
Marshall, J. A.; Eidam, P.; Eidam, H. S. J. Org. Chem. 2006, 71, 4840–4844. doi:10.1021/jo060542k
Return to citation in text: [1] -
Marshall, J. A.; Yanik, M. M.; Adams, N. D.; Ellis, K. C.; Chobanian, H. R. Org. Synth. 2005, 81, 157–170.
Return to citation in text: [1] -
Diederich, F.; Rubin, Y.; Chapman, O. L.; Goroff, N. S. Helv. Chim. Acta 1994, 77, 1441–1457. doi:10.1002/hlca.19940770522
Return to citation in text: [1] -
Myers, A. G.; Zheng, B.; Movassaghi, M. J. Org. Chem. 1997, 62, 7507. doi:10.1021/jo9710137
Return to citation in text: [1] -
Moura-Letts, G.; Curran, D. P. Org. Lett. 2007, 9, 5–8. doi:10.1021/ol062017d
Return to citation in text: [1] -
Denmark, S. E.; Regens, C. S.; Kobayashi, T. J. Am. Chem. Soc. 2007, 129, 2774–2776. doi:10.1021/ja070071z
Return to citation in text: [1] -
Devos, A.; Remion, J.; Frisque-Hesbain, A.-M.; Colens, A.; Ghosez, L. J. Chem. Soc., Chem. Commun. 1979, 1180–1181. doi:10.1039/C39790001180
Return to citation in text: [1] -
Paterson, I.; Wren, S. P. J. Chem. Soc., Chem. Commun. 1993, 1790–1792. doi:10.1039/C39930001790
Return to citation in text: [1] -
Mahoney, W. S.; Brestensky, D. M.; Stryker, J. M. J. Am. Chem. Soc. 1988, 110, 291–293. doi:10.1021/ja00209a048
Return to citation in text: [1] -
Chen, K.-M.; Hardtmann, G. E.; Prasad, K.; Repič, O.; Shapiro, M. J. Tetrahedron Lett. 1987, 28, 155–158. doi:10.1016/S0040-4039(00)95673-9
Return to citation in text: [1] -
Rychnovsky, S. D.; Rogers, B. N.; Richardson, T. I. Acc. Chem. Res. 1998, 31, 9–17. doi:10.1021/ar960223n
Return to citation in text: [1] [2] -
Zhan, W.; Jiang, Y.; Brodie, P. J.; Kingston, D. G. I.; Liotta, D. C.; Snyder, J. P. Org. Lett. 2008, 10, 1565–1568. doi:10.1021/ol800422q
Return to citation in text: [1] -
Stamos, D. P.; Sheng, X. C.; Chen, S. S.; Kishi, Y. Tetrahedron Lett. 1997, 38, 6355–6358. doi:10.1016/S0040-4039(97)01462-7
Return to citation in text: [1] -
Pilli, R. A.; Victor, M. M.; de Meijere, A. J. Org. Chem. 2000, 65, 5910–5916. doi:10.1021/jo000327i
Return to citation in text: [1]
| 39. | Moura-Letts, G.; Curran, D. P. Org. Lett. 2007, 9, 5–8. doi:10.1021/ol062017d |
| 40. | Denmark, S. E.; Regens, C. S.; Kobayashi, T. J. Am. Chem. Soc. 2007, 129, 2774–2776. doi:10.1021/ja070071z |
| 41. | Devos, A.; Remion, J.; Frisque-Hesbain, A.-M.; Colens, A.; Ghosez, L. J. Chem. Soc., Chem. Commun. 1979, 1180–1181. doi:10.1039/C39790001180 |
| 1. | Zhou, J.; Giannakakou, P. Curr. Med. Chem. - Anti-Cancer Agents 2005, 5, 65–71. doi:10.2174/1568011053352569 |
| 2. | Myles, D. C. Emerging microtubule stabilizing agents for cancer chemotherapy. In Annual Reports in Medicinal Chemistry; Doherty, A. M., Ed.; Academic Press: San Diego, CA, 2002; Vol. 37, pp 125–132. |
