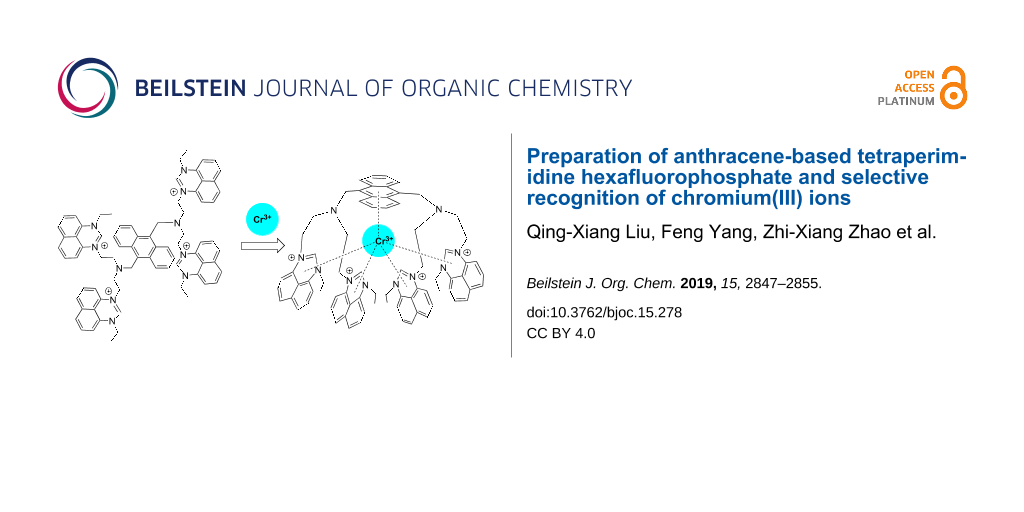
Abstract
A novel anthracene-based tetraperimidine hexafluorophosphate 3 was prepared, and its structure was determined through X-ray analysis, HRMS as well as 1H and 13C NMR spectroscopy. In the cationic moiety of 3, two (N-ethylperimidinyl–C2H4)2NCH2– arms were attached to the 9- and 10-positions of anthracene. In addition, compound 3 was used as a chemosensor to research the ability to recognize Cr3+ through fluorescence and UV titrations, HRMS, as well as 1H NMR and IR spectroscopy. The results indicate that 3 is an effective chemosensor for Cr3+.

Graphical Abstract
Introduction
Fluorescent chemosensors are an attractive and efficient tool for the detection of metal ions in environmental and biological science because of their high sensitivity, selectivity, and simple usage [1-8]. Among metal ions, the detection of Cr3+ ions occupies an important position. Chromium(III) is an essential microelement for humans and animals, and it plays an important role in glucose and lipid metabolism in the body [9,10]. The deficiency of chromium(III) in the human body leads to various diseases, such as diabetes as well as autoimmune and cardiovascular diseases [11]. On the other hand, the excessive incorporation of chromium(III) is toxic to humans, and can cause cancer through the oxidation of DNA and some proteins [12-14]. Therefore, the detection of chromium(III) has a vital practical significance for human health monitoring.
In recent years, some fluorescent chemosensors for the detection of chromium(III) have been developed [15-23]. Generally, chemosensors with fluorescence enhancement are more efficient than fluorescence turn-off chemosensors [24-29] because the paramagnetic nature of chromium(III) can cause fluorescence quenching of the fluorophore via the enhancement of spin–orbit coupling [30-35]. So far, only a few successful examples of fluorescence enhancement sensors for Cr3+ have been reported [36-40]. Thus, developing new and effective fluorescence turn-on chemosensors for Cr3+ is necessary.
In the process of our research, a tetradentate compound bearing a fluorophore aroused our interest. In this paper, we report the synthesis of a novel anthracene-based tetraperimidine hexafluorophosphate 3, and its structure was determined by X-ray analysis as well as 1H and 13C NMR spectroscopy. Particularly, compound 3 was tested as a chemosensor for the recognition of Cr3+ through fluorescence, UV, IR, and 1H NMR spectroscopy along with HRMS. Altogether, the results indicate the utility of 3 as an effective chemosensor for Cr3+.
Results and Discussion
Synthesis and characterization of 3
As displayed in Scheme 1, paraformaldehyde was reacted with anthracene to give 9,10-di(chloromethyl)anthracene in 82% yield, which reacted further with HN(CH2CH2OH)2 to form 9,10-bis{[N,N-di(2-hydroxyethyl)amino]methyl}anthracene (1) with a yield of 58% [41]. Compound 1 was then treated with SOCl2 to generate 9,10-bis{[N,N-di(2-chloroethyl)amino]methyl}anthracene (2) in 75% yield, which reacted with 1-ethylperimidine in the presence of KI to afford the analogous iodide salt to tetraperimidine 3. Subsequently, an anion exchange reaction with NH4PF6 was performed to generate tetraperimidine hexafluorophosphate 3 with a yield of 85%. Compound 3 was stable to heat, moisture, and air, and it had a good solubility in DCM, DMSO, and CH3CN. In turn, it had a poor solubility in benzene and petroleum ether. In the 1H NMR spectrum of 3, the proton signal corresponding to the NCHN motif in perimidine was present at δ = 8.69 ppm [42].
![[1860-5397-15-278-i1]](/bjoc/content/inline/1860-5397-15-278-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Structure elucidation of compound 3
As can be seen in the crystal structure of 3 in Figure 1, the cationic moiety of the complex contained two (N-ethylperimidinyl–C2H4)2NCH2– arms attached to the 9- and 10-positions of anthracene, and the dihedral angle between two perimidine units of each arm was determined to be 18.1(4)°. Two of the four perimidine groups were parallel to the anthracene plane, with intramolecular π–π interactions [43] being present in this setup (with a face-to-face distance of 3.566(1) Å between perimidine and anthracene and a center-to-center distance of 3.664(4) Å). The bond distances C(3)–N(1) and C(3)–N(2) were 1.310(5) and 1.315(5) Å and the dihedral angles N(2)–C(3)–N(1) and N(4)–C(18)–N(5) were 125.2(3) and 124.3(4)° [42]. Further, a 1D polymeric chain of 3 monomers was generated through intermolecular π–π interactions between perimidine moieties (with a face-to-face distance of 3.558(4) Å and a center-to-center distance of 3.566(1) Å), as shown in Supporting Information File 1, Figure S1a. Besides, a 2D supramolecular layer was formed by the 1D supramolecular chains through two types of C–H···F hydrogen bonds, namely C(3)–H(3A)···F(2) and C(17)–H(17A)···F(2) interactions (Supporting Information File 1, Figure S1b).
![[1860-5397-15-278-1]](/bjoc/content/figures/1860-5397-15-278-1.png?scale=1.8&max-width=1024&background=FFFFFF)
Figure 1: View of the molecular structure of the cationic moiety of 3 in the crystal. Selected bond angles and lengths are N(2)–C(3)–N(1): 125.2(3)°; N(5)–C(18)–N(4): 124.3(4)°; C(3)–N(1): 1.310(5) Å; and C(3)–N(2): 1.315(5) Å.
