Abstract
The synthesis of (4R,5R)-streptopyrrolidine (1), (4S,5R)-streptopyrrolidine (2) (4R,5S)-streptopyrrolidine (3) and (4S,5S)-streptopyrrolidine (4) have been achieved in a concise and highly efficient manner via a highly stereoselective aldol type reaction with the trimethylsilyl enolate of ethyl acetate and Lewis acid mediated lactamization as the key reactions in ≈42% yield over six steps starting from D-phenylalanine and L-phenylalanine, respectively. The absolute configuration of the natural product was shown to be (4S,5S) by comparing its spectral and analytical data with the reported values.

Graphical Abstract
Introduction
Cancer is at present the second most common cause of death, after cardiovascular diseases, and will become the primary cause in the next 10 to 20 years [1]. Traditional cancer therapies make use of chemotherapy at the maximum tolerated dose. This approach has generally considerable associated toxicity, often with limited success. Therefore, more universal, more effective, and less toxic therapeutic agents are desirable. Recently, inhibition of angiogenesis has been considered as a desirable pathway for preventing tumor growth and metastasis, primarily because of the low potential for toxicity or resistance [2], as well as the potential for treating a broad spectrum of tumor types, arthritis, and psoriasis [3-8]. For this reason, angiogenesis inhibition has become an active area of pharmaceutical research, and over 40 such agents are currently undergoing clinical trials [9]. In particular, efforts have been focused on small-molecules based on inhibitors isolated from natural products that can block tumor angiogenesis [10,11].
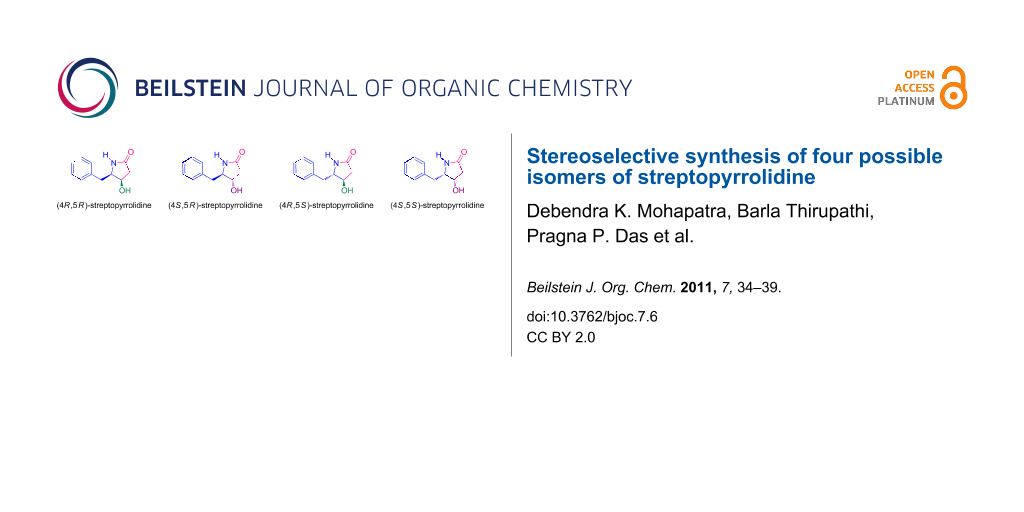
Recently, streptopyrrolidine (Figure 1), was isolated as an angiogenesis inhibitor from the fermentation broth of a marine Streptomyces sp. found in deep sea sediments [12]. The system is present in many biologically active compounds [13-21] and it could act as a versatile intermediate for the synthesis of a wide range of γ-amino acids as well as pyrrolidines [22,23]. The interesting chemical structure and potent anti-angiogenic activity at non-toxic threshold doses of streptopyrrolidine attracted our attention for developing a concise and efficient protocol for its synthesis in sizable amounts for further biological studies. To date, three syntheses [15,24,25] of (4S,5S)-streptopyrrolidine have been described, two of which [15,24] were reported only as a synthetic intermediate prior to its isolation from the natural source.
![[1860-5397-7-6-1]](/bjoc/content/figures/1860-5397-7-6-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structures of (4R,5R)-streptopyrrolidine (1), (4S,5R)-streptopyrrolidine (2) (4R,5S)-streptopyrrolidine (3) and (4S,5S)-streptopyrrolidine (4).
Figure 1: Structures of (4R,5R)-streptopyrrolidine (1), (4S,5R)-streptopyrrolidine (2) (4R,5S)-streptopyrroli...
Results and Discussion
In this paper, we report the syntheses of (4R,5R)-streptopyrrolidine (1), (4S,5R)-streptopyrrolidine (2), (4R,5S)-streptopyrrolidine (3) and (4S,5S)-streptopyrrolidine (4) in a concise and highly efficient manner via a highly stereoselective aldol type reaction and Lewis acid mediated lactamization as the key reactions. A retrosynthetic analysis for streptopyrrolidine is depicted in Scheme 1.
