Abstract
A new flexible anthracene linked benzimidazolium-based receptor 1 has been designed, synthesized and its binding properties have been studied by NMR, UV–vis and fluorescence spectroscopic techniques. While the receptor 1 exhibits a greater change in emission in the presence of tetrabutylammonium dihydrogenphosphate in CH3CN over the other anions studied, iodide is selectively preferred in CHCl3 containing 0.1% CH3CN. Upon complexation of dihydrogen phosphate and iodide, the emission of 1 gradually decreased without showing any other characteristic change in the spectra. Hydrogen bonding and charge–charge interactions interplay simultaneously in a cooperative manner for selectivity in the binding process.

Graphical Abstract
Introduction
The selective recognition of anionic species by artificial abiotic receptors is a rapidly growing area in supramolecular chemistry [1-6]. Anion recognition has gained significant importance as it plays an important role in a wide range of biological, environmental and chemical processes [7-10]. Sensors based on anion-induced changes in fluorescence appear to be particularly attractive in anion recognition due to the simplicity and high detection limit of fluorescence [11,12]. In devising such sensors, various functional sites with hydrogen bond donors and acceptors are of considerable importance. Among the different binding motifs for anions, the hydrogen bonding properties of NH groups in neutral amines [13], amides [14], ureas/thioureas [15,16], indoles [17-19] and pyrroles [20] as well as in guanidinium [21] and imidazolium [22] groups are well established. In addition, the use of the benzimidazolium motif [23,24] in anion recognition is also known. During the course of our work on anion recognition, we used this motif along with the other functionalities for selective recognition of carboxylates, and dihydrogen phosphate [25]. Selective recognition of dihydrogen phosphate is an important aspect of supramolecular chemistry and various receptors of different designs for this anion have been reported in the literature [26-33].
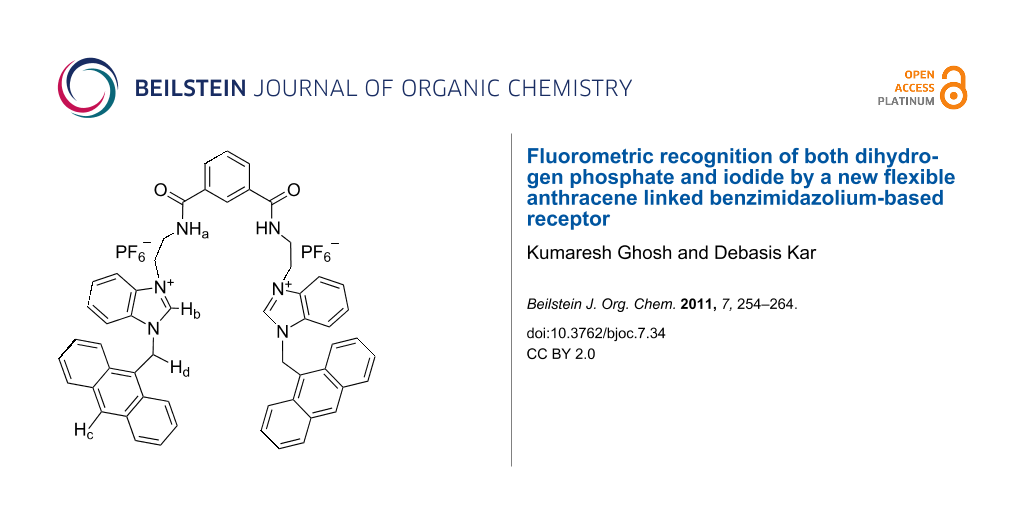
In an effort to investigate the anion recognition behavior of the benzimidazolium group in the presence of a flexible spacer, we report herein the design and synthesis of a new chemosensor 1 (Figure 1), which shows selective recognition of dihydrogen phosphate in CH3CN among the other anions studied. The selectivity is changed on changing the polarity of the solvent, and the receptor 1 exhibits a preference for iodide when CH3CN is replaced by CHCl3 containing 0.1% CH3CN. The results were compared with the monomeric unit 2 (Figure 1).
![[1860-5397-7-34-1]](/bjoc/content/figures/1860-5397-7-34-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Results and Discussion
The receptor 1 was obtained according to Scheme 1. Initially, 2-chloroethylamine was reacted with isophthaloyl dichloride to afford the diamide 4 in 80% yield. Subsequent reaction of 4 with anthracene coupled benzimidazole 3 (prepared in 64% yield from benzimidazole and 9-chloromethylanthracene in the presence of NaH in dry THF) gave the dichloride salt 5 in 55% yield. Anion exchange using NH4PF6 in warm aqueous CH3OH gave the desired compound 1 in 90% yield. Similarly, model compound 2 was obtained in 64% yield from the reaction between 6 and 3 in dry CH3CN followed by anion exchange using NH4PF6 in aqueous CH3OH. All the compounds were characterized unequivocally by 1H NMR, 13C, mass spectrometry and FTIR.
![[1860-5397-7-34-i1]](/bjoc/content/inline/1860-5397-7-34-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
In receptor 1, two amide NHs and two benzimidazolium protons (C+–H) are accessible for the complexation of anions. The residual charge on each benzimidazolium motif, in principle, will stabilize the complex by charge-charge interaction. It is quite rational that the binding group in each arm of flexible receptor 1 could either be involved cooperatively or functions individually for complexation of anions. Thus the selectivity in the binding process is related with the disposition of the binding groups around the isophthaloyl spacer. Figure 2, for example, shows the energy-optimized geometry of the complex of 1 with H2PO4− in the gas phase [34]. In the complex, benzimidazolium protons (C+–H) and amide protons are cooperatively involved in hydrogen bonding with H2PO4−. Anthracene, being a fluorophore in 1, has the advantage of being considered as a flat hydrophobic fluorophore probe for sensing of anions by the change in intensity due to the photo-induced electron transfer (PET) mechanism.
![[1860-5397-7-34-2]](/bjoc/content/figures/1860-5397-7-34-2.png?scale=2.4&max-width=1024&background=FFFFFF)
Figure 2: Energy optimized geometry of 1 with H2PO4− (E = 92.14 kcal/mol, a = 2.45 Å, b = 1.94 Å, c = 2.34 Å, d = 2.82 Å).
Figure 2: Energy optimized geometry of 1 with H2PO4− (E = 92.14 kcal/mol, a = 2.45 Å, b = 1.94 Å, c = 2.34 Å, ...