Figure 1: View of the molecular structure of the cationic moiety of 3 in the crystal. Selected bond angles an...
Chemosensing of cations by 3
Compound 3 was employed as a host to study its ability to detect some cations through fluorescence and UV titrations in CH3CN/DMSO, 9:1, v/v at room temperature. In its free form, three emission bands at 402, 423, and 447 nm were observed at c = 5.0⋅10−6 M, which were ascribed to the emission of anthracene (Figure 2). When adding 30 equiv of K+, Na+, Li+, Ag+, NH4+, Zn2+, Cd2+, Ca2+, Ni2+, Pb2+, Cu2+, Co2+, Al3+, Hg+, Hg2+, Rh3+, Ir3+, Cr2+, Ga3+, Ru3+, and Fe3+, respectively, the intensities of the emission bands did not change significantly. However, a strong enhancement of the emission intensity in the region of 388–500 nm was observed after the addition of 30 equiv of Cr3+. Moreover, the absorption peak of 3 at 258 nm (ε = 3.5⋅103 mol−1⋅dm3⋅cm−1) did not exhibit any remarkable response to the addition of these cations, except for Cr3+ (ε = 1.1⋅104 mol−1⋅dm3⋅cm−1) as a result of 3·Cr3+ formation (Supporting Information File 1, Figure S2). These results show that 3 is able to effectively distinguish Cr3+ from other cations.
![[1860-5397-15-278-2]](/bjoc/content/figures/1860-5397-15-278-2.png?scale=2.4&max-width=1024&background=FFFFFF)
Figure 2: Fluorescence spectra of 3 (c = 5.0⋅10−6 M) upon addition of 30 equiv of salts of K+, Na+, Li+, Ag+, NH4+, Zn2+, Cd2+, Ca2+, Ni2+, Pb2+, Cu2+, Co2+, Al3+, Cr3+, Hg+, Hg2+, Rh3+, Ir3+, Cr2+, Ga3+, Ru3+, and Fe3+, respectively (c = 1.5⋅10−4 M) in CH3CN/DMSO, 9:1, v/v at room temperature (excitation wavelength λex = 258 nm).
Figure 2: Fluorescence spectra of 3 (c = 5.0⋅10−6 M) upon addition of 30 equiv of salts of K+, Na+, Li+, Ag+,...
To further investigate the recognition of Cr3+ by 3, fluorescence titrations were carried out (Figure 3). The fluorescence intensity of 3 in the region of 388–500 nm increased gradually upon addition of Cr3+ (c = 5.0⋅10−6 M). Titration was continued until no more notable changes in emission intensity occurred. In the inset of Figure 3, it is shown that when the molar ratio of Cr3+ to 3, i.e., cCr3+/c3, was below 1:1, fluorescence intensity enhanced sharply. However, when the molar ratio exceeded 1:1, the rate of fluorescence enhancement gradually slowed down until no more changes were noticeable. The limit of detection (LOD) was calculated to be 2.33⋅10−7 M (Supporting Information File 1, Figure S3). This value is analogous to the lowest corresponding value that has been reported in the literature (9.40⋅10−7–5.55⋅10−6 M) [44-46]. The association constant KSV was calculated to be 6.6⋅104 M−1 (R = 0.998) for 3·Cr3+ using Equation 1 (Supporting Information File 1, Figure S4) [47].
![[1860-5397-15-278-i3]](/bjoc/content/inline/1860-5397-15-278-i3.svg?max-width=590&scale=1.18182)
In Equation 1, the fluorescence intensities of 3 in the presence of Cr3+ and in its free form are represented by F and F0.
In the UV titration experiments, the absorption band in the region of 240–265 nm increased gradually upon addition of Cr3+ to a solution of 3 (c = 5.0⋅10−6 M) in CH3CN/DMSO, 9:1, v/v at room temperature (Supporting Information File 1, Figure S5). To evaluate the stability of 3·Cr3+, the stability constant K for the complex was computed as 8.23⋅104 M−1 (R = 0.999) at 258 nm using Equation 2 (Supporting Information File 1, Figure S6) [48-51].
![[1860-5397-15-278-i4]](/bjoc/content/inline/1860-5397-15-278-i4.svg?max-width=590&scale=1.18182)
In Equation 2, the absorbances of 3 in presence and absence of Cr3+ are represented as A0 and A. The discrepancy in absorbance in the presence and absence of Cr3+ is represented through the expression A0 − A (i.e., ΔA). The molar extinction coefficients of Cr3+ and the complex 3·Cr3+ are represented by εr and εc.
![[1860-5397-15-278-3]](/bjoc/content/figures/1860-5397-15-278-3.png?scale=2.4&max-width=1024&background=FFFFFF)
Figure 3: Fluorescence spectra of 3 (c = 5⋅10−6 M) upon addition of various amounts of Cr3+. cCr3+ for curves 1–24 (from bottom to top) were (0.00, 0.04, 0.08, 0.16, 0.24, 0.35, 0.49, 0.67, 0.80, 1.00, 1.45, 1.85, 2.30, 3.70, 4.50, 7.00, 10.00, 16.00, 20.00, 25.00, 33.00, 38.00, 40.00, and 45.00)⋅10−5 M (λex = 258 nm).
Figure 3: Fluorescence spectra of 3 (c = 5⋅10−6 M) upon addition of various amounts of Cr3+. cCr3+ for curves...
The complexation stoichiometry between 3 and Cr3+ was established by using Job’s method (inset of Figure S5, Supporting Information File 1). When the molar fraction (χ) of 3 was 0.5, the ΔAχ value for 3·Cr3+ reached a maximum, which indicated that the complexation stoichiometry between 3 and Cr3+ was 1:1 in 3·Cr3+ [8,52,53].
To measure the selectivity of Cr3+ complexation by 3, displacement experiments were carried out (Supporting Information File 1, Figure S7). Firstly, 30 equiv of Cr3+ were added to solutions of 3 containing 30 equiv of K+, Na+, Li+, Ag+, NH4+, Zn2+, Cd2+, Ca2+, Ni2+, Pb2+, Cu2+, Co2+, Al3+, Hg+, Hg2+, Rh3+, Ir3+, Cr2+, Ga3+, Ru3+, and Fe3+, respectively. The emission intensities of the resulting mixtures were similar to that of a solution containing only 3 and Cr3+. These experimental results show that 3 can capture Cr3+ selectively while neglecting other cations, with no remarkable interference being caused in the presence of the latter.
In order to probe whether the anions of the chromium(III) salts had effects on the binding of 3 and Cr3+, other chromium(III) salts, CrCl3, CrBr3, Cr2(SO4)3, Cr(NO3)3, and Cr(OAc)3, were tested. As displayed in Figure S8, Supporting Information File 1, when 30 equiv of any of these were added to 3, similar fluorescence intensities could be detected. A reversible binding experiment was also carried out (Supporting Information File 1, Figure S9). Therein, 30 equiv of ethylenediaminetetraacetic acid (EDTA) were added to a solution of Cr3+ (c = 1.5⋅10−4 M) and 3 (c = 5.0⋅10−6 M), which led to a reduction of the fluorescence intensity at 388–500 nm. This reduced fluorescent intensity was analogous to that of free 3, displaying that 3 was regenerated in its uncomplexed form. When Cr3+ was added anew, the fluorescent intensity increased again. These results show that the binding process of 3 and Cr3+ has good reversibility and highlights the regenerative capacity of the 3⋅Cr3+ complex.
