

Abstract
Since Garner’s aldehyde has several drawbacks, first of all is prone to racemization, alternative three-carbon chirons would be of great value in enantioselective syntheses of natural compounds and/or drugs. This review article summarizes applications of N-(1-phenylethyl)aziridine-2-carboxylates, -carbaldehydes and -methanols in syntheses of approved drugs and potential medications as well as of natural products mostly alkaloids but also sphingoids and ceramides and their 1- and 3-deoxy analogues and several hydroxy amino acids and their precursors. Designed strategies provided new procedures to several drugs and alternative approaches to natural products and proved efficiency of a 2-substituted N-(1-phenylethyl)aziridine framework as chiron bearing a chiral auxiliary.
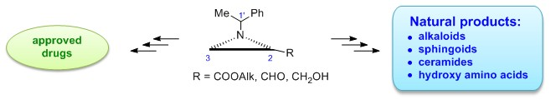
Graphical Abstract
Introduction
The synthesis of enantiomerically pure compounds belongs to the most challenging tasks in organic chemistry for several reasons, just to mention structural studies of natural products or preparation of chiral drugs. They become available by asymmetric synthesis frequently employing chiral synthons (chirons) [1].
Chirons contain functional groups for structural enlargement and at least one stereogenic center which is usually transferred into the final product. To assure the highest possible enantiomeric purity chirons are obtained in most instances from natural products like carbohydrates, amino acids, hydroxy acids or terpenes. The structurally simplest chirons containing three carbon atoms and one stereogenic center can be exemplified by derivatives of ᴅ-glyceraldehyde [2] (2,3-O-isopropylidene 1a [3,4] and 2,3-O-cyclohexylidene 1b [5,6]) and (2R,5R,6R)-5,6-dimethoxy-5,6-dimethyl-1,4-dioxane-2-carbaldehyde (2) [7] (Figure 1) which could be prepared from ᴅ-mannitol. While derivatives of glyceraldehyde are configurationally stable Garner’s aldehyde 3a [8,9], available from ʟ-serine, is configurationally labile and samples with ee as low as 75% can be obtained depending on the reaction conditions. Significant improvement in terms of chemical and configurational stability was achieved by introducing N,N-dibenzylserinals 4 [10].
![[1860-5397-15-168-1]](/bjoc/content/figures/1860-5397-15-168-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Examples of three-carbon chirons.
Figure 1: Examples of three-carbon chirons.
Another important strategy of asymmetric synthesis relies on chiral auxiliaries, i.e., a specially selected homochiral part of a starting material governing the stereoselectivity of subsequent reactions which is finally easily removed [11]. Among three-carbon chirons related to Garner’s aldehyde derivatives of N-(1-phenylethyl)aziridine-2-carboxylic acid 5–8 (Figure 2) play an important role in asymmetric synthesis as they function as a chiral synthon combined with a chiral auxiliary [(R)- or (S)-1-phenylethyl group].
![[1860-5397-15-168-2]](/bjoc/content/figures/1860-5397-15-168-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Structures of derivatives of N-(1-phenylethyl)aziridine-2-carboxylic acid 5–8.
Figure 2: Structures of derivatives of N-(1-phenylethyl)aziridine-2-carboxylic acid 5–8.
As the closest analogues of Garner’s aldehyde (3a) and other chiral α-aminoaldehydes, e.g., 4, or aziridine aldehydes 6 do not undergo epimerization during preparation as well as in further transformations conducted in the presence of basic reagents because of the high barrier to inversion at the nitrogen in the aziridine ring. The immediate starting materials, esters 5 and methyl ester 3b, are easier (one step and separation of diastereoisomers for 5 vs three steps for 3b) to prepare for the aziridine chiron while they are priced comparably. Although DIBAL-H was applied as a reagent of choice in the synthesis of Garner’s aldehyde it has several drawbacks, e.g., overreduction, tricky removal of aluminum salts, a two-step easy to perform sequence (ester 5 to alcohol 7 reduction and re-oxidation to 6) was generally adapted for aziridine-2-carbaldehydes 6.
Since the aziridine ring opening with nucleophiles, e.g., oxygen, can relatively easily be achieved [12], and for the structural framework of 5–8 occurs with high regioselectivity at the less substituted carbon atom, aldehydes (2R,1'R)- or (2R,1'S)-6 are considered as synthetic equivalents of (R)- or ᴅ-serinal while (2S,1'R)- or (2S,1'S)-6 correspond to (S)- or ʟ-serinal (Figure 3) [13].
![[1860-5397-15-168-3]](/bjoc/content/figures/1860-5397-15-168-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Synthetic equivalency of aziridine aldehydes 6.
Figure 3: Synthetic equivalency of aziridine aldehydes 6.
Furthermore, since the aziridine ring openings can be accomplished with other nucleophiles and the reductive cleavage is also known [14,15] the aziridines 5–8 offer a plethora of opportunities for enantioselective synthesis of structurally diversified compounds.
In this review we wish to focus attention on applications of 5–8 in syntheses of biologically important compounds having a 2-amino-1,3-disubstituted propane unit implanted into their structures because vicinal amino alcohol and 2-aminopropane-1,3-diol scaffolds are present in many natural products as well as compounds of commercial interest as medications.
Synthetic strategies to molecules as simple as amino alcohols to as complex as indolizine alkaloids will be discussed with a special focus on stereoselectivities of key transformations. Whenever possible biological activities of new compounds will be shown to underscore their biological potency. In a few cases mechanistic considerations will be presented to clarify the structural diversity of the products resulted from openings of the aziridine ring. This review was intended to cover the entire literature including patents on (1-phenylethyl)aziridine-2-carboxylic acid and its derivatives till the beginning of 2019. The usefulness of any chiron depends on its availability and to some extent on versatility of the protective groups. In case of the 1-phenylethyl group its removal at any stage of synthesis is not limited to a catalytic hydrogenation but metal-ammonia reduction (Birch reaction) and organic acid in anisole (vide infra) can be efficiently applied. We begin with a short presentation of syntheses of aziridine-2-carboxylates, the corresponding aldehydes and 2-methanols.
Review
Syntheses of starting materials
Synthesis of N-(1-phenylethyl)aziridine-2-carboxylates 5
In general, enantiomerically pure starting materials used in asymmetric syntheses are prepared from natural products. This does not hold for N-(1-phenylethyl)aziridine-2-carboxylates 5 since after synthesis in the Gabriel–Cromwell reaction between 3,3-dibromopropanoate and (R)- or (S)-1-phenylethylamine (Scheme 1) the separation of mixtures of diastereoisomers (2R,1'R)- and (2S,1'R)-5 as well as (2R,1'S)- and (2S,1'S)-5 has to be accomplished. Fortunately, chromatography appeared a method of choice for esters of aliphatic alcohols: methyl 5a [16,17], ethyl 5b [16,18,19], isopropyl 5c [16] and tert-butyl 5d [16,20] as well as for 4-methoxyphenyl ester 5e [21].
![[1860-5397-15-168-i1]](/bjoc/content/inline/1860-5397-15-168-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of N-(1-phenylethyl)aziridine-2-carboxylates 5. Reagents and conditions: a) TEA, toluene, reflux, 3 h [13]; b) silica gel chromatography or solvent-driven selective crystallization at low-temperature.
Scheme 1: Synthesis of N-(1-phenylethyl)aziridine-2-carboxylates 5. Reagents and conditions: a) TEA, toluene,...
However, the corresponding (−)-menthyl esters (2R,1'R)-5f and (2S,1'R)-5f were separated by solvent-driven selective crystallization at low-temperature from ethanol and hexane, respectively. The aziridine esters (2R,1'S)-5f and (2S,1'S)-5f were obtained in a similar manner. After transesterification methyl 5a and ethyl 5b esters were prepared. Thus, enantiomerically pure N-(1-phenylethyl)aziridine-2-carboxylates 5 became commercially available [22].
Other synthetic pathways to esters 5 were elaborated although they are more complex [23-25]. Enzymatically-catalyzed aminolysis of a mixture of (2R,1'R)-5a and (2S,1'R)-5a provided diastereoisomerically pure (de >99%) methyl ester (2R,1'R)-5a and amide (2S,1'R)-8 (Figure 2). The same selectivity was observed for (2R,1'S)-5a and (2S,1'S)-5a and both pairs of ethyl esters [26].
When a mixture of tert-butyl esters (2R,1'S)-5d and (2S,1'S)-5d was subjected to kinetic resolution in the presence of potassium tert-butoxide in tetrahydrofuran (2R,1'S)-5d was produced with low 40% de [27].
The absolute configuration at C2 in esters 5 was established by transforming enantiomerically pure (2R,1'S)-5a and (2S,1'S)-5a into (R)- and (S)-serine, respectively (Scheme 2) [13,16].
![[1860-5397-15-168-i2]](/bjoc/content/inline/1860-5397-15-168-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Absolute configuration at C2 in (2S,1'S)-5a. Reagents and conditions: a) 20% HClO4, 80 °C, 30 h then Dowex 50 (H+); b) H2, 20% Pd(OH)2/C, EtOH/H2O, rt, 24 h.
Scheme 2: Absolute configuration at C2 in (2S,1'S)-5a. Reagents and conditions: a) 20% HClO4, 80 °C, 30 h the...
Synthesis of N-(1-phenylethyl)aziridine-2-carbaldehydes 6
Aziridine aldehydes 6 are in most instances prepared from the corresponding alcohols 7 by Swern oxidation [28-33]. Procedures relying on DIBAL-H reduction of esters 5 are less frequent [34]. Although aziridine aldehydes 6 are chemically stable enough to be chromatographed on silica gel and can later be stored at −10 °C they are normally prepared before use and applied as crude materials.
Synthesis of N-(1-phenylethyl)aziridine-2-methanols 7
Aziridine alcohols 7 are usually prepared by LiAlH4 reduction of the corresponding esters 5 [22,35,36] although a milder method with a NaBH4 and LiCl mixture was also elaborated [37]. A multistep synthesis of (2S,1'R)-7 employing the aziridine ring closure as the last step has also been described [38].
Syntheses of biologically relevant compounds from N-(1-phenylethyl)aziridine-2-carboxylate esters
A 2-ketoaziridine scaffold present in esters 5 and aldehydes 6 can undergo a variety of transformations. Elongation together with further functionalization are possible employing ester and aldehyde groups and the stereochemical outcome of these reactions is controlled by configurations at C2 and at the chiral auxiliary (Scheme 3). The opening of the aziridine ring is expected to occur at the less substituted carbon atom and can be executed with nucleophiles to provide 9 or even by catalytic hydrogenation to form 10. Thus, biologically important fragments like vicinal amino alcohols 11 or 2-amino-1,3-propanediols 12a [Nu = OH] can be obtained in highly enantioselective procedures preserving the absolute configuration at C2. The latter compounds are useful precursors to amino acids. Installation of halogen atoms in 9 (Nu = Cl, Br, I) allows for extending of the carbon chain.
![[1860-5397-15-168-i3]](/bjoc/content/inline/1860-5397-15-168-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Major synthetic strategies for a 2-ketoaziridine scaffold [R* = (R)- or (S)-1-phenylethyl; R′ = Alk, Ar].
Scheme 3: Major synthetic strategies for a 2-ketoaziridine scaffold [R* = (R)- or (S)-1-phenylethyl; R′ = Alk...
Before we start reviewing the synthesis of biologically relevant compounds prepared via opening of the aziridine ring it should be mentioned that cyanides (2R,1'S)- and (2S,1'S)-13 prepared from the respective esters (Scheme 4) 5b appeared moderately active as immunostimulants [18].
![[1860-5397-15-168-i4]](/bjoc/content/inline/1860-5397-15-168-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of cyanide (2S,1'S)-13. Reagents and conditions: a) NH3, EtOH/H2O, rt, 72 h; b) Ph3P, CCl4, TEA, CH2Cl2, reflux.
Scheme 4: Synthesis of cyanide (2S,1'S)-13. Reagents and conditions: a) NH3, EtOH/H2O, rt, 72 h; b) Ph3P, CCl4...
Amines and amino alcohols
By functionalization at C2: Synthesis of enantiomerically pure amines from 2-substituted N-(1-phenylethyl)aziridines 5–7 requires a regioselective reductive aziridine ring opening at the less substituted carbon and effective deoxygenation performed at the C2 substituent [39].
This strategy was applied in the formal synthesis of (R,R)-formoterol (14) and (R)-tamsulosin (15) (Scheme 5) which have been used as therapeutic drugs for many years [40,41]. They are structurally related since they contain an (R)-1-aryl-2-propanamine moiety. The synthesis of the respective intermediates (R)-16 and (R)-17 commenced from the ester (2R,1'R)-5f and relied on arylation of Weinreb amide (2R,1'R)-18 to afford the aziridine ketone 19. Its highly stereoselective reduction with the NaBH4/ZnCl2 mixture (chelation controlled) gave the aziridine alcohol 20 as a major product. Reductive opening of the aziridine ring produced the amino alcohol 21 which was transformed into the substituted oxazolidin-2-one 22. Its catalytic hydrogenation effected deoxygenation at the benzylic position to supply (R)-16 [42]. To synthesize (R)-17 the aminosulfonyl group was introduced in a standard way before hydrogenation [43].
![[1860-5397-15-168-i5]](/bjoc/content/inline/1860-5397-15-168-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Synthesis of key intermediates (R)-16 and (R)-17 for (R,R)-formoterol (14) and (R)-tamsulosin (15). Reagents and conditions: a) MeONHMe, iPrMgCl, THF, 0 °C, 0.5 h; b) 4-MeOC6H4MgBr, THF, −10 °C, 1 h and 0 °C, 0.5 h; c) NaBH4, ZnCl2, MeOH, −78 °C, 0.5 h; d) H2, Pd(OH)2, Boc2O, EtOH, rt, 8 h; e) NaH, THF, rt, 24 h; f) H2, Pd/C, MeOH, rt, 1 h; g) ClSO3H, 0 °C, 0.5 h then NH3, THF.
Scheme 5: Synthesis of key intermediates (R)-16 and (R)-17 for (R,R)-formoterol (14) and (R)-tamsulosin (15)....
The synthesis of amino alcohols of general formula 11 (Scheme 3) from 2-substituted N-(1-phenylethyl)aziridines 5 and 6 can be achieved by a regioselective reductive aziridine ring opening combined with functionalization of the C2 substituent and optional alkylation or arylation of the nitrogen atom [44].
Following a similar regioselective aziridine opening, a mixture of epimeric amino alcohols (2R/S,1'R)-23 was prepared in two steps from the aziridine alcohol (2R/S,1'R)-7 (Scheme 6) which was found to be an effective inhibitor of the mitotic kinesin.
![[1860-5397-15-168-i6]](/bjoc/content/inline/1860-5397-15-168-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Synthesis of mitotic kinesin inhibitors (2R/S,1'R)-23. Reagents and conditions: a) H2, Pd(OH)2, EtOH, rt; b, 3-chloro-4-fluorophenylboronic acid, Cu(OAc)2, pyridine, CH2Cl2, MS 4 Å, rt, 24 h.
Scheme 6: Synthesis of mitotic kinesin inhibitors (2R/S,1'R)-23. Reagents and conditions: a) H2, Pd(OH)2, EtO...
The biologically active enantiomer of mexiletine (R)-24 was efficiently synthesized from the alcohol (2R,1'R)-7 (Scheme 7) [45]. When the respective tosylate (2R,1'R)-25 was treated with 2,6-dimethylphenoxide two compounds were obtained in a 84:16 ratio. The major ether (2R,1'R)-26, which emerged from the displacement of the tosyloxy group (path a), was accompanied by (2S,1'R)-26 which came from the aziridine ring opening at C3 (path b). After chromatographic purification and hydrogenolysis enantiomerically pure (R)-24 was obtained. On the other hand, alkylation of 2,6-dimethylphenol with the tosylate (2S,1'R)-25 proceeded regioselectively to give (2S,1'R)-26, a precursor to (S)-24.
![[1860-5397-15-168-i7]](/bjoc/content/inline/1860-5397-15-168-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Synthesis of (R)-mexiletine ((R)-24). Reagents and conditions: a) TsCl, TEA, DMAP, CH2Cl2, rt, 1 h; b) 2,6-dimethylphenol, K2CO3, DMF/acetone, reflux, 4 h; c) H2, Pd/C, MeOH, rt, 12 h.
Scheme 7: Synthesis of (R)-mexiletine ((R)-24). Reagents and conditions: a) TsCl, TEA, DMAP, CH2Cl2, rt, 1 h;...
(−)-Cathinone ((S)-27) is an alkaloid acting as a central nervous system stimulant found in leaves of Catha edulis. It was efficiently synthesized from the aldehyde (2S,1'R)-6 begining from the addition of phenylmagnesium bromide to give a 4:1 mixture of aziridine alcohols 28 (Scheme 8) [46]. They were subjected a stepwise hydrogenation to form first products of the aziridine ring opening and next N-Boc-protected diastereoisomeric amino alcohols (1R/S,2S)-29 after removal of a chiral auxiliary and finally oxidized to a ketone from which (−)-cathinone ((S)-27) was isolated as the hydrochloride salt. Starting from the aldehyde (2R,1'S)-6 its enantiomer was prepared in a similar way.
![[1860-5397-15-168-i8]](/bjoc/content/inline/1860-5397-15-168-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Synthesis of (−)-cathinone ((S)-27). Reagents and conditions: a) PhMgBr, ether, 0 °C; b) H2, 10% Pd(OH)2/C, AcOEt, rt, 14 h; c) H2, 20% Pd(OH)2/C, Boc2O, AcOEt, rt, 12 h; d) PCC, CH2Cl2, rt, 1.5 h; e) 3 N HCl in AcOEt, rt, 0.5 h.
Scheme 8: Synthesis of (−)-cathinone ((S)-27). Reagents and conditions: a) PhMgBr, ether, 0 °C; b) H2, 10% Pd...
Under optimized conditions the diastereoselectivity of the addition of phenyllithium to the aldehyde (2S,1'R)-6 exceeded 80% and the major aziridine alcohol (2S,1'S,1''R)-28 could be separated chromatographically [29]. When (2S,1'S,1''R)-28 was hydrogenated in the presence of Boc2O N-protected (−)-norpseudoephedrine (1S,2S)-(+)-29 was obtained.
To synthesize N-Boc-norephedrine ((1R,2S)-29) the epimeric aziridine alcohol (2S,1'R,1''R)-28 was needed (Scheme 9) [47]. To this end the aziridine ketone (2S,1'R)-30 prepared from Weinreb amide (2S,1'R)-18 was reduced with a NaBH4/ZnCl2 mixture to give almost enantiomerically pure (>99:1) alcohol (2S,1'R,1''R)-28. After reductive aziridine ring opening N-Boc-norephedrine ((1R,2S)-29) was formed.
![[1860-5397-15-168-i9]](/bjoc/content/inline/1860-5397-15-168-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Synthesis of N-Boc-norpseudoephedrine ((1S,2S)-(+)-29) and N-Boc-norephedrine ((1R,2S)-29). Reagents and conditions: a) PhBr, t-BuLi, THF, −78 °C, 2 h; b) H2, Pd(OH)2/C, AcOEt, rt, 6 h, then Boc2O, rt, 6 h; c) PhMgBr, THF, −78 °C, 0.5 h; d) NaBH4, ZnCl2, MeOH, −78 °C, 0.5 h.
Scheme 9: Synthesis of N-Boc-norpseudoephedrine ((1S,2S)-(+)-29) and N-Boc-norephedrine ((1R,2S)-29). Reagent...
The synthesis of (−)-ephedrine ((1R,2S)-31) required prior methylation of the nitrogen atom and for this reason it was performed on the benzyl ether (2S,1'R,1''R)-32 (Scheme 10) [48]. Its reaction with methyl triflate afforded the respective aziridinium ion which was regioselectively reduced to (1R,2S,1'R)-33, a protected form of (−)-ephedrine (1R,2S)-31.
![[1860-5397-15-168-i10]](/bjoc/content/inline/1860-5397-15-168-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Synthesis of (−)-ephedrine ((1R,2S)-31). Reagents and conditions: a) TfOMe, MeCN then NaBH3CN, rt; b) H2, Pd(OH)2, 50 psi, EtOH, rt.
Scheme 10: Synthesis of (−)-ephedrine ((1R,2S)-31). Reagents and conditions: a) TfOMe, MeCN then NaBH3CN, rt; ...
Xestoaminol C ((2S,3R)-34) and 3-epi-xestoaminol C ((2S,3S)-34) represent a large family of naturally occurring 1-deoxysphingoids [49]. To finally prove their stereochemistry, they were synthesized from the aziridine ketone (2S,1'R)-36 readily available from Weinreb amide (2S,1'R)-18 which already contained the required configuration at C2 (Scheme 11) [47]. Introduction of the 3R configuration in xestoaminol C and 3S in its epimer was achieved by stereoselective reductions with a NaBH4/ZnCl2 mixture or ʟ-Selectride®, respectively, to form aziridine alcohols (2S,1'R,1''R)-37 and (2S,1'S,1''R)-37. Reductive opening of the aziridine ring with concomitant removal of the chiral auxiliary produced protected (2S,3R)-35 and (2S,3S)-35 which were hydrolyzed to xestoaminol C ((2S,3R)-34) and 3-epi-xestoaminol C ((2S,3S)-34). Homologous N-Boc-spisulosine ((2S,3R)-38) was acquired following a similar procedure [47].
![[1860-5397-15-168-i11]](/bjoc/content/inline/1860-5397-15-168-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 11: Synthesis of xestoaminol C ((2S,3R)-35), 3-epi-xestoaminol C ((2S,3S)-35) and N-Boc-spisulosine ((2S,3R)-38). Reagents and conditions: a) C11H23Br, Mg, THF, 0 °C, 1 h; b) NaBH4, ZnCl2, MeOH, −78 °C, 2 h; c) ʟ-Selectride®, THF, −78 °C, 1 h; d) H2, Pd(OH)2, Boc2O, MeOH, rt 6 h; e) HCl, MeOH, reflux, 3 h.
Scheme 11: Synthesis of xestoaminol C ((2S,3R)-35), 3-epi-xestoaminol C ((2S,3S)-35) and N-Boc-spisulosine ((2S...
By functionalization at C3: In the presence of carbonyldiimidazole (CDI) and iodotrimethylsilane the aziridine alcohol (2S,1'S)-7 was transformed into the 4-(iodomethyl)oxazolidin-2-one (4R,1'S)-39 via a regioselective opening with iodide and cyclization (Scheme 12) [50]. As expected alkylation of the indole ring with (4R,1'S)-39 occurred at C3 to give (4S,1'S)-40 which was deprotected in two steps (Birch reduction and alkaline hydrolysis) to provide ʟ-tryptophanol ((S)-41).
![[1860-5397-15-168-i12]](/bjoc/content/inline/1860-5397-15-168-i12.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 12: Synthesis of ʟ-tryptophanol ((S)-41). Reagents and conditions: a) CDI, MeCN, rt, 1 h then TMSI, MeCN, rt, 3 h; b) indole, EtMgBr, toluene/THF, reflux, 3 h; c) Li, NH3, THF, −78 °C, 0.5 h; d) LiOH, EtOH/H2O, reflux, 2 h.
Scheme 12: Synthesis of ʟ-tryptophanol ((S)-41). Reagents and conditions: a) CDI, MeCN, rt, 1 h then TMSI, MeC...