As expected, we observed a change in emission of 1 (c = 5.78 × 10−5 M) in CH3CN upon the addition of anions as their tetrabutylammonium salts. Receptor 1 (c = 5.78 × 10−5 M) in CH3CN gave a structured emission band when excited at 369 nm. Upon the addition of 2 equiv of each particular anion to the receptor solution of 1, a large change in emission of the anthracene group was observed for H2PO4− (Figure 3): Other anions perturbed the emission of 1 only weakly. Upon the addition of 2 equiv of tetrabutylammonium salts of H2PO4−, F−, Br− and I−, the emission of 1 was quenched by 72, 18, 14 and 30%, respectively. During titration experiments, no other changes such as excimer or exciplex formation were observed. The large quenching in emission of 1 upon increasing H2PO4− concentration is illustrated in Figure 4.
![[1860-5397-7-34-3]](/bjoc/content/figures/1860-5397-7-34-3.png?scale=2.4&max-width=1024&background=FFFFFF)
Figure 3: Change in fluorescence emission of 1 (c = 5.78 × 10−5 M) in the presence of 2 equiv of tetrabutylammonium salts of different guests in CH3CN.
Figure 3: Change in fluorescence emission of 1 (c = 5.78 × 10−5 M) in the presence of 2 equiv of tetrabutylam...
![[1860-5397-7-34-4]](/bjoc/content/figures/1860-5397-7-34-4.png?scale=2.6&max-width=1024&background=FFFFFF)
Figure 4: Change in emission spectra of 1 (c = 5.78 × 10−5 M) in presence of increasing amounts of H2PO4− in CH3CN; Inset: Plot of fluorescence intensity vs concentration of H2PO4−.
Figure 4: Change in emission spectra of 1 (c = 5.78 × 10−5 M) in presence of increasing amounts of H2PO4− in ...
Interestingly, the emission of 1 is quenched up to the addition of an equivalent amount of H2PO4− ions. Further addition caused an increase in emission. This is presumably due to a change in conformation upon complexation or decomplexation of anion, although deprotonation of the bound H2PO4− cannot be ignored [35]. We believe that initially, the binding sites of 1 interact cooperatively to make a 1:1 complex according to the suggested mode A, which in turn, changes to mode B with 2:1 (guest:host) stoichiometry in the presence of excess H2PO4− (Figure 5).
![[1860-5397-7-34-5]](/bjoc/content/figures/1860-5397-7-34-5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Suggested modes of binding of the H2PO4− ion into the cleft of 1.
Figure 5: Suggested modes of binding of the H2PO4− ion into the cleft of 1.
The flexible nature of the binding site contributes to this aspect. For an example of a related system, see [36]. The stoichiometry of the complexes was confirmed by Job plots (see Supporting Information File 1) as well as from the break of the titration curves at [G]/[H] = 2 (Figure 6).
![[1860-5397-7-34-6]](/bjoc/content/figures/1860-5397-7-34-6.png?scale=2.6&max-width=1024&background=FFFFFF)
Figure 6: Plot of change in emission of 1 at 420 nm vs the ratio of guest to host concentration in CH3CN.
Figure 6: Plot of change in emission of 1 at 420 nm vs the ratio of guest to host concentration in CH3CN.
Figure 7, for example, represents the Job plot [37] for H2PO4−, which corresponds to a clear-cut case for 2:1 stoichiometry of the complex. In the binding process, cooperative interaction of the two benzimidazolium motifs in 1 are primarily necessary for a large change in emission. This was proved by considering the model compound 2, where only one binding site is present for interaction. Under similar experimental conditions, the emission of 2 was only slightly changed.
![[1860-5397-7-34-7]](/bjoc/content/figures/1860-5397-7-34-7.png?scale=2.6&max-width=1024&background=FFFFFF)
Figure 7: Fluorescence Job plot of 1 with H2PO4−.
Figure 7: Fluorescence Job plot of 1 with H2PO4−.
Figure 8 displays the change in emission of 2 upon addition of 1 equiv of the same anions in CH3CN. In the presence of excess H2PO4− ions, the change in emission of 2 was less compared to the case of 1 (see Supporting Information File 1).
![[1860-5397-7-34-8]](/bjoc/content/figures/1860-5397-7-34-8.png?scale=2.6&max-width=1024&background=FFFFFF)
Figure 8: Change in fluorescence emission of 2 (c = 3.93 × 10−5 M) in the presence of 1 equiv of tetrabutylammonium salts of different guests in CH3CN.
Figure 8: Change in fluorescence emission of 2 (c = 3.93 × 10−5 M) in the presence of 1 equiv of tetrabutylam...
The quenching of emission of 1 upon complexation is attributed to the activation of a PET process occurring between the binding site and the excited state of anthracene. The Stern–Volmer plot in Figure 9 illustrates the quenching phenomena with anions such as H2PO4−, F−-, Br− and I−. The non-linear nature of the curves in Figure 9 indicates that both static (hydrogen bonding effects) and dynamic quenching (bimolecular collision) take place during binding.
![[1860-5397-7-34-9]](/bjoc/content/figures/1860-5397-7-34-9.png?scale=2.6&max-width=1024&background=FFFFFF)
Figure 9: Stern–Volmer plots for 1 (c = 5.78 × 10−5 M) with H2PO4−, F−, Br− and I− at 420 nm (up to the addition of 2 equiv of guest) in CH3CN.
Figure 9: Stern–Volmer plots for 1 (c = 5.78 × 10−5 M) with H2PO4−, F−, Br− and I− at 420 nm (up to the addit...
Concurrent UV–vis studies of 1 with the anions exhibited only a small change in absorbance of anthracene and predicted 1 as a PET system [38]. In many cases, the change of absorbance during the titration with the anions was irregular. Figure 10 corroborates the irregular change in absorbance of 1 on titration with H2PO4− and importantly, in the ground state, all the anions showed 1:1 binding (Supporting Information File 1). However, the selectivity of 1 towards the anions studied was established by determining the binding constant values from fluorescence titration data (Table 1) [39]. As can be seen from Table 1, the receptor 1 shows a marginal selectivity for H2PO4−. The receptor 1 also binds the larger sized iodide ion with 2:1 (guest:host) stoichiometry. By comparison, the change in emission of 1 in the presence of the smaller sized F− is attributed to its greater charge density which causes strong hydrogen bonding followed by deprotonation. Although receptor 1 demonstrates a similar order of binding with F−, I− and H2PO4− in CH3CN, the greater fluorometric change of 1 in the presence of H2PO4− is quite worth mentioning for its fluorometric distinction from other anions in the present study.
![[1860-5397-7-34-10]](/bjoc/content/figures/1860-5397-7-34-10.png?scale=2.6&max-width=1024&background=FFFFFF)
Figure 10: Change in UV–vis spectra of 1 (c = 5.78 × 10−5 M) in presence of increasing amounts of H2PO4− in CH3CN.
Figure 10: Change in UV–vis spectra of 1 (c = 5.78 × 10−5 M) in presence of increasing amounts of H2PO4− in CH3...