Interactions between 3 and Cr3+
Looking at the structural characteristics of 3, the nitrogen atoms of the tertiary amines, and the π systems, were most likely the binding sites for Cr3+ through Cr3+···N and Cr3+···π interactions (Scheme 2). In order to obtain further information on the binding pattern between 3 and Cr3+, 1H NMR titration studies were done in DMSO-d6. The spectra are depicted in Figure 4.
Upon incremental addition of Cr3+ to 3 (from 0.0 to 1.0 equiv), the proton signals a and b, corresponding to anthracene, shifted downfield by 0.03 ppm in total while the proton signals f and l, corresponding to perimidine, shifted downfield between 0.02 and 0.07 ppm. Further, the proton signals c, d, and e of the CH2 groups beside perimidine and anthracence shifted downfield by 0.03 ppm while the chemical shifts of other protons did not undergo visible changes. These experimental results suggest Cr3+···π interactions as the most likely binding mode between Cr3+ and 3, as illustrated in Scheme 2. In 3, each perimidine moiety is electron-rich due to the existence of a ![[Graphic 1]](/bjoc/content/inline/1860-5397-15-278-i5.svg?max-width=637&scale=1.18182) bond (Supporting Information File 1, Figure S10), and the strong affinity of the π system of each perimidine and the anthracene motif towards Cr3+ resulted in the facile formation of Cr3+···π interactions. It is worth noting that Cr3+···π interactions are not uncommon, and they have been reported in diaryl chromium complexes [54,55]. Besides, Cr3+···N interactions in 3·Cr3+ did not appear relevant for complexation. The reasons were that (1) the signal m, corresponding to the CH2 fragment beside the nitrogen atom of the tertiary amine, did not shift discernibly during 1H NMR titration; (2) if the tertiary amine groups coordinated to one Cr3+ ion each, a 1:2 binding mode would have been determined for 3⋅Cr3+; and (3) from the molecular structure of 3, due to the distance, it is spatially impossible that one Cr3+ ion is bound by both tertiary amine functions at the same time. All these arguments underline the absence of Cr3+···N interactions. The selectivity of the Cr3+ binding process by 3 may be mainly due to the metal ion's size, which could have been particularly suitable for coordination between anthracence and four perimidine groups, whereas the sizes of other metal cations were unsuitable. The fact that chromium(III) is triply charged may not have been a key influence because otherwise, other metal cations M3+ would have also been bound by 3.
bond (Supporting Information File 1, Figure S10), and the strong affinity of the π system of each perimidine and the anthracene motif towards Cr3+ resulted in the facile formation of Cr3+···π interactions. It is worth noting that Cr3+···π interactions are not uncommon, and they have been reported in diaryl chromium complexes [54,55]. Besides, Cr3+···N interactions in 3·Cr3+ did not appear relevant for complexation. The reasons were that (1) the signal m, corresponding to the CH2 fragment beside the nitrogen atom of the tertiary amine, did not shift discernibly during 1H NMR titration; (2) if the tertiary amine groups coordinated to one Cr3+ ion each, a 1:2 binding mode would have been determined for 3⋅Cr3+; and (3) from the molecular structure of 3, due to the distance, it is spatially impossible that one Cr3+ ion is bound by both tertiary amine functions at the same time. All these arguments underline the absence of Cr3+···N interactions. The selectivity of the Cr3+ binding process by 3 may be mainly due to the metal ion's size, which could have been particularly suitable for coordination between anthracence and four perimidine groups, whereas the sizes of other metal cations were unsuitable. The fact that chromium(III) is triply charged may not have been a key influence because otherwise, other metal cations M3+ would have also been bound by 3.
![[1860-5397-15-278-i2]](/bjoc/content/inline/1860-5397-15-278-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Illustration of interactions between 3 and Cr3+ in 3⋅Cr3+.
Scheme 2: Illustration of interactions between 3 and Cr3+ in 3⋅Cr3+.
Furthermore, looking at Figure 4, the proton signals a to l remained unchanged after 1 equiv of Cr3+ had been added to 3. That is, the signals in spectra (iv) and (v) have the same positions. This again illustrates 1:1 complexation between 3 and Cr3+. This result is consistent with the conclusions obtained from Job’s plot. In addition, HRMS analysis of 3·Cr3+ (Supporting Information File 1, Figure S11) produced a distinctive signal at m/z = 587.1086, matching (3⋅Cr3+)/3, again indicating 1:1 complexation. In the IR spectra of 3 and 3⋅Cr3+ (Supporting Information File 1, Figure S12), the C–C absorption band of a benzene moiety in 3 shifted from 1170 to 1185 cm−1 upon complexation, and the C=N absorption band at 1664 cm−1 shifted to a value of 1672 cm−1.
![[1860-5397-15-278-4]](/bjoc/content/figures/1860-5397-15-278-4.png?scale=2.6&max-width=1024&background=FFFFFF)
Figure 4: 1H NMR spectra of 3 in the presence of Cr3+ in DMSO-d6. (i) 3. (ii) 3 + 0.25 equiv of Cr3+. (iii) 3 + 0.5 equiv of Cr3+. (iv) 3 + 1 equiv of Cr3+. (v) 3 + 1.5 equiv of Cr3+.
Figure 4: 1H NMR spectra of 3 in the presence of Cr3+ in DMSO-d6. (i) 3. (ii) 3 + 0.25 equiv of Cr3+. (iii) 3...
Conclusion
In summary, a new anthracene-based tetraperimidine hexafluorophosphate 3 was prepared, and its structure was determined through X-ray analysis and 1H and 13C NMR spectroscopy. Compound 3 was proved to be a highly sensitive and selective chemosensor for Cr3+, and it can effectively distinguish Cr3+ from other cations through fluorescence enhancement. Thus, complex 3 may have potential value for the application as a Cr3+ detector.
Experimental
Materials and instruments
The solvents and chemicals used for synthesis and analysis were analytical grade and obtained commercially. A RF-5301PC fluorescence spectrophotometer (Shimadzu) was used to record fluorescence spectra at room temperature. The excitation and emission slits were set to 10 nm. UV–vis absorption spectra were recorded at room temperature using a JASCO-V570 spectrometer. A Varian spectrometer was employed to record 1H and 13C NMR spectra. A Perkin-Elmer 2400C Elemental Analyzer was employed for elemental analyses. IR spectra were measured with a PerkinElmer Spectrum 100 FT-IR spectrophotometer. A Q-TOF LC/MS (Agilent) and a VG ZAB-HS (VG) mass spectrometer were applied for HRMS analysis. Melting points were recorded employing a Boetius Block apparatus.