![[1860-5397-7-6-i1]](/bjoc/content/inline/1860-5397-7-6-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
The synthesis was initiated from D-phenylalanine (7) which was converted to N-Boc-D-phenylalaninal (6) in 76% yield in three steps by a known protocol [26,27]. The aldehyde was treated with the lithium enolate of ethyl acetate [28,29] at −78 °C according to a modification of the procedure of Steulmann and Klostermeyer [30] to afford two diastereomers 8a and 8b in a 3:2 ratio (as determined by NMR) (Scheme 2).
![[1860-5397-7-6-i2]](/bjoc/content/inline/1860-5397-7-6-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Reagents and conditions: (a) LDA, EtOAc, THF, −78 °C, 4 h, 80% (8a:8b = 3:2); (b) BF3·OEt2, (1-ethoxyvinyloxy)trimethylsilane, CH2Cl2, −78 °C, 2 h, 85% (exclusively 8b); (c) SnCl4, (1-ethoxyvinyloxy)trimethylsilane, CH2Cl2, −78 °C, 2 h, 70% (8a:8b = 3:7); (d) LDA, EtOAc, ZnBr2, −78 °C, 1 h, 82%, (8a:8b = 95:5).
Scheme 2: Reagents and conditions: (a) LDA, EtOAc, THF, −78 °C, 4 h, 80% (8a:8b = 3:2); (b) BF3·OEt2, (1-etho...
Diastereomers 8a and 8b were easily separated by standard column chromatography on silica gel. The pioneering work on catalytic aldol reactions recently reported by Shibasaki [31] prompted us to investigate whether the ketene silyl acetal could be utilized for the aldol reaction in the presence of different Lewis acids to obtain a better selectivity particularly for (R)-tert-butyl (1-oxo-3-phenylpropan-2-yl)carbamate (6). To our surprise, when the reaction was carried out with the ketene silyl acetate at −78 °C, of the five Lewis acids investigated (SnCl4, BF3·OEt2, ZnI2, TiCl4, EtOAc/LDA/ZnBr2) BF3·OEt2 gave excellent selectivity (exclusively 8b) (Table 1).
Table 1: Addition reaction under different conditions to obtain 8a and 8b.
| Entry | Reagent | Additive (Lewis Acid) | Time (h) | Yielda (%) | 8a:8b |
|---|---|---|---|---|---|
| 1 | ketene silyl acetate | SnCl4 | 2 | 70 | 30:70 |
| 2 | ketene silyl acetate | BF3·OEt2 | 2 | 85 | 0:100 |
| 3 | ketene silyl acetate | TiCl4 | 10 | 42 | 35:65 |
| 4 | ketene silyl acetate | ZnI2 | 12 | 59 | 80:20 |
| 5 | EtOAC/LDA | ZnBr2 | 1 | 82 | 95:05 |
aisolated yield.
Following a modification of the Mosher method [32,33], the newly created stereogenic center in compound 8b bearing the hydroxyl group was assigned. The syntheses of both the (S)- and (R)-MTPA ester of 8b were achieved using MTPA acid with DCC as the coupling reagent. The chemical shifts of both the (S)- and (R)-MTPA esters of 8b were assigned by 1H NMR. From the equation given in Figure 2, the Δδ values were calculated for as many protons as possible. The carbon chain bearing protons showing Δδ negative values should be placed on the left hand side of the model (Figure 2) whilst that where Δδ has positive values should be placed on the right hand side. From this the center was found to have the S-configuration which thus establishes the absolute stereochemistry of 8a. With the absolute stereochemistry of both the isomers known, we were interested to develop a protocol to have control over the selectivity to obtain exclusively 8a. Stereoselective addition of the zinc enolate [34] of ethyl acetate to N-Boc-phenylalaninal (6) resulted in 8a as a 95:5 mixture of diastereomers (as determined by HPLC), which were easily separated by silica gel column chromatography. The stereochemical outcome in both the cases was explained by Crams’ rule, i.e., for a non-chelation model, the (R)-aldehyde would give rise to the (3S,4R) isomer. Alternatively, in a chelation model, the metal imposes a syn relationship between the formyl and neighboring nitrogen functionality which would lead to the (3R,4R) isomer [35].
![[1860-5397-7-6-2]](/bjoc/content/figures/1860-5397-7-6-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Δδ = (δS−δR) × 103 for (S)- and (R)-MTPA esters of compound 8b.
Figure 2: Δδ = (δS−δR) × 103 for (S)- and (R)-MTPA esters of compound 8b.
Deprotection of the Boc-group with TFA and CH2Cl2 was followed by evaporation and treatment of the crude product under different reaction conditions for lactamization (Table 2 is for lactamization of 8a, almost similar yields were obtained for 8b), afforded (4R,5R)-streptopyrrolidine (1) and (4S,5R)-streptopyrrolidine (2) in good to excellent overall yield (Scheme 3). From all of the conditions investigated, Zr(OtBu)4 in presence of 1-hydroxy-7-azabenzotriazole (HOAt) gave the best result [36], i.e., 82% yield over the two steps. The spectral and analytical data {[α]D25 +40.5 (c 1.8, MeOH); lit. [15,25] [α]D25 −43.5 (c 1.0, MeOH) for its enantiomer} of compound 1 [6] were in good agreement with the reported values for its enantiomer except the sign of the specific rotation, which confirmed the absolute configuration of the natural product as (4S,5S). To confirm further the absolute stereochemistry of natural streptopyrrolidine, (4R,5S)-streptopyrrolidine (3) and (4S,5S)-streptopyrrolidine (4) were prepared starting from L-phenylalanine. The spectral and analytical data of (4S,5S)-streptopyrrolidine were in good agreement with natural streptopyrrolidine.