Table 1: Binding constant values of 1 with the guests by fluorescence method.
| Guestsa | Ka (M−1)d in CH3CN | Ka (M−1)d in CHCl3 containing 0.1% CH3CN |
|---|---|---|
| Acetate | —b | —b |
| Benzoate | —b | —b |
| Dihydrogen phosphate | 5.56 × 103c | 1.29 × 104 |
| Hydrogen sulfate | —b | —b |
| Perchlorate | —b | —b |
| Fluoride | 4.06 × 103c | —b |
| Chloride | —b | —b |
| Bromide | —b | 5.05 × 103 |
| Iodide | 2.41 × 103c | 6.90 × 104 |
aTetrabutylammonium salts were used; bNot determined due to minor change; cConsidering K11; dError: ≤ ±10%.
For the application of this simple receptor in aqueous system, we carried out the complexation study of 1 in aq CH3OH (CH3OH:H2O = 4:1 v/v) with different phosphate salts. Surprisingly, the change in emission of 1 was found to be negligible (see Supporting Information File 1). This suggested weak or no interactions of 1 in aq CH3OH. However, on changing the polarity of the medium both 1 and 2 responded more efficiently and behaved differently. When CH3CN was replaced by CHCl3 containing 0.1% CH3CN, the change in emission of 1 upon complexation of the same anions was very sharp and found to be sharpest for the iodide ion. Figure 11 shows the change in emission of 1 in presence of particular anions. The large quenching of emission in the presence of I− is a characteristic feature of 1 for the fluorometric identification of I− among the other anions in CHCl3 containing 0.1% CH3CN. Iodide is an important halide that plays an important role in several biological processes such as neurological activity and thyroid function. The iodide content of urine and milk is often required to provide information for nutritional, metabolic, and epidemiological studies of thyroid disorder [40]. In relation to this, very few reports on iodide recognition are known in the literature [41-45].
![[1860-5397-7-34-11]](/bjoc/content/figures/1860-5397-7-34-11.png?scale=2.6&max-width=1024&background=FFFFFF)
Figure 11: Change in fluorescence emission of 1 (c = 5.78 × 10−5 M) in the presence of 2 equiv of tetrabutylammonium salts of different guests in CHCl3 containing 0.1% CH3CN.
Figure 11: Change in fluorescence emission of 1 (c = 5.78 × 10−5 M) in the presence of 2 equiv of tetrabutylam...
Iodide binding induced quenching of emission is attributed to the i) complementarity in size of iodide with the pseudocavity formed by the receptor-binding site and ii) heavy atom effect of iodide, which is also true for Br−. But the small quenching of emission in the presence of Br− suggests that I− induced quenching is not only due to the heavy atom effect but also involves some hydrogen bonding effects. Figure 12 is the Stern–Volmer plot of the quenching process. It was also noted that while the monomeric benzimidazolium unit 2 was ineffective in CH3CN, it showed measurable changes in emission in CHCl3 containing 0.1% CH3CN (Supporting Information File 1). These observations thus intimate that solvent polarity is an important aspect for monitoring the sensing behavior of benzimidazolium-based receptors. In our opinion, CH3CN in the present case participates in H-bonding with the polar C+–H bond of benzimidazolium motif and reduces the possibility of host–guest interactions [46]. This is clearly reflected in the binding constant values in Table 1. Due to the presence of a minimum amount of CH3CN in CHCl3 the binding constant values for the selected anions are greater in magnitude. In the series I− shows a higher value of Ka. We presume that it is due to the dimension of the open cavity of 1 in CHCl3 containing 0.1% CH3CN for which I− anion fits sterically with 1:1 stoichiometry. Other complexes in CHCl3 containing 0.1% CH3CN had also 1:1 stoichiometry.
![[1860-5397-7-34-12]](/bjoc/content/figures/1860-5397-7-34-12.png?scale=2.6&max-width=1024&background=FFFFFF)
Figure 12: Stern–Volmer plots for 1 (c = 5.78 × 10−5 M) with H2PO4−, F−, Br− and I− at 420 nm (up to the addition of 2 equiv of guest) in CHCl3 containing 0.1% CH3CN.
Figure 12: Stern–Volmer plots for 1 (c = 5.78 × 10−5 M) with H2PO4−, F−, Br− and I− at 420 nm (up to the addit...
Time resolved fluorescence measurements were additionally carried out to study the dynamics of the flexible receptor 1 (λexc = 369 nm) in the presence and absence of the anions such as H2PO4− in CH3CN. The emission decay profile of 1 monitored at 420 nm could be fitted bi-exponentially with two constants τ1 = 3.42 ns (12.22%), τ2 = 7.76 ns (87.78%). The faster decay component (3.42 ns) is due to the anthracene moiety [47] and the relatively stable component with longer lifetime (7.76 ns) is attributed to the benzimidazolium motif of 1. However, in the presence of 1 equiv of H2PO4−, the lifetime of both the components marginally decreases (Table 2). In the presence of 2 equiv of H2PO4−, the lifetime of the anthracene component increases without showing any marked change in the pre-exponential factor. This was also true for the benzimidazolium component. Figure 13 shows the decay profiles. The change in the lifetimes of the components in 1 in the presence of H2PO4− ions can be correlated with the change in emission intensity in Figure 4. The fluorescence dynamics of 1 was also carried out for 1 in the presence and absence of I− in CHCl3 containing 0.1% CH3CN. On changing the solvent combination, the lifetimes of the anthracene and benzimidazolium components of 1 changed (Table 2). Interestingly, while the faster decay component (1.45 ns) due to anthracene moiety contributed a small pre-exponential factor (2%), the relatively stable benzimidazolium component (8.90 ns) had a larger contribution (98%) to the total emission of the molecule. However, upon addition of equivalent amounts of I− the decay profile followed a tri-exponential fitting that indicated three emitting species with lifetimes τ1 = 2.15 ns (5.82%), τ2 = 8.95 ns (92.86%) and τ3 = 0.28 ns (1.32%) (Figure 14). A small increase in the lifetimes of both the anthracene and benzimidazolium moieties in 1 is attributed to the formation of hydrogen bonds with I− in the open cavity of 1. The faster decay component (0.28 ns) is assumed to be either due to a very short-lived species or an artifact or for tunneling of extra energy to the bulk by a non-radiative pathway [47,48].
Table 2: Fluorescence decay times (τ) and pre-exponential factors (c) for 1 in the presence and absence of anions.
| τ1 (c) | τ2 (c) | τ3 (c) | χ2 | |
|---|---|---|---|---|
| Receptor 1a | 3.42 ns (12.22%) | 7.76 ns (87.78%) | — | 1.13 |
| 1 + Bu4N+H2PO4− (1:1)a | 3.36 ns (11.40%) | 7.72 ns (88.60%) | — | 1.18 |
| 1 + Bu4N+H2PO4− (1:2)a | 3.49 ns (11.74%) | 7.75 ns (88.26%) | — | 1.13 |
| Receptor 1b | 1.45 ns (2%) | 8.90 ns (98%) | — | 1.12 |
| 1 + Bu4N+I− (1:1)b | 2.15 ns (5.82%) | 8.95 ns (92.86%) | 0.28 ns (1.32%) | 1.10 |
aIn CH3CN; bIn CHCl3 containing 0.1% CH3CN.