The successful transformation of the aziridine alcohol (2S,1'R)-7 into ʟ-homophenylalaninol ((S)-42) depended on two crucial steps: the aziridine ring opening in the presence of phosgene to form the 4-(chloromethyl)oxazolidin-2-one (4R,1'R)-43 and Wittig olefination to add the benzyl moiety (Scheme 13) [51]. Before conversion of the chloromethyl group into a phosphonium salt the chiral auxiliary was removed. The reaction of the ylide (R)-45 prepared from the chloride (R)-44 with benzaldehyde afforded (E)-alkene (S)-46 which after hydrogenation and basic hydrolysis gave ʟ-homophenylalaninol ((S)-42).
![[1860-5397-15-168-i13]](/bjoc/content/inline/1860-5397-15-168-i13.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 13: Synthesis of ʟ-homophenylalaninol ((S)-42). Reagents and conditions: a) NaH, THF, 0 °C to −78 °C, 1 h then phosgene/toluene, −78 °C, 2 h; b) anisole, MeSO3H, hexane, reflux, 4 h; c) Ph3P, NaI, DMF, 100 °C, 24 h; d) LiHMDS, THF, −78 °C then PhCHO, −78 °C to rt, 2 h; e) H2, 5% Pd/C, EtOH, rt, 10 h; f) LiOH, EtOH, reflux, 2 h.
Scheme 13: Synthesis of ʟ-homophenylalaninol ((S)-42). Reagents and conditions: a) NaH, THF, 0 °C to −78 °C, 1...
ᴅ-Homophenylalaninol ((R)-42) and its homolog containing a 4-octylphenyl group (R)-47 are of interest in the synthesis of 3-deoxysphingoid analogues [52,53]. For this reason their syntheses began from the 4-(iodomethyl)oxazolidin-2-one (4S,1'R)-39 prepared from the aziridine alcohol (2R,1'R)-7 as shown on Scheme 12 and involved formation of the amino alcohols (R)-42 and (R)-47 employing procedures described in Scheme 14. Both (R)-47 and (R)-48 appeared moderate inhibitors of human sphingosine kinases hSphK1 and hSphK2 at 50 μM concentrations as compared to N,N-dimethylsphingosine (DMS) [52].
![[1860-5397-15-168-i14]](/bjoc/content/inline/1860-5397-15-168-i14.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 14: Synthesis of ᴅ-homo(4-octylphenyl)alaninol ((R)-47) and a sphingolipid analogue (R)-48. Reagents and conditions: a) see Scheme 13, steps b–f; b) C12H25COCl, NaOH, H2O, rt.
Scheme 14: Synthesis of ᴅ-homo(4-octylphenyl)alaninol ((R)-47) and a sphingolipid analogue (R)-48. Reagents an...
By functionalization at C2 and C3: Florfenicol ((1R,2S)-49) is used as antimicrobial in veterinary medicine [54]. Recently another strategy which applied the (1-phenylethyl)aziridine chemistry has been disclosed (Scheme 15) [55]. The aziridine ketone (2R,1'S)-51 (de 98.8%) was obtained when ketone 50 was reacted with (S)-1-phenylethylamine. Its reduction with a NaBH4/ZnCl2 mixture gave the alcohol (2R,1'S,1''S)-52 (de 92%) which was subjected to the aziridine ring opening with HF followed by the removal of the chiral auxiliary to afford (1S,2S)-53 (de 99.1%). Dichloroacetylation led to the formation of (1S,2S)-49 and provided a basis for the inversion of configuration at C2 by O-mesylation and intramolecular displacement to yield the 2-oxazoline (4S,5R)-54 readily hydrolyzed to florfenicol ((1R,2S)-49).
![[1860-5397-15-168-i15]](/bjoc/content/inline/1860-5397-15-168-i15.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 15: Synthesis of florfenicol ((1R,2S)-49). Reagents and conditions: a) (S)-1-phenylethylamine, TEA, MeOH, rt, 3 h then crystallization; b) NaBH4, ZnCl2, MeOH, −40 °C, 4 h; c) Et3N·3HF, ClCH2CH2Cl, reflux, 3 h; d) H2, 10% Pd/C, MeOH, 1.2 atm, 40 °C; e) Cl2CHCOOMe, TEA, MeOH, 50 °C, 5 h; f) MsCl, TEA, CH2Cl2, rt, overnight; g) H2O, iPrOH, 80 °C, 1 h.
Scheme 15: Synthesis of florfenicol ((1R,2S)-49). Reagents and conditions: a) (S)-1-phenylethylamine, TEA, MeO...
Naturally occurring tyroscherin ((2S,3R,6E,8R,10R)-55) was recognized for its anticancer activity [56]. Alkylation of Weinreb amide (2S,1'S)-18 and stereoselective reduction of the corresponding ketone (2S,1'S)-56 with the NaBH4/ZnCl2 mixture gave the aziridine alcohol (2S,1'R,1''S)-57 already containing exact absolute configurations (2S and 3R) of the final product (Scheme 16) [57]. A two-step aziridine ring opening was performed on the silylated alcohol (2S,1'R,1''S)-58 and included N-methylation and reaction with the protected phenylmagnesium bromide to form (2S,3R)-59. The last step opened the way to synthesis of tyroscherin analogues [58]. The alcohol (2S,3R)-60 was obtained after selective desilylation and N-debenzylation with simultaneous N-Boc protection and served as a key intermediate for installation of a dimethylhexylene fragment in Julia–Kocienski olefination [56].
![[1860-5397-15-168-i16]](/bjoc/content/inline/1860-5397-15-168-i16.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 16: Synthesis of natural tyroscherin ((2S,3R,6E,8R,10R)-55). Reagents and conditions: a) I(CH2)3OTIPS, t-BuLi, ether, −78 °C to rt, 2 h; b) NaBH4, ZnCl2, MeOH, −78 °C, 0.5 h; c) TBDMSCl, DMAP, CH2Cl2, rt, 12 h; d) TfOMe, CuI, dioxane, rt, 0.5 h then 4-MOMOC6H4MgBr, THF, 0 °C, 10 min.; e) Et3N·HF, pyridine, rt, 26 h; f) H2, Pd(OH)2, EtOH, rt, 3 h then Boc2O, rt, 1 h; g) see ref. [56].
Scheme 16: Synthesis of natural tyroscherin ((2S,3R,6E,8R,10R)-55). Reagents and conditions: a) I(CH2)3OTIPS, t...
Pyrrolidine alkaloids (−)-hygrine ((S)-61) and (−)-hygroline ((2S,2'S)-62) were isolated from many natural sources and are known as precursors to tropane alkaloids [59]. Together with (−)-pseudohygroline (2S,2'R)-62 they were synthesized from a common intermediate (2S,1'R)-63 prepared from the aldehyde (2R,1'R)-6 by Wittig olefination [60] and a regioselective C=C bond reduction (Scheme 17) [61]. N-Methylation was followed by the aziridinium ion opening with cyanide to access compound 64 which was first transformed into diester and later after hydrogenolysis into the γ-lactam (S)-65 [62]. The methoxycarbonyl to acetyl group conversion was accomplished via Weinreb amide to give the pyrrolidin-2-one (S)-66. Acetal formation, reduction of the amide function and deprotection completed synthesis of (−)-hygrine (S)-61. To synthesize (−)-hygroline (2S,2'S)-62 and (−)-pseudohygroline (2S,2'R)-62 the carbonyl group in (S)-66 was reduced and the diastereoisomeric alcohols (2S,2'S)-67 and (2S,2'R)-67 were separated as tert-butyldiphenylsilyl ethers, individually transformed into 2-(3-hydroxypropyl)pyrrolidines by LiAlH4 reduction and deprotected.
![[1860-5397-15-168-i17]](/bjoc/content/inline/1860-5397-15-168-i17.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 17: Syntheses of (−)-hygrine (S)-61, (−)-hygroline (2S,2'S)-62 and (−)-pseudohygroline (2S,2'R)-62. Reagents and conditions: a) Ph3P=CHCOOMe, MeOH, 0 °C, 1 h; b) 2-nitrobenzenesulfonyl hydrazide (NBSH), TEA, CH2Cl2, 0 °C to rt, 12 h; c) TfOMe, NaCN, MeCN, 0 °C to rt, 1 h; d) HCl, reflux, 1 h then MeOH, H2SO4, reflux, 8 h; e) H2, Pd(OH)2, MeOH, rt, 6 h; f) MeONHMe, Me3Al, CH2Cl2, 0 °C, 1 h; g) MeMgBr, THF, −78 °C to −45 °C, 4 h; h) HC(OMe)3, PTSA, MeOH, 40 °C, 1.5 h; i) LiAlH4, THF, reflux, 1 h then 6 M HCl; j) NaBH4, CoCl2, MeOH, −78 °C, 1.5 h and rt, 1 h; k) TBDPSCl, imidazole, DMAP, CH2Cl2, rt, 1 h; l) LiAlH4, THF, reflux, 1 h; m) see ref. [63].
Scheme 17: Syntheses of (−)-hygrine (S)-61, (−)-hygroline (2S,2'S)-62 and (−)-pseudohygroline (2S,2'R)-62. Rea...
Diamines
PF-00951966 (Scheme 18) belongs to a fluoroquinolone family of antibiotics which is substituted at C7 with a (3R)-3-[(1S)-2-cyano-1-(methylamino)ethyl]pyrrolidin-1-yl group [64]. The synthesis of the corresponding pyrrolidine (3S,3'R)-68 began from the aziridine (E)-acrylate (2R,1'S)-69a (Scheme 18) readily prepared from the aldehyde (2S,1'S)-6 [31]. Michael addition of nitromethane to the acrylate (2R,1'S)-69a gave an inseparable mixture (70:30) of aziridine esters 70 which were subjected to a selective reduction of the nitro group to afford the major pyrrolidine-2-one (2R,4'R,1''S)-71 after chromatographic purification [65]. The reduction of the amide bond gave (2R,3'R,1''S)-72 thus providing a cyclic part of (3S,3'R)-68. The transformation of the aziridine moiety into the 2-cyano-1-(methylamino)ethyl group was carried out after N-Boc protection and involved methylation of (2R,3'R,1''S)-73 with simultaneous opening of the aziridinium ion with cyanide to form (3S,3'R,1''S)-74 with high (97:3) regioselectivity. The removal of the chiral auxiliary afforded a stable di-N-Boc derivative (3S,3'R)-75 which was transformed into (3S,3'R)-68 in the presence of acids.
![[1860-5397-15-168-i18]](/bjoc/content/inline/1860-5397-15-168-i18.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 18: Synthesis of pyrrolidine (3S,3'R)-68, a fragment of the fluoroquinolone antibiotic PF-00951966. Reagents and conditions: a) MeNO2, TBAF, THF, rt, 4 h; b) H2, 10% PtO2, MeOH, rt, 48 h; c) LiAlH4, THF, reflux, 1 h; d) Boc2O, EtOH, rt, 6 h; e) TfOMe, NaCN, MeCN, 0 °C to rt; f) H2, 10% Pd/C, Boc2O, THF, rt; g) CF3COOH, CH2Cl2, rt, 2 h.
Scheme 18: Synthesis of pyrrolidine (3S,3'R)-68, a fragment of the fluoroquinolone antibiotic PF-00951966. Rea...
In search for a sphingolipid analogue synthesis of compounds of general formula (R)-76 was undertaken (Scheme 19) [53]. The aziridine ketone (2S,1'R)-77 obtained from Weinreb amide (2S,1'R)-18 was subjected to a three-step deoxygenation (reduction, mesylation of the alcohol (2S,1'R)-78 and reduction of the mesylate) to locate the 2-phenylethyl substituent at C2. Installation of the pyrrolidine ring at C3 was achieved after opening of the aziridine ring with iodotrimethylsilane to give a protected diamine (R)-80a. Removal of the chiral auxiliary yielded the diamine (R)-80b which after acylation provided analogues (R)-76. Compound (R)-76 (R = C9H19) appeared inactive as inhibitor of human sphingosine kinases 1 and 2.
![[1860-5397-15-168-i19]](/bjoc/content/inline/1860-5397-15-168-i19.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 19: Synthesis of sphingolipid analogues (R)-76. Reagents and conditions: a) BnBr, Mg, THF, reflux, 6 h; b) LiAlH4, THF, 0 °C, 1 h; c) MsCl, TEA, CH2Cl2, 0 °C, 1 h; d) LiAlH4; e) TMSCl, NaI, MeCN, rt, 2 h then pyrrolidine, reflux, 2 h; f) H2, Pd(OH)2, AcOH, EtOH, rt; g) RC(O)Cl, NaOH, THF/H2O, rt.
Scheme 19: Synthesis of sphingolipid analogues (R)-76. Reagents and conditions: a) BnBr, Mg, THF, reflux, 6 h;...
1,2-Diamino-3-hydroxy derivatives
The interest in sphingoid analogues stems from the involvement of sphingolipid metabolites in an array of important cell processes. ᴅ-threo-PDMP (1R,2R)-81 is a ceramide analogue identified as an inhibitor of glucosylceramide synthase (GCS) at micromolar concentrations [66,67]. It was efficiently synthesized [68,69] employing the alcohol (2R,1′R,1''S)-28 prepared from the aziridine aldehyde (2R,1′S)-6 which provided the 2R absolute configuration of the final product while the configuration at C1' was created by a stereoselective addition of phenylmagnesium bromide (Scheme 20) [29]. The aziridine ring opening in (2R,1′R,1''S)-28 with morpholine first required the reaction with iodotrimethylsilane and furnished diamino alcohol 82 which after N-debenzylation and acylation gave (1R,2R)-81.
![[1860-5397-15-168-i20]](/bjoc/content/inline/1860-5397-15-168-i20.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 20: Synthesis of ᴅ-threo-PDMP (1R,2R)-81. Reagents and conditions: a) TMSCl, NaI, MeCN, rt, 1 h 50 min, then morpholine, reflux, 2 h; b) H2, 10% Pd(OH)2, AcOH, 40 °C, 4 h; c) Me(CH2)8COCl, NaOH, THF/H2O, rt.
Scheme 20: Synthesis of ᴅ-threo-PDMP (1R,2R)-81. Reagents and conditions: a) TMSCl, NaI, MeCN, rt, 1 h 50 min,...
In a similar way other GCS inhibitors, e.g., ᴅ-threo-P4 (1R,2R)-83a and ᴅ-threo-4'-hydroxy-P4 (1R,2R)-83b (Scheme 20) were obtained and examined as potential medications for treating cognitive disorders [70], chronic pain [71] or as immunostimulants [72].
Homologues of ʟ-threo-P4, e.g., compound (2S,3S)-84 were synthesized following the strategy depicted on a previous scheme [52,53]. Reduction of the aziridine ketone (2S,1′R)-77 with LiAlH4 afforded a mixture of diastereoisomeric alcohols (2S,1'R,1''R)-78 and (2S,1'S,1''R)-78 with low (up to 33%) diastereoselectivity readily separable chromatographically. The sphingolipid analogue SG-14 (2S,3S)-84 was obtained from (2S,1'S,1''R)-78 as already described (Scheme 21) and was found to inhibit hSphK2 with the same potency as N,N-dimethylsphingosine (DMS) being completely inactive toward hSphK1.
![[1860-5397-15-168-i21]](/bjoc/content/inline/1860-5397-15-168-i21.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 21: Synthesis of the sphingolipid analogue SG-14 (2S,3S)-84. Reagents and conditions: a) LiAlH4, THF, 0 °C, 1 h then separation of diastereoisomers; b) TMSCl, NaI, MeCN, rt, 2 h then pyrrolidine, reflux, 2 h; c) H2, Pd(OH)2, AcOH, EtOH, rt; d) Me(CH2)14COCl, NaOH, THF/H2O, rt.
Scheme 21: Synthesis of the sphingolipid analogue SG-14 (2S,3S)-84. Reagents and conditions: a) LiAlH4, THF, 0...
2-Amino-1,3-diols
A 2-amino-1,3-dihydroxypropyl fragment 12 of sphingosine and ceramides of the required 2S,3R configuration can also originate from the aziridine alcohol, e.g., (2S,1'R,1''R)-87 prepared from the ketone (2S,1'R)-86 via Weinreb amide (2S,1'R)-18 [52,53]. The aziridine ring opening in (2S,1'R,1''R)-87 with acetic acid followed by hydrolysis and hydrogenolytic removal of the chiral auxiliary gave the sphingosine analogue SG-12 (2S,3R)-88 (Scheme 22) as selective and potent as the ceramide analogue SG-14 (2S,3S)-84 in inhibiting hSphK2.
![[1860-5397-15-168-i22]](/bjoc/content/inline/1860-5397-15-168-i22.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 22: Synthesis of the sphingolipid analogue SG-12 (2S,3R)-88. Reagents and conditions: a) 1-(bromomethyl)-4-octylbenzene, Mg, THF, 50 °C, 6 h; b) LiAlH4, THF, 0 °C, 1 h then separation of diastereoisomers; c) AcOH, CH2Cl2, rt, 18 h then KOH, MeOH, rt, 2 h; d) H2, Pd(OH)2, EtOH, 100 psi, rt.
Scheme 22: Synthesis of the sphingolipid analogue SG-12 (2S,3R)-88. Reagents and conditions: a) 1-(bromomethyl...
Sphingosine-1-phosphate (S1P) analogues (2S,3R)-89a and (2S,3R)-89b with 1,4- and 1,3-disubstituted benzene rings incorporated into the alkyl chain were obtained from aziridine ketones (2S,1'R)-90a and (2S,1'R)-90b (Scheme 23) readily prepared using Weinreb amide (2S,1'R)-18 which introduced the correct 2S configuration into the final product [73]. The highly stereoselective reduction of ketones 90a and 90b with a NaBH4/ZnCl2 mixture gave aziridine alcohols 91a and 91b having the required 3R configuration of the final product. The terminal hydroxymethyl group was acquired as shown earlier to provide (2S,3R)-92a and (2S,3R)-92b as N-Boc derivatives after N-debenzylation. Analogues (2S,3R)-89a (DS-SG-44) and (2S,3R)-89b (DS-SG-45) were formed after a regioselective phosphorylation and final treatment with bromotrimethylsilane. DS-SG-44 emerged as an agonist of S1P receptors while DS-SG-45 was found inactive.
![[1860-5397-15-168-i23]](/bjoc/content/inline/1860-5397-15-168-i23.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 23: Synthesis of sphingosine-1-phosphate analogues DS-SG-44 and DS-SG-45 (2S,3R)-89a and (2S,3R)-89a. Reagents and conditions: a) NaBH4, ZnCl2, MeOH, −78 °C; b) AcOH, CH2Cl2, rt then KOH, EtOH, rt; c) H2, Pd(OH)2, 100 psi, Boc2O, rt; d) P(OMe)3, CBr4, pyridine, 0 °C; e) TMSBr, CH2Cl2, rt then H2O, rt.
Scheme 23: Synthesis of sphingosine-1-phosphate analogues DS-SG-44 and DS-SG-45 (2S,3R)-89a and (2S,3R)-89a. R...
Dihydrosphingosines, e.g., safingol and sphinganine itself or as components of dihydroceramides are of interest as enzyme inhibitors [74,75]. Their common vicinal aminohydroxy fragment was efficiently synthesized from the aziridine ketone (2S,1'R)-93 readily prepared from the ester (2S,1'R)-5f [47]. To secure the 3S configuration in N-Boc-safingol the ketone (2S,1'R)-93 was reduced with ʟ-Selectride® giving the (S)-alcohol (2S,1'S,1''R)-94 stereoselectively (Scheme 24). On the other hand, the 3R configuration in ᴅ-erythro-sphinganine was assured when a NaBH4/ZnCl2 mixture was applied in a chelation-controlled reduction of the ketone (2S,1'R)-93 to provide the (R)-alcohol (2S,1'R,1''R)-94 as a major (94:6) product. Alcohols (2S,1'S,1''R)- and (2S,1'R,1''R)-94 were transformed into N-Boc-safingol (2S,3S)-95 and N-Boc-ᴅ-erythro-sphinganine (2S,3R)-95 in three standard steps.
![[1860-5397-15-168-i24]](/bjoc/content/inline/1860-5397-15-168-i24.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 24: Synthesis of N-Boc-safingol ((2S,3S)-95) and N-Boc-ᴅ-erythro-sphinganine ((2S,3R)-95). Reagents and conditions: a) ʟ-Selectride®, THF, −70 °C, 0.5 h; b) NaBH4, ZnCl2, MeOH, −30 °C, 0.5 h; c) AcOH, CH2Cl2, rt, 14 h; d) KOH, EtOH, rt, 3 h; e) H2, Pd(OH)2/C, Boc2O, MeOH, rt, 15 h.
Scheme 24: Synthesis of N-Boc-safingol ((2S,3S)-95) and N-Boc-ᴅ-erythro-sphinganine ((2S,3R)-95). Reagents and...
Ceramide and other sphingolipids are mainly localized in cell membranes and are involved in important cell processes. A series of constrained analogues of ceramide (2S,3R)-96 modified by converting the terminal aminohydroxy fragment into oxazolidin-2-one and acylation of the nitrogen atom with 13 selected acyl chlorides was synthesized starting from the aziridine ketone (2S,1'R)-97 (Scheme 25) [76]. To introduce the R absolute configuration at C3 (ceramide numbering) the aziridine alcohol (2S,1'R,1''R)-98 was stereoselectively obtained by reduction of the corresponding ketone with a NaBH4/ZnCl2 mixture. Treatment of (2S,1'R,1''R)-98 with LiAlH4 to secure the trans-configured alkene, protection of the hydroxy group and the regioselective aziridine ring opening led to the formation of the acetate (2S,3R,1'R)-99. After basic hydrolysis the oxazolidin-2-one (2S,3R,1'R)-100 was produced which was later transformed into the ceramide analogues (2S,3R)-96 in three standard steps. From a series of 13 modified ceramides the analogue containing the N-cyclopentylcarbonyl group appeared more active against human leukemia HL-60 cells than natural N-acetylceramide. This observation prompted to synthesize a next series of analogues retaining the N-cyclopentycarbonyl moiety while modifying the alkyl chain. It appeared that an analogue having a shorter chain (C10H21 instead of C13H27) was slightly less active than (2S,3R)-96 (R = cyclopentyl) but still more active than N-acetylceramide.
![[1860-5397-15-168-i25]](/bjoc/content/inline/1860-5397-15-168-i25.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 25: Synthesis of ceramide analogues (2S,3R)-96. Reagents and conditions: a) NaBH4, ZnCl2, MeOH, −78 °C, 0.5 h; b) LiAlH4, THF, rt, 3 h; c) TBDMSCl, DMAP, CH2Cl2, rt, 17 h; d) AcOH, CH2Cl2, rt, 10 h; e) KOH, EtOH, rt, 1 h; f) CDI, DBU, CH2Cl2, rt, overnight; g) Li, NH3 liquid, t-BuOH, THF, −78 °C, 0.5 h; h) RCOCl, NaHMDS, THF, −78 °C; i) TBAF, THF, 0 °C, 4 h.
Scheme 25: Synthesis of ceramide analogues (2S,3R)-96. Reagents and conditions: a) NaBH4, ZnCl2, MeOH, −78 °C,...