Table 2: Lactamization under different reaction conditions to obtain 1 and 2.
| Entry | Reagent | Additive/ Solvent | Temp. (°C) | Time (h) | Yielda (%) |
|---|---|---|---|---|---|
| 1 | Py | none/CH2Cl2 | 50 | 12 | 41 |
| 2 | Et3N | none/CH2Cl2 | 50 | 12 | 46 |
| 3 | Py | none/toluene | 100 | 6 | 55 |
| 4 | Et3N | none/toluene | 100 | 6 | 58 |
| 5 | KOtBu | HOAt/toluene | 60 | 6 | 42 |
| 6 | Zr(OtBu)4 | HOAt/toluene | 60 | 6 | 82 |
ayield over two steps.
![[1860-5397-7-6-i3]](/bjoc/content/inline/1860-5397-7-6-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Reagents and conditions: (a) (1) TFA, CH2Cl2, rt, 4 h; (2) Zr(OtBu)4, HOAt, toluene, 60 °C, 12 h, 82% over two steps.
Scheme 3: Reagents and conditions: (a) (1) TFA, CH2Cl2, rt, 4 h; (2) Zr(OtBu)4, HOAt, toluene, 60 °C, 12 h, 8...
Conclusion
In conclusion, the total synthesis of all four possible isomers of streptopyrrolidine (1–4) has been achieved in ≈42% yield over 6 steps starting from either D- or L-phenylalanine which further confirmed the absolute configuration of natural streptopyrrolidine. Our protocol is highly flexible for the synthesis of all four possible isomers of streptopyrrolidine compared to previous reports.
Experimental
General information
All reactions were carried out under an inert atmosphere, unless otherwise stated. Solvents were dried and purified by standard methods prior to use. The progress of all reactions was monitored by TLC using glass plates pre-coated with silica gel 60 F254 with a thickness of 0.5 mm. Column chromatography was performed on silica gel (60 mesh) with ethyl acetate and hexane the eluent. Optical rotations were measured with a Perkin Elmer P241 polarimeter and a JASCO DIP-360 digital polarimeter at 25 °C. IR spectra were recorded on a Perkin-Elmer FT-IR spectrometer. 1H and 13C NMR spectra were recorded on a Variant Gemini 200 MHz, Bruker Avance 300 MHz, or Varian Inova 500 MHz spectrometer with TMS as an internal standard in CDCl3, CD3OD etc. Mass spectra were recorded on a Micromass VG-7070H for EI.
(3R,4R)-Ethyl-4-(tert-butoxycarbonylamino)-3-hydroxy-5-phenylpentanoate (8a) and (3S,4R)-ethyl-4-(tert-butoxycarbonylamino)-3-hydroxy-5-phenylpentanoate (8b): To a stirred solution of the aldehyde 6 (0.2 g, 0.8 mmol) in CH2Cl2 (5 mL) under a nitrogen atmosphere and cooled to −78 °C, was added 1 M solution of SnCl4 in CH2Cl2 (0.1 mL, 0.81 mmol). After 10 min, (1-ethoxyvinyloxy)trimethylsilane (0.26 g, 1.6 mmol) was added at the same temperature. The reaction mixture was stirred for 1 h at −78 °C. After completion of the reaction (as determined by TLC), the reaction mixture was quenched with 1 N KOH (3 mL). The reaction mixture was extracted with ethyl acetate (3 × 25 mL). The combined organic layers were washed with brine, dried over Na2SO4 and concentrated under reduced pressure to obtain pale yellow liquid which on purification by silica gel column chromatography afforded 8a and 8b in a ratio of 3:7 (0.19 g, 70%) as colorless viscous liquid.
To a stirred solution of the aldehyde 6 (0.1 g, 0.4 mmol) in CH2Cl2 (5 mL) under a nitrogen atmosphere and cooled to −78 °C, was added BF3·OEt2 (0.03 g, 0.2 mmol). After 30 min, (1-ethoxy vinyloxy)trimethylsilane (0.26 g, 1.6 mmol) was added at the same temperature. The reaction mixture was stirred for 1 h at −78 °C when TLC showed completion of the reaction. The reaction mixture was quenched with saturated aqueous Na2CO3, extracted with ethyl acetate (3 × 25 mL). The combined organic layers were washed with brine, dried over Na2SO4 and concentrated under reduced pressure to give a pale yellow liquid. The crude product was purified by silica gel column chromatography with ethyl acetate and hexane (1:3) as eluent to yield 8b as a colorless viscous liquid as the sole product (0.115 g, 85%).