![[1860-5397-7-34-13]](/bjoc/content/figures/1860-5397-7-34-13.png?scale=2.6&max-width=1024&background=FFFFFF)
Figure 13: Fluorescence decays (at λmax = 420 nm) of receptor 1 upon the addition of H2PO4− ion ([H] = 5.78 × 10−5 M, [G] = 4.33 × 10−3 M) in CH3CN solvent.
Figure 13: Fluorescence decays (at λmax = 420 nm) of receptor 1 upon the addition of H2PO4− ion ([H] = 5.78 × ...
![[1860-5397-7-34-14]](/bjoc/content/figures/1860-5397-7-34-14.png?scale=2.6&max-width=1024&background=FFFFFF)
Figure 14: Fluorescence decays (at λmax = 420 nm) of receptor 1 upon the addition of I− ion ([H] = 5.78 × 10−5 M, [G] = 4.55 × 10−3 M) in CHCl3 containing 0.1% CH3CN.
Figure 14: Fluorescence decays (at λmax = 420 nm) of receptor 1 upon the addition of I− ion ([H] = 5.78 × 10−5...
The expected strong interaction of 1 with H2PO4− was established additionally by 1H NMR. Although the guest is primarily bound by the benzimidazolium motif on account of a charge–charge interaction, there is undoubtedly a degree of cooperation from the amide protons as evidenced by the downfield change in their chemical shift positions in 1H NMR. Figure 15 indicates the change in chemical shift of the interacting amides and benzimidazolium protons upon complexation of H2PO4− ions and shows the broadening of the signals of 1 in the 1H NMR spectrum. A precipitate appeared during the course of the study due to insolubility in the NMR concentration range and this was one reason for not determining the binding constant values by the NMR method. However, both the amide and benzimidazolium protons (0.51 ppm) were found to move downfield and thereby supported our binding proposition as indicated in Figure 5. The exact position of amide protons Ha was difficult to determine upon complexation. Hydrogen bonding and deprotonation of 1 in the presence of F− was also evidenced from 1H NMR (Supporting Information File 1, Figure 11S). In the presence of 1 equiv of F− the benzimidazolium protons in 1 moved downfield by 0.30 ppm which became 0.59 ppm when 2 equiv of F− were added to the receptor solution.
![[1860-5397-7-34-15]](/bjoc/content/figures/1860-5397-7-34-15.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 15: Partial 1H NMR (300 MHz, CDCl3 containing 4% CD3CN) spectra of (a) 1 (c = 1.38 × 10−3 M), (b) 1:1 and (c) 2:1 (guest:host) complexes with H2PO4− [see labeled structure 1].
Figure 15: Partial 1H NMR (300 MHz, CDCl3 containing 4% CD3CN) spectra of (a) 1 (c = 1.38 × 10−3 M), (b) 1:1 a...
Conclusion
In conclusion, we have designed a new type of flexible receptor 1, which uniquely responses the recognition of H2PO4− and I− by exhibiting large quenching of emission of anthracene group in solvents of different polarities. Two benzimidazolium motifs with an isophthaloyl spacer group in 1, act cooperatively for successful detection of H2PO4− in CH3CN. In comparison, under the same experimental condition, a single benzimidazolium unit in 2 is inefficient in sensing H2PO4− ion. By contrast, in the less polar solvent CHCl3 containing 0.1% CH3CN, the receptor shows a greater fluorescence quenching for I−. Such differential quenching of 1 in the presence of H2PO4− and I− in different solvent combinations is presumably due to the activation of the PET process occurring between the binding site and the excited state of anthracene function at the different rates. We believe at present that the different degrees of solvation of the anions as well as the effective dimension of the binding site which in turn, controls the selectivity in binding process in the excited state are the key factors that regulate the PET to different extents. The binding affinity and selectivity of this simple fluororeceptor are associated with the combined effects of semi-rigid structures of receptor, charge-charge interactions, and the involvement of both N–H---O and C–H---O hydrogen bonds. Further work is underway in our laboratory.
Experimental
General methods
Solvents were distilled prior to use, and dried according to the literature procedure when required. Chromatographic separations were performed on silica gel (60–120 mesh). All melting points were determined in open capillaries and are uncorrected. 1H NMR spectra were recorded on Bruker 400 and 300 MHz spectrometers. 13C NMR spectra were recorded on a Bruker 400 MHz spectrometer. FT IR spectra were recorded on a Perkin-Elmer L120-00A spectrometer as KBr discs. UV–vis spectra were recorded on Perkin-Elmer Lambda-25 and fluorescence spectra on a Perkin-Elmer LS 55 spectrofluorometer, respectively.
Synthesis of 3 [49]
To a solution of benzimidazole (0.300 g, 2.54 mmol) in dry THF (15 mL), NaH (0.14 g) was added and the mixture refluxed for 1 h under a nitrogen atmosphere. The reaction mixture was then cooled to room temperature, 9-chloromethylanthracene (0.700 g, 3.09 mmol) in THF (15 mL) added and then heated under reflux for 10 h. The THF was removed, water added and the mixture extracted with CHCl3 (3 × 30 mL). The organic layer was dried over anhydrous Na2SO4 and concentrated on a rotary evaporator. Purification of the crude product by silica gel column chromatography with 20% ethyl acetate in petroleum ether as eluent gave compound 3 (0.500 g, yield 64%). 1H NMR in CDCl3 (400 MHz) δ 8.61 (s, 2H), 8.10 (d, 4H, J = 8 Hz), 7.82 (d, 1H, J = 8 Hz), 7.71 (d, 1H, J = 8 Hz), 7.51 (m, 4H), 7.42 (t, 1H, J = 8 Hz), 7.35(t, 1H, J = 8 Hz), 6.19 (s, 2H); 13C NMR (CDCl3, 100 MHz) δ 144.0, 142.2, 134.2, 131.4, 131.0, 129.7, 129.5, 127.5, 125.4, 123.6, 123.1, 123.0, 122.5, 120.6, 109.5, 41.3; m/z (ES+): 308.9 [M]+.