| 8. | Paterson, I.; Britton, R.; Delgado, O.; Meyer, A.; Poullennec, K. G. Angew. Chem., Int. Ed. 2004, 43, 4629–4633. doi:10.1002/anie.200460589 |
| 9. | Shin, Y.; Fournier, J.-H.; Fukui, Y.; Brückner, A. M.; Curran, D. P. Angew. Chem., Int. Ed. 2004, 43, 4634–4637. doi:10.1002/anie.200460593 |
| 11. | Paterson, I.; Gardner, N. M.; Poullenneca, K. G.; Wright, A. E. Bioorg. Med. Chem. Lett. 2007, 17, 2443–2447. doi:10.1016/j.bmcl.2007.02.031 |
| 19. | Shin, Y.; Fournier, J.-H.; Balachandran, R.; Madiraju, C.; Raccor, B. S.; Zhu, G.; Edler, M. C.; Hamel, E.; Day, B. W.; Curran, D. P. Org. Lett. 2005, 7, 2873–2876. doi:10.1021/ol050808u |
| 48. | Pilli, R. A.; Victor, M. M.; de Meijere, A. J. Org. Chem. 2000, 65, 5910–5916. doi:10.1021/jo000327i |
| 7. | Paterson, I.; Britton, R.; Delgado, O.; Wright, A. E. Chem. Commun. 2004, 632–633. doi:10.1039/b316390c |
| 30. | Vollmer, L. L.; Jiménez, M.; Camarco, D. P.; Zhu, W.; Daghestani, H. N.; Balachandran, R.; Reese, C. E.; Lazo, J. S.; Hukriede, N. A.; Curran, D. P.; Day, B. W.; Vogt, A. Mol. Cancer Ther. 2011, 10, 994–1006. doi:10.1158/1535-7163.MCT-10-1048 |
| 45. | Rychnovsky, S. D.; Rogers, B. N.; Richardson, T. I. Acc. Chem. Res. 1998, 31, 9–17. doi:10.1021/ar960223n |
| 5. | Pettit, G. R.; Cichacz, Z. A.; Gao, F.; Boyd, M. R.; Schmidt, J. M. J. Chem. Soc., Chem. Commun. 1994, 1111–1112. doi:10.1039/C39940001111 |
| 6. | Isbrucker, R. A.; Cummins, J.; Pomponi, S. A.; Longley, R. E.; Wright, A. E. Biochem. Pharmacol. 2003, 66, 75–82. doi:10.1016/S0006-2952(03)00192-8 |
| 28. | Eiseman, J. L.; Bai, L.; Jung, W.-H.; Moura-Letts, G.; Day, B. W.; Curran, D. P. J. Med. Chem. 2008, 51, 6650–6653. doi:10.1021/jm800979v |
| 46. | Zhan, W.; Jiang, Y.; Brodie, P. J.; Kingston, D. G. I.; Liotta, D. C.; Snyder, J. P. Org. Lett. 2008, 10, 1565–1568. doi:10.1021/ol800422q |
| 3. | Kingston, D. G. I. J. Nat. Prod. 2009, 72, 507–515. doi:10.1021/np800568j |
| 4. | Dalby, S. M.; Paterson, I. Curr. Opin. Drug Discovery Dev. 2010, 13, 777–794. |
| 29. | Zhu, W.; Jiménez, M.; Jung, W.-H.; Camarco, D. P.; Balachandran, R.; Vogt, A.; Day, B. W.; Curran, D. P. J. Am. Chem. Soc. 2010, 132, 9175–9187. doi:10.1021/ja103537u |
| 47. | Stamos, D. P.; Sheng, X. C.; Chen, S. S.; Kishi, Y. Tetrahedron Lett. 1997, 38, 6355–6358. doi:10.1016/S0040-4039(97)01462-7 |
| 24. | Jogalekar, A. S.; Damodaran, K.; Kriel, F. H.; Jung, W.-H.; Alcaraz, A. A.; Zhong, S.; Curran, D. P.; Snyder, J. P. J. Am. Chem. Soc. 2011, 133, 2427–2436. doi:10.1021/ja1023817 |