Analytical data
Synthesis of 1-ethylperimidine: Through a dropping funnel, a solution of perimidine (1.43 g, 8.5 mmol) in dry THF (30 mL) was added to a suspension of NaH (0.479 g, 20 mmol) in dry THF (10 mL), followed by stirring at room temperature for 1 h. Subsequently, bromoethane (1.308 g, 12 mmol) was added to the suspension, and the mixture was reacted for 24 h at ambient temperature. After filtration, the solvent was removed at reduced pressure, and water (50 mL) was added to the residue. This was extracted with CHCl3 (3 × 20 mL), the organic layer was rinsed with water (3 × 30 mL), and dried over anhydrous MgSO4. 1-Ethylperimidine was obtained as a yellow-green solid (0.825 g, 49%) after removal of the solvent. mp 198–200 °C; 1H NMR (400 MHz, DMSO-d6) δ 1.49 (t, J = 7.2 Hz, 3H, CH3), 4.27 (q, J = 7.2 Hz, 2H, CH2), 7.31 (d, J = 8.8 Hz, 2H, ArH), 7.76 (d, J = 8.8 Hz, 2H, ArH), 8.04 (s, 1H, ArH), 8.25 (s, 1H, ArH), 9.73 ppm (s, 1H, NCHN).
Synthesis of 9,10-bis(chloromethyl)anthracene: A suspension of paraformaldehyde (6.155 g, 205 mmol) and anthracene (17.823 g, 100 mmol) in a mixture of acetic acid (50 mL) and hydrochloric acid (36%, 20 mL) was heated to 100 °C for 5 h. Then, 200 mL of water were added to precipitate a yellow solid. After filtration, 9,10-bis(chloromethyl)anthracene was obtained as a yellow powder (22.452 g, 82%). mp 246–248 °C; 1H NMR (400 MHz, CDCl3) δ 5.62 (s, 4H, CH2), 7.68 (q, J = 1.6 Hz, 4H, ArH), 8.41 ppm (q, J = 1.6 Hz, 4H, ArH); 13C NMR (100 MHz, CDCl3) δ 130.2 (ArC), 129.7 (ArC), 126.7 (ArC), 124.3 (ArC), 67.1 ppm (CH2).
Synthesis of 9,10-bis{[N,N-di(2-hydroxyethyl)amino]methyl}anthracene (1): A suspension of diethanolamine (9.988 g, 95 mmol) and K2CO3 (25.015 g, 181 mmol) in 100 mL of CH3CN/CHCl3, 1:1, v/v was stirred under reflux for 1 h. Subsequently, 9,10-bis(chloromethyl)anthracene (8.255 g, 30 mmol) and KI (0.914 g, 5.5 mmol) were added to the solution, and the mixture was reacted for 30 h at 35 °C. Then, the solvent was evaporated in vacuo to give a yellow oil. After rinsing with water, a yellow solid, 9,10-bis{[N,N-di(2-hydroxyethyl)amino]methyl}anthracene (1), was obtained (7.189 g, 58%). mp 153–155 °C; 1H NMR (400 MHz, CDCl3) δ 1.96 (s, 4H, OH), 2.77 (t, J = 5.3 Hz, 8H, CH2), 3.43 (t, J = 5.3 Hz, 8H, CH2), 4.74 (s, 4H, CH2), 7.55 (q, J = 3.3 Hz, 4H, ArH), 8.51 ppm (q, J = 3.3 Hz, 4H, ArH); 13C NMR (100 MHz, CDCl3) δ 130.9 (ArC), 130.7 (ArC), 125.8 (ArC), 125.1 (ArC), 59.8 (CH2), 56.1 (CH2), 51.6 ppm (CH2).
Synthesis of 9,10-bis{[N,N-di(2-chloroethyl)amino]methyl}anthracene (2): A solution of SOCl2 (9.517 g, 80 mmol) in dioxane (30 mL) was added dropwise to a solution of compound 1 (4.125 g, 10 mmol) in dioxane (50 mL). The reaction mixture was stirred at 30 °C for 3 days, during which the formation of a yellow precipitate occurred. After filtration, the hydrochloride of 9,10-bis{[N,N-di(2-chloroethyl)amino]methyl}anthracene (2) was obtained as a yellow solid. This was rinsed with 200 mL of a NaOH solution (20%) for neutralization and extracted with CHCl3 (3 × 60 mL). The organic layer was washed with water (3 × 20 mL) and dried over anhydrous MgSO4. A yellow powder of 9,10-bis{[N,N-di(2-chloroethyl)amino]methyl}anthracene (2) was obtained (3.67 g, 75%) after removal of CHCl3. mp 131–133 °C; 1H NMR (400 MHz, DMSO-d6) δ 2.92 (t, J = 6.7 Hz, 8H, CH2), 3.52 (q, J = 6.1 Hz, 8H, CH2), 4.74 (s, 4H, CH2), 7.55 (q, J =3.4 Hz, 4H, ArH), 8.60 ppm (q, J = 3.4 Hz, 4H, ArH); 13C NMR (100 MHz, DMSO-d6) δ 130.9 (ArC), 130.8 (ArC), 126.0 (ArC), 125.8 (ArC), 55.4 (CH2), 50.3 (CH2), 42.7 ppm (CH2).
Synthesis of 3,3',3'',3'''-(((anthracene-9,10-diylbis(methylene))bis(azanetriyl))tetrakis(ethane-2,1-diyl))tetrakis(1-ethyl-1H-perimidin-3-ium) tetra(hexafluorophosphate) (3): A mixture of 1-ethylperimidine (1.177 g, 6 mmol), 9,10-bis{[N,N-di(2-chloroethyl)amino]methyl}anthracene (2, 0.389 g, 0.8 mmol), and KI (0.5 g, 3 mmol) in 30 mL of DMF/dioxane, 1:4, v/v was stirred under reflux for 5 days. After the solvent was removed, water (50 mL) was added to the residue. This was extracted with CHCl3 (3 × 30 mL), the organic layer was washed with water (3 × 20 mL), and dried over anhydrous MgSO4. A yellow powder of the tetraperimidine iodide species was gained after removal of CHCl3.
A solution of NH4PF6 (0.978 g, 6 mmol) and the tetraperimidine iodide compound (1.309 g, 0.8 mmol) in MeOH (100 mL) was stirred at room temperature for 3 days to give a yellow precipitate. This precipitate was collected through filtration and washed with methanol (2⋅10 mL) to afford tetraperimidine hexafluorophosphate 3 (1.158 g, 85%). mp 220–222 °C; anal calcd for C76H76N10P4F24, C, 53.40; H, 4.48; N, 8.19%; found, C, 53.53; H, 4.26; N, 8.21%; HRESIMS (m/z): [M − 2PF6−]2+/2 calcd for C38H38F6N5P, 709.2769; found, 709.3631; 1H NMR (400 MHz, DMSO-d6) δ 1.37 (s, 12H, CH3), 3.18 (s, 8H, CH2), 3.87 (d, J = 6.4 Hz, 8H, CH2), 4.08 (t, J = 3.2 Hz, 12H, CH2), 6.50 (q, J = 2.0 Hz, 4H, ArH), 6.94 (d, J = 4.4 Hz, 4H, ArH), 7.05 (s, 4H, ArH), 7.23 (m, 4H, ArH), 7.40 (t, J = 8.4 Hz, 8H, ArH), 7.45 (s, 4H, ArH), 7.51 (s, 4H, ArH), 8.69 ppm (s, 4H, NCHN); 13C NMR (100 MHz, DMSO-d6) δ 152.2 (NCN), 134.1 (ArC), 130.7 (ArC), 130.6 (ArC), 129.1 (ArC), 128.1 (ArC), 127.6 (ArC), 124.8 (ArC), 124.4 (ArC), 123.4 (ArC), 120.5 (ArC), 107.9 (ArC), 106.8 (ArC), 48.5 (CH2), 46.7 (CH2), 46.6 (CH2), 38.9 (CH2), 11.9 ppm (CH2).