Synthesis of 4
A mixture of 2-chloroethylamine hydrochloride (704 mg, 6 mmol) in dry CH2Cl2 (30 mL) containing triethylamine (1.35 mL) was stirred for 1 h to give the free amine. To this solution, isophthaloyl dichloride (600 mg, 2.9 mmol) was added and the reaction mixture stirred at room temperature for 8 h. The solvent was evaporated and water added. The aqueous layer was extracted with CHCl3 (3 × 50 mL), the combined organic phases was washed with water, dried over anhydrous Na2SO4 and concentrated in vacuo. The resulting crude solid was purified by column chromatography with 50% ethyl acetate in petroleum ether as eluent to give pure 4 (0.67 g, yield 78%), mp 145 °C, 1H NMR (CDCl3, 400 MHz): δ 8.25 (s, 1H), 7.94 (dd, 2H, J1 = 8 Hz, J2 = 4 Hz), 7.53 (t, 1H, J = 8 Hz), 6.84 (br t, 2H, -NH-), 3.84–3.79 (m, 4H), 3.75–3.71 (m, 4H); 13C NMR (CDCl3 containing few drops DMSO-d6, 100 MHz) δ 166.0, 134.21, 129.8, 128.3, 126.2, 42.8, 41.3; FTIR: ν cm−1 (KBr): 3288, 3066, 2915, 2863, 1639, 1539; m/z (LCMS): 289 (M)+.
Synthesis of 1
To a stirred solution of 4 (0.1 g, 3.8 mmol) in dry CH3CN (20 mL), compound 3 (0.236 g, 7.6 mmol) was added and the reaction mixture heated under reflux for 3 days. During the heating the dichloride salt 5 precipitated and was removed by filtration. Repeated recrystallization of the salt from CH3CN gave almost pure 5 (0.184 g, 59.7% yield). Compound 5 (0.184 g) was then dissolved in hot CH3OH (20 mL) followed by addition of NH4PF6. After stirring the reaction mixture for 30 min, the CH3OH was evaporated to reduce the volume. The precipitated salt was filtered, washed with cold water and dried. Repeated recrystallization of 1 from CH3CN afforded the pure 1 (0.21 g, 90% yield), mp 210 °C, 1H NMR (DMSO-d6, 400 MHz): δ 8.94 (s, 2H), 8.83 (s, 2H), 8.50–8.10 (m, 14H), 7.78–7.72 (m, 4H), 7.59–7.49 (m, 12H), 6.63 (s, 4H), 4.46 (br s, 4H), 3.59 (br s, 4H) [due to solubility problem signals were broad in nature]; 13C NMR (DMSO-d6, 100 MHz): 141.8, 134.3, 132.2, 132.0, 131.5, 131.47, 131.43, 131.1, 130.8, 129.7, 128.2, 128.1, 127.3, 127.1, 126.0, 125.9, 123.7, 122.0, 114.6, 114.1, 47.0, 43.7, 38.7; FTIR: ν cm−1 (KBr): 3433, 3146, 2954, 1721, 1652, 1565, 1531; m/z (ES+): 1123.7 (M − 1)+, 979.5 [(M − PF6−) ]+.
Synthesis of 2
A mixture of 2-chloroethylamine hydrochloride (460 mg, 3.9 mmol) in dry CH2Cl2 (20 mL) containing triethylamine (1 mL) was stirred at room temperature for 1 h to give the free amine. Acetyl chloride (0.582 mL, 5.9 mmol) was added to the reaction mixture which was then stirred at room temperature for 8 h. After completion of the reaction, the solvent was evaporated and the residue extracted with CHCl3 (3 × 20 mL). The organic extracts were washed with NaHCO3 solution (3 × 15 mL) and dried over anhydrous Na2SO4. The solvent was removed under vacuum and the residue purified by silica gel column chromatography with 80% ethyl acetate in petroleum ether as eluent to afford the compound 6 (0.309 g, 64% yield). The chloro-amide 6 (150 mg, 1.2 mmol) was heated under reflux in dry CH3CN (20 mL) with the anthracene-coupled benzimidazole 3 (570 mg, 1.85 mmol) for 4 days. The precipitated chloride salt was filtered, washed with water and dried (0.51 g, 64.4%). The compound 6 in MeOH was subsequently treated with aqueous NH4PF6 solution to carry out the anion exchange reaction. The solution was heated with stirring for 20 min until a precipitate appeared. Filtration of the precipitate followed by thorough washing with ether afforded compound 2 in 95% yield (0.61 g), mp 203 °C (decomposition); 1H NMR (DMSO-d6, 400 MHz): δ 8.93 (s, 1H), 8.82 (s, 1H), 8.41 (d, 1H, J = 8 Hz), 8.33 (d, 2H, J = 8 Hz), 8.27 (d, 2H, J = 8 Hz), 8.08 (d, 1H, J = 8 Hz) 7.84–7.78 (m, 2H), 7.63 (br m, 5H), 6.67 (s, 2H), 4.32 (br s, 2H), 3.50 (br s, 2H), 2.32 (s, 3H); 13C NMR (DMSO-d6, 100 MHz): 169.4, 141.2, 131.8, 131.4, 131.1, 131.0, 130.4, 129.3, 127.8, 126.8, 126.6, 125.6, 123.3, 121.5, 114.2, 113.5, 57.4, 46.4, 43.2, 37.2; FTIR: ν cm−1 (KBr): 3436, 1718, 1654, 1563, 1453. m/z (LCMS): 394.2 [(M − PF6−) ]+.
General procedure of fluorescence and UV–vis titrations: Stock solutions of the receptors were prepared in different solvents such as CH3CN and CHCl3 containing 0.1% CH3CN and 2.5 ml of the individual receptor solution was placed in the cuvette. Stock solutions of anions were prepared in the same solvents, and were added individually in different amounts to the receptor solution. For fluorescence, the receptor solutions prepared in dry CH3CN were irradiated at the excitation wavelength 369 nm maintaining the excitation and emission slits 8 and 8, respectively and the filter was 1% attenuated. Upon the addition of anions, the change in emission and absorbance of the receptors were noted. Similarly, for emission, the receptors dissolved in CHCl3 containing 0.1% CH3CN were irradiated at the excitation wavelength 369 nm maintaining the excitation and emission slits 10 and 5, respectively.
Method for Job plots: The stoichiometry was determined by the continuous variation method (Job Plot) [35]. In this method, solutions of host and guests of equal concentrations were prepared in dry CH3CN and CHCl3 containing 0.1% CH3CN. Then the host and guest solutions were mixed in different proportions maintaining a total volume of 3 mL of the mixture. The related compositions for host:guest (v/v) were 3:0, 2.8:0.2, 2.5:0.5, 2.2:0.8, 2:1, 1.8:1.2, 1.5:1.5, 1:2, 0.8:2.2, 0.5:2.5, 0.2:2.8. All the prepared solutions were kept for 1 h at room temperature with occasional shaking. Then the emission and absorbance of the solutions of different compositions was recorded. The concentration of the complex, i.e., [HG] was calculated using the equation [HG] = ΔI/I0 × [H] or [HG] = ΔA/A0 × [H] where ΔI/I0 and ΔA/A0 indicate the relative emission and absorbance intensities. [H] corresponds to the concentration of pure host. The mole fraction of the host (XH) was plotted against the concentration of the complex [HG]. In the plot, the mole fraction of the host at which the concentration of the host–guest complex concentration [HG] is maximum, gives the stoichiometry of the complex.