A 2-amino-1,3-dihydroxypropyl fragment 12 can also be found in serinol and 3-phenylserinol, potential precursors to the respective amino acids. Orthogonally protected serinols (S)-101 and (R)-102 were synthesized from the benzyl ether 103 utilizing the aziridine alcohol (2S,1'S)-7 (Scheme 26) [77]. The aziridine ring opening with acetic acid and saponification led to the formation of (S)-101. Protection of the vicinal aminohydroxy fragment as an oxazolidin-2-one provided (R)-104 which after selective debenzylation produced (R)-102.
![[1860-5397-15-168-i26]](/bjoc/content/inline/1860-5397-15-168-i26.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 26: Synthesis of orthogonally protected serinols, (S)-101 and (R)-102. Reagents and conditions: a) BnBr, NaH, TBAI, THF, rt, 22 h; b) AcOH, CHCl3, reflux, 6 h; c) KOH, EtOH, reflux, 0.5 h; d) CDI, CHCl3, 50 °C, 17 h; e) H2, Pd(OH)2, AcOEt/MeOH, rt, 2 h.
Scheme 26: Synthesis of orthogonally protected serinols, (S)-101 and (R)-102. Reagents and conditions: a) BnBr...
The synthesis of N-acetyl-3-phenylserinol ((1R,2R)-105) was readily accomplished [78] from the aziridine alcohol (2R,1'R,1''S)-28 available by addition of phenyllithium to the aldehyde (2R,1'S)-6 [30]. Opening of the aziridine ring in (2R,1'R,1''S)-28 with acetic acid yielded the acetate (1R,2R,1'S)-106 while catalytic hydrogenolysis led to the formation of (1R,2R)-105 (Scheme 27) [78]. Application of other organometallics paved the way to synthesis of a variety of 3-substituted 2-amino-1,3-propanodiols.
![[1860-5397-15-168-i27]](/bjoc/content/inline/1860-5397-15-168-i27.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 27: Synthesis of N-acetyl-3-phenylserinol ((1R,2R)-105). Reagents and conditions: a) AcOH, CH2Cl2, reflux, 4 h; b) H2, Pd(OH)2, AcOEt/MeOH, rt, 48 h.
Scheme 27: Synthesis of N-acetyl-3-phenylserinol ((1R,2R)-105). Reagents and conditions: a) AcOH, CH2Cl2, refl...
1,3-Diamino-2-hydroxy derivatives
Linezolid ((S)-107) represents a new class of 1,3-oxazolidin-2-one antibiotics and contains the (2S)-1,3-diamino-2-hydroxypropyl fragment which could also be derived from the aziridine amide (2R,1'S)-8 (Scheme 28) [19]. N-Boc-protected amine (2S,1'S)-108 was converted to a key 1,3-oxazolidin-2-one (5S,1'S)-109 by trifluoroborate-catalyzed regioselective and stereospecific (SN2 displacement) cyclization. N-Arylation and debenzylation followed by acylation provided enantiomerically pure (S)-linezolid ((S)-107).
![[1860-5397-15-168-i28]](/bjoc/content/inline/1860-5397-15-168-i28.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 28: Synthesis of (S)-linezolid (S)-107. Reagents and conditions: a) LiAlH4, THF, 0 °C to reflux; b) Boc2O, MeOH, rt, overnight; c) BF3·OEt2, THF, 0 °C to reflux, 2 h; d) 1-bromo-3-fluoro-4-morpholinobenzene, CuI, K2CO3, MeNHCH2CH2NHMe, toluene, 115 °C, 27 h; e) HCOONH4, 10% Pd/C, MeOH, overnight then AcCl, Et3N, CH2Cl2, rt, overnight.
Scheme 28: Synthesis of (S)-linezolid (S)-107. Reagents and conditions: a) LiAlH4, THF, 0 °C to reflux; b) Boc2...
2-Amino-1,3,4-triols
ᴅ-ribo-Phytosphingosine ((2S,3S,4R)-2-aminooctadecane-1,3,4-triol, (2S,3S,4R)-110) appears to be the most common from other stereoisomeric phytosphingosines which are present in many species and show diversified biological activity. From several synthetic approaches to ᴅ-ribo-phytosphingosine [79] application of the aziridine aldehyde (2S,1'R)-6 provided (2S,3S,4R)-110 in four steps with full control of stereochemistry (Scheme 29) [80]. Since the 2S absolute configuration of the final product was already secured in the starting aldehyde introduction of the two others required cis-dihydroxylation of the Z-alkene 111. The highest diastereoselectivity (99:1) was observed at −10 °C providing the aziridine diol (2S,1'S,2'R,1''R)-112 as a major diastereoisomer. The regioselective aziridine ring opening combined with ester hydrolysis and debenzylation gave enantiomerically pure (2S,3S,4R)-110 and two stable derivatives (2S,3S,4R)-114 and (2S,3S,4R)-115 (Scheme 29).
![[1860-5397-15-168-i29]](/bjoc/content/inline/1860-5397-15-168-i29.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 29: Synthesis of (2S,3S,4R)-2-aminooctadecane-1,3,4-triol (ᴅ-ribo-phytosphingosine) (2S,3S,4R)-110. Reagents and conditions: a) Ph3P+CH2C14H29 Brˉ, LiHMDS, THF, −78 °C to rt, 2 h; b) OsO4, NMO, Me2O/H2O, −10 °C, 10 h; c) AcOH, CH2Cl2, rt, 8 h then KOH, EtOH, rt, 3 h; d) H2, Pd(OH)2, EtOH, 100 psi, rt; e) H2, Pd(OH)2, Boc2O, EtOH, rt, 100 psi; f) Ac2O, pyridine, rt, overnight.
Scheme 29: Synthesis of (2S,3S,4R)-2-aminooctadecane-1,3,4-triol (ᴅ-ribo-phytosphingosine) (2S,3S,4R)-110. Rea...
Amino acids and derivatives
α-Amino acids: Homochiral amino alcohols of general formula RCH(NH2)CH2OH as precursors of α-amino acids can be synthesized from aziridine-2-methanols either by functionalization at C3 (Scheme 12 and Scheme 13) or by opening of the aziridine ring to form 2-amino-1,3-diols 12 (Schemes 22–24) combined with the removal of the secondary hydroxy group when simple amino acids (R = alkyl, aryl) are to be prepared. For the latter case the known strategies will be illustrated by syntheses of ᴅ-phenylalanine (R)-116 [81,82]. The acetate (1S,2S,1'R)-106 obtained from the aziridine alcohol (2S,1'S,1''R)-28 was subjected to mesylation to yield cis-2,3-disubstituted aziridine (2R,3R,1'R)-117 (Scheme 30). The hydrogenolytic cleavage of the aziridine ring occurred regiospecifically at the N–C3 bond (benzylic position) to provide N-Boc-ᴅ-phenylalaninol ((R)-118) after saponification. When mesylation of (2S,1'R/S,1''R)-28 was followed by LiAlH4 reduction the aziridine (2R,1'R)-119 was produced from which the acetate (2R,1'R)-120 and next (R)-118 were formed. The aziridine ring opening in (2S,1'R/S,1''R)-28 gave the acetate (1R/S,2S,1'R)-106 which was transformed into the 1,3-oxazolidin-2-one (4S,5R/S,1'R)-121 a precursor to (R)-118. Catalytic ruthenium tetroxide oxidation of (R)-118 followed by hydrolysis gave ᴅ-phenylalanine (R)-116 as the hydrochloride salt [83]. Other α-amino acids of general formula RCH2CH(NH2)COOH, e.g., R = Me, iPr, PhCH2, were prepared in a similar way [76].
![[1860-5397-15-168-i30]](/bjoc/content/inline/1860-5397-15-168-i30.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 30: Syntheses of ᴅ-phenylalanine (R)-116. Reagents and conditions: a) AcOH, CH2Cl2, reflux, 4 h; b) MsCl, TEA, CH2Cl2, −78 °C to rt, 6 h; c) H2, Pd(OH)2, Boc2O, MeOH, rt, 7 h; d) KOH, EtOH, rt, 10 min; e) MsCl, TEA, CH2Cl2, 0 °C, 2 h; f) LiAlH4, ether, rt, 4 h; g) CDI, CH2Cl2, rt, 24 h; h) RuCl3, NaIO4, CCl4/MeCN/H2O, rt, 7 h; i) HCl, reflux, 4 h.
Scheme 30: Syntheses of ᴅ-phenylalanine (R)-116. Reagents and conditions: a) AcOH, CH2Cl2, reflux, 4 h; b) MsC...
A straightforward synthesis of N-Boc-ᴅ-3,3-diphenylalanine ((R)-122) was carried out from the aziridine menthyl ester (2S,1'R)-5f [84]. Phenyl groups were introduced by Grignard reaction while the hydroxymethyl fragment was derived from the aziridine ring opening to produce (2S,1'R)-123. N-Debenzylation was accompanied with deoxygenation at the hydroxydiphenylmethyl site to give N-Boc-3,3-diphenylalaninol ((R)-124) which was oxidized with Jones reagent to enantiomerically pure (R)-122 (Scheme 31) useful in pseudopeptide synthesis.
![[1860-5397-15-168-i31]](/bjoc/content/inline/1860-5397-15-168-i31.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 31: Synthesis of N-Boc-ᴅ-3,3-diphenylalanine ((R)-122). Reagents and conditions: a) PhMgBr, THF, −78 °C, 2 h; b) AcOH, CH2Cl2, rt, 4 h; c) KOH, EtOH, rt, 2 h; d) H2, 20% Pd(OH)2/C, HCOOH, 100 psi, rt, 6 h then Boc2O, NaOH, AcOEt, rt, 4 h; e) Jones reagent (CrO3, H2SO4, Me2CO/H2O), 0 °C, 4 h.
Scheme 31: Synthesis of N-Boc-ᴅ-3,3-diphenylalanine ((R)-122). Reagents and conditions: a) PhMgBr, THF, −78 °C...
Derivatives of nonproteinogenic ʟ-2,3-diaminopropanoic acid, e.g., (S)-125 were synthesized from the aziridine ester (2R,1'R)-5b employing the aluminum chloride catalyzed opening of the aziridine ring with azide (Scheme 32) [85]. The reaction involved a nucleophilic displacement at more hindered C2 with inversion of configuration to form the azido ester (2S,1'R)-126 which was transformed into (S)-125 in usual way. However, since amines can be obtained from azides by a Staudinger reaction the ester (2S,1'R)-126 can serve as the starting material to a variety of orthogonally protected derivatives of 2,3-diaminopropanoic acid.
![[1860-5397-15-168-i32]](/bjoc/content/inline/1860-5397-15-168-i32.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 32: Synthesis of ethyl N,N’-di-Boc-ʟ-2,3-diaminopropanoate ((S)-125). Reagents and conditions: a) NaN3, AlCl3, EtOH/H2O, pH 4, rt; b) H2, Pd/C, Boc2O, rt.
Scheme 32: Synthesis of ethyl N,N’-di-Boc-ʟ-2,3-diaminopropanoate ((S)-125). Reagents and conditions: a) NaN3,...
The bicyclic amino acid (S)-127 is a major metabolite of isazofos in corn grain and for toxicological studies both enantiomers were required [17]. To this end the aziridine ester (2S,1′R)-5a was reacted with 3-bromo-5-methoxy-1H-1,2,4-triazole (128) to give N-protected bicyclic amino ester 129 which was next converted into (S)-(+)-127 in two standard steps (Scheme 33) [17]. Its enantiomer was prepared from (2R,1′S)-5a.
![[1860-5397-15-168-i33]](/bjoc/content/inline/1860-5397-15-168-i33.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 33: Synthesis of the bicyclic amino acid (S)-(+)-127. Reagents and conditions: a) BF3·OEt2, THF, 60 °C, 3 h; b) KOH, MeOH/H2O, rt, 1 h; c) H2, 10% Pd/C, dioxane, 35 °C, 5 bar, 13 h.
Scheme 33: Synthesis of the bicyclic amino acid (S)-(+)-127. Reagents and conditions: a) BF3·OEt2, THF, 60 °C,...
Lacosamide ((R)-130) is a derivative of ᴅ-serine and has found application as an anticonvulsant medication [86]. Under optimized conditions the aziridine ring opening in the ester (2R,1'S)-5b with methanol gave a 94:6 mixture of regioisomeric methoxy amino esters with formation of (R)-131 as a major product (Scheme 34). Catalytic hydrogenation of this mixture in the presence of acetic anhydride produced a crude acetamido ester (R)-132 which was transformed into the final N-benzyl amide (R)-130 with of 99.9% ee after crystallization.
![[1860-5397-15-168-i34]](/bjoc/content/inline/1860-5397-15-168-i34.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 34: Synthesis of lacosamide, (R)-2-acetamido-N-benzyl-3-methoxypropanamide (R)-130. Reagents and conditions: a) MeOH, BF3·OEt2, MeCN, 90 °C, 3 h; b) H2, 20% Pd(OH)2/C, Ac2O, EtOH, rt, 14 h; c) BnNH2, Me3Al, CH2Cl2, rt, 3 h.
Scheme 34: Synthesis of lacosamide, (R)-2-acetamido-N-benzyl-3-methoxypropanamide (R)-130. Reagents and condit...
ʟ-(+)-Furanomycin (Scheme 35) is a natural nonproteinogenic amino acid which showed pronounced antibacterial activity. From several syntheses of this compound the general approach which employs the aziridine ester (2R,1'R)-5b as a starting material allows to also obtain its 5'-epimer and norfuranomycin (2S,2'R)-133 [87]. To construct the 2,5-dihydrofurane ring the aziridine alcohol (2R,1'R,1''R)-134 (Scheme 35) [88] was converted to the allyl ether (2R,1'R,1''R)-135 which in the presence of Grubbs 1st generation catalyst produced (2R,1'R,1''R)-136. Transformation of the aziridine portion of 136 into an amino acid fragment of norfuranomycin (2S,2'R)-133 was accomplished starting from the hydrolytic opening of the aziridine ring and was followed by protection of the amino alcohol and Birch debenzylation to give 137. After basic hydrolysis and N-protection to form 138 a two-step hydroxymethyl to carboxyl oxidation was performed to yield N-Boc-norfuranomycin ((2S,2'R)-133). Installation of the 1-methylprop-2-en-1-yl group in (2R,1'R,1''R)-135 instead of the allyl residue opened the way to synthesis of ʟ-(+)-furanomycin and its 5'-epimer.
![[1860-5397-15-168-i35]](/bjoc/content/inline/1860-5397-15-168-i35.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 35: Synthesis of N-Boc-norfuranomycin ((2S,2'R)-133). Reagents and conditions: a) H2C=CHCH2I, NaH, THF, 0 °C to rt, 3 h; b) Grubbs' 1st, CH2Cl2, rt, 20 h; c) H2O, BF3·OEt2, MeCN, reflux, 3 h; d) CDI, DBU, CH2Cl2, rt, 12 h; e) Na, NH3 liquid, THF, −78 °C, 0.5 h; f) KOH, EtOH/H2O, reflux, 2 h; g) Boc2O, MeOH, rt, 3 h; h) Dess–Martin periodinane, CH2Cl2, rt, 1 h; i) NaClO2, t-BuOH/H2O, Me2C=CHMe, NaH2PO4, 0 °C to rt.
Scheme 35: Synthesis of N-Boc-norfuranomycin ((2S,2'R)-133). Reagents and conditions: a) H2C=CHCH2I, NaH, THF,...
MeBmt (2S,3R,4R,6E)-139 is a nonproteinogenic amino acid found as a constituent of the naturally occurring cyclic peptide cyclosporine currently in medical use as immunosuppressant [89]. Syntheses of MeBmt are rather challenging endeavor since these amino acids in addition to an E-configured C=C bond has three neighboring stereogenic centers. The starting aziridine aldehyde (2R,1'R)-6 already introduces the required configuration at C2 and the two other centers of chirality were created by the stereospecific crotylation with a homochiral boronate to give the aziridine alcohol (2R,1'R,2'R,1''R)-140 (Scheme 36) [90]. After O-benzylation and N-methylation the ring opening in the intermediate aziridinium ion was tried. It appeared that the best regioselectivity (87:13) was achieved with cesium acetate and the major product (2R,3R,4R,1'R)-141 was separated chromatographically. To install two lacking carbon atoms the vinyl moiety was transformed into the respective aldehyde 143 (via a primary alcohol 142) which when subjected to Julia–Kocienski reaction furnished the E-olefinic terminus. Since under these conditions the acetate function was also hydrolyzed the carboxy group was formed by oxidation of the hydroxymethyl residue. To complete the synthesis N- and O-benzylic protecting groups were removed during the Birch reaction.
![[1860-5397-15-168-i36]](/bjoc/content/inline/1860-5397-15-168-i36.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 36: Synthesis of MeBmt (2S,3R,4R,6E)-139. Reagents and conditions: a) diisopropyl (S,S)-tartrate (E)-crotylboronate, toluene, MS 4 Å, −78 °C, 3 h; b) BnBr, NaH, THF, 0 °C to rt, 3 h; c) TfOMe, MeCN, rt, 10 min then AcOCs, rt, 1 h; d) catecholborane, (Ph3P)3RhCl, THF, 0 °C, 3 h then NaOH, MeOH, H2O2, rt, 1 h; e) Swern oxidation; f) 5-(ethylsulfonyl)-1-phenyl-1H-tetrazole, KHMDS, DME, −78 °C, 40 min; g) PDC, DMF, rt, 15 h; h) Na, NH3 liquid/THF, −78 °C, 15 min.
Scheme 36: Synthesis of MeBmt (2S,3R,4R,6E)-139. Reagents and conditions: a) diisopropyl (S,S)-tartrate (E)-cr...
A polyoxamic structural framework was found in polyoxins, natural peptidyl nucleosides with primarily antifungal properties. The stereoselective synthesis of (+)-polyoxamic acid ((2S,3S,4S)-144) was successfully carried out starting with Horner–Wadsworth–Emmons olefination of the aziridine aldehyde (2R,1'R)-6 which provided a 98:2 mixture of trans- and cis-acrylates 69b (Scheme 37) [32]. The major product (2S,1'R)-69b was subjected to Sharpless asymmetric dihydroxylation in the presence of AD-mix-α to give the diol 145a as a major (10:1) diastereoisomer. The ester moiety in 145a was reduced and hydroxy groups were protected to give the tribenzyloxy aziridine (2R,1'S,2'S,1''R)-146. After treatment with acetic acid and ester hydrolysis the intermediary amino alcohol (2R,3S,4S)-147 was oxidized and hydrogenolytic removal of benzyl groups completed the synthesis.
![[1860-5397-15-168-i37]](/bjoc/content/inline/1860-5397-15-168-i37.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 37: Synthesis of (+)-polyoxamic acid (2S,3S,4S)-144. Reagents and conditions: a) AD-mix-α, MeSO2NH2, t-BuOH/H2O, 0 °C, 36 h; b) LiAlH4, THF, 0 °C, 20 min; c) BnBr, NaH, TBAI, THF, rt, 12 h; d) AcOH, CH2Cl2, rt, 18 h; e) KOH, EtOH, rt, 2 h; f) Jones reagent, Me2O/H2O, 0 °C, 4 h; g) H2, Pd/C, MeOH, rt, 12 h.
Scheme 37: Synthesis of (+)-polyoxamic acid (2S,3S,4S)-144. Reagents and conditions: a) AD-mix-α, MeSO2NH2, t-...
The orthogonally protected 3-hydroxy-ʟ-glutamic acid (2S,3R)-148 was obtained from the aziridine ester (2R,1′S)-5b (Scheme 38) [91]. A two-carbon fragment came from tert-butyl acetate while the required R configuration at C3 in the final product was secured by stereoselective reduction (10:1) of the ketone (2R,1′S)-149 to give the aziridine alcohol (2R,1′R,1''S)-150 as the major product. Silylation of the hydroxy group preceded the aziridine ring opening with acetic acid while hydrogenation in the presence of Boc2O led to the formation of (3R,4R)-151. To conclude the synthesis of (2S,3R)-148 the hydroxymethyl group was recovered after basic deacetylation and it was further oxidized and esterified. The same methodology was applied in the preparation of (2S,3S)-148.
![[1860-5397-15-168-i38]](/bjoc/content/inline/1860-5397-15-168-i38.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 38: Synthesis of the protected 3-hydroxy-ʟ-glutamic acid (2S,3R)-148. Reagents and conditions: a) LiHMDS, AcOt-Bu, THF, −78 °C, 0.5 h; b) NaBH4, iPrOH, −40 °C, 1 h; c) TBDMSCl, TEA, DMAP, CH2Cl2, rt, 17 h; d) AcOH, CH2Cl2, rt, 10 h; e) H2, Pd(OH)2, Boc2O, MeOH, rt, 3 h; f) KOH, EtOH, 0 °C, 10 min; g) NaIO4, RuCl3, CCl4/MeCN/H2O, rt, 7 h; h) MeI, KHCO3, DMF, rt, 5 h.
Scheme 38: Synthesis of the protected 3-hydroxy-ʟ-glutamic acid (2S,3R)-148. Reagents and conditions: a) LiHMD...
β-Amino acids: (+)-Isoserine ((R)-152) was synthesized from the aziridine ester (2R,1'R)-5b in three simple steps (Scheme 39) [92]. Treatment of (2R,1'R)-5b with acetyl chloride led to N-acetylation with concomitant opening of the corresponding aziridinium ion with the chloride anion at C2 to give an unstable intermediate 153 which in the presence of aqueous base was transformed into the protected ethyl (R)-isoserinate 154. N-Debenzylation and hydrolysis completed the synthesis of (+)-isoserine ((R)-152).
![[1860-5397-15-168-i39]](/bjoc/content/inline/1860-5397-15-168-i39.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 39: Synthesis of (+)-isoserine (R)-152. Reagents and conditions: a) AcCl, MeCN, rt, 0.5 h then Na2CO3, CH2Cl2/H2O, rt; b) H2, Pd/C, Boc2O, MeOH, rt, 12 h; c) HCl, MeOH, reflux, 1 h then Amberlite IR-120 H.
Scheme 39: Synthesis of (+)-isoserine (R)-152. Reagents and conditions: a) AcCl, MeCN, rt, 0.5 h then Na2CO3, ...
Enantiomerically pure (3R,4S)-N3-Boc-3,4-diaminopentanoic acid ((3R,4S)-155) was synthesized from the Z-acrylate (2R,1'R)-156a prepared in a highly (88:12) stereoselective olefination of the aziridine aldehyde (2S,1'R)-6 (Scheme 40) [60]. Michael addition of (S)-1-phenylethylamine to 156a gave almost pure (>99:1) β-amino ester 157 which when subjected to the reductive opening of the aziridine ring was transformed into the pyrrolidin-2-one (4R,5S)-158. Basic hydrolysis produced (3R,4S)-155.
![[1860-5397-15-168-i40]](/bjoc/content/inline/1860-5397-15-168-i40.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 40: Synthesis of (3R,4S)-N3-Boc-3,4-diaminopentanoic acid (3R,4S)-155. Reagents and conditions: a) Ph3P=CHCOOMe, MeOH, 0 °C, 1 h; b) (S)-PhMeCHNH2, MeOH, 50 °C, 7 h; c) H2, Pd(OH)2, MeOH, rt, 4 h then Boc2O, MeOH, rt, 4 h; d) LiOH, MeOH/H2O, reflux, 6 h then Dowex 50W X2-200.
Scheme 40: Synthesis of (3R,4S)-N3-Boc-3,4-diaminopentanoic acid (3R,4S)-155. Reagents and conditions: a) Ph3P...