Fluorescence titrations
The concentration of 3 and the guest ions was 5.0⋅10−6 and 0.0–45.0⋅10−5 M, respectively, in the sample solutions. The excitation wavelength was set to λex = 258 nm and the widths of the emission and excitation spectral lines were adjusted to 5 and 10 nm. The emission spectra were recorded in the range of 375–500 nm. The program Origin 8.0 was employed for data processing.
UV–vis titrations
For UV–vis titrations, the sample solutions were prepared analogously to fluorescence titrations. The concentration of 3 was adjusted to 1.0⋅10−6 M while the concentration of Cr3+ ranged between 0.0 and 36.0⋅10−6 M. The absorption spectra were recorded at 240–300 nm. Origin 8.0 was employed for data processing.
X-ray analysis
Diffraction data of 3 were collected by a Bruker Apex II CCD diffractometer [56]. SHELXS was used to solve the structure of 3 [57]. Other crystallographic data are shown in Supporting Information File 1, Table S1.
Supporting Information
| Supporting Information File 1: Supporting crystallographic data, fluorescence, UV, HRMS, and IR spectra of 3 and 3·Cr3+, general considerations, characterization data, and copies of the 1H and 13C NMR spectra of all compounds. | ||
| Format: PDF | Size: 1.7 MB | Download |
References
-
Lee, M. H.; Kim, J. S.; Sessler, J. L. Chem. Soc. Rev. 2015, 44, 4185–4191. doi:10.1039/c4cs00280f
Return to citation in text: [1] -
Carter, K. P.; Young, A. M.; Palmer, A. E. Chem. Rev. 2014, 114, 4564–4601. doi:10.1021/cr400546e
Return to citation in text: [1] -
Zhang, X.; Yin, J.; Yoon, J. Chem. Rev. 2014, 114, 4918–4959. doi:10.1021/cr400568b
Return to citation in text: [1] -
Kim, H. N.; Ren, W. X.; Kim, J. S.; Yoon, J. Chem. Soc. Rev. 2012, 41, 3210–3244. doi:10.1039/c1cs15245a
Return to citation in text: [1] -
Li, Z.; Yu, M.; Zhang, L.; Yu, M.; Liu, J.; Wei, L.; Zhang, H. Chem. Commun. 2010, 46, 7169–7171. doi:10.1039/c0cc01687j
Return to citation in text: [1] -
Jiao, X.; Huang, K.; He, S.; Liu, C.; Zhao, L.; Zeng, X. Org. Biomol. Chem. 2019, 17, 108–114. doi:10.1039/c8ob02583e
Return to citation in text: [1] -
Jiao, X.; Liu, C.; He, S.; Zhao, L.; Zeng, X. Dyes Pigm. 2019, 160, 86–92. doi:10.1016/j.dyepig.2018.07.040
Return to citation in text: [1] -
Cheng, J.; Yang, E.; Ding, P.; Tang, J.; Zhang, D.; Zhao, Y.; Ye, Y. Sens. Actuators, B 2015, 221, 688–693. doi:10.1016/j.snb.2015.07.003
Return to citation in text: [1] [2] -
Arakawa, H.; Ahmad, R.; Naoui, M.; Tajmir-Riahi, H.-A. J. Biol. Chem. 2000, 275, 10150–10153. doi:10.1074/jbc.275.14.10150
Return to citation in text: [1] -
Anderson, R. A. Regul. Toxicol. Pharmacol. 1997, 26, S35–S41. doi:10.1006/rtph.1997.1136
Return to citation in text: [1] -
Vincent, J. B. Nutr. Rev. 2000, 58, 67–72. doi:10.1111/j.1753-4887.2000.tb01841.x
Return to citation in text: [1] -
Vincent, J. B. Proc. Nutr. Soc. 2004, 63, 41–47. doi:10.1079/pns2003315
Return to citation in text: [1] -
Costa, M.; Klein, C. Crit. Rev. Toxicol. 2006, 36, 779. doi:10.1080/10408440600932169
Return to citation in text: [1] -
Dai, R.; Yu, C.; Liu, J.; Lan, Y.; Deng, B. Environ. Sci. Technol. 2010, 44, 6959–6964. doi:10.1021/es100902y
Return to citation in text: [1] -
Tang, Y.; Liu, L.; Zhang, Y.; Zhao, H.; Kong, L.; Gao, S. New J. Chem. 2018, 42, 19340–19343. doi:10.1039/c8nj04398a
Return to citation in text: [1] -
Raju, V.; Selva Kumar, R.; Ashok Kumar, S. K.; Tharakeswar, Y.; Sahoo, S. K. Inorg. Chem. Commun. 2019, 101, 74–80. doi:10.1016/j.inoche.2019.01.011
Return to citation in text: [1] -
Wu, S.; Zhang, K.; Wang, Y.; Mao, D.; Liu, X.; Yu, J.; Wang, L. Tetrahedron Lett. 2014, 55, 351–353. doi:10.1016/j.tetlet.2013.11.024
Return to citation in text: [1] -
Shahim, S.; Sukesan, R.; Sarangadharan, I.; Wang, Y.-L. Sensors 2019, 19, 1969–1982. doi:10.3390/s19091969
Return to citation in text: [1] -
Yin, H.; Zhao, B.; Kan, W.; Liu, T.; Wang, W.; Yin, G.; Wang, L.; Gao, Y.; Wang, J. Spectrochim. Acta, Part A 2019, 217, 18–26. doi:10.1016/j.saa.2019.03.060
Return to citation in text: [1] -
Zhang, M.; Gong, L.; Sun, C.; Li, W.; Chang, Z.; Qi, D. Spectrochim. Acta, Part A 2019, 214, 7–13. doi:10.1016/j.saa.2019.01.089
Return to citation in text: [1] -
Sutariya, P. G.; Soni, H.; Gandhi, S. A.; Pandya, A. J. Lumin. 2019, 208, 6–17. doi:10.1016/j.jlumin.2018.12.009
Return to citation in text: [1] -
Koonrugsa, N.; Fuangswasdi, S. Spectrochim. Acta, Part A 2019, 215, 15–23. doi:10.1016/j.saa.2019.02.052
Return to citation in text: [1] -
Zhang, T.; Mu, L.; She, G.; Shi, W. J. Lumin. 2019, 209, 267–273. doi:10.1016/j.jlumin.2019.01.055
Return to citation in text: [1] -
Zhu, H.; Fan, J.; Wang, B.; Peng, X. Chem. Soc. Rev. 2015, 44, 4337–4366. doi:10.1039/c5cs90058a
Return to citation in text: [1] -
Shirinfar, B.; Ahmed, N.; Park, Y. S.; Cho, G.-S.; Youn, I. S.; Han, J.-K.; Nam, H. G.; Kim, K. S. J. Am. Chem. Soc. 2013, 135, 90–93. doi:10.1021/ja3112274
Return to citation in text: [1] -
Lim, N. C.; Pavlova, S. V.; Brückner, C. Inorg. Chem. 2009, 48, 1173–1182. doi:10.1021/ic801322x