Supporting Information
| Supporting Information File 1: Supplementary Data. | ||
| Format: PDF | Size: 1.5 MB | Download |
References
-
Spichiger-Keller, U. E. Chemical Sensors and Biosensors for Medical and Biological Applications; Wiley-VCH: Weinheim, Germany, 1998. doi:10.1002/9783527612284
Return to citation in text: [1] -
Steed, J. W. Chem. Commun. 2006, 2637–2649. doi:10.1039/b601511e
Return to citation in text: [1] -
Martínez-Máñez, R.; Sancenón, F. Chem. Rev. 2003, 103, 4419–4476. doi:10.1021/cr010421e
Return to citation in text: [1] -
Caltagirone, C.; Gale, P. A. Chem. Soc. Rev. 2009, 38, 520–538. doi:10.1039/b806422a
Return to citation in text: [1] -
Gale, P. A.; García-Garrido, S. E.; Garric, J. Chem. Soc. Rev. 2008, 37, 151–190. doi:10.1039/b715825d
Return to citation in text: [1] -
Sessler, J. L.; Barkey, N. M.; Pantos, G. D.; Lynch, V. M. New J. Chem. 2007, 31, 646–654. doi:10.1039/b615673h
Return to citation in text: [1] -
Ullmann’s encyclopedia of industrial chemistry, 6th ed.; Wiley-VCH: Weinheim, Germany, 2002.
Return to citation in text: [1] -
Kirk, K. L. Biochemistry of the Elemental Halogens and Inorganic Halides; Plenum Press: New York, NY, 1991; p 591.
Return to citation in text: [1] -
Rurack, K.; Resch-Genger, U. Chem. Soc. Rev. 2002, 31, 116–127. doi:10.1039/b100604p
Return to citation in text: [1] -
Tajc, S. G.; Miller, B. L. J. Am. Chem. Soc. 2006, 128, 2532–2533. doi:10.1021/ja058126p
Return to citation in text: [1] -
James, T. D.; Sandanayake, K. R. A. S.; Shinkai, S. Angew. Chem., Int. Ed. Engl. 1994, 33, 2207–2209. doi:10.1002/anie.199422071
Return to citation in text: [1] -
Khalil, M. M. H.; De Schryver, F. C.; Keller, H.; Lehn, J.-M. Supramol. Sci. 1995, 2, 175–182. doi:10.1016/0968-5677(96)89673-8
Return to citation in text: [1] -
Vance, D. H.; Czarnik, A. W. J. Am. Chem. Soc. 1994, 116, 9397–9398. doi:10.1021/ja00099a094
Return to citation in text: [1] -
Lankshear, M. D.; Beer, P. D. Coord. Chem. Rev. 2006, 250, 3142–3160. doi:10.1016/j.ccr.2006.04.018
Return to citation in text: [1] -
Blondean, P.; Benet-Buchholz, J.; de Mendoza, J. New J. Chem. 2007, 31, 736–740. doi:10.1039/b616409a
and references cited therein.
Return to citation in text: [1] -
Ghosh, K.; Adhikari, S. Tetrahedron Lett. 2006, 47, 8165–8169. doi:10.1016/j.tetlet.2006.09.035
and references cited therein.
Return to citation in text: [1] -
Chang, K.-J.; Moon, D.; Lah, M. S.; Jeong, K.-S. Angew. Chem., Int. Ed. 2005, 44, 7926–7929. doi:10.1002/anie.200503121
Return to citation in text: [1] -
Caltagirone, C.; Gale, P. A.; Hiscock, J. R.; Brooks, S. J.; Hursthouse, M. S.; Light, M. E. Chem. Commun. 2008, 3007–3009. doi:10.1039/b806238b
Return to citation in text: [1] -
Caltagirone, C.; Hiscock, J. R.; Hursthouse, M. B.; Light, M. E.; Gale, P. A. Chem.–Eur. J. 2008, 14, 10236–10243. doi:10.1002/chem.200801639
Return to citation in text: [1] -
Gale, P. A. Chem. Commun. 2005, 3761–3772. doi:10.1039/b504596g
Return to citation in text: [1] -
Schmuck, C.; Machon, U. Eur. J. Org. Chem. 2006, 4385–4392. doi:10.1002/ejoc.200600324
Return to citation in text: [1] -
Yoon, J.; Kim, S. K.; Singh, N. J.; Kim, K. S. Chem. Soc. Rev. 2006, 35, 355–360. doi:10.1039/b513733k
Return to citation in text: [1] -
Bai, Y.; Zhang, B.-G.; Xu, J.; Duan, C.-Y.; Dang, D.-B.; Liu, D.-J.; Meng, Q.-J. New J. Chem. 2005, 29, 777–779. doi:10.1039/b500252d
Return to citation in text: [1] -
Bai, Y.; Zhang, B.-G.; Duan, C.-Y.; Dang, D.-B.; Meng, Q.-J. New J. Chem. 2006, 30, 266–271. doi:10.1039/b508173d
Return to citation in text: [1] -
Ghosh, K.; Saha, I.; Patra, A. Tetrahedron Lett. 2009, 50, 2392–2397. doi:10.1016/j.tetlet.2009.02.215
and references cited therein.
Return to citation in text: [1] -
Gong, W.; Hiratani, K. Tetrahedron Lett. 2008, 49, 5655–5657. doi:10.1016/j.tetlet.2008.07.078
Return to citation in text: [1] -
Kondo, S.-I.; Hiraoka, Y.; Kurumatani, N.; Yano, Y. Chem. Commun. 2005, 1720–1722. doi:10.1039/B417304J
Return to citation in text: [1] -
Guo, D.-S.; Liu, Z.-P.; Ma, J.-P.; Huang, R.-Q. Tetrahedron Lett. 2007, 48, 1221–1224. doi:10.1016/j.tetlet.2006.12.047
Return to citation in text: [1] -
Konodo, S.-I.; Hiraoka, Y.; Kurumatani, N.; Yano, Y. Chem. Commun. 2005, 1720–1722. doi:10.1039/b417304j
Return to citation in text: [1] -
Ihm, H.; Yun, S.; Kim, H. G.; Kim, J. K.; Kim, K. S. Org. Lett. 2002, 4, 2897–2900. doi:10.1021/ol026373h
Return to citation in text: [1] -
Choi, K.; Hamilton, A. D. Angew. Chem., Int. Ed. 2001, 40, 3912–3915. doi:10.1002/1521-3773(20011015)40:20<3912::AID-ANIE3912>3.0.CO;2-R
Return to citation in text: [1] -
Kwon, T. H.; Jeong, K.-S. Tetrahedron Lett. 2006, 47, 8539–8541. doi:10.1016/j.tetlet.2006.09.105
Return to citation in text: [1] -
Xu, Z.; Kim, S.; Lee, K.-H.; Yoon, J. Tetrahedron Lett. 2007, 48, 3797–3800. doi:10.1016/j.tetlet.2007.03.159
Return to citation in text: [1] -
PC Mode; Serena Software, 1993.