Other amino acids: Calyculins were isolated from marine sponges and they are of interest because of possible applications as protein phosphatase 1 and 2A inhibitors [93]. An interesting structural feature of calyculins is a C33–C37 fragment γ-amino acid (2S,3S,4S)-159. Its stereocontrolled synthesis involved cis-dihydroxylation of the aziridine cis-acrylate (2R,1'R)-156a which led to the formation of a 91:9 mixture of diastereoisomeric diols with 160 preponderating (Scheme 41) [34]. The regioselective aziridine ring opening with methanol and catalytic hydrogenation in the presence of formalin gave the final product (2S,3S,4S)-159.
![[1860-5397-15-168-i41]](/bjoc/content/inline/1860-5397-15-168-i41.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 41: Synthesis of methyl (2S,3S,4S)-4-(dimethylamino)-2,3-dihydroxy-5-methoxypentanoate (2S,3S,4S)-159. Reagents and conditions: a) OsO4, NMO, acetone, 0 °C to rt, 5 h; b) MeOH, BF3·OEt2, reflux, 3 h; c) H2, HCHO, 10% Pd/C, MeOH, rt, 12 h.
Scheme 41: Synthesis of methyl (2S,3S,4S)-4-(dimethylamino)-2,3-dihydroxy-5-methoxypentanoate (2S,3S,4S)-159. ...
The innovative application of the aldehyde (2S,1'R)-6 in syntheses of nonproteinogenic γ-amino hydroxy acids and their cyclic forms (pyrrolidin-2-ones) takes advantage of the stereoselective epoxidation of the aziridine acrylaldehyde 161 to predominantly (98:2) form the aziridine epoxide 162 when (S)-[diphenyl(trimethylsilyloxy)methyl]pyrrolidine was used as a catalyst (Scheme 42) [94]. A key β-hydroxyester 163 was produced from the epoxide 162 employing N-heterocyclic carbene catalysis. Openings of the aziridine ring in 163a with azide or in 163b with in acetic acid provided enantiomerically pure methyl (3S,4S)-4,5-di-N-Boc-amino-3-hydroxypentanoate 164 or 4-N-Boc-amino-3,5-dihydroxypentanoate 165, respectively. The latter compound as an unprotected acid was identified as a component of immunosuppressive thalassospiramide A [95] and siderophores called crochelins [96]. On the other hand, the former one was used in the synthesis of edenine A and D analogues [97] to study their biological properties. When the O-protected ester 163b was subjected to methylation and the corresponding aziridinium ion was treated with phenylmagnesium bromide a regioselective opening of the aziridine ring occurred at the less substituted carbon atom to give the protected pentanoate (3S,4S)-166. After catalytic debenzylation it was transformed into a silylated (4S,5S)-5-benzyl-4-hydroxy-1-methylpyrrolidin-2-one 167, a molecule having a structural core of antifungal (+)-preussin [98].
![[1860-5397-15-168-i42]](/bjoc/content/inline/1860-5397-15-168-i42.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 42: Syntheses of methyl (3S,4S) 4,5-di-N-Boc-amino-3-hydroxypentanoate ((3S,4S)-164), methyl (3S,4S)-4-N-Boc-amino-3,5-dihydroxypentanoate ((3S,4S)-165) and (4S,5S)-5-benzyl-4-hydroxy-1-methylpyrrolidin-2-one (4S,5S)-167. Reagents and conditions: a) Ph3P=CHCHO, toluene, 60 °C, 18 h; b) 35% H2O2, (S)-[diphenyl(trimethylsilyloxy)methyl]pyrrolidine, EtOH, rt, 6 h; c) 3-benzyl-4,5-dimethylthiazolium chloride, DIPEA, MeOH, CH2Cl2, rt, 24 h; d) TBDMSCl, DMAP, CH2Cl2, 0 °C to rt, 6 h; e) NaN3, BF3·OEt2, MeCN, 50 °C, 6 h; f) H2, 50% Pd(OH)2/C, Boc2O, MeOH, rt, 6 h; g) AcOH neat, rt, 6 h; h) K2CO3, MeOH, rt, 0.5 h then HF•pyridine, MeCN, 0 °C, 0.5 h; i) TfOMe, dioxane, 0 °C, 10 min then CuI, PhMgBr, THF, 0 °C, 10 min; j) H2, 50% Pd(OH)2/C, MeOH, rt, 6 h.
Scheme 42: Syntheses of methyl (3S,4S) 4,5-di-N-Boc-amino-3-hydroxypentanoate ((3S,4S)-164), methyl (3S,4S)-4-N...
(3R,5S)-5-(Aminomethyl)-3-(4-methoxyphenyl)dihydrofuran-2(3H)-one ((3R,5S)-168) was discovered as a potential medication in Parkinson’s disease [99]. Synthesis of enantiomerically pure (3R,5S)-168 could be accomplished from the aziridine aldehyde (2S,1'R)-6 because the butyrolactone ring formation would proceed with inversion of configuration at C2 in the aziridine ring (Scheme 43) [100]. Thus, condensation of the aldehyde (2S,1'R)-6 with lithium enolate of methyl 4-methoxyphenylacetate would give the β-hydroxyester 169 which when treated with Lewis acid experienced the aziridine ring cleavage with simultaneous dihydrofuran-2-one ring closure to produce 170 contaminated with small amounts of the corresponding furan-2(5H)-one 171. Dehydration of 170 was completed in the presence of acid. Catalytic hydrogenation of the C=C bond in 171 took place preferentially (9:1) from the less hindered side to finally give (3R,5S)-168 as the hydrochloride salt.
![[1860-5397-15-168-i43]](/bjoc/content/inline/1860-5397-15-168-i43.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 43: Syntheses of (3R,5S)-5-(aminomethyl)-3-(4-methoxyphenyl)dihydrofuran-2(3H)-one ((3R,5S)-168). Reagents and conditions: a) 4-MeOC6H4CH2COOMe, LiHMDS, THF, −78 °C, 0.5 h; b) BF3·OEt2, MeCN, reflux, 2 h; c) H2SO4, THF, 70 °C, 48 h; d) H2, Pd(OH)2, Boc2O, EtOH, 1 h; e) HCl/MeOH, rt, 12 h.
Scheme 43: Syntheses of (3R,5S)-5-(aminomethyl)-3-(4-methoxyphenyl)dihydrofuran-2(3H)-one ((3R,5S)-168). Reage...
Substituted imidazolin-2-ones are of interest as potential aminoacyl-tRNA synthase inhibitors. When the aziridine-aldehyde (2R,1'R)-6 was subjected to the reductive amination with 4 dipeptides secondary amines 172 (a R' = iBu, R''= sec-Bu; b R' = R'' = iBu; c R' = sec-Bu, R'' = iBu; d R' = R'' = sec-Bu) were produced (Scheme 44) [101]. In the presence of triphosgene a series of imidazolin-2-ones having a 2-chloromethyl substituent 173 was formed. Removal of the 1-phenylethyl moiety and a subsequent replacement of the chlorine atom by the amino group via azide gave imidazolin-2-one dipeptides 174 which were transformed into 12 derivatives 175–177 after hydrolysis of the corresponding methyl esters. Although docking simulation predicted binding of these compounds to isoleucyl-tRNA synthetase (IleRS) none of them showed inhibitory activity.
![[1860-5397-15-168-i44]](/bjoc/content/inline/1860-5397-15-168-i44.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 44: Syntheses of a series of imidazolin-2-one dipeptides 175–177 (for R' and R'' see text). Reagents and conditions: a) H2NCHR'C(O)NHCHR''COOMe, MgSO4, CH2Cl2, rt, 5 h; b) NaBH3CN, MeOH, rt, overnight; c, triphosgene, NaH, THF, −10 °C, 2 h; d) MsOH, anisole, hexane, reflux, 4 h; e) NaN3, DMF, 80 °C, overnight; f) H2, Pd/C, MeOH, rt, overnight; g) PhNCO, THF, rt, 3 h or phenyl (pyrimidin-2-yl)carbamate, MeCN, reflux, 3 h or PhSO2Cl, TEA, THF, reflux, 1 h; h) NaOH 1N, MeOH/H2O, rt, 1 h.
Scheme 44: Syntheses of a series of imidazolin-2-one dipeptides 175–177 (for R' and R'' see text). Reagents an...
Alkaloids
Pyrrolidines: Pyrrolidine alkaloids and among them polyhydroxypyrrolidines like 1,4-dideoxy-1,4-imino-ʟ-ribitol (2S,3S,4R)-182 were found as components of a variety of plants and exhibit a wide range of biological properties including inhibition of glucosidases [102]. When the aziridine epoxide 162 was treated with acetic acid the aziridine ring cleavage was observed to give the epoxyaldehyde 178 (Scheme 45) [94]. Catalytic hydrogenation followed by saponification converted 178 into (2S,3S)-N-Boc-3-hydroxy-2-hydroxymethylpyrrolidine (2S,3S)-179, one of the simplest members of the iminosugar family.
![[1860-5397-15-168-i45]](/bjoc/content/inline/1860-5397-15-168-i45.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 45: Syntheses of (2S,3S)-N-Boc-3-hydroxy-2-hydroxymethylpyrrolidine ((2S,3S)-179). Reagents and conditions: a) AcOH, CH2Cl2, rt, 6 h; b) H2, 50% Pd(OH)2/C, Boc2O, MeOH, rt, 12 h; c) KOH, EtOH, rt, 0.5 h.
Scheme 45: Syntheses of (2S,3S)-N-Boc-3-hydroxy-2-hydroxymethylpyrrolidine ((2S,3S)-179). Reagents and conditi...
Reaction of aziridine 4-methoxyphenyl esters either (2R,1′S)-5e or (2S,1′S)-5e with vinylene carbonate at 280 °C gave a mixture of four stereoisomers with low (ca. 3:1) diastereoselectivity and in a non-enantioselective manner (Scheme 46) [21]. Undoubtedly, it was the 1,3-dipolar cycloaddition of the azomethine ylide 180 with an electron-rich alkene. Fortunately, cycloadducts (2R,3R,4S)-, (2S,3S,4R)-, (2R,3S,4R)- and (2S,3R,4S)-181 were efficiently separated chromatographically and after reduction and catalytic removal of the chiral auxiliary enantiomerically pure 1,4-dideoxy-1,4-imino-ʟ- and -ᴅ-lyxitols, (2S,3R,4S)-182 and (2R,3S,4R)-182, and 1,4-dideoxy-1,4-imino-ʟ- and -ᴅ-ribitols, (2S,3S,4R)-182 and (2R,3R,4S)-182 were obtained as hydrochloride salts.
![[1860-5397-15-168-i46]](/bjoc/content/inline/1860-5397-15-168-i46.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 46: Syntheses of enantiomers of 1,4-dideoxy-1,4-imino-ʟ- and -ᴅ-lyxitols (2S,3R,4S)-182 and (2R,3S,4R)-182, and 1,4-dideoxy-1,4-imino-ʟ- and -ᴅ-ribitol (2S,3S,4R)-182 and (2R,3R,4S)-182. Reagents and conditions: a) vinylene carbonate, toluene, 280 °C, 0.5 h; b) LiAlH4, THF, rt; c) H2, Pd(OH)2, HCl, MeOH/H2O, rt.
Scheme 46: Syntheses of enantiomers of 1,4-dideoxy-1,4-imino-ʟ- and -ᴅ-lyxitols (2S,3R,4S)-182 and (2R,3S,4R)-...
Since the pyrrolidine (2S,3S,4R)-182 has three stereogenic centers of the same configuration as in (2S,3S,4S)-159 its synthesis started from the common intermediate diol 160 (Scheme 41) with the aziridine ring opening to produce the pyrrolidin-2-one (3S,4S,5S,1'R)-183a (Scheme 47) [34]. The amide bond reduction and N-debenzylation gave 1,4-dideoxy-1,4-imino-ʟ-ribitol (2S,3S,4R)-182 which was isolated as the hydrochloride salt.
![[1860-5397-15-168-i47]](/bjoc/content/inline/1860-5397-15-168-i47.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 47: Synthesis of 1,4-dideoxy-1,4-imino-ʟ-ribitol (2S,3S,4R)-182. Reagents and conditions: a) AcOH, CH2Cl2, reflux, 3 h; b) BH3•SMe2, THF, 0 °C, 10 h; c) H2, Pd(OH)2, MeOH, 50 psi, rt, 12 h; d) HCl, MeOH, 0 °C, 5 h.
Scheme 47: Synthesis of 1,4-dideoxy-1,4-imino-ʟ-ribitol (2S,3S,4R)-182. Reagents and conditions: a) AcOH, CH2Cl...
Syntheses of 1,4-dideoxy-1,4-imino-ᴅ-arabinitol ((2R,3R,4R)-182) and 1,4-dideoxy-1,4-imino-ᴅ-xylitol ((2R,3S,4S)-182) were accomplished starting from the aziridine (E)-acrylate (2S,1'R)-69b readily prepared from the aldehyde (2R,1'R)-6 (Scheme 48) [31]. The (E)-acrylate was subjected to cis-dihydroxylation to give a 1:1 mixture of the diastereoisomeric diols 145a and 145b [103]. Their separation could be achieved after transformation into pyrrolidin-2-ones (3S,4R,5R,1'R)-183b (by crystallization from cold ethanol) and (3R,4S,5R,1'R)-183b (by flash chromatography). After reduction and hydrogenolysis they were converted into 1,4-dideoxy-1,4-imino-ᴅ-arabinitol and ((2R,3R,4R)-182) and 1,4-dideoxy-1,4-imino-ᴅ-xylitol ((2R,3S,4S)-182), respectively. Their enantiomers were prepared from the aziridine aldehyde (2S,1'R)-6 in an analogous manner.
![[1860-5397-15-168-i48]](/bjoc/content/inline/1860-5397-15-168-i48.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 48: Syntheses of 1,4-dideoxy-1,4-imino-ᴅ-arabinitol (2R,3R,4R)-182 and 1,4-dideoxy-1,4-imino-ᴅ-xylitol (2R,3S,4S)-182. Reagents and conditions: a) OsO4, NMO, THF/H2O, rt, 12 h; b) AcOH, CH2Cl2, rt, 18 h, then toluene, 50 °C, 12 h; c) separation of diastereoisomers; d) BH3·SMe2, THF, 0 °C to rt, 12 h, e) H2, Pd(OH)2, MeOH, 3 h.
Scheme 48: Syntheses of 1,4-dideoxy-1,4-imino-ᴅ-arabinitol (2R,3R,4R)-182 and 1,4-dideoxy-1,4-imino-ᴅ-xylitol ...
2,5-Imino-2,5,6-trideoxy-ʟ-gulo-heptitol ((2S,3R,4R,5R)-184) an iminosugar isolated from Hyacintus orientalis was recognized as inhibitor of glycosidases [104]. The efficient syntheses of (2S,3R,4R,5R)-184 and its C4 epimer (2S,3R,4S,5R)-184 were accomplished from the aziridine aldehyde (2R,1'S)-6 (Scheme 49) [105]. The Lewis acid-catalyzed nucleophilic anti-addition of 2-trimethylsilyloxyfuran to (2R,1'S)-6 furnished diastereoisomerically pure secondary alcohol 185 since chelation-controlled transition state was involved. Acid-induced aziridine ring openings and subsequent conjugate additions to the α,β-unsaturated lactone led to the formation of cis-fused [5,5']bicyclic compounds 186a or 186b. Reduction of the lactone moiety in 186a and subsequent deprotection gave (2S,3R,4S,5R)-184. In order to synthesize the natural (2S,3R,4R,5R)-184 the lactone 186b was subjected to Mitsunobu reaction followed by the ester reduction and the hydrogenolytic cleavage of the 1-phenylethyl group.
![[1860-5397-15-168-i49]](/bjoc/content/inline/1860-5397-15-168-i49.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 49: Syntheses of natural 2,5-imino-2,5,6-trideoxy-ʟ-gulo-heptitol ((2S,3R,4R,5R)-184) and its C4 epimer (2S,3R,4S,5R)-184. Reagents and conditions: a) 2-trimethylsilyloxyfuran, ZnBr2, THF, 0 °C, 12 h; b) TFA, THF/H2O, rt, 15 h; c) AcOH, CH2Cl2, rt, 15 h; d) BH3·SMe2, THF, rt, 4 h; e) H2, Pd(OH)2/C, MeOH, rt, 7 h; f) Ph3P, DIAD, 4-O2NC6H4COOH, toluene, 100 °C, 5 h; g) BH3·SMe2, THF, 50 °C, 4 h.
Scheme 49: Syntheses of natural 2,5-imino-2,5,6-trideoxy-ʟ-gulo-heptitol ((2S,3R,4R,5R)-184) and its C4 epimer...
Piperidines: A cis-disubstituted piperidine scaffold was identified in several piperidine alkaloids of diverse biological activities [106]. (−)-Dihydropinidine (2S,6R)-187a, isosolenopsin (2S,6R)-187b, and isosolenopsins A (2S,6R)-187c and B (2S,6R)-187d were synthesized as hydrochloride salts from the aldehyde (2S,1'R)-6 applying a reductive amination as a key step (Scheme 50) [33]. To this end the aldehyde (2S,1'R)-6 was transformed into its higher homolog (2R,1'R)-188 employing Wittig olefination, the C=C bond reduction and Swern oxidation. The required alkyl chains were introduced by Grignard reagents and mixtures of diastereoisomeric alcohols were oxidized to ketones 189a–d. Catalytic hydrogenation allowed for the aziridine ring opening and the removal of the 1-phenylethyl group and also for the reduction of the intermediate cyclic imine which appeared stereospecific.
![[1860-5397-15-168-i50]](/bjoc/content/inline/1860-5397-15-168-i50.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 50: Syntheses of (−)-dihydropinidine ((2S,6R)-187a) (R = C3H7) and (2S,6R)-isosolenopsins (2S,6R)-187b (R = C9H19), (2S,6R)-187c (R = C11H23) and (2S,6R)-187d (R = C15H31). Reagents and conditions: a) Ph3P+(CH2)3OH Brˉ, BuLi, THF, rt, 12 h; b) 2-O2NC6H4SO2NHNH2, Et3N, rt, 12 h; c) Swern oxidation; d) RMgBr, THF, 0 °C to rt, 1 h; e) Dess–Martin periodinane, CH2Cl2, 0 °C to rt, 1 h; f) H2, 20% Pd(OH)2/C, MeOH, rt, 12 h; g) 1 M HCl, MeOH.
Scheme 50: Syntheses of (−)-dihydropinidine ((2S,6R)-187a) (R = C3H7) and (2S,6R)-isosolenopsins (2S,6R)-187b ...
Two other piperidine alkaloids, (+)-deoxocassine ((2S,3S,6R)-190a) and (+)-spectaline ((2S,3S,6R)-190b) were synthesized as hydrochloride salts in a similar way [33]. However, to introduce the hydroxy group at C3 of the piperidine ring addition of a lithium acetylide to the aziridine aldehyde (2S,1'R)-6 was performed (Scheme 51) instead of alkylation (Scheme 50). Two diastereoisomeric acetylenic alcohols were formed in the reaction with lithium ethyl propiolate in an 8:2 ratio, and the major product 191 had the required configuration. After protection of the hydroxy group in 191 Weinreb amide 192 was prepared to facilitate the installation of the respective alkyl chains in ketones 193a and 193b. They were transformed into (+)-deoxocassine ((2S,3S,6R)-190a) and (+)-spectaline ((2S,3S,6R)-190b) as already described.
![[1860-5397-15-168-i51]](/bjoc/content/inline/1860-5397-15-168-i51.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 51: Syntheses of (+)-deoxocassine ((2S,3S,6R)-190a, R = C12H25) and (+)-spectaline ((2S,3S,6R)-190b, R = C12H24C(O)Me). Reagents and conditions: a) HC≡CCOOEt, LiHMDS, THF, −78 °C, 2 h; b) TfOTBDMS, 2,6-lutidine, CH2Cl2, 0 °C, 10 min.; c) MeONHMe, iPrCl, THF, −10 °C, 1 h; d) C12H25MgBr, THF, 0 °C to rt, 1 h; e) HC≡C(CH2)10C(OCH2)2CH3, LiHMDS, THF, −78 °C, 1 h; f) H2, 20% Pd(OH)2/C, MeOH, rt, 12 h; g) 1 M HCl, THF, rt, 24 h.
Scheme 51: Syntheses of (+)-deoxocassine ((2S,3S,6R)-190a, R = C12H25) and (+)-spectaline ((2S,3S,6R)-190b, R ...
(+)-Microgrewiapine A ((2R,3S,6R)-194a) [107] and (+)-microcosamine A ((2R,3S,6R)-194b) [108] have recently been isolated and exhibited interesting biological activities, for example cytotoxicity [107]. The total synthesis of (−)-microgrewiapine A was initiated from the aziridine ketone 195 readily prepared from the ester (2S,1'R)-5b (Scheme 52) [109]. After chelation-controlled reduction of 195 with the NaBH4/ZnCl2 mixture and protection of a secondary alcohol the asymmetric dihydroxylation of the terminal C=C bond with AD-mix-β provided an inseparable 82:18 mixture of diastereoisomers with the diol 196 as a major product. Silylation of the terminal hydroxy group was followed by mesylation of a secondary one to facilitate the piperidine ring closure triggered by hydrogenolytic removal of the chiral auxiliary to form a cis-2,6-disubstituted piperidine framework in (2S,3R,6S)-197. N-Methylation of (2S,3R,6S)-197 was accomplished by reductive amination while a selective deprotection provided the hydroxymethyl group in (2S,3R,6S)-198. Swern oxidation, Julia–Kocienski olefination and desilylation gave (−)-(2S,3R,6S)-194a which appeared to be the enantiomer of natural microgrewiapine A although its structure was identical with the originally proposed. When the total synthesis started from the ester (2R,1'S)-5b and AD-mix-α was used the natural (+)-microgrewiapine A (2R,3S,6R)-194a was obtained. To synthesize the (+)-microcosamine A the protected piperidine (2S,3R,6S)-197 served as a starting material and was first converted into the N-Boc derivative while selective desilylation exposed the hydroxymethyl group to give (2S,3R,6S)-199. Oxidation to the respective aldehyde was accomplished with Dess–Martin periodinane and olefination was carried out as already described. Acidic hydrolysis of N-Boc and O-TBDMS groups afforded (2S,3R,6S)-194b which was identical with the natural (+)-microcosamine A.
![[1860-5397-15-168-i52]](/bjoc/content/inline/1860-5397-15-168-i52.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 52: Synthesis of (−)-microgrewiapine A ((2S,3R,6S)-194a) and (+)-microcosamine A ((2S,3R,6S)-194b). Reagents and conditions: a) MeONHMe, iPrMgCl, THF, −10 °C, 1 h; b) H2C=CHCH2CH2MgBr, THF, 0 °C to rt, 1 h; c) NaBH4, ZnCl2, MeOH, −78 °C, 1 h; d) TfOTBDMS, lutidine, CH2Cl2, 0 °C, 0.5 h; e) AD-mix-β, (DHQD)2PHAL, MeSO2NH2, NaHCO3, t-BuOH/H2O, 0 °C, 4 h; f) TBDMSCl, imidazole, CH2Cl2, rt, 3 h; g) MsCl, TEA, DMAP, rt, 0,5 h; h) H2, 20% Pd(OH)2/C, MeOH, rt, 12 h; i) 37% HCHO, NaBH3CN, AcOH, rt, 1 h; j) HF·pyridine, THF, 0 °C to rt, 12 h; k) Swern oxidation; l) 5-[(2E,4E)-nona-2,4-dien-1-ylsulfonyl]-1-phenyl-1H-tetrazole, KHMDS, 18-crown-6, DME/THF, −78 °C, 2 h and rt overnight; m) TBAF, THF, rt, 1 h; n) Boc2O, NaHCO3, MeOH, 0 °C to rt, 3 h; o) Dess–Martin periodinane, CH2Cl2, 0 °C to rt, 4 h; p) 3 M HCl, MeOH, rt, 12 h.