Return to citation in text: [1] -
Xu, M.; Wu, S.; Zeng, F.; Yu, C. Langmuir 2010, 26, 4529–4534. doi:10.1021/la9033244
Return to citation in text: [1] -
Weerasinghe, A. J.; Schmiesing, C.; Varaganti, S.; Ramakrishna, G.; Sinn, E. J. Phys. Chem. B 2010, 114, 9413–9419. doi:10.1021/jp1034568
Return to citation in text: [1] -
Goswami, S.; Aich, K.; Das, S.; Das, A. K.; Sarkar, D.; Panja, S.; Mondal, T. K.; Mukhopadhyay, S. Chem. Commun. 2013, 49, 10676–10678. doi:10.1039/c3cc46860g
Return to citation in text: [1] -
Mahato, P.; Saha, S.; Suresh, E.; Di Liddo, R.; Parnigotto, P. P.; Conconi, M. T.; Kesharwani, M. K.; Ganguly, B.; Das, A. Inorg. Chem. 2012, 51, 1769–1777. doi:10.1021/ic202073q
Return to citation in text: [1] -
Liu, C.; Pan, J.; Li, S.; Zhao, Y.; Wu, L. Y.; Berkman, C. E.; Whorton, A. R.; Xian, M. Angew. Chem., Int. Ed. 2011, 50, 10327–10329. doi:10.1002/anie.201104305
Return to citation in text: [1] -
Choi, M. G.; Cha, S.; Lee, H.; Jeon, H. L.; Chang, S.-K. Chem. Commun. 2009, 7390–7392. doi:10.1039/b916476f
Return to citation in text: [1] -
Sasakura, K.; Hanaoka, K.; Shibuya, N.; Mikami, Y.; Kimura, Y.; Komatsu, T.; Ueno, T.; Terai, T.; Kimura, H.; Nagano, T. J. Am. Chem. Soc. 2011, 133, 18003–18005. doi:10.1021/ja207851s
Return to citation in text: [1] -
Zhang, L.; Lou, X.; Yu, Y.; Qin, J.; Li, Z. Macromolecules 2011, 44, 5186–5193. doi:10.1021/ma200777e
Return to citation in text: [1] -
Cao, X.; Lin, W.; He, L. Org. Lett. 2011, 13, 4716–4719. doi:10.1021/ol201932c
Return to citation in text: [1] -
Zhang, H.; Sun, T.; Ruan, Q.; Zhao, J.-L.; Mu, L.; Zeng, X.; Jin, Z.; Su, S.; Luo, Q.; Yan, Y.; Redshaw, C. Dyes Pigm. 2019, 162, 257–265. doi:10.1016/j.dyepig.2018.10.025
Return to citation in text: [1] -
Mabhai, S.; Dolai, M.; Dey, S. K.; Dhara, A.; Choudhury, S. M.; Das, B.; Dey, S.; Jana, A. Spectrochim. Acta, Part A 2019, 219, 319–332. doi:10.1016/j.saa.2019.04.056
Return to citation in text: [1] -
Hosseini, M.; Khoobi, M.; Tarasi, R.; Ganjali, M. R. Res. Chem. Intermed. 2018, 44, 5031–5042. doi:10.1007/s11164-018-3407-z
Return to citation in text: [1] -
Jang, H. J.; Kang, J. H.; Yun, D.; Kim, C. J. Fluoresc. 2018, 28, 785–794. doi:10.1007/s10895-018-2240-5
Return to citation in text: [1] -
Singh, J.; Kaur, V.; Singh, R.; Bhardwaj, V. K. Spectrochim. Acta, Part A 2018, 201, 46–53. doi:10.1016/j.saa.2018.04.056
Return to citation in text: [1] -
Miller, M. W.; Amidon, R. W.; Tawney, P. O. J. Am. Chem. Soc. 1955, 77, 2845–2848. doi:10.1021/ja01615a053
Return to citation in text: [1] -
Verlinden, K.; Ganter, C. J. Organomet. Chem. 2014, 750, 23–29. doi:10.1016/j.jorganchem.2013.10.047
Return to citation in text: [1] [2] -
Pickering, A. L.; Seeber, G.; Long, D.-L.; Cronin, L. CrystEngComm 2005, 7, 504–510. doi:10.1039/b506718a
Return to citation in text: [1] -
Upadhyay, Y.; Paira, P.; Ashok Kumar, S. K.; Choi, H.-J.; Kumar, R.; Sahoo, S. K. Inorg. Chim. Acta 2019, 489, 198–203. doi:10.1016/j.ica.2019.02.028
Return to citation in text: [1] -
Adhikari, S.; Ta, S.; Ghosh, A.; Guria, S.; Pal, A.; Ahir, M.; Adhikary, A.; Hira, S. K.; Manna, P. P.; Das, D. J. Photochem. Photobiol., A 2019, 372, 49–58. doi:10.1016/j.jphotochem.2018.12.010
Return to citation in text: [1] -
Jin, H.; Xu, J.; Zhang, L.; Ma, B.; Shi, X.; Fan, Y.; Wang, L. J. Solid State Chem. 2018, 268, 168–174. doi:10.1016/j.jssc.2018.08.035
Return to citation in text: [1] -
Liu, Q.-X.; Yao, Z.-Q.; Zhao, X.-J.; Zhao, Z.-X.; Wang, X.-G. Organometallics 2013, 32, 3493–3501. doi:10.1021/om400277z
Return to citation in text: [1] -
Goswami, S.; Hazra, A.; Chakrabarty, R.; Fun, H.-K. Org. Lett. 2009, 11, 4350–4353. doi:10.1021/ol901737s
Return to citation in text: [1] -
Ulatowski, F.; Dąbrowa, K.; Bałakier, T.; Jurczak, J. J. Org. Chem. 2016, 81, 1746–1756. doi:10.1021/acs.joc.5b02909
Return to citation in text: [1] -
Arunkumar, E.; Ajayaghosh, A.; Daub, J. J. Am. Chem. Soc. 2005, 127, 3156–3164. doi:10.1021/ja045760e
Return to citation in text: [1] -
Thordarson, P. Chem. Soc. Rev. 2011, 40, 1305–1323. doi:10.1039/c0cs00062k
Return to citation in text: [1] -
Caballero, A.; Martínez, R.; Lloveras, V.; Ratera, I.; Vidal-Gancedo, J.; Wurst, K.; Tárraga, A.; Molina, P.; Veciana, J. J. Am. Chem. Soc. 2005, 127, 15666–15667. doi:10.1021/ja0545766
Return to citation in text: [1] -
Li, S.; Zhang, D.; Xie, X.; Ma, S.; Liu, Y.; Xu, Z.; Gao, Y.; Ye, Y. Sens. Actuators, B 2016, 224, 661–667. doi:10.1016/j.snb.2015.10.086
Return to citation in text: [1] -
Braga, D.; Costa, A. L.; Grepioni, F.; Scaccianoce, L.; Tagliavini, E. Organometallics 1997, 16, 2070–2079. doi:10.1021/om9603878
Return to citation in text: [1] -
Foster, N. R.; Grieves, G. A.; Buchanan, J. W.; Flynn, N. D.; Duncan, M. A. J. Phys. Chem. A 2000, 104, 11055–11062. doi:10.1021/jp002131s
Return to citation in text: [1] -
SAINT Software Reference Manual (SMART 5.0 and SAINT 4.0 for Windows NT); Bruker Analytical X-ray Systems Inc.: Madison, WI, USA, 1998.