Energy minimization: MMX using standard constants and dielectric constant of 1.5.
Return to citation in text: [1] -
Gale, P. A.; Hiscock, J. R.; Moore, S. J.; Caltagirone, C.; Hursthouse, M. B.; Light, M. E. Chem.–Asian J. 2009, 5, 555–561. doi:10.1002/asia.200900230
Return to citation in text: [1] [2] -
Lowe, A. J.; Dyson, G. A.; Pfeffer, F. M. Org. Biomol. Chem. 2007, 5, 1343–1346. doi:10.1039/b703626b
Return to citation in text: [1] -
Job, P. Ann. Chim. Appl. 1928, 9, 113–203.
Return to citation in text: [1] -
Bissel, R. A.; de Silva, A. P.; Gunaratne, H. Q. N.; Lynch, P. L. M.; Maguire, G. E. M.; Sandanayake, K. R. A. S. Chem. Soc. Rev. 1992, 21, 187–195. doi:10.1039/cs9922100187
Return to citation in text: [1] -
Chou, P.-T.; Wu, G.-R.; Wei, C.-Y.; Cheng, C.-C.; Chang, C.-P.; Hung, F.-T. J. Phys. Chem. B 2000, 104, 7818–7829. doi:10.1021/jp001001g
Return to citation in text: [1] -
Haldimann, M.; Zimmerli, B.; Als, C.; Gerber, H. Clin. Chem. 1998, 44, 817–824.
and references therein.
Return to citation in text: [1] -
Kim, H.; Kang, J. Tetrahedron Lett. 2005, 46, 5443–5445. doi:10.1016/j.tetlet.2005.06.068
Return to citation in text: [1] -
Singh, N.; Jang, D. O. Org. Lett. 2007, 9, 1991–1994. doi:10.1021/ol070592r
and references cited therein.
Return to citation in text: [1] -
Singh, N.; Jung, H. J.; Jang, D. O. Tetrahedron Lett. 2009, 50, 71–74. doi:10.1016/j.tetlet.2008.10.088
Return to citation in text: [1] -
Ghosh, K.; Sen, T. Tetrahedron Lett. 2008, 49, 7204–7208. doi:10.1016/j.tetlet.2008.10.009
Return to citation in text: [1] -
Ghosh, K.; Saha, I. Supramol. Chem. 2010, 22, 311–317. doi:10.1080/10610270903469773
Return to citation in text: [1] -
Singh, N. J.; Jun, E. J.; Chellappan, K.; Thangadurai, D.; Chandran, R. P.; Hwang, I.-C.; Yoon, J.; Kim, K. S. Org. Lett. 2007, 9, 485–488. doi:10.1021/ol062849b
Return to citation in text: [1] -
Shiraishi, Y.; Kohno, Y.; Hirai, T. J. Phys. Chem. B 2005, 109, 19139–19147. doi:10.1021/jp052645x
Return to citation in text: [1] [2] -
Inoue, H.; Hida, M.; Nakashima, N.; Yoshihara, K. J. Phys. Chem. 1982, 86, 3184–3188. doi:10.1021/j100213a024
Return to citation in text: [1] -
Ghosh, K.; Saha, I. Tetrahedron Lett. 2008, 49, 4591–4595. doi:10.1016/j.tetlet.2008.05.096
Return to citation in text: [1]
| 1. | Spichiger-Keller, U. E. Chemical Sensors and Biosensors for Medical and Biological Applications; Wiley-VCH: Weinheim, Germany, 1998. doi:10.1002/9783527612284 |
| 2. | Steed, J. W. Chem. Commun. 2006, 2637–2649. doi:10.1039/b601511e |
| 3. | Martínez-Máñez, R.; Sancenón, F. Chem. Rev. 2003, 103, 4419–4476. doi:10.1021/cr010421e |
| 4. | Caltagirone, C.; Gale, P. A. Chem. Soc. Rev. 2009, 38, 520–538. doi:10.1039/b806422a |
| 5. | Gale, P. A.; García-Garrido, S. E.; Garric, J. Chem. Soc. Rev. 2008, 37, 151–190. doi:10.1039/b715825d |
| 6. | Sessler, J. L.; Barkey, N. M.; Pantos, G. D.; Lynch, V. M. New J. Chem. 2007, 31, 646–654. doi:10.1039/b615673h |
| 14. | Lankshear, M. D.; Beer, P. D. Coord. Chem. Rev. 2006, 250, 3142–3160. doi:10.1016/j.ccr.2006.04.018 |
| 35. | Gale, P. A.; Hiscock, J. R.; Moore, S. J.; Caltagirone, C.; Hursthouse, M. B.; Light, M. E. Chem.–Asian J. 2009, 5, 555–561. doi:10.1002/asia.200900230 |
| 13. | Vance, D. H.; Czarnik, A. W. J. Am. Chem. Soc. 1994, 116, 9397–9398. doi:10.1021/ja00099a094 |
| 36. | Lowe, A. J.; Dyson, G. A.; Pfeffer, F. M. Org. Biomol. Chem. 2007, 5, 1343–1346. doi:10.1039/b703626b |
| 11. | James, T. D.; Sandanayake, K. R. A. S.; Shinkai, S. Angew. Chem., Int. Ed. Engl. 1994, 33, 2207–2209. doi:10.1002/anie.199422071 |
| 12. | Khalil, M. M. H.; De Schryver, F. C.; Keller, H.; Lehn, J.-M. Supramol. Sci. 1995, 2, 175–182. doi:10.1016/0968-5677(96)89673-8 |
| 26. | Gong, W.; Hiratani, K. Tetrahedron Lett. 2008, 49, 5655–5657. doi:10.1016/j.tetlet.2008.07.078 |
| 27. | Kondo, S.-I.; Hiraoka, Y.; Kurumatani, N.; Yano, Y. Chem. Commun. 2005, 1720–1722. doi:10.1039/B417304J |
| 28. | Guo, D.-S.; Liu, Z.-P.; Ma, J.-P.; Huang, R.-Q. Tetrahedron Lett. 2007, 48, 1221–1224. doi:10.1016/j.tetlet.2006.12.047 |
| 29. | Konodo, S.-I.; Hiraoka, Y.; Kurumatani, N.; Yano, Y. Chem. Commun. 2005, 1720–1722. doi:10.1039/b417304j |
| 30. | Ihm, H.; Yun, S.; Kim, H. G.; Kim, J. K.; Kim, K. S. Org. Lett. 2002, 4, 2897–2900. doi:10.1021/ol026373h |
| 31. | Choi, K.; Hamilton, A. D. Angew. Chem., Int. Ed. 2001, 40, 3912–3915. doi:10.1002/1521-3773(20011015)40:20<3912::AID-ANIE3912>3.0.CO;2-R |
| 32. | Kwon, T. H.; Jeong, K.-S. Tetrahedron Lett. 2006, 47, 8539–8541. doi:10.1016/j.tetlet.2006.09.105 |
| 33. | Xu, Z.; Kim, S.; Lee, K.-H.; Yoon, J. Tetrahedron Lett. 2007, 48, 3797–3800. doi:10.1016/j.tetlet.2007.03.159 |
| 7. | Ullmann’s encyclopedia of industrial chemistry, 6th ed.; Wiley-VCH: Weinheim, Germany, 2002. |
| 8. | Kirk, K. L. Biochemistry of the Elemental Halogens and Inorganic Halides; Plenum Press: New York, NY, 1991; p 591. |
| 9. | Rurack, K.; Resch-Genger, U. Chem. Soc. Rev. 2002, 31, 116–127. doi:10.1039/b100604p |
| 10. | Tajc, S. G.; Miller, B. L. J. Am. Chem. Soc. 2006, 128, 2532–2533. doi:10.1021/ja058126p |
| 34. |
PC Mode; Serena Software, 1993.