| 26. | Ramachandran, P. V.; Srivastava, A.; Hazra, D. Org. Lett. 2007, 9, 157–160. doi:10.1021/ol062737k |
| 44. | Chen, K.-M.; Hardtmann, G. E.; Prasad, K.; Repič, O.; Shapiro, M. J. Tetrahedron Lett. 1987, 28, 155–158. doi:10.1016/S0040-4039(00)95673-9 |
| 18. | Shin, Y.; Choy, N.; Turner, T. R.; Balachandran, R.; Madiraju, C.; Day, B. W.; Curran, D. P. Org. Lett. 2002, 4, 4443–4446. doi:10.1021/ol026942l |
| 19. | Shin, Y.; Fournier, J.-H.; Balachandran, R.; Madiraju, C.; Raccor, B. S.; Zhu, G.; Edler, M. C.; Hamel, E.; Day, B. W.; Curran, D. P. Org. Lett. 2005, 7, 2873–2876. doi:10.1021/ol050808u |
| 20. | Fukui, Y.; Brückner, A. M.; Shin, Y.; Balachandran, R.; Day, B. W.; Curran, D. P. Org. Lett. 2006, 8, 301–304. doi:10.1021/ol0526827 |
| 21. | Jung, W.-H.; Harrison, C.; Shin, Y.; Fournier, J.-H.; Balachandran, R.; Raccor, B. S.; Sikorski, R. P.; Vogt, A.; Curran, D. P.; Day, B. W. J. Med. Chem. 2007, 50, 2951–2966. doi:10.1021/jm061385k |
| 22. | Shin, Y.; Fournier, J.-H.; Brückner, A.; Madiraju, C.; Balachandran, R.; Raccor, B. S.; Edler, M. C.; Hamel, E.; Sikorski, R. P.; Vogt, A.; Day, B. W.; Curran, D. P. Tetrahedron 2007, 63, 8537–8562. doi:10.1016/j.tet.2007.05.033 |
| 23. | Raccor, B. S.; Vogt, A.; Sikorski, R. P.; Madiraju, C.; Balachandran, R.; Montgomery, K.; Shin, Y.; Fukui, Y.; Jung, W.-H.; Curran, D. P.; Day, B. W. Mol. Pharmacol. 2008, 73, 718–726. doi:10.1124/mol.107.042598 |
| 27. | Zanato, C.; Pignataro, L.; Ambrosi, A.; Hao, Z.; Trigili, C.; Díaz, J. F.; Barasoain, I.; Gennari, C. Eur. J. Org. Chem. 2011, 2643–2661. doi:10.1002/ejoc.201100244 |
| 45. | Rychnovsky, S. D.; Rogers, B. N.; Richardson, T. I. Acc. Chem. Res. 1998, 31, 9–17. doi:10.1021/ar960223n |
| 11. | Paterson, I.; Gardner, N. M.; Poullenneca, K. G.; Wright, A. E. Bioorg. Med. Chem. Lett. 2007, 17, 2443–2447. doi:10.1016/j.bmcl.2007.02.031 |
| 12. | Paterson, I.; Gardner, N. M.; Guzmán, E.; Wright, A. E. Bioorg. Med. Chem. Lett. 2008, 18, 6268–6272. doi:10.1016/j.bmcl.2008.09.109 |
| 13. | Paterson, I.; Gardner, N. M.; Poullennec, K. G.; Wright, A. E. J. Nat. Prod. 2008, 71, 364–369. doi:10.1021/np070547s |
| 14. | Paterson, I.; Naylor, G. J.; Wright, A. E. Chem. Commun. 2008, 4628–4630. doi:10.1039/b811575c |
| 15. | Paterson, I.; Gardner, N. M.; Guzmán, E.; Wright, A. E. Bioorg. Med. Chem. 2009, 17, 2282–2289. doi:10.1016/j.bmc.2008.10.084 |
| 16. | Paterson, I.; Naylor, G. J.; Fujita, T.; Guzmán, E.; Wright, A. E. Chem. Commun. 2010, 46, 261–263. doi:10.1039/b921237j |
| 17. | Paterson, I.; Naylor, G. J.; Gardner, N. M.; Guzmán, E.; Wright, A. E. Chem.–Asian J. 2011, 6, 459–473. doi:10.1002/asia.201000541 |