Return to citation in text: [1] -
Sheldrick, G. M. Acta Crystallogr., Sect. C: Struct. Chem. 2015, 71, 3–8. doi:10.1107/s2053229614024218
Return to citation in text: [1]
| 57. | Sheldrick, G. M. Acta Crystallogr., Sect. C: Struct. Chem. 2015, 71, 3–8. doi:10.1107/s2053229614024218 |
| 54. | Braga, D.; Costa, A. L.; Grepioni, F.; Scaccianoce, L.; Tagliavini, E. Organometallics 1997, 16, 2070–2079. doi:10.1021/om9603878 |
| 55. | Foster, N. R.; Grieves, G. A.; Buchanan, J. W.; Flynn, N. D.; Duncan, M. A. J. Phys. Chem. A 2000, 104, 11055–11062. doi:10.1021/jp002131s |
| 56. | SAINT Software Reference Manual (SMART 5.0 and SAINT 4.0 for Windows NT); Bruker Analytical X-ray Systems Inc.: Madison, WI, USA, 1998. |
| 1. | Lee, M. H.; Kim, J. S.; Sessler, J. L. Chem. Soc. Rev. 2015, 44, 4185–4191. doi:10.1039/c4cs00280f |
| 2. | Carter, K. P.; Young, A. M.; Palmer, A. E. Chem. Rev. 2014, 114, 4564–4601. doi:10.1021/cr400546e |
| 3. | Zhang, X.; Yin, J.; Yoon, J. Chem. Rev. 2014, 114, 4918–4959. doi:10.1021/cr400568b |
| 4. | Kim, H. N.; Ren, W. X.; Kim, J. S.; Yoon, J. Chem. Soc. Rev. 2012, 41, 3210–3244. doi:10.1039/c1cs15245a |
| 5. | Li, Z.; Yu, M.; Zhang, L.; Yu, M.; Liu, J.; Wei, L.; Zhang, H. Chem. Commun. 2010, 46, 7169–7171. doi:10.1039/c0cc01687j |
| 6. | Jiao, X.; Huang, K.; He, S.; Liu, C.; Zhao, L.; Zeng, X. Org. Biomol. Chem. 2019, 17, 108–114. doi:10.1039/c8ob02583e |
| 7. | Jiao, X.; Liu, C.; He, S.; Zhao, L.; Zeng, X. Dyes Pigm. 2019, 160, 86–92. doi:10.1016/j.dyepig.2018.07.040 |
| 8. | Cheng, J.; Yang, E.; Ding, P.; Tang, J.; Zhang, D.; Zhao, Y.; Ye, Y. Sens. Actuators, B 2015, 221, 688–693. doi:10.1016/j.snb.2015.07.003 |
| 15. | Tang, Y.; Liu, L.; Zhang, Y.; Zhao, H.; Kong, L.; Gao, S. New J. Chem. 2018, 42, 19340–19343. doi:10.1039/c8nj04398a |
| 16. | Raju, V.; Selva Kumar, R.; Ashok Kumar, S. K.; Tharakeswar, Y.; Sahoo, S. K. Inorg. Chem. Commun. 2019, 101, 74–80. doi:10.1016/j.inoche.2019.01.011 |
| 17. | Wu, S.; Zhang, K.; Wang, Y.; Mao, D.; Liu, X.; Yu, J.; Wang, L. Tetrahedron Lett. 2014, 55, 351–353. doi:10.1016/j.tetlet.2013.11.024 |
| 18. | Shahim, S.; Sukesan, R.; Sarangadharan, I.; Wang, Y.-L. Sensors 2019, 19, 1969–1982. doi:10.3390/s19091969 |
| 19. | Yin, H.; Zhao, B.; Kan, W.; Liu, T.; Wang, W.; Yin, G.; Wang, L.; Gao, Y.; Wang, J. Spectrochim. Acta, Part A 2019, 217, 18–26. doi:10.1016/j.saa.2019.03.060 |
| 20. | Zhang, M.; Gong, L.; Sun, C.; Li, W.; Chang, Z.; Qi, D. Spectrochim. Acta, Part A 2019, 214, 7–13. doi:10.1016/j.saa.2019.01.089 |
| 21. | Sutariya, P. G.; Soni, H.; Gandhi, S. A.; Pandya, A. J. Lumin. 2019, 208, 6–17. doi:10.1016/j.jlumin.2018.12.009 |
| 22. | Koonrugsa, N.; Fuangswasdi, S. Spectrochim. Acta, Part A 2019, 215, 15–23. doi:10.1016/j.saa.2019.02.052 |
| 23. | Zhang, T.; Mu, L.; She, G.; Shi, W. J. Lumin. 2019, 209, 267–273. doi:10.1016/j.jlumin.2019.01.055 |
| 48. | Goswami, S.; Hazra, A.; Chakrabarty, R.; Fun, H.-K. Org. Lett. 2009, 11, 4350–4353. doi:10.1021/ol901737s |
| 49. | Ulatowski, F.; Dąbrowa, K.; Bałakier, T.; Jurczak, J. J. Org. Chem. 2016, 81, 1746–1756. doi:10.1021/acs.joc.5b02909 |
| 50. | Arunkumar, E.; Ajayaghosh, A.; Daub, J. J. Am. Chem. Soc. 2005, 127, 3156–3164. doi:10.1021/ja045760e |
| 51. | Thordarson, P. Chem. Soc. Rev. 2011, 40, 1305–1323. doi:10.1039/c0cs00062k |
| 12. | Vincent, J. B. Proc. Nutr. Soc. 2004, 63, 41–47. doi:10.1079/pns2003315 |
| 13. | Costa, M.; Klein, C. Crit. Rev. Toxicol. 2006, 36, 779. doi:10.1080/10408440600932169 |
| 14. | Dai, R.; Yu, C.; Liu, J.; Lan, Y.; Deng, B. Environ. Sci. Technol. 2010, 44, 6959–6964. doi:10.1021/es100902y |
| 8. | Cheng, J.; Yang, E.; Ding, P.; Tang, J.; Zhang, D.; Zhao, Y.; Ye, Y. Sens. Actuators, B 2015, 221, 688–693. doi:10.1016/j.snb.2015.07.003 |
| 52. | Caballero, A.; Martínez, R.; Lloveras, V.; Ratera, I.; Vidal-Gancedo, J.; Wurst, K.; Tárraga, A.; Molina, P.; Veciana, J. J. Am. Chem. Soc. 2005, 127, 15666–15667. doi:10.1021/ja0545766 |