Energy minimization: MMX using standard constants and dielectric constant of 1.5. |
| 21. | Schmuck, C.; Machon, U. Eur. J. Org. Chem. 2006, 4385–4392. doi:10.1002/ejoc.200600324 |
| 23. | Bai, Y.; Zhang, B.-G.; Xu, J.; Duan, C.-Y.; Dang, D.-B.; Liu, D.-J.; Meng, Q.-J. New J. Chem. 2005, 29, 777–779. doi:10.1039/b500252d |
| 24. | Bai, Y.; Zhang, B.-G.; Duan, C.-Y.; Dang, D.-B.; Meng, Q.-J. New J. Chem. 2006, 30, 266–271. doi:10.1039/b508173d |
| 25. |
Ghosh, K.; Saha, I.; Patra, A. Tetrahedron Lett. 2009, 50, 2392–2397. doi:10.1016/j.tetlet.2009.02.215
and references cited therein. |
| 17. | Chang, K.-J.; Moon, D.; Lah, M. S.; Jeong, K.-S. Angew. Chem., Int. Ed. 2005, 44, 7926–7929. doi:10.1002/anie.200503121 |
| 18. | Caltagirone, C.; Gale, P. A.; Hiscock, J. R.; Brooks, S. J.; Hursthouse, M. S.; Light, M. E. Chem. Commun. 2008, 3007–3009. doi:10.1039/b806238b |
| 19. | Caltagirone, C.; Hiscock, J. R.; Hursthouse, M. B.; Light, M. E.; Gale, P. A. Chem.–Eur. J. 2008, 14, 10236–10243. doi:10.1002/chem.200801639 |
| 15. |
Blondean, P.; Benet-Buchholz, J.; de Mendoza, J. New J. Chem. 2007, 31, 736–740. doi:10.1039/b616409a
and references cited therein. |
| 16. |
Ghosh, K.; Adhikari, S. Tetrahedron Lett. 2006, 47, 8165–8169. doi:10.1016/j.tetlet.2006.09.035
and references cited therein. |
| 22. | Yoon, J.; Kim, S. K.; Singh, N. J.; Kim, K. S. Chem. Soc. Rev. 2006, 35, 355–360. doi:10.1039/b513733k |
| 39. | Chou, P.-T.; Wu, G.-R.; Wei, C.-Y.; Cheng, C.-C.; Chang, C.-P.; Hung, F.-T. J. Phys. Chem. B 2000, 104, 7818–7829. doi:10.1021/jp001001g |
| 38. | Bissel, R. A.; de Silva, A. P.; Gunaratne, H. Q. N.; Lynch, P. L. M.; Maguire, G. E. M.; Sandanayake, K. R. A. S. Chem. Soc. Rev. 1992, 21, 187–195. doi:10.1039/cs9922100187 |
| 35. | Gale, P. A.; Hiscock, J. R.; Moore, S. J.; Caltagirone, C.; Hursthouse, M. B.; Light, M. E. Chem.–Asian J. 2009, 5, 555–561. doi:10.1002/asia.200900230 |
| 47. | Shiraishi, Y.; Kohno, Y.; Hirai, T. J. Phys. Chem. B 2005, 109, 19139–19147. doi:10.1021/jp052645x |
| 48. | Inoue, H.; Hida, M.; Nakashima, N.; Yoshihara, K. J. Phys. Chem. 1982, 86, 3184–3188. doi:10.1021/j100213a024 |
| 49. | Ghosh, K.; Saha, I. Tetrahedron Lett. 2008, 49, 4591–4595. doi:10.1016/j.tetlet.2008.05.096 |
| 46. | Singh, N. J.; Jun, E. J.; Chellappan, K.; Thangadurai, D.; Chandran, R. P.; Hwang, I.-C.; Yoon, J.; Kim, K. S. Org. Lett. 2007, 9, 485–488. doi:10.1021/ol062849b |
| 47. | Shiraishi, Y.; Kohno, Y.; Hirai, T. J. Phys. Chem. B 2005, 109, 19139–19147. doi:10.1021/jp052645x |
| 40. |
Haldimann, M.; Zimmerli, B.; Als, C.; Gerber, H. Clin. Chem. 1998, 44, 817–824.
and references therein. |
| 41. | Kim, H.; Kang, J. Tetrahedron Lett. 2005, 46, 5443–5445. doi:10.1016/j.tetlet.2005.06.068 |
| 42. |
Singh, N.; Jang, D. O. Org. Lett. 2007, 9, 1991–1994. doi:10.1021/ol070592r
and references cited therein. |
| 43. | Singh, N.; Jung, H. J.; Jang, D. O. Tetrahedron Lett. 2009, 50, 71–74. doi:10.1016/j.tetlet.2008.10.088 |
| 44. | Ghosh, K.; Sen, T. Tetrahedron Lett. 2008, 49, 7204–7208. doi:10.1016/j.tetlet.2008.10.009 |
| 45. | Ghosh, K.; Saha, I. Supramol. Chem. 2010, 22, 311–317. doi:10.1080/10610270903469773 |
© 2011 Ghosh and Kar; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)