Scheme 52: Synthesis of (−)-microgrewiapine A ((2S,3R,6S)-194a) and (+)-microcosamine A ((2S,3R,6S)-194b). Rea...
1-Deoxynojirimycin was discovered in several natural species and later found as a potent inhibitor of glycosidases [110]. ʟ-1-Deoxynojirimycin ((2S,3S,4S,5R)-200) and five of its stereoisomers were synthesized in a unique approach using (2S,1'R)-5b as a starting material [88]. When the aziridine ketone (2S,1'R)-201 was treated with a NaBH4/ZnCl2 mixture the aziridine alcohol (2S,1'R,1''R)-202 was stereoselectively formed in a chelation-controlled reduction establishing the absolute configurations at C2 and C3 in three final products (2S,3S,4S,5R)-200, (2S,3S,4S,5S)-200, (2S,3S,4R,5S)-200. A triple bond reduction and a protection of the hydroxy group preceded the aziridine ring opening to provide 203 after basic hydrolysis. Installation of the allyl group at the nitrogen atom required prior derivatization as an oxazolidin-2-one and Birch reduction to furnish 204. To construct a properly functionalized piperidine ring olefin metathesis was performed to supply the key intermediate 205. Taking advantage of the steric bulkiness of the tert-butyldimethylsilyloxy group in 205 three different approaches to stereoselective dihydroxylations of the C=C bond were elaborated. Thus, treatment with Oxone® gave the anti-epoxide 206 which was regioselectively opened in a boron trifluoride-catalyzed isopropylidenation to yield 207. A two-step removal of protecting groups led to the formation of (2S,3S,4S,5R)-2-(hydroxymethyl)piperidine-3,4,5-triol ((2S,3S,4S,5R)-200, ʟ-1-deoxynojirimycin). Direct dihydroxylation of 205 with osmium tetroxide introduced a cis-diol moiety oriented anti to the tert-butyldimethylsilyloxy group and basic hydrolysis of 208 gave (2S,3S,4S,5S)-2-(hydroxymethyl)piperidine-3,4,5-triol ((2S,3S,4S,5S)-200, ʟ-1-deoxymannojirimycin). After removal of the silyl protection from 205 the epoxidation with MCPBA produced the syn-epoxide 209 which was converted into (2S,3S,4R,5S)-2-(hydroxymethyl)piperidine-3,4,5-triol ((2S,3S,4R,5S)-200, ʟ-1-deoxyaltronojirimycin) as described earlier [111].
To synthesize stereoisomeric ʟ-deoxynojirimycins having the R configuration at C3 [(2S,3R,4R,5S)-200 (ʟ-1-deoxyidonojirimycin), (2S,3R,4R,5R)-200 (ʟ-1-deoxygulonojirimycin) and (2S,3R,4S,5R)-200 (ʟ-1-deoxygalactonojirimycin)] the aziridine alcohol (2S,1'S,1''R)-202 prepared by reduction of the ketone (2S,1'R)-201 with ʟ-Selectride® was transformed into the bicyclic intermediate 210, the epimer of 205, as already shown (Scheme 53) [88]. Furthermore, syntheses of six stereoisomers of ᴅ-1-deoxynojirimycins were accomplished starting from the ester (2R,1'R)-5b and proceeding as already shown in Scheme 53 [88].
![[1860-5397-15-168-i53]](/bjoc/content/inline/1860-5397-15-168-i53.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 53: Syntheses of ʟ-1-deoxynojirimycin ((2S,3S,4S,5R)-200), ʟ-1-deoxymannojirimycin ((2S,3S,4S,5S)-200) and ʟ-1-deoxyaltronojirimycin ((2S,3S,4R,5S)-200). Reagents and conditions: a) MeONHMe, iPrMgCl, THF, 0 °C, 1 h; b) MeC≡CMgBr, THF, −78 °C, 1 h and rt, 1 h; c) NaBH4, ZnCl2, MeOH, −78 °C, 1 h; d) LiAlH4, THF, 0 °C, 0.5 h and reflux, 8 h; e) TBDMSCl, TEA, DMAP, CH2Cl2, rt, 17 h; f) AcOH, CH2Cl2, rt, 15 h; g) KOH, EtOH, rt, 0.5 h; h) CDI, DBU, CH2Cl2, 0 °C, 1 h and rt, 24 h; i) Na, NH3 liquid, THF, −78 °C, 0.5 h; j) H2CCH=CH2I, NaH, ClCH2CH2Cl, reflux, 10 h; k) Grubbs catalyst (1st generation), CH2Cl2, rt, 24 h; l) Oxone®, NaHCO3, Me2CO/H2O, rt, 0.5 h; m) Me2CO, BF3·OEt2, CH2Cl2, 0 °C, 2 h; n) LiOH, EtOH/H2O, reflux, 4 h; o) HCl, MeOH, reflux, 4 h, then Amberlite IRA-410 OH¯; p) OsO4, NMO, MeCN/H2O, 0 °C to rt, 3 h; r) MCPBA, NaHPO4, CH2Cl2, 0 °C to rt, 72 h.
Scheme 53: Syntheses of ʟ-1-deoxynojirimycin ((2S,3S,4S,5R)-200), ʟ-1-deoxymannojirimycin ((2S,3S,4S,5S)-200) ...
Enantioselective syntheses of 1-deoxy-ᴅ-galacto-homonojirimycin ((2R,3S,4R,5S)-211) as well as pyrrolizidine alkaloids were completed from the cis-acrylate (2S,1'S)-156a (Scheme 54) [34]. Dihydroxylation of Weinreb amide (2S,1'S)-212 gave the diol 213 with excellent diastereoselectivity (>99:1) which already had the correct configurations at C3 and C4 of the final product (3S,4R). After silylation of the hydroxy groups the protected three-carbon moiety was attached by the enantiospecific addition of the Grignard reagent to the respective aldehyde to give the aziridine alcohol (S)-214. Sequential treatment of (S)-214 with mesyl chloride and cesium acetate followed by catalytic hydrogenation furnished substituted piperidines 215. Hydrolysis of the dioxolane acetal in 215 in the presence of a reducing agent provided 1-deoxy-ᴅ-galacto-homonojirimycin ((2R,3S,4R,5S)-211). Undoubtedly after mesylation of the alcohol (S)-214 the intermediary bicyclic aziridinium ion (R)-216 was formed which can equilibrate with a 2-chloromethylpyrrolidine (R)-217. For steric reasons in the presence of cesium acetate ring expansion in (R)-216 took place to produce 215.
![[1860-5397-15-168-i54]](/bjoc/content/inline/1860-5397-15-168-i54.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 54: Syntheses of 1-deoxy-ᴅ-galacto-homonojirimycin (2R,3S,4R,5S)-211. Reagents and conditions: a) MeONHMe, iPrMgCl, THF, 0 °C, 15 min; b) OsO4, NMO, acetone, 0 °C to rt, 6 h; c) TfOTBDMS, 2,6-lutidine, CH2Cl2, −78 °C to rt, 2 h; d) DIBAL-H, hexane, −40 °C, 8.5 h; e) 2-(2-bromoethyl)-1,3-dioxolane, Mg, THF, rt, 0.5 h; f) MsCl, TEA, DMAP, CH2Cl2, 0 °C to reflux, 2 h; g) AcOCs, DMF, 100 °C, 3 h; h) H2, 20% Pd/C, Boc2O, MeOH, rt, 12 h; i) Et3SiH, TFA/CH2Cl2/H2O, rt, 12 h; j) HCl, MeOH, reflux, 2 h then Amberlite 410 Cl.
Scheme 54: Syntheses of 1-deoxy-ᴅ-galacto-homonojirimycin (2R,3S,4R,5S)-211. Reagents and conditions: a) MeONH...
Pyrrolizidines: Pyrrolizidine alkaloids [112], e.g., (+)-hyacinthacine A1 [113] were found in many plants and they are primarily recognized as inhibitors of glycosidases [113,114]. To synthesize a hyacinthacine A1 framework the aziridine ketone 218 was stereoselectively reduced to the alcohol (R)-214 which accommodated three stereogenic centers (1S,2R,3R) of the final pyrrolizidine [34]. When (R)-214 was first reacted with mesyl chloride then with cesium acetate and finally was subjected to catalytic hydrogenation the pyrrolidine (2S,3S,4R,5R)-219 was obtained. The formation of the pyrrolizidine skeleton was accomplished as already shown (Scheme 55) to produce (+)-7a-epi-hyacinthacine A1 ((1S,2R,3R,7aS)-220). As for the alcohol (S)-214 (Scheme 54) mesylation of the alcohol (R)-214 gave the bicyclic aziridinium ion (S)-216 which for steric reasons was transformed into a stable chloromethylpyrrolidine (S)-217. In the presence of cesium acetate the chloride for acetate displacement occurred to yield the pyrrolidine (2S,3S,4R,5R)-219.
![[1860-5397-15-168-i55]](/bjoc/content/inline/1860-5397-15-168-i55.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 55: Syntheses of 7a-epi-hyacinthacine A1 (1S,2R,3R,7aS)-220. Reagents and conditions: a) TfOTBDMS, 2,6-lutidine, CH2Cl2, −78 °C to rt, 2 h; b) 2-(2-bromoethyl)-1,3-dioxolane, Mg, THF, rt, 1 h; c) ʟ-Selectride®, CH2Cl2, −10 °C, 1.5 h; d) MsCl, TEA, DMAP, CH2Cl2, 0 °C to reflux, 2 h; e) AcOCs, DMF, 100 °C, 3 h; f) H2, 20% Pd/C, Boc2O, MeOH, rt, 12 h; g) Et3SiH, TFA/CH2Cl2/H2O, rt, 12 h; h) HCl, MeOH, reflux, 2 h then Amberlite 410 Cl.
Scheme 55: Syntheses of 7a-epi-hyacinthacine A1 (1S,2R,3R,7aS)-220. Reagents and conditions: a) TfOTBDMS, 2,6-...
The enantiospecific synthesis of 8-deoxyhyacinthacine A1 ((1S,2R,3R,7aR)-221) started from the aziridine ketone 218 (Scheme 56) which already had the correct configurations at C1, C2 and C3 of the final product (1S,2R,3R) [34]. Catalytic hydrogenation of 218 in the presence of strong acids combined with treatment with triethylsilane and final acidic hydrolysis gave 8-deoxyhyacinthacine A1 ((1S,2R,3R,7aR)-221) since even in the presence of acetic acid the Me–C3 group was formed by the reductive opening of the aziridine ring and the first reductive amination appeared to be stereospecific.
![[1860-5397-15-168-i56]](/bjoc/content/inline/1860-5397-15-168-i56.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 56: Syntheses of 8-deoxyhyacinthacine A1 ((1S,2R,3R,7aR)-221). Reagents and conditions: a) H2, Pd/C, PTSA, AcOH, rt, 12 h then TFA, rt, 2 h; b) Et3SiH, TFA/CH2Cl2/H2O, rt, 12 h; c) HCl, MeOH, reflux, 2 h then Amberlite 410 Cl; d) TFA/H2O, rt, 48 h; e) polymethylhydrosiloxane (PMHS), Pd/C, MeOH, reflux, 1 h.
Scheme 56: Syntheses of 8-deoxyhyacinthacine A1 ((1S,2R,3R,7aR)-221). Reagents and conditions: a) H2, Pd/C, PT...
Indolizidines: Indolizidine alkaloids represent another large group of natural products of manifold biological activities [115]. For example, swainsonine is known as mannosidase inhibitor and it also has a potential in chemotherapy [116]. Castanospermine inhibits several glucosidases and also shows anticancer and antiviral activities [117,118]. (+)-Lentiginosine revealed a potent and selective amyloglucosidase and Hsp90 inhibitory activity [119]. Several synthetic strategies to polyhydroxyindolizidines have been elaborated including procedures employing 2-substituted (1-phenylethyl)aziridines. Thus, in the presence of a strong acid the aziridine ketone 226 was transformed into the piperidine 222 because water attacked C2 in the intermediate bicyclic aziridinium ion 223 from the less hindered side (Scheme 56) [34]. N-Debenzylation caused a five-membered ring closure which occurred in a stereospecific manner to form (R)-3-hydroxy-1-deoxy-8-epi-castanospermine ((3R,6S,7R,8S,8aR)-224).
(3R)-3-Methyl-8-deoxyswainsonine ((1S,2R,3R,8aR)-225) was obtained from the aziridine ketone 226 following strategy applied in synthesis of (1S,2R,3R,7aR)-221 (Scheme 56) [34].
To construct a five-membered ring of (+)-lentiginosine ((1S,2S,8aS)-227) the aziridine aldehyde (2S,1'R)-6 was subjected to HEW olefination followed by asymmetric Sharpless dihydroxylation to give the major (11:1) ester 228 which was transformed into the pyrrolidin-2-one (3R,4S,5S,1'R)-183a in two standard steps (Scheme 57) [32]. Before installation of a three-carbon moiety hydroxy groups in (3R,4S,5S,1'R)-183a were protected, the acetate hydrolyzed, an amide carbonyl reduced and the hydroxymethyl residue oxidized to provide the pyrrolidine aldehyde 229. After Wittig reaction and hydrogenation, the 4-hydroxybutyl chain was installed in the protected pyrrolidine (2S,3S,4S)-230. The piperidine ring closure was carried out with an Appel reaction while treatment with acids afforded (1S,2S,8aS)-octahydroindolizine-1,2-diol (1S,2S,8aS)-227.
![[1860-5397-15-168-i57]](/bjoc/content/inline/1860-5397-15-168-i57.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 57: Syntheses of (+)-lentiginosine ((1S,2S,8aS)-227). Reagents and conditions: a) (EtO)2P(O)CH2COOEt, LiHMDS, THF, rt, 1 h; b) AD-mix-α, MeSO2NH2, t-BuOH/H2O, 0 °C, 24 h; c) AcOH, CH2Cl2, rt, 12 h, then toluene, 90 °C, 12 h; d) TfOTBDMS, 2,6-lutidine, CH2Cl2, 0 °C, 1 h; e) KOH, EtOH, rt, 1 h; f) BH3·SMe2, THF, 0 °C to 70 °C, 3 h; g) Swern oxidation; h) Ph3P+(CH2)3OBn Brˉ, t-BuOK, THF, 0 °C to rt, 1 h; i) H2, Pd(OH)2, CF3COOH, MeOH, rt, 24 h; j) CBr4, Ph3P, TEA, CH2Cl2, 0 °C to rt, 6 h; k) HCl, MeCN, rt, 4 h.
Scheme 57: Syntheses of (+)-lentiginosine ((1S,2S,8aS)-227). Reagents and conditions: a) (EtO)2P(O)CH2COOEt, L...
The elegant approach to 8-epi-swainsonine ((1S,2R,8S,8aR)-231) started from cis-acrylate (2S,1'R)-156a readily separable from a 4:1 cis:trans mixture prepared in Wittig reaction from the aldehyde (2R,1'R)-6 (Scheme 58) [120]. Sharpless asymmetric dihydroxylation of (2S,1'R)-156a provided a 10:1 mixture of diols with the diastereoisomer 232 predominating. Opening of the aziridine ring with acetic acid caused cyclization to the substituted pyrrolidin-2-one (3S,4S,5R,1'R)-183a. Protection of the diol moiety, ester hydrolysis and Dess–Martin oxidation afforded the aldehyde 233 which was used as a starting material in asymmetric Brown allylation to form a 4:1 mixture of diastereoisomers and the major one 234 was separated chromatographically. Silylation preceded treatment with borane to execute reduction of the carbonyl group and to form a primary alcohol and was followed by the removal of the chiral auxiliary to give 235. A six-membered ring closure in the alcohol 235 was carried out by Appel reaction to produce 8-epi-swainsonine ((1S,2R,8S,8aR)-231) after acidic deprotection.
![[1860-5397-15-168-i58]](/bjoc/content/inline/1860-5397-15-168-i58.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 58: Syntheses of 8-epi-swainsonine (1S,2R,8S,8aR)-231. Reagents and conditions: a) Ph3P=CHCOOMe, MeOH, 0 °C, 1 h; b) AD-mix-β, MeSO2NH2, t-BuOH/H2O, rt, 24 h; c) AcOH, CH2Cl2, rt, 12 h, then toluene, 90 °C, 12 h; d) Me2C(OMe)2, PTSA, CH2Cl2, rt, 1 h; e) KOH, MeOH, rt, 1 h; f) Dess–Martin periodinane, CH2Cl2, rt, 2 h; g) H2C=CHCH2MgBr, (−)-Ipc2BOMe, CH2Cl2/ether, −78 °C to 0 °C, 3 h; h) TfOTBDMS, 2,6-lutidine, CH2Cl2, 0 °C to rt, 2 h; i) BH3·SMe2, THF, 0 °C to rt, 3 h then H2O2, NaOH, MeOH, 0 °C; j) H2, Pd(OH)2, MeOH, rt, 6 h; k) CBr4, Ph3P, CH2Cl2, NEt3, rt, 3 h; l) TFA, CH2Cl2, 40 °C, 4 h.
Scheme 58: Syntheses of 8-epi-swainsonine (1S,2R,8S,8aR)-231. Reagents and conditions: a) Ph3P=CHCOOMe, MeOH, ...
Several syntheses of (−)-swainsonine ((1S,2R,8R,8aR)-231) have been described including an approach relying on the cis-dihydroxylation of the intermediate 236 which was prepared from the vinylpiperidine (2S,3R)-237 (Scheme 59) [121]. Synthesis of (2S,3R)-237 started with a stereoselective reduction (>99:1) of the aziridine ketone (2S,1'R)-238 to give a protected aziridine diol (2S,1'R,1''R)-239 [122]. After the aziridine ring opening with acetic acid and selective desilylation to form (2S,3R,1'R)-240 the piperidine ring closure was achieved by mesylation producing (2S,3R,1'R)-241. The final steps to the key intermediate (2S,3R)-237 included removal of the chiral auxiliary, deacetylation, Boc protection, Swern oxidation of the hydroxymethyl group and methylenation with Wittig reagent.
![[1860-5397-15-168-i59]](/bjoc/content/inline/1860-5397-15-168-i59.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 59: Synthesis of a protected vinylpiperidine (2S,3R)-237, a key intermediate in the synthesis of (−)-swainsonine ((1S,2R,8R,8aR)-231). Reagents and conditions: a) Mg, Br(CH2)3OTBDMS, THF, reflux, 8 h; b) NaBH4, ZnCl2, MeOH, −78 °C, 1.5 h; c) TBDMSCl, DMAP, CH2Cl2, 0 °C to rt, 12 h; d) AcOH, CH2Cl2, rt, overnight; e) AcOH, THF/H2O, rt, 48 h; f) MsCl, TEA, CH2Cl2, 0 °C to rt, 24 h; g) H2, Pd(OH)2, Boc2O, MeOH, 100 psi, rt, 5 h; h) KOH, MeOH, rt, 0.5 h; i) Swern oxidation; j) Ph3PMe+Br¯, LiHMDS, THF, 0 °C to rt, 1 h.
Scheme 59: Synthesis of a protected vinylpiperidine (2S,3R)-237, a key intermediate in the synthesis of (−)-sw...
Miscellaneous
FDA-approved meropenem and R-82301 belong to a class of carbapenem antibiotics containing a substituted exocyclic 3-mercaptopyrrolidine scaffold. In search for new drugs this fragment was replaced by a 5-methyl-4-mercaptopyrrolidin-2-one moiety which was synthesized from aziridine esters 5 [123]. Thus, a five-carbon framework was assembled from (2S,1′S)-5b and ethyl acetate followed by reduction of an intermediary ketone which produced a diastereoisomeric pair of aziridine alcohols readily separable chromatographically. Hydrogenation of, e.g., the alcohol 242 led to the formation of the pyrrolidine-2-one (4R,5S)-243 (Scheme 60). The hydroxy for mercapto replacement was achieved in Mitsunobu reaction to produce (4S,5S)-244 after deacetylation. Three other enantiomers were prepared accordingly. Modified carbapenems, e.g., 245, were obtained by coupling of the thiol (4S,5S)-244 with the respective enolphosphate and some of them appeared as active as meropenem and R-82301 towards selected bacterial strains.
![[1860-5397-15-168-i60]](/bjoc/content/inline/1860-5397-15-168-i60.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 60: Synthesis of a modified carbapenem 245. Reagents and conditions: a) AcOEt, LiHMDS, THF, −78 °C, 1.5 h; b) NaBH4, NH4Cl, EtOH/H2O, rt, 1 h; c) H2, Pd(OH)2, EtOH, rt, 4.1 bar, 12 h; d) AcSH, Ph3P, DEAD, THF/DMF, rt, 2 h; e) NaOH, MeOH, 0 °C, 3 min; f) 4-nitrobenzyl (1R,5S,6S)-2-diphenylphosphoryloxy-6-[(R)-1-hydroxyethyl]-1-methyl-1-carbapen-2-em-3-carboxylate, DIPEA, MeCN, 0–5 °C, 2 h; g) H2, Pd/C, THF/EtOH/Mops buffer, rt, 15 h.
Scheme 60: Synthesis of a modified carbapenem 245. Reagents and conditions: a) AcOEt, LiHMDS, THF, −78 °C, 1.5...
Conclusion
The current body of literature on N-(1-phenylethyl)aziridine-2-carboxylate esters and their derivatives revealed their successful applications in syntheses of over a dozen of approved drugs and potential medications as well as over 40 important natural products mostly alkaloids including ephedrine and related compounds, polyhydroxy pyrrolidines, piperidines, pyrrolizidines and indolizidines but also various sphingoids and ceramides and their 1- and 3-deoxy analogues and several hydroxy amino acids and their precursors. Designed strategies provided new procedures to several drugs and alternative approaches to natural products. This was possible because three-carbon aziridine chirons 5–7 are configurationally stable, in majority of cases the openings of the aziridine ring took place at the less substituted C3 atom, either alkoxycarbonyl or hydroxymethyl but especially formyl functionalities could be further elaborated in a highly stereoselective manner while the cleavage of the chiral auxiliary could be easily accomplished with three different reagents depending on a particular stage of multi-step sequences.
Since a vicinal amino alcohol unit and various amino(dihydroxy)propyl and (diamino)hydroxypropyl combinations are crucial for biologically active compounds (2R)- or (2S)-aziridine-2-carboxylates, -carbaldehydes and -methanols appeared perfectly suited for introducing the required stereochemistry into a C*–N moiety of the target compound without fear of racemization. The next stereogenic center was best created by reductions of 2-acyl-N-(1-phenylethyl)aziridines which proceeded in almost diastereospecific manner when a NaBH4/ZnCl2 mixture or ʟ-Selectride® were used to give alcohols of opposite configurations. To install at C2 the 1,2-dihydroxyalkyl substituent of required configurations the cis-dihydroxylation of E- or Z-olefins obtained in Wittig or HWE reactions appeared a method of choice. Although Michael reaction on the respective E- or Z-acylates could introduce various nucleophiles (C, N, O), the stereoselectivity of additions in most cases was not satisfactory. Summing up, a majority of strategies relied on specific functionalization at the C2 site of starting aziridines.