| 53. | Li, S.; Zhang, D.; Xie, X.; Ma, S.; Liu, Y.; Xu, Z.; Gao, Y.; Ye, Y. Sens. Actuators, B 2016, 224, 661–667. doi:10.1016/j.snb.2015.10.086 |
| 11. | Vincent, J. B. Nutr. Rev. 2000, 58, 67–72. doi:10.1111/j.1753-4887.2000.tb01841.x |
| 44. | Upadhyay, Y.; Paira, P.; Ashok Kumar, S. K.; Choi, H.-J.; Kumar, R.; Sahoo, S. K. Inorg. Chim. Acta 2019, 489, 198–203. doi:10.1016/j.ica.2019.02.028 |
| 45. | Adhikari, S.; Ta, S.; Ghosh, A.; Guria, S.; Pal, A.; Ahir, M.; Adhikary, A.; Hira, S. K.; Manna, P. P.; Das, D. J. Photochem. Photobiol., A 2019, 372, 49–58. doi:10.1016/j.jphotochem.2018.12.010 |
| 46. | Jin, H.; Xu, J.; Zhang, L.; Ma, B.; Shi, X.; Fan, Y.; Wang, L. J. Solid State Chem. 2018, 268, 168–174. doi:10.1016/j.jssc.2018.08.035 |
| 9. | Arakawa, H.; Ahmad, R.; Naoui, M.; Tajmir-Riahi, H.-A. J. Biol. Chem. 2000, 275, 10150–10153. doi:10.1074/jbc.275.14.10150 |
| 10. | Anderson, R. A. Regul. Toxicol. Pharmacol. 1997, 26, S35–S41. doi:10.1006/rtph.1997.1136 |
| 47. | Liu, Q.-X.; Yao, Z.-Q.; Zhao, X.-J.; Zhao, Z.-X.; Wang, X.-G. Organometallics 2013, 32, 3493–3501. doi:10.1021/om400277z |
| 41. | Miller, M. W.; Amidon, R. W.; Tawney, P. O. J. Am. Chem. Soc. 1955, 77, 2845–2848. doi:10.1021/ja01615a053 |
| 43. | Pickering, A. L.; Seeber, G.; Long, D.-L.; Cronin, L. CrystEngComm 2005, 7, 504–510. doi:10.1039/b506718a |
| 36. | Zhang, H.; Sun, T.; Ruan, Q.; Zhao, J.-L.; Mu, L.; Zeng, X.; Jin, Z.; Su, S.; Luo, Q.; Yan, Y.; Redshaw, C. Dyes Pigm. 2019, 162, 257–265. doi:10.1016/j.dyepig.2018.10.025 |
| 37. | Mabhai, S.; Dolai, M.; Dey, S. K.; Dhara, A.; Choudhury, S. M.; Das, B.; Dey, S.; Jana, A. Spectrochim. Acta, Part A 2019, 219, 319–332. doi:10.1016/j.saa.2019.04.056 |
| 38. | Hosseini, M.; Khoobi, M.; Tarasi, R.; Ganjali, M. R. Res. Chem. Intermed. 2018, 44, 5031–5042. doi:10.1007/s11164-018-3407-z |
| 39. | Jang, H. J.; Kang, J. H.; Yun, D.; Kim, C. J. Fluoresc. 2018, 28, 785–794. doi:10.1007/s10895-018-2240-5 |
| 40. | Singh, J.; Kaur, V.; Singh, R.; Bhardwaj, V. K. Spectrochim. Acta, Part A 2018, 201, 46–53. doi:10.1016/j.saa.2018.04.056 |
| 42. | Verlinden, K.; Ganter, C. J. Organomet. Chem. 2014, 750, 23–29. doi:10.1016/j.jorganchem.2013.10.047 |
| 30. | Mahato, P.; Saha, S.; Suresh, E.; Di Liddo, R.; Parnigotto, P. P.; Conconi, M. T.; Kesharwani, M. K.; Ganguly, B.; Das, A. Inorg. Chem. 2012, 51, 1769–1777. doi:10.1021/ic202073q |
| 31. | Liu, C.; Pan, J.; Li, S.; Zhao, Y.; Wu, L. Y.; Berkman, C. E.; Whorton, A. R.; Xian, M. Angew. Chem., Int. Ed. 2011, 50, 10327–10329. doi:10.1002/anie.201104305 |
| 32. | Choi, M. G.; Cha, S.; Lee, H.; Jeon, H. L.; Chang, S.-K. Chem. Commun. 2009, 7390–7392. doi:10.1039/b916476f |
| 33. | Sasakura, K.; Hanaoka, K.; Shibuya, N.; Mikami, Y.; Kimura, Y.; Komatsu, T.; Ueno, T.; Terai, T.; Kimura, H.; Nagano, T. J. Am. Chem. Soc. 2011, 133, 18003–18005. doi:10.1021/ja207851s |
| 34. | Zhang, L.; Lou, X.; Yu, Y.; Qin, J.; Li, Z. Macromolecules 2011, 44, 5186–5193. doi:10.1021/ma200777e |
| 35. | Cao, X.; Lin, W.; He, L. Org. Lett. 2011, 13, 4716–4719. doi:10.1021/ol201932c |
| 24. | Zhu, H.; Fan, J.; Wang, B.; Peng, X. Chem. Soc. Rev. 2015, 44, 4337–4366. doi:10.1039/c5cs90058a |
| 25. | Shirinfar, B.; Ahmed, N.; Park, Y. S.; Cho, G.-S.; Youn, I. S.; Han, J.-K.; Nam, H. G.; Kim, K. S. J. Am. Chem. Soc. 2013, 135, 90–93. doi:10.1021/ja3112274 |
| 26. | Lim, N. C.; Pavlova, S. V.; Brückner, C. Inorg. Chem. 2009, 48, 1173–1182. doi:10.1021/ic801322x |
| 27. | Xu, M.; Wu, S.; Zeng, F.; Yu, C. Langmuir 2010, 26, 4529–4534. doi:10.1021/la9033244 |
| 28. | Weerasinghe, A. J.; Schmiesing, C.; Varaganti, S.; Ramakrishna, G.; Sinn, E. J. Phys. Chem. B 2010, 114, 9413–9419. doi:10.1021/jp1034568 |
| 29. | Goswami, S.; Aich, K.; Das, S.; Das, A. K.; Sarkar, D.; Panja, S.; Mondal, T. K.; Mukhopadhyay, S. Chem. Commun. 2013, 49, 10676–10678. doi:10.1039/c3cc46860g |
| 42. | Verlinden, K.; Ganter, C. J. Organomet. Chem. 2014, 750, 23–29. doi:10.1016/j.jorganchem.2013.10.047 |
© 2019 Liu et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)