A stirred solution of LDA (4.9 mL, 2 N, 9.8 mmol) in anhydrous THF (10 mL) was cooled to −78 °C under nitrogen atmosphere. Ethyl acetate (1.05 mL, 9.8 mmol) was then added followed by a 0 °C solution of anhydrous ZnBr2 (2.17 g, 9.8 mmol) in anhydrous THF (5 mL). A −78 °C solution of N-Boc-protected aldehyde 6 (0.35 g, 1.41 mmol) in anhydrous THF (4 mL) was added and the mixture stirred at −78 °C for 30 min then allowed to warm to room temperature. The reaction mixture was stirred at room temperature for 10 h. After completion of the reaction (as determined by TLC), saturated NH4Cl/acetic acid (9:1) (20 mL) was added to the reaction mixture which was then extracted with ethyl acetate (3 × 30 mL). The combined organic layers were washed with brine (2 × 50 mL), dried over Na2SO4, concentrated under reduced pressure, and the crude product purified by silica gel column chromatography to give 8a (0.378 g, 82%) as a colorless viscous liquid. Analytical and spectral data of 8a: [α]D25 +34.9 (c 1.2, MeOH); IR (KBr): 3553, 3496, 3376, 2979, 2934, 1727, 1683 cm−1; 1H NMR (300 MHz, CDCl3): 7.28–7.20 (m, 5H, ArH), 5.07 (d, J = 9.6 Hz, 1H, NH), 4.16–4.09 (q, J = 6.9, 14.1 Hz, 2H, OCH2CH3), 3.98 (d, J = 9.6 Hz, 1H, CHOH), 3.74 (q, J = 8.1, 16.2 Hz, 1H, CHNH), 2.91 (d, J = 7.5 Hz, 2H, PhCH2), 2.59 (dd, J = 10.1, 16.8 Hz, 1H, CH'2COO), 2.38 (dd, J = 2.3, 16.9 Hz, 1H, CH2COO), 1.41 (s, 9H, t-Bu), 1.23 (t, J = 6.9 Hz, 3H, OCH2CH3); 13C NMR (75 MHz, CDCl3): 173.4, 155.8, 138.1, 129.3, 128.4, 126.3, 79.3, 67.0, 60.7, 55.3, 38.5, 29.6, 28.3, 14.0; ESI-MS: m/z = 338 [M + H]+; ESI-HRMS: Calcd. for C18H27NNaO5, 360.1781; found: 360.1789. Analytical and spectral data of 8b: [α]D25 +11.6 (c 0.6, MeOH); IR (KBr): 3354, 2982, 2936, 1735, 1684 cm−1; 1H NMR (300 MHz, CDCl3) 7.31–7.20 (m, 5H, ArH), 4.61 (d, J = 8.4 Hz, 1H, NH), 4.20–4.13 (q, J = 7.1, 14.1 Hz, 2H, OCH2CH3), 3.99–3.86 (m, 2H, CHOH, CHNH), 2.96 (dd, J = 3.8, 13.7 Hz, 1H, PhCH'2), 2.82 (m, 1H, PhCH2), 2.64–2.45 (m, 2H, CH2COO), 1.34 (s, 9H, t-Bu), 1.25 (t, J = 7.1 Hz, 3H, OCH2CH3); 13C NMR (75 MHz, CDCl3): 172.9, 155.7, 137.6, 129.4, 128.4, 126.4, 79.6, 70.1, 60.8, 55.1, 38.2, 35.8, 28.2, 14.1; ESI-MS: m/z = 360 [M + Na]+; ESI-HRMS: Calcd. for C18H27NNaO5, 360.1781; found: 360.1794.
(4R,5R)-streptopyrrolidine (1), (4S,5R)-streptopyrrolidine (2), (4R,5S)-streptopyrrolidine (3), (4S,5S)-streptopyrrolidine (4): A solution of TFA/H2O (2.8 mL, 0.28 mL) was added to 8a or 8b (200 mg, 0.59 mmol) and the resulting mixture stirred at room temperature for 3 h. After completion of the reaction (as determined by TLC), the solvent was evaporated under reduced pressure and the resulting reddish oil dissolved in ethyl acetate (10 mL). The organic phase was washed with NaHCO3, dried over Na2SO4 and concentrated under reduced pressure. The red oil so obtained was dissolved in toluene. Zr(OtBu)4 (22 mg, 0.059 mmol) followed by HOAt (160 mg, 0.11 mmol) were added and the reaction mixture allowed to stir at 60 °C for 12 h. After completion of the reaction (as determined by TLC), the toluene was evaporated under reduced pressure. Water was added and the reaction mixture extracted with CH2Cl2 (3 × 20 mL). The combined organic layers were dried over Na2SO4 and concentrated under reduced pressure to give a brown liquid which on purification by silica gel column chromatography furnished 1 (98 mg, 82%) as a white sticky solid. Analytical and spectral data of 1: [α]D25 +40.5 (c 1.8, MeOH); IR (KBr): 3452, 3234, 2924, 1690 cm−1; 1H NMR (500 MHz, DMSO-d6): 7.51 (s, 1H, NH), 7.27 (m, 4H, ArH), 7.17 (m, 1H, ArH), 5.14 (d, J = 4.1 Hz, 1H, OH), 4.10 (s, 1H, H-4), 3.67 (q, J = 5.6 Hz, 1H, H-5), 2.96 (dd, J = 7.9, 13.4 Hz, 1H, NCHCH'2), 2.65 (dd, J = 6.2, 13.4 Hz, 1H, NCHCH2), 2.38 (dd, J = 6.1, 16.4 Hz, 1H, H-3'), 1.96 (dd, J = 2.6, 16.4 Hz, 1H, H-3); 13C NMR (75 MHz, DMSO-d6): 174.8, 138.6, 129.2, 128.1, 125.9, 66.9, 60.0, 40.8, 34.5; EIMS: 192 [M+H]+; ESI-HRMS: Calcd. for C11H14NO2, 192.1019; found: 192.1026. Analytical and spectral data of 2: [α]D25 −13.2 (c 1.6, MeOH); IR (neat): 3384, 2925, 2855, 1679 cm−1; 1H NMR (500 MHz, DMSO-d6): 7.68 (s, 1H, NH), 7.29–7.21 (m, 5H, ArH), 5.13 (d, J = 3.9 Hz, 1H, OH), 3.95 (s, 1H, H-4), 3.51 (t, J = 6.8 Hz, 1H, H-5), 2.69 (m, 2H, NCHCH2), 2.27 (dd, J = 6.8, 17.5 Hz, 1H, H-3'), 1.80 (dd, J = 1.9, 16.5 Hz, 1H, H-3); 13C NMR (75 MHz, DMSO-d6): 174.8, 137.6, 129.4, 128.4, 126.2, 69.3, 63.4, 40.0, 38.9; ESI-HRMS: Calcd. for C11H14NO2, 192.1019; found: 192.1024. Analytical and spectral data of 3: [α]D25 +12.6 (c 1.2, MeOH); IR (KBr): 3462, 2928, 1784, 1730,1450, 1147, 1069, 744.8 cm−1; 1H NMR (300 MHz, DMSO-d6): δ 7.70 (s, 1H, NH), 7.18–7.32 (m, 5H, ArH), 5.14 (d, J = 3.7 Hz, 1H, OH), 3.95 (s, 1H, H-4), 3.51 (q, J = 6.0, 13.2 Hz, 1H, H-5), 2.67 (d, J = 2.2 Hz, 2H, NCHCH2), 2.27 (dd, 1H, J = 6.0, 16.6 Hz, 1H, H-3'), 1.82 (dd, J = 2.2, 16.6 Hz, 1H, H-3); 13C NMR (75 MHz, DMSO-d6): 174.8, 137.6, 129.4, 128.2, 126.0, 69.2, 63.4, 39.9, 39.1; ESI-HRMS: Calcd. for C11H14NO2, 192.1019; found: 192.1026. Analytical and spectral data of 4: [α]D25 −41.2 (c 1.0, MeOH); IR (KBr): 3451, 1785, 1453, 1185, 1147, 1058, 786, 698 cm−1; 1H NMR (300 MHz, DMSO-d6): δ 7.55 (s, 1H, NH), 7.08–7.37 (m, 5H, ArH), 5.18 (d, J = 3.8 Hz, 1H, OH), 4.09(s, 1H, H-4), 3.67 (s, 1H, H-5), 2.96 (dd, J = 7.1, 12.6 Hz, 1H, NCHCH2'), 2.66 (dd, J = 8.0, 16.9 Hz, 1H, NCHCH2), 2.38 (dd, J = 6.1, 15.6 Hz, 1H, H-3'), 1.99 (dd, J = 3.0, 15.6 Hz, 1H, H-3); 13C NMR (75 MHz, DMSO-d6): 174.9, 138.6, 129.2, 128.2, 126.8, 66.9, 60.1, 40.8, 34.5; ESI-HRMS: Calcd. for C11H14NO2, 192.1019; found: 192.1026.
Supporting Information
Supporting Information features 1H and 13C NMR spectra of all intermediates.
| Supporting Information File 1: 1H and 13C NMR spectra of all intermediates. | ||
| Format: PDF | Size: 444.7 KB | Download |
References
-
Cancer Facts & Figures. http://www.cancer.org/Research/CancerFactsFigures/CancerFactsFigures/index (accessed Sept 1, 2010).
Return to citation in text: [1] -
Folkman, J. Nat. Med. 1995, 1, 27–30. doi:10.1038/nm0195-27
Return to citation in text: [1] -
Folkman, J. J. Natl. Cancer Inst. 1990, 82, 4–6. doi:10.1093/jnci/82.1.4
Return to citation in text: [1] -
Folkman, J. Breast Cancer Res. Treat. 1995, 36, 109–118. doi:10.1007/BF00666033
Return to citation in text: [1] -
Hanahan, D.; Folkman, J. Cell 1996, 86, 353–364. doi:10.1016/S0092-8674(00)80108-7
Return to citation in text: [1] -
Ferrara, N.; Alitalo, K. Nat. Med. 1999, 5, 1359–1364. doi:10.1038/70928
Return to citation in text: [1] [2] -
Yancopouls, G. D.; Davis, S.; Gale, N. W.; Rudge, J. S.; Wiegand, S. J.; Holash, J. Nature 2000, 407, 242–248. doi:10.1038/35025215
Return to citation in text: [1] -
Carmeliet, P.; Jain, R. K. Nature 2000, 407, 249–257. doi:10.1038/35025220
Return to citation in text: [1] -
Clinical Trials Home Page - National Cancer Institute. http://www.cancer.gov/clinicaltrials (accessed Sept 1, 2010).