| 42. | Paterson, I.; Wren, S. P. J. Chem. Soc., Chem. Commun. 1993, 1790–1792. doi:10.1039/C39930001790 |
| 10. | Madiraju, C.; Edler, M. C.; Hamel, E.; Raccor, B. S.; Balachandran, R.; Zhu, G.; Giuliano, K. A.; Vogt, A.; Shin, Y.; Fournier, J.-H.; Fukui, Y.; Brückner, A. M.; Curran, D. P.; Day, B. W. Biochemistry 2005, 44, 15053–15063. doi:10.1021/bi050685l |
| 25. | O'Neil, G. W.; Phillips, A. J. J. Am. Chem. Soc. 2006, 128, 5340–5341. doi:10.1021/ja0609708 |
| 43. | Mahoney, W. S.; Brestensky, D. M.; Stryker, J. M. J. Am. Chem. Soc. 1988, 110, 291–293. doi:10.1021/ja00209a048 |
| 31. | Wessjohann, L. A.; Scheid, G. Synthesis 1999, 1–36. doi:10.1055/s-1999-3672 |
| 32. | Takai, K. Org. React. 2004, 64, 253–612. |
| 29. | Zhu, W.; Jiménez, M.; Jung, W.-H.; Camarco, D. P.; Balachandran, R.; Vogt, A.; Day, B. W.; Curran, D. P. J. Am. Chem. Soc. 2010, 132, 9175–9187. doi:10.1021/ja103537u |
| 30. | Vollmer, L. L.; Jiménez, M.; Camarco, D. P.; Zhu, W.; Daghestani, H. N.; Balachandran, R.; Reese, C. E.; Lazo, J. S.; Hukriede, N. A.; Curran, D. P.; Day, B. W.; Vogt, A. Mol. Cancer Ther. 2011, 10, 994–1006. doi:10.1158/1535-7163.MCT-10-1048 |
| 8. | Paterson, I.; Britton, R.; Delgado, O.; Meyer, A.; Poullennec, K. G. Angew. Chem., Int. Ed. 2004, 43, 4629–4633. doi:10.1002/anie.200460589 |
| 9. | Shin, Y.; Fournier, J.-H.; Fukui, Y.; Brückner, A. M.; Curran, D. P. Angew. Chem., Int. Ed. 2004, 43, 4634–4637. doi:10.1002/anie.200460593 |
| 37. | Diederich, F.; Rubin, Y.; Chapman, O. L.; Goroff, N. S. Helv. Chim. Acta 1994, 77, 1441–1457. doi:10.1002/hlca.19940770522 |
| 38. | Myers, A. G.; Zheng, B.; Movassaghi, M. J. Org. Chem. 1997, 62, 7507. doi:10.1021/jo9710137 |
| 35. | Marshall, J. A.; Eidam, P.; Eidam, H. S. J. Org. Chem. 2006, 71, 4840–4844. doi:10.1021/jo060542k |
| 36. | Marshall, J. A.; Yanik, M. M.; Adams, N. D.; Ellis, K. C.; Chobanian, H. R. Org. Synth. 2005, 81, 157–170. |
| 17. | Paterson, I.; Naylor, G. J.; Gardner, N. M.; Guzmán, E.; Wright, A. E. Chem.–Asian J. 2011, 6, 459–473. doi:10.1002/asia.201000541 |
| 34. | Myers, A. G.; Yang, B. H.; Kopecky, D. J. Tetrahedron Lett. 1996, 37, 3623–3626. doi:10.1016/0040-4039(96)00652-1 |
| 29. | Zhu, W.; Jiménez, M.; Jung, W.-H.; Camarco, D. P.; Balachandran, R.; Vogt, A.; Day, B. W.; Curran, D. P. J. Am. Chem. Soc. 2010, 132, 9175–9187. doi:10.1021/ja103537u |
| 33. | Myers, A. G.; Yang, B. H.; Chen, H.; McKinstry, L.; Kopecky, D. J.; Gleason, J. L. J. Am. Chem. Soc. 1997, 119, 6496–6511. doi:10.1021/ja970402f |
| 14. | Paterson, I.; Naylor, G. J.; Wright, A. E. Chem. Commun. 2008, 4628–4630. doi:10.1039/b811575c |
© 2011 Jiménez et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)