Expansions of a three-carbon framework of aziridines 5–8 were also carried out starting from regioselective openings at less substituted C3 with appropriate nucleophiles combined with further functionalizations but were less common and no stereogenic center was generated. Furthermore, hydrogenolytic cleavage of the aziridine ring always occurred at C3 and allowed for the one-step introduction of the terminal methyl group. Amines also reacted preferentially at C3 leading to formation of 1,2-diamino- or 1,2-diamino-3-propyl fragments of designed stereochemistry extremely useful in the synthesis of new sphingoid and ceramide analogues for biological studies. Although hydroxylations also took place at C3, they were carried out as a two-step process (acetylation and saponification).
Openings of the aziridine ring at C2 in 2-substituted N-(1-phenylethyl)aziridines were observed in a few cases and were limited to a Lewis acid-catalyzed five-membered ring closure and the aziridine ring opening sequence as well as to reactions at C2–COOEt.
Regardless of regioselectivity the aziridine ring opening in N-(1-phenylethyl)aziridines required prior activation to form respective aziridinium ions. Besides protic (AcOH, HF) and Lewis acids (aluminum chloride, boron trifluoride etherate), chlorotrimethylsilane–amine pairs, iodotrimethylsilane, phosgene and triphosgene were applied to mention frequently used. However, when bicyclic aziridinium ions are formed their further reactivity appeared to be under steric and/or electronic control and followed rules recently described [124,125].
On a few occasions only epimers of naturally occurring compounds were obtained without any comments on modification of the synthetic strategy to reach the target molecule. With a collection of reliable transformations of 2-substituted N-(1-phenylethyl)aziridines in hands new applications in enantioselective syntheses of medications and natural products will appear soon and we hope this review will stimulate further research in this area.
References
-
Hanessian, S. Total synthesis of natural products: The ”chiron” approach; Pergamon Press: Elmsford, NY, 1983.
Return to citation in text: [1] -
Dibello, E.; Gamenara, D.; Seoane, G. Org. Prep. Proced. Int. 2015, 47, 415–442. doi:10.1080/00304948.2015.1088753
Return to citation in text: [1] -
Jurczak, J.; Pikul, S.; Bauer, T. Tetrahedron 1986, 42, 447–488. doi:10.1016/s0040-4020(01)87445-7
Return to citation in text: [1] -
Schmidt, C. R.; Bryant, J. D. Org. Synth. 1995, 72, 6–13. doi:10.15227/orgsyn.072.0006
Return to citation in text: [1] -
Sugiyama, T.; Sugawara, H.; Watanabe, M.; Yamashita, K. Agric. Biol. Chem. 1984, 48, 1841–1844. doi:10.1080/00021369.1984.10866392
Return to citation in text: [1] -
Grauert, M.; Schöllkopf, U. Liebigs Ann. Chem. 1985, 1817–1824. doi:10.1002/jlac.198519850909
Return to citation in text: [1] -
Michel, P.; Ley, S. V. Synthesis 2003, 1598–1602. doi:10.1055/s-2003-40519
Return to citation in text: [1] -
Garner, P. Tetrahedron Lett. 1984, 25, 5855–5858. doi:10.1016/s0040-4039(01)81703-2
Return to citation in text: [1] -
Passiniemi, M.; Koskinen, A. M. P. Beilstein J. Org. Chem. 2013, 9, 2641–2659. doi:10.3762/bjoc.9.300
Return to citation in text: [1] -
Reetz, M. T. Chem. Rev. 1999, 99, 1121–1162. doi:10.1021/cr980417b
Return to citation in text: [1] -
Roos, G. Key Chiral Auxiliary Applications, 2nd ed.; Academic Press: Amsterdam, Netherlands, 2014. doi:10.1016/c2011-1-05800-9
Return to citation in text: [1] -
Yudin, A. K., Ed. Aziridines and Epoxides in Organic Synthesis; Wiley-VCH: Weinheim, Germany, 2006. doi:10.1002/3527607862
Return to citation in text: [1] -
Häner, R.; Olano, B.; Seebach, D. Helv. Chim. Acta 1987, 70, 1676–1693. doi:10.1002/hlca.19870700704
Return to citation in text: [1] [2] [3] -
Ha, H.-J.; Jung, J.-H.; Lee, W. K. Asian J. Org. Chem. 2014, 3, 1020–1035. doi:10.1002/ajoc.201402098
Return to citation in text: [1] -
Akhtar, R.; Naqvi, S. A. R.; Zahoor, A. F.; Saleem, S. Mol. Diversity 2018, 22, 447–501. doi:10.1007/s11030-018-9829-0
Return to citation in text: [1] -
Harada, K.; Nakamura, I. J. Chem. Soc., Chem. Commun. 1978, 522–523. doi:10.1039/c39780000522
Return to citation in text: [1] [2] [3] [4] [5] -
Farooa, S.; Swain, W. E., Jr.; Daeppen, R.; Rihs, G. Tetrahedron: Asymmetry 1992, 3, 51–63. doi:10.1016/s0957-4166(00)82312-5
Return to citation in text: [1] [2] [3] -
Bosies, E.; Berger, H.; Kampe, W.; Bicker, U.; Grafe, A. N-Substituted aziridine-2-carboxylic acid derivatives and their immune-stimulation compositions and methods. U.S. Pat. Appl. US4410532A, Oct 18, 1983.
Return to citation in text: [1] [2] -
Morán-Ramallal, R.; Liz, R.; Gotor, V. Org. Lett. 2008, 10, 1935–1938. doi:10.1021/ol800443p
Return to citation in text: [1] [2] -
Alezra, V.; Bonin, M.; Micouin, L.; Policar, C.; Husson, H.-P. Eur. J. Org. Chem. 2001, 2589–2594. doi:10.1002/1099-0690(200107)2001:13<2589::aid-ejoc2589>3.0.co;2-y
Return to citation in text: [1] -
Takano, S.; Moriya, M.; Ogasawara, K. Tetrahedron: Asymmetry 1992, 3, 681–684. doi:10.1016/s0957-4166(00)80499-1
Return to citation in text: [1] [2] -
Lee, W. K.; Park, C. S.; Lim, Y. H.; Ha, H.-J. Optically active aziridine-2-carboxylate derivatives and process for preparing them. PCT Int. Pat. Appl. WO2002012186A1, Feb 14, 2002.
Return to citation in text: [1] [2] -
Ha, H.-J.; Suh, J.-M.; Kang, K.-H.; Ahn, Y.-G.; Han, O. Tetrahedron 1998, 54, 851–858. doi:10.1016/s0040-4020(97)10358-1
Return to citation in text: [1] -
Tranchant, M.-J.; Dalla, V.; Jabin, I.; Decroix, B. Tetrahedron 2002, 58, 8425–8432. doi:10.1016/s0040-4020(02)01031-1
Return to citation in text: [1] -
Bew, S. P.; Hughes, D. L.; Savic, V.; Soapi, K. M.; Wilson, M. A. Chem. Commun. 2006, 3513–3515. doi:10.1039/b606033a
Return to citation in text: [1] -
Park, J.-H.; Ha, H.-J.; Lee, W. K.; Généreux-Vincent, T.; Kazlauskas, R. J. ChemBioChem 2009, 10, 2213–2222. doi:10.1002/cbic.200900343
Return to citation in text: [1] -
Alezra, V.; Bouchet, C.; Micouin, L.; Bonin, M.; Husson, H.-P. Tetrahedron Lett. 2000, 41, 655–658. doi:10.1016/s0040-4039(99)02157-7
Return to citation in text: [1] -
Mancuso, A. J.; Huang, S.-L.; Swern, D. J. Org. Chem. 1978, 43, 2480–2482. doi:10.1021/jo00406a041
Return to citation in text: [1] -
Hwang, G.-I.; Chung, J.-H.; Lee, W. K. J. Org. Chem. 1996, 61, 6183–6188. doi:10.1021/jo9603183
Return to citation in text: [1] [2] [3] -
Kim, B. C.; Lee, W. K. Tetrahedron 1996, 52, 12117–12124. doi:10.1016/0040-4020(96)00703-x
Return to citation in text: [1] [2] -
Yoon, H.; Sim, T. Synthesis 2013, 45, 3276–3280. doi:10.1055/s-0033-1338545
Return to citation in text: [1] [2] [3] -
Yoon, H.; Cho, K. S.; Sim, T. Tetrahedron: Asymmetry 2014, 25, 497–502. doi:10.1016/j.tetasy.2014.02.009
Return to citation in text: [1] [2] [3] -
Yadav, N. N.; Choi, J.; Ha, H.-J. Org. Biomol. Chem. 2016, 14, 6426–6434. doi:10.1039/c6ob00806b
Return to citation in text: [1] [2] [3] -
Eum, H.; Choi, J.; Cho, C.-G.; Ha, H.-J. Asian J. Org. Chem. 2015, 4, 1399–1409. doi:10.1002/ajoc.201500285
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] -
Lim, Y.; Lee, W. K. Tetrahedron Lett. 1995, 36, 8431–8434. doi:10.1016/0040-4039(95)01814-x
Return to citation in text: [1] -
Sugiyama, S.; Fukuchi, H.; Ishii, K. Tetrahedron 2007, 63, 12047–12057. doi:10.1016/j.tet.2007.09.004
Return to citation in text: [1] -
Wróblewski, A. E.; Drozd, J. Tetrahedron: Asymmetry 2007, 18, 1134–1141. doi:10.1016/j.tetasy.2007.04.021
Return to citation in text: [1] -
Sugiyama, S.; Inoue, T.; Ishii, K. Tetrahedron: Asymmetry 2003, 14, 2153–2160. doi:10.1016/s0957-4166(03)00410-5
Return to citation in text: [1] -
Kim, Y.; Kang, L.-S.; Ha, H.-J.; Ko, S. W.; Lee, W. K. Heterocycles 2007, 71, 2243–2248. doi:10.1016/s0385-5414(07)81100-2
Return to citation in text: [1] -
Anderson, G. P. Life Sci. 1993, 52, 2145–2160. doi:10.1016/0024-3205(93)90729-m
Return to citation in text: [1] -
Laxman Prabhu, G. G.; Prajapati, H.; Chaturvedi, A.; Dave, N.; Jain, A. B. Drugs Ther. Perspect. 2019, 35, 181–184. doi:10.1007/s40267-019-00611-1
Return to citation in text: [1] -
Hett, R.; Fang, Q. K.; Gao, Y.; Wald, S. A.; Senanayake, C. H. Org. Process Res. Dev. 1998, 2, 96–99. doi:10.1021/op970116o
Return to citation in text: [1] -
Minoru, O.; Koichi, Y.; Kiyoshi, T. Process for producing optically active benzene-sulfonamide derivatives. Eur. Pat. Appl. EP 0257787 A1, July 21, 1987.
Return to citation in text: [1] -
Garbaccio, R. M.; Olson, C. M.; Tasber, E. S.; Torrent, M. Mitotic kinesin inhibitors. PCT Int. Appl. WO2005/092011 A2, Oct 6, 2005.
Return to citation in text: [1] -
Han, S.-M.; Ma, S.-h.; Ha, H.-J.; Lee, W. K. Tetrahedron 2008, 64, 11110–11114. doi:10.1016/j.tet.2008.09.068
Return to citation in text: [1] -
Hwang, G. I.; Chung, J.-H.; Lee, W. K. Tetrahedron 1996, 52, 12111–12116. doi:10.1016/0040-4020(96)00702-8
Return to citation in text: [1] -
Yun, J. M.; Sim, T. B.; Hahm, H. S.; Lee, W. K.; Ha, H.-J. J. Org. Chem. 2003, 68, 7675–7680. doi:10.1021/jo034755a
Return to citation in text: [1] [2] [3] [4] -
Kim, Y.; Ha, H.-J.; Yun, S. Y.; Lee, W. K. Chem. Commun. 2008, 4363–4365. doi:10.1039/b809124b
Return to citation in text: [1] -
Choi, J.; Ha, H.-J. J. Korean Chem. Soc. 2015, 59, 203–204. doi:10.5012/jkcs.2015.59.3.203
Return to citation in text: [1] -
Pyun, D. K.; Lee, C. H.; Ha, H.-J.; Park, C. S.; Chang, J.-W.; Lee, W. K. Org. Lett. 2001, 3, 4197–4199. doi:10.1021/ol016861+
Return to citation in text: [1] -
Park, C. S.; Kim, M. S.; Sim, T. B.; Pyun, D. K.; Lee, C. H.; Choi, D.; Lee, W. K.; Chang, J.-W.; Ha, H.-J. J. Org. Chem. 2003, 68, 43–49. doi:10.1021/jo025545l
Return to citation in text: [1] -
Kim, J.-W.; Kim, Y.-W.; Inagaki, Y.; Hwang, Y.-A.; Mitsutake, S.; Ryu, Y.-W.; Lee, W. K.; Ha, H.-J.; Park, C.-S.; Igarashi, Y. Bioorg. Med. Chem. 2005, 13, 3475–3485. doi:10.1016/j.bmc.2005.02.053
Return to citation in text: [1] [2] [3] [4] -
Kim, J.; Park, S.; Lee, H.-J.; Park, C.; Kim, Y.-W.; Lee, W.-K.; Ha, H.-J. Sphingolipid derivatives and the composition for anti-cancer containing the same. PCT Int. Appl. WO2006/004359 A1, Jan 12, 2006.
Return to citation in text: [1] [2] [3] [4] -
Dowling, P. M. Chloramphenicol, Thiamphenicol, and Florfenicol. In Antimicrobial Therapy in Veterinary Medicine, 5th ed.; Giguère, S.; Prescott, J. F.; Dowling, P. M., Eds.; Wiley, 2013; pp 269–277. doi:10.1002/9781118675014.ch16
Return to citation in text: [1] -
Peng, Y.; Tian, W.; Ye, Q.; Fang, Z. Florfenicol synthesizing method. U.S. Patent US2016/0318859 A1, Nov 3, 2016.
Return to citation in text: [1] -
Ishigami, K.; Katsuta, R.; Shibata, C.; Hayakawa, Y.; Watanabe, H.; Kitahara, T. Tetrahedron 2009, 65, 3629–3638. doi:10.1016/j.tet.2009.03.003
Return to citation in text: [1] [2] [3] -
Yoon, D.-H.; Kang, P.; Lee, W. K.; Kim, Y.; Ha, H.-J. Org. Lett. 2012, 14, 429–431. doi:10.1021/ol202683k
Return to citation in text: [1] -
Yoon, D.-H.; Ji, M.-K.; Ha, H.-J.; Park, J.; Kang, P.; Lee, W. K. Bull. Korean Chem. Soc. 2013, 34, 1899–1902. doi:10.5012/bkcs.2013.34.6.1899
Return to citation in text: [1] -
Milen, M.; Abranyi-Balogh, P.; Keglevich, G. Curr. Org. Synth. 2014, 11, 889–901. doi:10.2174/1570179411666140818210247
Return to citation in text: [1] -
Yoon, D.-H.; Ha, H.-J.; Kim, B. C.; Lee, W. K. Tetrahedron Lett. 2010, 51, 2181–2183. doi:10.1016/j.tetlet.2010.02.087
Return to citation in text: [1] [2] -
Lee, B.-K.; Sung, B.-J.; Lee, W.-K.; Yoon, D.-H.; Ha, H.-J. Bull. Korean Chem. Soc. 2009, 30, 3123–3126. doi:10.5012/bkcs.2009.30.12.3123
Return to citation in text: [1] -
Lee, J.; Lee, J. E.; Ha, H.-J.; Son, S. I.; Lee, W. K. Tetrahedron Lett. 2015, 56, 856–858. doi:10.1016/j.tetlet.2014.12.133
Return to citation in text: [1] -
Kocieński, P. J. Protecting groups, 3rd ed.; Georg Thieme Verlag: Stuttgart, Germany, 2005.
Return to citation in text: [1] -
Murphy, S. T.; Case, H. L.; Ellsworth, E.; Hagen, S.; Huband, M.; Joannides, T.; Limberakis, C.; Marotti, K. R.; Ottolini, A. M.; Rauckhorst, M.; Starr, J.; Stier, M.; Taylor, C.; Zhu, T.; Blaser, A.; Denny, W. A.; Lu, G.-L.; Smaill, J. B.; Rivault, F. Bioorg. Med. Chem. Lett. 2007, 17, 2150–2155. doi:10.1016/j.bmcl.2007.01.090
Return to citation in text: [1] -
Jung, J.-H.; Kim, S.; Eum, H.; Lee, W. K.; Ha, H.-J. Tetrahedron 2017, 73, 5993–5999. doi:10.1016/j.tet.2017.08.043
Return to citation in text: [1] -
Inokuchi, J.; Radin, N. S. J. Lipid Res. 1987, 28, 565–571. doi:10.1007/bf03026693
Return to citation in text: [1] -
Liu, J.; Beckman, B. S.; Foroozesh, M. Future Med. Chem. 2013, 5, 1405–1421. doi:10.4155/fmc.13.107
Return to citation in text: [1] -
Shin, S.-H.; Han, E. Y.; Park, C. S.; Lee, W. K.; Ha, H.-J. Tetrahedron: Asymmetry 2000, 11, 3293–3301. doi:10.1016/s0957-4166(00)00319-0
Return to citation in text: [1] -
Leblond, B.; Beausoleil, E.; Taverne, T.; Donello, J. E. 3-Heteroaryl-3-hydroxy-2-aminopropylamines and related compounds having analgesic and/or immunostimulant activity. U.S. Patent US8013000 B2, Sept 6, 2011.
Return to citation in text: [1] -
Donello, J. E.; Schweighoffer, F. J.; Luhrs, L. M. Methods for treating cognitive disorders using 1-benzyl-1-hydroxy-2,3-diamino-propylamines, 3-benzyl-3-hydroxy-2-amino-propionic acid amides and related compounds. PCT Int. Appl. WO2008/109287 A1, Sept 12, 2008.
Return to citation in text: [1] -
Donello, J. E.; Schweighoffer, F. J.; Leblond, B. Method for treating chronic pain using 1-benzyl-1-hydroxy-2,3-diamino-propylamines, 3-benzyl-3-hydroxy-2-amino-propionic acid amides and related compounds. PCT Int. Appl. WO2008/011487 A2, Jan 24, 2008.
Return to citation in text: [1] -
Leblond, B.; Beusoleil, E.; Taverne, T.; Donello, J. E. 3-Heteroaryl-3-hydroxy-2-amino-propylamines and related compounds having analgesic and/or immunostimulant activity. PCT Int. Appl. WO2006/081276 A1, Aug 3, 2006.
Return to citation in text: [1] -
Kim, K.; Kim, Y.-L.; Sacket, S. J.; Kim, H.-L.; Han, M.; Park, D. S.; Lee, B. K.; Lee, W. K.; Ha, H.-J.; Im, D.-S. J. Pharm. Pharmacol. 2007, 59, 1035–1041. doi:10.1211/jpp.59.7.0017
Return to citation in text: [1] -
Sachs, C. W.; Safa, A. R.; Harrison, S. D.; Fine, R. L. J. Biol. Chem. 1995, 270, 26639–26648. doi:10.1074/jbc.270.44.26639
Return to citation in text: [1] -
Pruett, S. T.; Bushnev, A.; Hagedorn, K.; Adiga, M.; Haynes, C. A.; Sullards, M. C.; Liotta, D. C.; Merrill, A. H., Jr. J. Lipid Res. 2008, 49, 1621–1639. doi:10.1194/jlr.r800012-jlr200
Return to citation in text: [1] -
Singh, A.; Ha, H.-J.; Park, J.; Kim, J. H.; Lee, W. K. Bioorg. Med. Chem. 2011, 19, 6174–6181. doi:10.1016/j.bmc.2011.09.022
Return to citation in text: [1] [2] -
Choi, S.-K.; Lee, W.-K. Heterocycles 1998, 48, 1917–1921. doi:10.3987/com-98-8129
Return to citation in text: [1] -
Choi, S.-K.; Lee, J.-S.; Kim, J.-H.; Lee, W. K. J. Org. Chem. 1997, 62, 743–745. doi:10.1021/jo961168z
Return to citation in text: [1] [2] -
Gao, Y.; He, X.; Ding, F.; Zhang, Y. Synthesis 2016, 48, 4017–4037. doi:10.1055/s-0036-1588311
Return to citation in text: [1] -
Yoon, H. J.; Kim, Y.-W.; Lee, B. K.; Lee, W. K.; Kim, Y.; Ha, H.-J. Chem. Commun. 2007, 79–81. doi:10.1039/b612740a
Return to citation in text: [1] -
Chang, J.-W.; Bae, J. H.; Shin, S.-H.; Park, C. S.; Choi, D.; Lee, W. K. Tetrahedron Lett. 1998, 39, 9193–9196. doi:10.1016/s0040-4039(98)02095-4
Return to citation in text: [1] -
Lee, W. K.; Yoo, H.-J.; Ha, H.-J. Process for preparing alpha-amino acids and their derivatives including phenylalanine and homophenylalanine and their intermediates. PCT Int. Appl. WO2002/012184 A1, Feb 14, 2002.