(See for information on current cancer clinical trials.)
Return to citation in text: [1] -
McDermott, L. A.; Higgins, B.; Simcox, M.; Luk, K.-C.; Nevins, T.; Kolinsky, K.; Smith, M.; Yang, H.; Li, J. K.; Chen, Y.; Ke, J.; Mallalieu, N.; Egan, T.; Kolis, S.; Railkar, A.; Gerber, L.; Liu, J.-J.; Konzelmann, F.; Zhang, Z.; Flynn, T.; Morales, O.; Chen, Y. Bioorg. Med. Chem. Lett. 2006, 16, 1950–1953. doi:10.1016/j.bmcl.2005.12.092
Return to citation in text: [1] -
Eskens, F. A. L. M.; Verweij, J. Eur. J. Cancer 2006, 42, 3127–3139. doi:10.1016/j.ejca.2006.09.015
Return to citation in text: [1] -
Shin, H. J.; Kim, T. S.; Lee, H.-S.; Park, J. Y.; Choi, I.-K.; Kwon, H. J. Phytochemistry 2008, 69, 2363–2366. doi:10.1016/j.phytochem.2008.05.020
Return to citation in text: [1] -
Vedejs, E.; Campbell, J. B., Jr.; Gadwood, R. C.; Rodgers, J. D.; Spear, K. L.; Watanabe, Y. J. Org. Chem. 1982, 47, 1534–1546. doi:10.1021/jo00347a034
Return to citation in text: [1] -
Midland, M. M.; Afonso, M. M. J. Am. Chem. Soc. 1989, 111, 4368–4371. doi:10.1021/ja00194a033
Return to citation in text: [1] -
Poncet, J.; Jouin, P.; Castro, B.; Nicolas, J.; Boutar, M.; Gaudemer, A. J. Chem. Soc., Perkin Trans. 1 1990, 611–616. doi:10.1039/P19900000611
Return to citation in text: [1] [2] [3] [4] -
Ohta, T.; Shiokawa, S.; Sakamoto, R.; Nozoe, S. Tetrahedron Lett. 1990, 31, 7329–7332. doi:10.1016/S0040-4039(00)88557-3
Return to citation in text: [1] -
Koot, W.-J.; van Ginkel, R.; Kranenburg, M.; Hiemstra, H.; Louwrier, S.; Moolenaar, M. J.; Speckamp, W. M. Tetrahedron Lett. 1991, 32, 401–404. doi:10.1016/S0040-4039(00)92639-X
Return to citation in text: [1] -
Rinehart, K. L.; Sakai, R.; Kishore, V.; Sullins, D. W.; Li, K.-M. J. Org. Chem. 1992, 57, 3007–3013. doi:10.1021/jo00037a012
Return to citation in text: [1] -
Galeotti, N.; Poncet, J.; Chiche, L.; Jouin, P. J. Org. Chem. 1993, 58, 5370–5376. doi:10.1021/jo00072a018
Return to citation in text: [1] -
Reddy, G. V.; Rao, G. V.; Iyengar, D. S. Tetrahedron Lett. 1999, 40, 775–776. doi:10.1016/S0040-4039(98)02409-5
Return to citation in text: [1] -
Lennartz, M.; Sadakane, M.; Steckhan, E. Tetrahedron 1999, 55, 14407–14420. doi:10.1016/S0040-4020(99)00904-7
Return to citation in text: [1] -
Huang, P. G.; Zheng, X.; Wang, S. L.; Ye, J. L.; Jin, L. R.; Chen, Z. Tetrahedron: Asymmetry 1999, 10, 3309–3317. doi:10.1016/S0957-4166(99)00321-3
Return to citation in text: [1] -
Park, T. H.; Paik, S.; Lee, S. H. Bull. Korean Chem. Soc. 2003, 24, 1227–1228.
Return to citation in text: [1] -
Kondekar, N. B.; Kandula, S. R. V.; Kumar, P. Tetrahedron Lett. 2004, 45, 5477–5479. doi:10.1016/j.tetlet.2004.05.057
Return to citation in text: [1] [2] -
Xiang, S.-H.; Yuan, H.-Q.; Huang, P.-Q. Tetrahedron: Asymmetry 2009, 20, 2021–2026. doi:10.1016/j.tetasy.2009.08.018
Return to citation in text: [1] [2] -
McKennon, M. J.; Meyers, A. I.; Drauz, K.; Schwarm, M. J. Org. Chem. 1993, 58, 3568–3571. doi:10.1021/jo00065a020
Return to citation in text: [1] -
Myers, A. G.; Zhong, B.; Movassaghi, M.; Kung, D. W.; Lanman, B. A.; Kwon, S. Tetrahedron Lett. 2000, 41, 1359–1362. doi:10.1016/S0040-4039(99)02293-5
Return to citation in text: [1] -
Rich, D. H.; Sun, E. T.; Boparia, A. S. J. Org. Chem. 1978, 43, 3624–3626. doi:10.1021/jo00412a053
Return to citation in text: [1] -
McConnel, R. M.; Frizzell, D.; Camp, A.; Evans, A.; Jones, W.; Cagle, C. J. Med. Chem. 1991, 34, 2298–2300. doi:10.1021/jm00111a054
Return to citation in text: [1] -
Steulmann, R.; Klostermeyer, H. Justus Liebigs Ann. Chem. 1975, 2245–2247.
Return to citation in text: [1] -
Oisaki, K.; Suto, Y.; Kanai, M.; Shibasaki, M. J. Am. Chem. Soc. 2003, 125, 5644–5645. doi:10.1021/ja034993n
Return to citation in text: [1] -
Ohtani, I.; Kusumi, J.; Kashman, Y.; Kakisawa, H. J. Am. Chem. Soc. 1991, 113, 4092–4096. doi:10.1021/ja00011a006
Return to citation in text: [1] -
Yoshida, W. Y.; Bryan, P. J.; Baker, B. J.; McClintock, J. B. J. Org. Chem. 1995, 60, 780–782. doi:10.1021/jo00108a057
Return to citation in text: [1] -
Dell'Agli, M.; Parapini, S.; Galli, G.; Vaiana, N.; Taramelli, D.; Sparatore, A.; Liu, P.; Dunn, B. M.; Bosisio, E.; Romeo, S. J. Med. Chem. 2006, 49, 7440–7449. doi:10.1021/jm061033d
Return to citation in text: [1] -
Danishefsky, S.; Kobayashi, S.; Kerwin, J. F., Jr. J. Org. Chem. 1982, 47, 1981–1983. doi:10.1021/jo00349a037
And references therein.
Return to citation in text: [1] -
Han, C.; Lee, J. P.; Lobkovsky, E.; Porco, J. A., Jr. J. Am. Chem. Soc. 2005, 127, 10039–10044. doi:10.1021/ja0527976
Return to citation in text: [1]
| 15. | Poncet, J.; Jouin, P.; Castro, B.; Nicolas, J.; Boutar, M.; Gaudemer, A. J. Chem. Soc., Perkin Trans. 1 1990, 611–616. doi:10.1039/P19900000611 |
| 25. | Xiang, S.-H.; Yuan, H.-Q.; Huang, P.-Q. Tetrahedron: Asymmetry 2009, 20, 2021–2026. doi:10.1016/j.tetasy.2009.08.018 |
| 35. |
Danishefsky, S.; Kobayashi, S.; Kerwin, J. F., Jr. J. Org. Chem. 1982, 47, 1981–1983. doi:10.1021/jo00349a037
And references therein. |
| 36. | Han, C.; Lee, J. P.; Lobkovsky, E.; Porco, J. A., Jr. J. Am. Chem. Soc. 2005, 127, 10039–10044. doi:10.1021/ja0527976 |
| 1. | Cancer Facts & Figures. http://www.cancer.org/Research/CancerFactsFigures/CancerFactsFigures/index (accessed Sept 1, 2010). |
| 10. | McDermott, L. A.; Higgins, B.; Simcox, M.; Luk, K.-C.; Nevins, T.; Kolinsky, K.; Smith, M.; Yang, H.; Li, J. K.; Chen, Y.; Ke, J.; Mallalieu, N.; Egan, T.; Kolis, S.; Railkar, A.; Gerber, L.; Liu, J.-J.; Konzelmann, F.; Zhang, Z.; Flynn, T.; Morales, O.; Chen, Y. Bioorg. Med. Chem. Lett. 2006, 16, 1950–1953. doi:10.1016/j.bmcl.2005.12.092 |
| 11. | Eskens, F. A. L. M.; Verweij, J. Eur. J. Cancer 2006, 42, 3127–3139. doi:10.1016/j.ejca.2006.09.015 |
| 32. | Ohtani, I.; Kusumi, J.; Kashman, Y.; Kakisawa, H. J. Am. Chem. Soc. 1991, 113, 4092–4096. doi:10.1021/ja00011a006 |
| 33. | Yoshida, W. Y.; Bryan, P. J.; Baker, B. J.; McClintock, J. B. J. Org. Chem. 1995, 60, 780–782. doi:10.1021/jo00108a057 |
| 9. |
Clinical Trials Home Page - National Cancer Institute. http://www.cancer.gov/clinicaltrials (accessed Sept 1, 2010).