Return to citation in text: [1] -
Carlsen, P. H. J.; Katsuki, T.; Martin, V. S.; Sharpless, K. B. J. Org. Chem. 1981, 46, 3936–3938. doi:10.1021/jo00332a045
Return to citation in text: [1] -
Chang, J.-W.; Ha, H.-J.; Park, C. S.; Kim, M. S.; Lee, W. K. Heterocycles 2002, 57, 1143–1148. doi:10.3987/com-02-9489
Return to citation in text: [1] -
Kim, Y.; Ha, H.-J.; Han, K.; Ko, S. W.; Yun, H.; Yoon, H. J.; Kim, M. S.; Lee, W. K. Tetrahedron Lett. 2005, 46, 4407–4409. doi:10.1016/j.tetlet.2005.04.039
Return to citation in text: [1] -
Jeong, H.; Yadav, N. N.; Ha, H.-J. Synthesis 2017, 49, 1264–1272. doi:10.1055/s-0036-1588093
Return to citation in text: [1] -
Jung, J.-H.; Yoon, D.-H.; Lee, K.; Shin, H.; Lee, W. K.; Yook, C.-M.; Ha, H.-J. Org. Biomol. Chem. 2015, 13, 8187–8195. doi:10.1039/c5ob00375j
Return to citation in text: [1] -
Singh, A.; Kim, B.; Lee, W. K.; Ha, H.-J. Org. Biomol. Chem. 2011, 9, 1372–1380. doi:10.1039/c0ob00730g
Return to citation in text: [1] [2] [3] [4] -
Faulds, D.; Goa, K. L.; Benfield, P. Drugs 1993, 45, 953–1040. doi:10.2165/00003495-199345060-00007
Return to citation in text: [1] -
Kim, Y.; Yoon, D.-H.; Ha, H.-J.; Kang, K. Y.; Lee, W. K. Tetrahedron Lett. 2011, 52, 5918–5920. doi:10.1016/j.tetlet.2011.08.048
Return to citation in text: [1] -
Park, C. S.; Choi, H. G.; Lee, H.; Lee, W. K.; Ha, H.-J. Tetrahedron: Asymmetry 2000, 11, 3283–3292. doi:10.1016/s0957-4166(00)00311-6
Return to citation in text: [1] -
Kim, Y.; Ha, H.-J.; Yun, H.; Lee, B. K.; Lee, W. K. Tetrahedron 2006, 62, 8844–8849. doi:10.1016/j.tet.2006.06.025
Return to citation in text: [1] -
Fagerholm, A. E.; Habrant, D.; Koskinen, A. M. P. Mar. Drugs 2010, 8, 122–172. doi:10.3390/md80100122
Return to citation in text: [1] -
Mao, H.; Jeong, H.; Yang, J.; Ha, H.-J.; Yang, J. W. Chem. – Eur. J. 2018, 24, 2370–2374. doi:10.1002/chem.201706161
Return to citation in text: [1] [2] -
Oh, D.-C.; Strangman, W. K.; Kauffman, C. A.; Jensen, P. R.; Fenical, W. Org. Lett. 2007, 9, 1525–1528. doi:10.1021/ol070294u
Return to citation in text: [1] -
Baars, O.; Zhang, X.; Gibson, M. I.; Stone, A. T.; Morel, F. M. M.; Seyedsayamdost, M. R. Angew. Chem., Int. Ed. 2018, 57, 536–541. doi:10.1002/anie.201709720
Return to citation in text: [1] -
Czajgucki, Z.; Sowiński, P.; Andruszkiewicz, R. Amino Acids 2003, 24, 289–291. doi:10.1007/s00726-002-0409-2
Return to citation in text: [1] -
Huang, P.-Q.; Geng, H.; Tian, Y.-S.; Peng, Q.-R.; Xiao, K.-J. Sci. China: Chem. 2015, 58, 478–482. doi:10.1007/s11426-014-5270-0
Return to citation in text: [1] -
Erdem, S. S.; Özpınar, G. A.; Boz, Ü. J. Enzyme Inhib. Med. Chem. 2014, 29, 81–86. doi:10.3109/14756366.2012.753882
Return to citation in text: [1] -
Kim, S.; Lee, W. K.; Ha, H.-J. Synthesis 2019, 51, 885–888. doi:10.1055/s-0037-1610667
Return to citation in text: [1] -
Eum, H.-S.; Lee, Y.-N.; Kim, S.-M.; Baek, A.-Y.; Son, M.-K.; Lee, K.-W.; Ko, S.-W.; Kim, S.-H.; Yun, S.-Y.; Lee, W.-K.; Ha, H.-J. Bull. Korean Chem. Soc. 2010, 31, 611–614. doi:10.5012/bkcs.2010.31.03.611
Return to citation in text: [1] -
Dhudmal, C. N.; Biradar, D. O.; Satyanarayana, M. V.; Reddy, B. V. S. Nat. Prod. Commun. 2018, 13, 1011–1012. doi:10.1177/1934578x1801300821
Return to citation in text: [1] -
Choi, H. G.; Park, D.-S.; Lee, W. K.; Sim, T. Tetrahedron Lett. 2013, 54, 5775–5777. doi:10.1016/j.tetlet.2013.08.040
Return to citation in text: [1] -
Asano, N.; Kato, A.; Miyauchi, M.; Kizu, H.; Kameda, Y.; Watson, A. A.; Nash, R. J.; Fleet, G. W. J. J. Nat. Prod. 1998, 61, 625–628. doi:10.1021/np9705726
Return to citation in text: [1] -
Lee, H.; Kim, J. H.; Lee, W. K.; Cho, J.; Nam, W.; Lee, J.; Ha, H.-J. Org. Biomol. Chem. 2013, 11, 3629–3634. doi:10.1039/c3ob27390c
Return to citation in text: [1] -
Jones, T. H.; Blum, M. S.; Fales, H. M. Tetrahedron 1982, 38, 1949–1958. doi:10.1016/0040-4020(82)80044-6
Return to citation in text: [1] -
Still, P. C.; Yi, B.; González-Cestari, T. F.; Pan, L.; Pavlovicz, R. E.; Chai, H.-B.; Ninh, T. N.; Li, C.; Soejarto, D. D.; McKay, D. B.; Kinghorn, A. D. J. Nat. Prod. 2013, 76, 243–249. doi:10.1021/np3007414
Return to citation in text: [1] [2] -
Feng, S.-X.; Lin, L.-D.; Xu, H.-H.; Wei, X.-Y. J. Asian Nat. Prod. Res. 2008, 10, 1155–1158. doi:10.1080/10286020802361289
Return to citation in text: [1] -
Macha, L.; Ha, H.-J. J. Org. Chem. 2019, 84, 94–103. doi:10.1021/acs.joc.8b02342
Return to citation in text: [1] -
Gao, K.; Zheng, C.; Wang, T.; Zhao, H.; Wang, J.; Wang, Z.; Zhai, X.; Jia, Z.; Chen, J.; Zhou, Y.; Wang, W. Molecules 2016, 21, 1600. doi:10.3390/molecules21111600
Return to citation in text: [1] -
Asano, K.; Hakogi, T.; Iwama, S.; Katsumura, S. Chem. Commun. 1999, 41–42. doi:10.1039/a807532h
Return to citation in text: [1] -
Moreira, R.; Pereira, D. M.; Valentão, P.; Andrade, P. B. Int. J. Mol. Sci. 2018, 19, 1668. doi:10.3390/ijms19061668
Return to citation in text: [1] -
Ritthiwigrom, T.; Au, C. W. G.; Pyne, S. G. Curr. Org. Synth. 2012, 9, 583–612. doi:10.2174/157017912803251765
Return to citation in text: [1] [2] -
Asano, N.; Kuroi, H.; Ikeda, K.; Kizu, H.; Kameda, Y.; Kato, A.; Adachi, I.; Watson, A. A.; Nash, R. J.; Fleet, G. W. J. Tetrahedron: Asymmetry 2000, 11, 1–8. doi:10.1016/s0957-4166(99)00508-x
Return to citation in text: [1] -
Michael, J. P. Simple Indolizidine and Quinolizidine Alkaloids. The Alkaloids: Chemistry and Biology; Elsevier: Amsterdam, Netherlands, 2016; Vol. 75, pp 1–498. doi:10.1016/bs.alkal.2014.12.001
Return to citation in text: [1] -
Li, Z.; Huang, Y.; Dong, F.; Li, W.; Ding, L.; Yu, G.; Xu, D.; Yang, Y.; Xu, X.; Tong, D. J. Biosci. (New Delhi, India) 2012, 37 (Suppl. 1), 1005–1016. doi:10.1007/s12038-012-9265-8
Return to citation in text: [1] -
Humphries, M. J.; Matsumoto, K.; White, S. L.; Olden, K. Cancer Res. 1986, 46, 5215–5222.
Return to citation in text: [1] -
Whitby, K.; Pierson, T. C.; Geiss, B.; Lane, K.; Engle, M.; Zhou, Y.; Doms, R. W.; Diamond, M. S. J. Virol. 2005, 79, 8698–8706. doi:10.1128/jvi.79.14.8698-8706.2005
Return to citation in text: [1] -
Cardona, F.; Goti, A.; Brandi, A. Eur. J. Org. Chem. 2007, 1551–1565. doi:10.1002/ejoc.200600633
Return to citation in text: [1] -
Lee, B. K.; Choi, H. G.; Roh, E. J.; Lee, W. K.; Sim, T. Tetrahedron Lett. 2013, 54, 553–556. doi:10.1016/j.tetlet.2012.11.087
Return to citation in text: [1] -
Bates, R. W.; Dewey, M. R. Org. Lett. 2009, 11, 3706–3708. doi:10.1021/ol901094h
Return to citation in text: [1] -
Choi, H. G.; Kwon, J. H.; Kim, J. C.; Lee, W. K.; Eum, H.; Ha, H.-J. Tetrahedron Lett. 2010, 51, 3284–3285. doi:10.1016/j.tetlet.2010.04.069
Return to citation in text: [1] -
Pyun, D. K.; Kim, B. J.; Jung, H. J.; Kim, J. H.; Lee, J. S.; Lee, W. K.; Lee, C. H. Chem. Pharm. Bull. 2002, 50, 415–418. doi:10.1248/cpb.50.415
Return to citation in text: [1] -
Boydas, E. B.; Tanriver, G.; D'hooghe, M.; Ha, H.-J.; Van Speybroeck, V.; Catak, S. Org. Biomol. Chem. 2018, 16, 796–806. doi:10.1039/c7ob02253k
Return to citation in text: [1] -
Ji, M.-K.; Hertsen, D.; Yoon, D.-H.; Eum, H.; Goossens, H.; Waroquier, M.; Van Speybroeck, V.; D'hooghe, M.; De Kimpe, N.; Ha, H.-J. Chem. – Asian J. 2014, 9, 1060–1067. doi:10.1002/asia.201301551
Return to citation in text: [1]
| 76. | Singh, A.; Ha, H.-J.; Park, J.; Kim, J. H.; Lee, W. K. Bioorg. Med. Chem. 2011, 19, 6174–6181. doi:10.1016/j.bmc.2011.09.022 |
| 77. | Choi, S.-K.; Lee, W.-K. Heterocycles 1998, 48, 1917–1921. doi:10.3987/com-98-8129 |
| 74. | Sachs, C. W.; Safa, A. R.; Harrison, S. D.; Fine, R. L. J. Biol. Chem. 1995, 270, 26639–26648. doi:10.1074/jbc.270.44.26639 |
| 75. | Pruett, S. T.; Bushnev, A.; Hagedorn, K.; Adiga, M.; Haynes, C. A.; Sullards, M. C.; Liotta, D. C.; Merrill, A. H., Jr. J. Lipid Res. 2008, 49, 1621–1639. doi:10.1194/jlr.r800012-jlr200 |
| 47. | Yun, J. M.; Sim, T. B.; Hahm, H. S.; Lee, W. K.; Ha, H.-J. J. Org. Chem. 2003, 68, 7675–7680. doi:10.1021/jo034755a |
| 73. | Kim, K.; Kim, Y.-L.; Sacket, S. J.; Kim, H.-L.; Han, M.; Park, D. S.; Lee, B. K.; Lee, W. K.; Ha, H.-J.; Im, D.-S. J. Pharm. Pharmacol. 2007, 59, 1035–1041. doi:10.1211/jpp.59.7.0017 |
| 79. | Gao, Y.; He, X.; Ding, F.; Zhang, Y. Synthesis 2016, 48, 4017–4037. doi:10.1055/s-0036-1588311 |
| 78. | Choi, S.-K.; Lee, J.-S.; Kim, J.-H.; Lee, W. K. J. Org. Chem. 1997, 62, 743–745. doi:10.1021/jo961168z |
| 19. | Morán-Ramallal, R.; Liz, R.; Gotor, V. Org. Lett. 2008, 10, 1935–1938. doi:10.1021/ol800443p |
| 78. | Choi, S.-K.; Lee, J.-S.; Kim, J.-H.; Lee, W. K. J. Org. Chem. 1997, 62, 743–745. doi:10.1021/jo961168z |
| 30. | Kim, B. C.; Lee, W. K. Tetrahedron 1996, 52, 12117–12124. doi:10.1016/0040-4020(96)00703-x |
| 84. | Chang, J.-W.; Ha, H.-J.; Park, C. S.; Kim, M. S.; Lee, W. K. Heterocycles 2002, 57, 1143–1148. doi:10.3987/com-02-9489 |
| 101. | Eum, H.-S.; Lee, Y.-N.; Kim, S.-M.; Baek, A.-Y.; Son, M.-K.; Lee, K.-W.; Ko, S.-W.; Kim, S.-H.; Yun, S.-Y.; Lee, W.-K.; Ha, H.-J. Bull. Korean Chem. Soc. 2010, 31, 611–614. doi:10.5012/bkcs.2010.31.03.611 |
| 85. | Kim, Y.; Ha, H.-J.; Han, K.; Ko, S. W.; Yun, H.; Yoon, H. J.; Kim, M. S.; Lee, W. K. Tetrahedron Lett. 2005, 46, 4407–4409. doi:10.1016/j.tetlet.2005.04.039 |
| 100. | Kim, S.; Lee, W. K.; Ha, H.-J. Synthesis 2019, 51, 885–888. doi:10.1055/s-0037-1610667 |
| 83. | Carlsen, P. H. J.; Katsuki, T.; Martin, V. S.; Sharpless, K. B. J. Org. Chem. 1981, 46, 3936–3938. doi:10.1021/jo00332a045 |
| 94. | Mao, H.; Jeong, H.; Yang, J.; Ha, H.-J.; Yang, J. W. Chem. – Eur. J. 2018, 24, 2370–2374. doi:10.1002/chem.201706161 |
| 76. | Singh, A.; Ha, H.-J.; Park, J.; Kim, J. H.; Lee, W. K. Bioorg. Med. Chem. 2011, 19, 6174–6181. doi:10.1016/j.bmc.2011.09.022 |
| 102. | Dhudmal, C. N.; Biradar, D. O.; Satyanarayana, M. V.; Reddy, B. V. S. Nat. Prod. Commun. 2018, 13, 1011–1012. doi:10.1177/1934578x1801300821 |
| 80. | Yoon, H. J.; Kim, Y.-W.; Lee, B. K.; Lee, W. K.; Kim, Y.; Ha, H.-J. Chem. Commun. 2007, 79–81. doi:10.1039/b612740a |
| 97. | Czajgucki, Z.; Sowiński, P.; Andruszkiewicz, R. Amino Acids 2003, 24, 289–291. doi:10.1007/s00726-002-0409-2 |
| 81. | Chang, J.-W.; Bae, J. H.; Shin, S.-H.; Park, C. S.; Choi, D.; Lee, W. K. Tetrahedron Lett. 1998, 39, 9193–9196. doi:10.1016/s0040-4039(98)02095-4 |
| 82. | Lee, W. K.; Yoo, H.-J.; Ha, H.-J. Process for preparing alpha-amino acids and their derivatives including phenylalanine and homophenylalanine and their intermediates. PCT Int. Appl. WO2002/012184 A1, Feb 14, 2002. |
| 96. | Baars, O.; Zhang, X.; Gibson, M. I.; Stone, A. T.; Morel, F. M. M.; Seyedsayamdost, M. R. Angew. Chem., Int. Ed. 2018, 57, 536–541. doi:10.1002/anie.201709720 |
| 99. | Erdem, S. S.; Özpınar, G. A.; Boz, Ü. J. Enzyme Inhib. Med. Chem. 2014, 29, 81–86. doi:10.3109/14756366.2012.753882 |
| 98. | Huang, P.-Q.; Geng, H.; Tian, Y.-S.; Peng, Q.-R.; Xiao, K.-J. Sci. China: Chem. 2015, 58, 478–482. doi:10.1007/s11426-014-5270-0 |
| 95. | Oh, D.-C.; Strangman, W. K.; Kauffman, C. A.; Jensen, P. R.; Fenical, W. Org. Lett. 2007, 9, 1525–1528. doi:10.1021/ol070294u |
| 86. | Jeong, H.; Yadav, N. N.; Ha, H.-J. Synthesis 2017, 49, 1264–1272. doi:10.1055/s-0036-1588093 |
| 87. | Jung, J.-H.; Yoon, D.-H.; Lee, K.; Shin, H.; Lee, W. K.; Yook, C.-M.; Ha, H.-J. Org. Biomol. Chem. 2015, 13, 8187–8195. doi:10.1039/c5ob00375j |
| 17. | Farooa, S.; Swain, W. E., Jr.; Daeppen, R.; Rihs, G. Tetrahedron: Asymmetry 1992, 3, 51–63. doi:10.1016/s0957-4166(00)82312-5 |
| 17. | Farooa, S.; Swain, W. E., Jr.; Daeppen, R.; Rihs, G. Tetrahedron: Asymmetry 1992, 3, 51–63. doi:10.1016/s0957-4166(00)82312-5 |
| 21. | Takano, S.; Moriya, M.; Ogasawara, K. Tetrahedron: Asymmetry 1992, 3, 681–684. doi:10.1016/s0957-4166(00)80499-1 |
| 56. | Ishigami, K.; Katsuta, R.; Shibata, C.; Hayakawa, Y.; Watanabe, H.; Kitahara, T. Tetrahedron 2009, 65, 3629–3638. doi:10.1016/j.tet.2009.03.003 |
| 33. | Yadav, N. N.; Choi, J.; Ha, H.-J. Org. Biomol. Chem. 2016, 14, 6426–6434. doi:10.1039/c6ob00806b |
| 56. | Ishigami, K.; Katsuta, R.; Shibata, C.; Hayakawa, Y.; Watanabe, H.; Kitahara, T. Tetrahedron 2009, 65, 3629–3638. doi:10.1016/j.tet.2009.03.003 |
| 33. | Yadav, N. N.; Choi, J.; Ha, H.-J. Org. Biomol. Chem. 2016, 14, 6426–6434. doi:10.1039/c6ob00806b |
| 108. | Feng, S.-X.; Lin, L.-D.; Xu, H.-H.; Wei, X.-Y. J. Asian Nat. Prod. Res. 2008, 10, 1155–1158. doi:10.1080/10286020802361289 |
| 58. | Yoon, D.-H.; Ji, M.-K.; Ha, H.-J.; Park, J.; Kang, P.; Lee, W. K. Bull. Korean Chem. Soc. 2013, 34, 1899–1902. doi:10.5012/bkcs.2013.34.6.1899 |
| 107. | Still, P. C.; Yi, B.; González-Cestari, T. F.; Pan, L.; Pavlovicz, R. E.; Chai, H.-B.; Ninh, T. N.; Li, C.; Soejarto, D. D.; McKay, D. B.; Kinghorn, A. D. J. Nat. Prod. 2013, 76, 243–249. doi:10.1021/np3007414 |
| 1. | Hanessian, S. Total synthesis of natural products: The ”chiron” approach; Pergamon Press: Elmsford, NY, 1983. |
| 104. | Asano, N.; Kato, A.; Miyauchi, M.; Kizu, H.; Kameda, Y.; Watson, A. A.; Nash, R. J.; Fleet, G. W. J. J. Nat. Prod. 1998, 61, 625–628. doi:10.1021/np9705726 |
| 103. | Choi, H. G.; Park, D.-S.; Lee, W. K.; Sim, T. Tetrahedron Lett. 2013, 54, 5775–5777. doi:10.1016/j.tetlet.2013.08.040 |
| 106. | Jones, T. H.; Blum, M. S.; Fales, H. M. Tetrahedron 1982, 38, 1949–1958. doi:10.1016/0040-4020(82)80044-6 |
| 105. | Lee, H.; Kim, J. H.; Lee, W. K.; Cho, J.; Nam, W.; Lee, J.; Ha, H.-J. Org. Biomol. Chem. 2013, 11, 3629–3634. doi:10.1039/c3ob27390c |
| 5. | Sugiyama, T.; Sugawara, H.; Watanabe, M.; Yamashita, K. Agric. Biol. Chem. 1984, 48, 1841–1844. doi:10.1080/00021369.1984.10866392 |
| 6. | Grauert, M.; Schöllkopf, U. Liebigs Ann. Chem. 1985, 1817–1824. doi:10.1002/jlac.198519850909 |
| 3. | Jurczak, J.; Pikul, S.; Bauer, T. Tetrahedron 1986, 42, 447–488. doi:10.1016/s0040-4020(01)87445-7 |
| 4. | Schmidt, C. R.; Bryant, J. D. Org. Synth. 1995, 72, 6–13. doi:10.15227/orgsyn.072.0006 |
| 63. | Kocieński, P. J. Protecting groups, 3rd ed.; Georg Thieme Verlag: Stuttgart, Germany, 2005. |
| 2. | Dibello, E.; Gamenara, D.; Seoane, G. Org. Prep. Proced. Int. 2015, 47, 415–442. doi:10.1080/00304948.2015.1088753 |
| 64. | Murphy, S. T.; Case, H. L.; Ellsworth, E.; Hagen, S.; Huband, M.; Joannides, T.; Limberakis, C.; Marotti, K. R.; Ottolini, A. M.; Rauckhorst, M.; Starr, J.; Stier, M.; Taylor, C.; Zhu, T.; Blaser, A.; Denny, W. A.; Lu, G.-L.; Smaill, J. B.; Rivault, F. Bioorg. Med. Chem. Lett. 2007, 17, 2150–2155. doi:10.1016/j.bmcl.2007.01.090 |
| 34. | Eum, H.; Choi, J.; Cho, C.-G.; Ha, H.-J. Asian J. Org. Chem. 2015, 4, 1399–1409. doi:10.1002/ajoc.201500285 |
| 12. | Yudin, A. K., Ed. Aziridines and Epoxides in Organic Synthesis; Wiley-VCH: Weinheim, Germany, 2006. doi:10.1002/3527607862 |
| 61. | Lee, B.-K.; Sung, B.-J.; Lee, W.-K.; Yoon, D.-H.; Ha, H.-J. Bull. Korean Chem. Soc. 2009, 30, 3123–3126. doi:10.5012/bkcs.2009.30.12.3123 |
| 11. | Roos, G. Key Chiral Auxiliary Applications, 2nd ed.; Academic Press: Amsterdam, Netherlands, 2014. doi:10.1016/c2011-1-05800-9 |
| 62. | Lee, J.; Lee, J. E.; Ha, H.-J.; Son, S. I.; Lee, W. K. Tetrahedron Lett. 2015, 56, 856–858. doi:10.1016/j.tetlet.2014.12.133 |
| 59. | Milen, M.; Abranyi-Balogh, P.; Keglevich, G. Curr. Org. Synth. 2014, 11, 889–901. doi:10.2174/1570179411666140818210247 |
| 8. | Garner, P. Tetrahedron Lett. 1984, 25, 5855–5858. doi:10.1016/s0040-4039(01)81703-2 |
| 9. | Passiniemi, M.; Koskinen, A. M. P. Beilstein J. Org. Chem. 2013, 9, 2641–2659. doi:10.3762/bjoc.9.300 |
| 60. | Yoon, D.-H.; Ha, H.-J.; Kim, B. C.; Lee, W. K. Tetrahedron Lett. 2010, 51, 2181–2183. doi:10.1016/j.tetlet.2010.02.087 |
| 66. | Inokuchi, J.; Radin, N. S. J. Lipid Res. 1987, 28, 565–571. doi:10.1007/bf03026693 |
| 67. | Liu, J.; Beckman, B. S.; Foroozesh, M. Future Med. Chem. 2013, 5, 1405–1421. doi:10.4155/fmc.13.107 |
| 112. | Moreira, R.; Pereira, D. M.; Valentão, P.; Andrade, P. B. Int. J. Mol. Sci. 2018, 19, 1668. doi:10.3390/ijms19061668 |
| 68. | Shin, S.-H.; Han, E. Y.; Park, C. S.; Lee, W. K.; Ha, H.-J. Tetrahedron: Asymmetry 2000, 11, 3293–3301. doi:10.1016/s0957-4166(00)00319-0 |
| 69. | Leblond, B.; Beausoleil, E.; Taverne, T.; Donello, J. E. 3-Heteroaryl-3-hydroxy-2-aminopropylamines and related compounds having analgesic and/or immunostimulant activity. U.S. Patent US8013000 B2, Sept 6, 2011. |
| 34. | Eum, H.; Choi, J.; Cho, C.-G.; Ha, H.-J. Asian J. Org. Chem. 2015, 4, 1399–1409. doi:10.1002/ajoc.201500285 |
| 65. | Jung, J.-H.; Kim, S.; Eum, H.; Lee, W. K.; Ha, H.-J. Tetrahedron 2017, 73, 5993–5999. doi:10.1016/j.tet.2017.08.043 |
| 53. | Kim, J.; Park, S.; Lee, H.-J.; Park, C.; Kim, Y.-W.; Lee, W.-K.; Ha, H.-J. Sphingolipid derivatives and the composition for anti-cancer containing the same. PCT Int. Appl. WO2006/004359 A1, Jan 12, 2006. |
| 113. | Ritthiwigrom, T.; Au, C. W. G.; Pyne, S. G. Curr. Org. Synth. 2012, 9, 583–612. doi:10.2174/157017912803251765 |
| 111. | Asano, K.; Hakogi, T.; Iwama, S.; Katsumura, S. Chem. Commun. 1999, 41–42. doi:10.1039/a807532h |
| 88. | Singh, A.; Kim, B.; Lee, W. K.; Ha, H.-J. Org. Biomol. Chem. 2011, 9, 1372–1380. doi:10.1039/c0ob00730g |
| 88. | Singh, A.; Kim, B.; Lee, W. K.; Ha, H.-J. Org. Biomol. Chem. 2011, 9, 1372–1380. doi:10.1039/c0ob00730g |
| 88. | Singh, A.; Kim, B.; Lee, W. K.; Ha, H.-J. Org. Biomol. Chem. 2011, 9, 1372–1380. doi:10.1039/c0ob00730g |
| 107. | Still, P. C.; Yi, B.; González-Cestari, T. F.; Pan, L.; Pavlovicz, R. E.; Chai, H.-B.; Ninh, T. N.; Li, C.; Soejarto, D. D.; McKay, D. B.; Kinghorn, A. D. J. Nat. Prod. 2013, 76, 243–249. doi:10.1021/np3007414 |
| 52. | Kim, J.-W.; Kim, Y.-W.; Inagaki, Y.; Hwang, Y.-A.; Mitsutake, S.; Ryu, Y.-W.; Lee, W. K.; Ha, H.-J.; Park, C.-S.; Igarashi, Y. Bioorg. Med. Chem. 2005, 13, 3475–3485. doi:10.1016/j.bmc.2005.02.053 |
| 53. | Kim, J.; Park, S.; Lee, H.-J.; Park, C.; Kim, Y.-W.; Lee, W.-K.; Ha, H.-J. Sphingolipid derivatives and the composition for anti-cancer containing the same. PCT Int. Appl. WO2006/004359 A1, Jan 12, 2006. |
| 110. | Gao, K.; Zheng, C.; Wang, T.; Zhao, H.; Wang, J.; Wang, Z.; Zhai, X.; Jia, Z.; Chen, J.; Zhou, Y.; Wang, W. Molecules 2016, 21, 1600. doi:10.3390/molecules21111600 |
| 52. | Kim, J.-W.; Kim, Y.-W.; Inagaki, Y.; Hwang, Y.-A.; Mitsutake, S.; Ryu, Y.-W.; Lee, W. K.; Ha, H.-J.; Park, C.-S.; Igarashi, Y. Bioorg. Med. Chem. 2005, 13, 3475–3485. doi:10.1016/j.bmc.2005.02.053 |
| 53. | Kim, J.; Park, S.; Lee, H.-J.; Park, C.; Kim, Y.-W.; Lee, W.-K.; Ha, H.-J. Sphingolipid derivatives and the composition for anti-cancer containing the same. PCT Int. Appl. WO2006/004359 A1, Jan 12, 2006. |
| 109. | Macha, L.; Ha, H.-J. J. Org. Chem. 2019, 84, 94–103. doi:10.1021/acs.joc.8b02342 |
| 71. | Donello, J. E.; Schweighoffer, F. J.; Leblond, B. Method for treating chronic pain using 1-benzyl-1-hydroxy-2,3-diamino-propylamines, 3-benzyl-3-hydroxy-2-amino-propionic acid amides and related compounds. PCT Int. Appl. WO2008/011487 A2, Jan 24, 2008. |
| 72. | Leblond, B.; Beusoleil, E.; Taverne, T.; Donello, J. E. 3-Heteroaryl-3-hydroxy-2-amino-propylamines and related compounds having analgesic and/or immunostimulant activity. PCT Int. Appl. WO2006/081276 A1, Aug 3, 2006. |
| 29. | Hwang, G.-I.; Chung, J.-H.; Lee, W. K. J. Org. Chem. 1996, 61, 6183–6188. doi:10.1021/jo9603183 |
| 70. | Donello, J. E.; Schweighoffer, F. J.; Luhrs, L. M. Methods for treating cognitive disorders using 1-benzyl-1-hydroxy-2,3-diamino-propylamines, 3-benzyl-3-hydroxy-2-amino-propionic acid amides and related compounds. PCT Int. Appl. WO2008/109287 A1, Sept 12, 2008. |
| 18. | Bosies, E.; Berger, H.; Kampe, W.; Bicker, U.; Grafe, A. N-Substituted aziridine-2-carboxylic acid derivatives and their immune-stimulation compositions and methods. U.S. Pat. Appl. US4410532A, Oct 18, 1983. |
| 39. | Kim, Y.; Kang, L.-S.; Ha, H.-J.; Ko, S. W.; Lee, W. K. Heterocycles 2007, 71, 2243–2248. doi:10.1016/s0385-5414(07)81100-2 |
| 32. | Yoon, H.; Cho, K. S.; Sim, T. Tetrahedron: Asymmetry 2014, 25, 497–502. doi:10.1016/j.tetasy.2014.02.009 |
| 40. | Anderson, G. P. Life Sci. 1993, 52, 2145–2160. doi:10.1016/0024-3205(93)90729-m |
| 41. | Laxman Prabhu, G. G.; Prajapati, H.; Chaturvedi, A.; Dave, N.; Jain, A. B. Drugs Ther. Perspect. 2019, 35, 181–184. doi:10.1007/s40267-019-00611-1 |
| 34. | Eum, H.; Choi, J.; Cho, C.-G.; Ha, H.-J. Asian J. Org. Chem. 2015, 4, 1399–1409. doi:10.1002/ajoc.201500285 |
| 117. | Humphries, M. J.; Matsumoto, K.; White, S. L.; Olden, K. Cancer Res. 1986, 46, 5215–5222. |
| 118. | Whitby, K.; Pierson, T. C.; Geiss, B.; Lane, K.; Engle, M.; Zhou, Y.; Doms, R. W.; Diamond, M. S. J. Virol. 2005, 79, 8698–8706. doi:10.1128/jvi.79.14.8698-8706.2005 |
| 116. | Li, Z.; Huang, Y.; Dong, F.; Li, W.; Ding, L.; Yu, G.; Xu, D.; Yang, Y.; Xu, X.; Tong, D. J. Biosci. (New Delhi, India) 2012, 37 (Suppl. 1), 1005–1016. doi:10.1007/s12038-012-9265-8 |
| 34. | Eum, H.; Choi, J.; Cho, C.-G.; Ha, H.-J. Asian J. Org. Chem. 2015, 4, 1399–1409. doi:10.1002/ajoc.201500285 |
| 119. | Cardona, F.; Goti, A.; Brandi, A. Eur. J. Org. Chem. 2007, 1551–1565. doi:10.1002/ejoc.200600633 |
| 47. | Yun, J. M.; Sim, T. B.; Hahm, H. S.; Lee, W. K.; Ha, H.-J. J. Org. Chem. 2003, 68, 7675–7680. doi:10.1021/jo034755a |
| 34. | Eum, H.; Choi, J.; Cho, C.-G.; Ha, H.-J. Asian J. Org. Chem. 2015, 4, 1399–1409. doi:10.1002/ajoc.201500285 |
| 48. | Kim, Y.; Ha, H.-J.; Yun, S. Y.; Lee, W. K. Chem. Commun. 2008, 4363–4365. doi:10.1039/b809124b |
| 113. | Ritthiwigrom, T.; Au, C. W. G.; Pyne, S. G. Curr. Org. Synth. 2012, 9, 583–612. doi:10.2174/157017912803251765 |
| 114. | Asano, N.; Kuroi, H.; Ikeda, K.; Kizu, H.; Kameda, Y.; Kato, A.; Adachi, I.; Watson, A. A.; Nash, R. J.; Fleet, G. W. J. Tetrahedron: Asymmetry 2000, 11, 1–8. doi:10.1016/s0957-4166(99)00508-x |
| 46. | Hwang, G. I.; Chung, J.-H.; Lee, W. K. Tetrahedron 1996, 52, 12111–12116. doi:10.1016/0040-4020(96)00702-8 |
| 115. | Michael, J. P. Simple Indolizidine and Quinolizidine Alkaloids. The Alkaloids: Chemistry and Biology; Elsevier: Amsterdam, Netherlands, 2016; Vol. 75, pp 1–498. doi:10.1016/bs.alkal.2014.12.001 |
| 29. | Hwang, G.-I.; Chung, J.-H.; Lee, W. K. J. Org. Chem. 1996, 61, 6183–6188. doi:10.1021/jo9603183 |
| 34. | Eum, H.; Choi, J.; Cho, C.-G.; Ha, H.-J. Asian J. Org. Chem. 2015, 4, 1399–1409. doi:10.1002/ajoc.201500285 |
| 44. | Garbaccio, R. M.; Olson, C. M.; Tasber, E. S.; Torrent, M. Mitotic kinesin inhibitors. PCT Int. Appl. WO2005/092011 A2, Oct 6, 2005. |
| 45. | Han, S.-M.; Ma, S.-h.; Ha, H.-J.; Lee, W. K. Tetrahedron 2008, 64, 11110–11114. doi:10.1016/j.tet.2008.09.068 |
| 42. | Hett, R.; Fang, Q. K.; Gao, Y.; Wald, S. A.; Senanayake, C. H. Org. Process Res. Dev. 1998, 2, 96–99. doi:10.1021/op970116o |
| 43. | Minoru, O.; Koichi, Y.; Kiyoshi, T. Process for producing optically active benzene-sulfonamide derivatives. Eur. Pat. Appl. EP 0257787 A1, July 21, 1987. |
| 49. | Choi, J.; Ha, H.-J. J. Korean Chem. Soc. 2015, 59, 203–204. doi:10.5012/jkcs.2015.59.3.203 |
| 47. | Yun, J. M.; Sim, T. B.; Hahm, H. S.; Lee, W. K.; Ha, H.-J. J. Org. Chem. 2003, 68, 7675–7680. doi:10.1021/jo034755a |
| 47. | Yun, J. M.; Sim, T. B.; Hahm, H. S.; Lee, W. K.; Ha, H.-J. J. Org. Chem. 2003, 68, 7675–7680. doi:10.1021/jo034755a |
| 56. | Ishigami, K.; Katsuta, R.; Shibata, C.; Hayakawa, Y.; Watanabe, H.; Kitahara, T. Tetrahedron 2009, 65, 3629–3638. doi:10.1016/j.tet.2009.03.003 |
| 122. | Choi, H. G.; Kwon, J. H.; Kim, J. C.; Lee, W. K.; Eum, H.; Ha, H.-J. Tetrahedron Lett. 2010, 51, 3284–3285. doi:10.1016/j.tetlet.2010.04.069 |
| 57. | Yoon, D.-H.; Kang, P.; Lee, W. K.; Kim, Y.; Ha, H.-J. Org. Lett. 2012, 14, 429–431. doi:10.1021/ol202683k |
| 121. | Bates, R. W.; Dewey, M. R. Org. Lett. 2009, 11, 3706–3708. doi:10.1021/ol901094h |
| 54. | Dowling, P. M. Chloramphenicol, Thiamphenicol, and Florfenicol. In Antimicrobial Therapy in Veterinary Medicine, 5th ed.; Giguère, S.; Prescott, J. F.; Dowling, P. M., Eds.; Wiley, 2013; pp 269–277. doi:10.1002/9781118675014.ch16 |
| 124. | Boydas, E. B.; Tanriver, G.; D'hooghe, M.; Ha, H.-J.; Van Speybroeck, V.; Catak, S. Org. Biomol. Chem. 2018, 16, 796–806. doi:10.1039/c7ob02253k |
| 125. | Ji, M.-K.; Hertsen, D.; Yoon, D.-H.; Eum, H.; Goossens, H.; Waroquier, M.; Van Speybroeck, V.; D'hooghe, M.; De Kimpe, N.; Ha, H.-J. Chem. – Asian J. 2014, 9, 1060–1067. doi:10.1002/asia.201301551 |
| 55. | Peng, Y.; Tian, W.; Ye, Q.; Fang, Z. Florfenicol synthesizing method. U.S. Patent US2016/0318859 A1, Nov 3, 2016. |
| 123. | Pyun, D. K.; Kim, B. J.; Jung, H. J.; Kim, J. H.; Lee, J. S.; Lee, W. K.; Lee, C. H. Chem. Pharm. Bull. 2002, 50, 415–418. doi:10.1248/cpb.50.415 |
| 52. | Kim, J.-W.; Kim, Y.-W.; Inagaki, Y.; Hwang, Y.-A.; Mitsutake, S.; Ryu, Y.-W.; Lee, W. K.; Ha, H.-J.; Park, C.-S.; Igarashi, Y. Bioorg. Med. Chem. 2005, 13, 3475–3485. doi:10.1016/j.bmc.2005.02.053 |
| 53. | Kim, J.; Park, S.; Lee, H.-J.; Park, C.; Kim, Y.-W.; Lee, W.-K.; Ha, H.-J. Sphingolipid derivatives and the composition for anti-cancer containing the same. PCT Int. Appl. WO2006/004359 A1, Jan 12, 2006. |
| 52. | Kim, J.-W.; Kim, Y.-W.; Inagaki, Y.; Hwang, Y.-A.; Mitsutake, S.; Ryu, Y.-W.; Lee, W. K.; Ha, H.-J.; Park, C.-S.; Igarashi, Y. Bioorg. Med. Chem. 2005, 13, 3475–3485. doi:10.1016/j.bmc.2005.02.053 |
| 50. | Pyun, D. K.; Lee, C. H.; Ha, H.-J.; Park, C. S.; Chang, J.-W.; Lee, W. K. Org. Lett. 2001, 3, 4197–4199. doi:10.1021/ol016861+ |
| 120. | Lee, B. K.; Choi, H. G.; Roh, E. J.; Lee, W. K.; Sim, T. Tetrahedron Lett. 2013, 54, 553–556. doi:10.1016/j.tetlet.2012.11.087 |
| 51. | Park, C. S.; Kim, M. S.; Sim, T. B.; Pyun, D. K.; Lee, C. H.; Choi, D.; Lee, W. K.; Chang, J.-W.; Ha, H.-J. J. Org. Chem. 2003, 68, 43–49. doi:10.1021/jo025545l |
| 92. | Kim, Y.; Ha, H.-J.; Yun, H.; Lee, B. K.; Lee, W. K. Tetrahedron 2006, 62, 8844–8849. doi:10.1016/j.tet.2006.06.025 |
| 60. | Yoon, D.-H.; Ha, H.-J.; Kim, B. C.; Lee, W. K. Tetrahedron Lett. 2010, 51, 2181–2183. doi:10.1016/j.tetlet.2010.02.087 |
| 32. | Yoon, H.; Cho, K. S.; Sim, T. Tetrahedron: Asymmetry 2014, 25, 497–502. doi:10.1016/j.tetasy.2014.02.009 |
| 91. | Park, C. S.; Choi, H. G.; Lee, H.; Lee, W. K.; Ha, H.-J. Tetrahedron: Asymmetry 2000, 11, 3283–3292. doi:10.1016/s0957-4166(00)00311-6 |
| 89. | Faulds, D.; Goa, K. L.; Benfield, P. Drugs 1993, 45, 953–1040. doi:10.2165/00003495-199345060-00007 |
| 90. | Kim, Y.; Yoon, D.-H.; Ha, H.-J.; Kang, K. Y.; Lee, W. K. Tetrahedron Lett. 2011, 52, 5918–5920. doi:10.1016/j.tetlet.2011.08.048 |
| 88. | Singh, A.; Kim, B.; Lee, W. K.; Ha, H.-J. Org. Biomol. Chem. 2011, 9, 1372–1380. doi:10.1039/c0ob00730g |
| 16. | Harada, K.; Nakamura, I. J. Chem. Soc., Chem. Commun. 1978, 522–523. doi:10.1039/c39780000522 |
| 20. | Alezra, V.; Bonin, M.; Micouin, L.; Policar, C.; Husson, H.-P. Eur. J. Org. Chem. 2001, 2589–2594. doi:10.1002/1099-0690(200107)2001:13<2589::aid-ejoc2589>3.0.co;2-y |
| 21. | Takano, S.; Moriya, M.; Ogasawara, K. Tetrahedron: Asymmetry 1992, 3, 681–684. doi:10.1016/s0957-4166(00)80499-1 |
| 16. | Harada, K.; Nakamura, I. J. Chem. Soc., Chem. Commun. 1978, 522–523. doi:10.1039/c39780000522 |
| 18. | Bosies, E.; Berger, H.; Kampe, W.; Bicker, U.; Grafe, A. N-Substituted aziridine-2-carboxylic acid derivatives and their immune-stimulation compositions and methods. U.S. Pat. Appl. US4410532A, Oct 18, 1983. |
| 19. | Morán-Ramallal, R.; Liz, R.; Gotor, V. Org. Lett. 2008, 10, 1935–1938. doi:10.1021/ol800443p |
| 16. | Harada, K.; Nakamura, I. J. Chem. Soc., Chem. Commun. 1978, 522–523. doi:10.1039/c39780000522 |
| 14. | Ha, H.-J.; Jung, J.-H.; Lee, W. K. Asian J. Org. Chem. 2014, 3, 1020–1035. doi:10.1002/ajoc.201402098 |
| 15. | Akhtar, R.; Naqvi, S. A. R.; Zahoor, A. F.; Saleem, S. Mol. Diversity 2018, 22, 447–501. doi:10.1007/s11030-018-9829-0 |
| 94. | Mao, H.; Jeong, H.; Yang, J.; Ha, H.-J.; Yang, J. W. Chem. – Eur. J. 2018, 24, 2370–2374. doi:10.1002/chem.201706161 |
| 16. | Harada, K.; Nakamura, I. J. Chem. Soc., Chem. Commun. 1978, 522–523. doi:10.1039/c39780000522 |
| 17. | Farooa, S.; Swain, W. E., Jr.; Daeppen, R.; Rihs, G. Tetrahedron: Asymmetry 1992, 3, 51–63. doi:10.1016/s0957-4166(00)82312-5 |
| 93. | Fagerholm, A. E.; Habrant, D.; Koskinen, A. M. P. Mar. Drugs 2010, 8, 122–172. doi:10.3390/md80100122 |
| 13. | Häner, R.; Olano, B.; Seebach, D. Helv. Chim. Acta 1987, 70, 1676–1693. doi:10.1002/hlca.19870700704 |
| 34. | Eum, H.; Choi, J.; Cho, C.-G.; Ha, H.-J. Asian J. Org. Chem. 2015, 4, 1399–1409. doi:10.1002/ajoc.201500285 |
| 23. | Ha, H.-J.; Suh, J.-M.; Kang, K.-H.; Ahn, Y.-G.; Han, O. Tetrahedron 1998, 54, 851–858. doi:10.1016/s0040-4020(97)10358-1 |
| 24. | Tranchant, M.-J.; Dalla, V.; Jabin, I.; Decroix, B. Tetrahedron 2002, 58, 8425–8432. doi:10.1016/s0040-4020(02)01031-1 |
| 25. | Bew, S. P.; Hughes, D. L.; Savic, V.; Soapi, K. M.; Wilson, M. A. Chem. Commun. 2006, 3513–3515. doi:10.1039/b606033a |
| 13. | Häner, R.; Olano, B.; Seebach, D. Helv. Chim. Acta 1987, 70, 1676–1693. doi:10.1002/hlca.19870700704 |
| 22. | Lee, W. K.; Park, C. S.; Lim, Y. H.; Ha, H.-J. Optically active aziridine-2-carboxylate derivatives and process for preparing them. PCT Int. Pat. Appl. WO2002012186A1, Feb 14, 2002. |
| 37. | Wróblewski, A. E.; Drozd, J. Tetrahedron: Asymmetry 2007, 18, 1134–1141. doi:10.1016/j.tetasy.2007.04.021 |
| 38. | Sugiyama, S.; Inoue, T.; Ishii, K. Tetrahedron: Asymmetry 2003, 14, 2153–2160. doi:10.1016/s0957-4166(03)00410-5 |
| 34. | Eum, H.; Choi, J.; Cho, C.-G.; Ha, H.-J. Asian J. Org. Chem. 2015, 4, 1399–1409. doi:10.1002/ajoc.201500285 |
| 22. | Lee, W. K.; Park, C. S.; Lim, Y. H.; Ha, H.-J. Optically active aziridine-2-carboxylate derivatives and process for preparing them. PCT Int. Pat. Appl. WO2002012186A1, Feb 14, 2002. |
| 35. | Lim, Y.; Lee, W. K. Tetrahedron Lett. 1995, 36, 8431–8434. doi:10.1016/0040-4039(95)01814-x |
| 36. | Sugiyama, S.; Fukuchi, H.; Ishii, K. Tetrahedron 2007, 63, 12047–12057. doi:10.1016/j.tet.2007.09.004 |
| 13. | Häner, R.; Olano, B.; Seebach, D. Helv. Chim. Acta 1987, 70, 1676–1693. doi:10.1002/hlca.19870700704 |
| 16. | Harada, K.; Nakamura, I. J. Chem. Soc., Chem. Commun. 1978, 522–523. doi:10.1039/c39780000522 |
| 28. | Mancuso, A. J.; Huang, S.-L.; Swern, D. J. Org. Chem. 1978, 43, 2480–2482. doi:10.1021/jo00406a041 |
| 29. | Hwang, G.-I.; Chung, J.-H.; Lee, W. K. J. Org. Chem. 1996, 61, 6183–6188. doi:10.1021/jo9603183 |
| 30. | Kim, B. C.; Lee, W. K. Tetrahedron 1996, 52, 12117–12124. doi:10.1016/0040-4020(96)00703-x |
| 31. | Yoon, H.; Sim, T. Synthesis 2013, 45, 3276–3280. doi:10.1055/s-0033-1338545 |
| 32. | Yoon, H.; Cho, K. S.; Sim, T. Tetrahedron: Asymmetry 2014, 25, 497–502. doi:10.1016/j.tetasy.2014.02.009 |
| 33. | Yadav, N. N.; Choi, J.; Ha, H.-J. Org. Biomol. Chem. 2016, 14, 6426–6434. doi:10.1039/c6ob00806b |
| 26. | Park, J.-H.; Ha, H.-J.; Lee, W. K.; Généreux-Vincent, T.; Kazlauskas, R. J. ChemBioChem 2009, 10, 2213–2222. doi:10.1002/cbic.200900343 |
| 27. | Alezra, V.; Bouchet, C.; Micouin, L.; Bonin, M.; Husson, H.-P. Tetrahedron Lett. 2000, 41, 655–658. doi:10.1016/s0040-4039(99)02157-7 |
© 2019 Głowacka et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)