(See for information on current cancer clinical trials.) |
| 34. | Dell'Agli, M.; Parapini, S.; Galli, G.; Vaiana, N.; Taramelli, D.; Sparatore, A.; Liu, P.; Dunn, B. M.; Bosisio, E.; Romeo, S. J. Med. Chem. 2006, 49, 7440–7449. doi:10.1021/jm061033d |
| 3. | Folkman, J. J. Natl. Cancer Inst. 1990, 82, 4–6. doi:10.1093/jnci/82.1.4 |
| 4. | Folkman, J. Breast Cancer Res. Treat. 1995, 36, 109–118. doi:10.1007/BF00666033 |
| 5. | Hanahan, D.; Folkman, J. Cell 1996, 86, 353–364. doi:10.1016/S0092-8674(00)80108-7 |
| 6. | Ferrara, N.; Alitalo, K. Nat. Med. 1999, 5, 1359–1364. doi:10.1038/70928 |
| 7. | Yancopouls, G. D.; Davis, S.; Gale, N. W.; Rudge, J. S.; Wiegand, S. J.; Holash, J. Nature 2000, 407, 242–248. doi:10.1038/35025215 |
| 8. | Carmeliet, P.; Jain, R. K. Nature 2000, 407, 249–257. doi:10.1038/35025220 |
| 31. | Oisaki, K.; Suto, Y.; Kanai, M.; Shibasaki, M. J. Am. Chem. Soc. 2003, 125, 5644–5645. doi:10.1021/ja034993n |
| 15. | Poncet, J.; Jouin, P.; Castro, B.; Nicolas, J.; Boutar, M.; Gaudemer, A. J. Chem. Soc., Perkin Trans. 1 1990, 611–616. doi:10.1039/P19900000611 |
| 24. | Kondekar, N. B.; Kandula, S. R. V.; Kumar, P. Tetrahedron Lett. 2004, 45, 5477–5479. doi:10.1016/j.tetlet.2004.05.057 |
| 25. | Xiang, S.-H.; Yuan, H.-Q.; Huang, P.-Q. Tetrahedron: Asymmetry 2009, 20, 2021–2026. doi:10.1016/j.tetasy.2009.08.018 |
| 26. | McKennon, M. J.; Meyers, A. I.; Drauz, K.; Schwarm, M. J. Org. Chem. 1993, 58, 3568–3571. doi:10.1021/jo00065a020 |
| 27. | Myers, A. G.; Zhong, B.; Movassaghi, M.; Kung, D. W.; Lanman, B. A.; Kwon, S. Tetrahedron Lett. 2000, 41, 1359–1362. doi:10.1016/S0040-4039(99)02293-5 |
| 22. | Huang, P. G.; Zheng, X.; Wang, S. L.; Ye, J. L.; Jin, L. R.; Chen, Z. Tetrahedron: Asymmetry 1999, 10, 3309–3317. doi:10.1016/S0957-4166(99)00321-3 |
| 23. | Park, T. H.; Paik, S.; Lee, S. H. Bull. Korean Chem. Soc. 2003, 24, 1227–1228. |
| 28. | Rich, D. H.; Sun, E. T.; Boparia, A. S. J. Org. Chem. 1978, 43, 3624–3626. doi:10.1021/jo00412a053 |
| 29. | McConnel, R. M.; Frizzell, D.; Camp, A.; Evans, A.; Jones, W.; Cagle, C. J. Med. Chem. 1991, 34, 2298–2300. doi:10.1021/jm00111a054 |
| 13. | Vedejs, E.; Campbell, J. B., Jr.; Gadwood, R. C.; Rodgers, J. D.; Spear, K. L.; Watanabe, Y. J. Org. Chem. 1982, 47, 1534–1546. doi:10.1021/jo00347a034 |
| 14. | Midland, M. M.; Afonso, M. M. J. Am. Chem. Soc. 1989, 111, 4368–4371. doi:10.1021/ja00194a033 |
| 15. | Poncet, J.; Jouin, P.; Castro, B.; Nicolas, J.; Boutar, M.; Gaudemer, A. J. Chem. Soc., Perkin Trans. 1 1990, 611–616. doi:10.1039/P19900000611 |
| 16. | Ohta, T.; Shiokawa, S.; Sakamoto, R.; Nozoe, S. Tetrahedron Lett. 1990, 31, 7329–7332. doi:10.1016/S0040-4039(00)88557-3 |
| 17. | Koot, W.-J.; van Ginkel, R.; Kranenburg, M.; Hiemstra, H.; Louwrier, S.; Moolenaar, M. J.; Speckamp, W. M. Tetrahedron Lett. 1991, 32, 401–404. doi:10.1016/S0040-4039(00)92639-X |
| 18. | Rinehart, K. L.; Sakai, R.; Kishore, V.; Sullins, D. W.; Li, K.-M. J. Org. Chem. 1992, 57, 3007–3013. doi:10.1021/jo00037a012 |
| 19. | Galeotti, N.; Poncet, J.; Chiche, L.; Jouin, P. J. Org. Chem. 1993, 58, 5370–5376. doi:10.1021/jo00072a018 |
| 20. | Reddy, G. V.; Rao, G. V.; Iyengar, D. S. Tetrahedron Lett. 1999, 40, 775–776. doi:10.1016/S0040-4039(98)02409-5 |
| 21. | Lennartz, M.; Sadakane, M.; Steckhan, E. Tetrahedron 1999, 55, 14407–14420. doi:10.1016/S0040-4020(99)00904-7 |
| 12. | Shin, H. J.; Kim, T. S.; Lee, H.-S.; Park, J. Y.; Choi, I.-K.; Kwon, H. J. Phytochemistry 2008, 69, 2363–2366. doi:10.1016/j.phytochem.2008.05.020 |
| 15. | Poncet, J.; Jouin, P.; Castro, B.; Nicolas, J.; Boutar, M.; Gaudemer, A. J. Chem. Soc., Perkin Trans. 1 1990, 611–616. doi:10.1039/P19900000611 |
| 24. | Kondekar, N. B.; Kandula, S. R. V.; Kumar, P. Tetrahedron Lett. 2004, 45, 5477–5479. doi:10.1016/j.tetlet.2004.05.057 |
© 2011 Mohapatra et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)