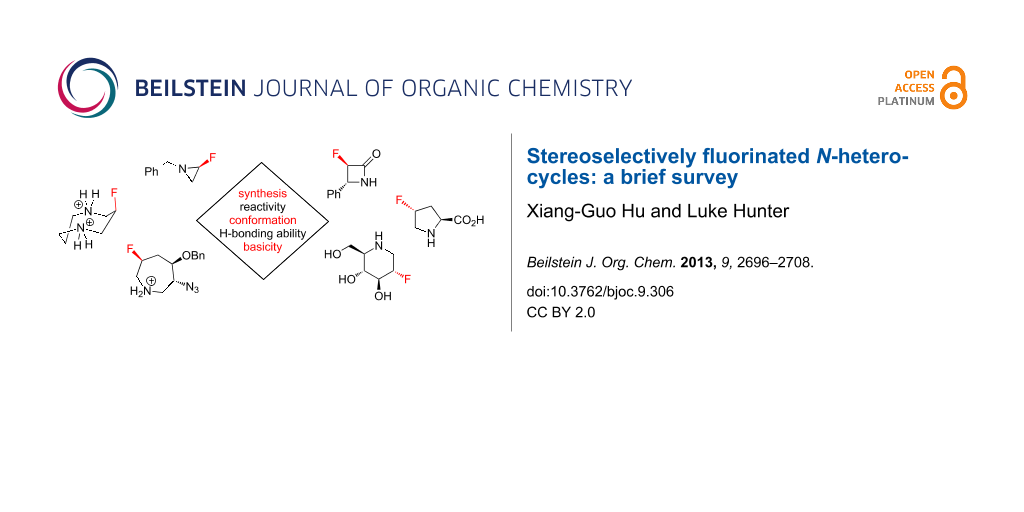
Abstract
The stereoselective incorporation of fluorine atoms into N-heterocycles can lead to dramatic changes in the molecules’ physical and chemical properties. These changes can be rationally exploited for the benefit of diverse fields such as medicinal chemistry and organocatalysis. This brief review will examine some of the effects that fluorine substitution can have in N-heterocycles, including changes to the molecules’ stability, their conformational behaviour, their hydrogen bonding ability, and their basicity. Finally, some methods for the synthesis of stereoselectively fluorinated N-heterocycles will also be reviewed.

Graphical Abstract
Review
1. Introduction
A cursory inspection of the medicinal chemistry literature will reveal two obvious themes in the structures of current drug candidates: the ubiquity of nitrogen heterocycles, and the popularity of organofluorine moieties. Therefore, it seems natural that a combination of these two features will offer rich possibilities in the future of drug development. To date, the introduction of fluorine into medicinal entities [1,2] has mostly taken the form of aryl fluorination [3,4] or trifluoromethylation [5,6], and fascinating developments in synthetic methodology of this type are continuing to occur [7-9]. However, with the advent of stereoselective fluorination methods [10,11] it seems clear that the subset of stereoselectively fluorinated N-heterocycles [12] offers particularly rich possibilities. We therefore felt that it would be worthwhile to examine in a brief review some of the unique features of this emerging class of molecules.
We have not attempted to cover this topic comprehensively; rather, in the following pages we aim to provide selected examples of the ways that fluorine can influence N-heterocycles’ stability and their conformational behaviour; we will see that fluorine can be used as a tool to probe the importance of hydrogen bonding in bioactive molecules; and we will observe how fluorine can affect the basicity of N-heterocycles. Finally, we will survey some of the various ways in which stereoselectively fluorinated N-heterocycles can be synthesised. Throughout, it will become clear that medicinal chemistry is not the only field that stands to benefit from a deeper understanding of these fascinating molecules: for example, attractive prospects are also clear in the field of organocatalysis [13].
2. Fluorination can influence N-heterocycles’ stability and reactivity
If a highly-polarised C–F bond is incorporated into a nitrogen heterocycle, it can be expected to have a dramatic influence on the molecules’ physical and chemical properties [14]. The influence that fluorine can have on chemical reactivity is illustrated by considering the smallest N-heterocycles, the aziridines. Aziridines (1, Figure 1) are generally very stable, in marked contrast with their oxygenated counterparts, the epoxides. However, if one or two fluorine atoms are attached to the aziridine backbone, the resulting molecule is much more susceptible to hydrolysis. De Kimpe and co-workers have investigated the reactivity of mono- and difluoroaziridines 2 and 3 (Figure 1) [15,16]. As well as the enhanced reactivity that 2 and 3 both show towards nucleophilic ring opening, there is an additional subtlety regarding the regioselectivity. While ab initio calculations predict that both 2 and 3 should favour nucleophilic ring opening at C3 [17], preliminary experiments showed that the mono- and difluorinated aziridines actually behave differently in the presence of nucleophiles, with monofluorinated aziridines 2 experiencing C2 attack and the difluorinated counterparts 3 favouring C3 attack.
![[1860-5397-9-306-1]](/bjoc/content/figures/1860-5397-9-306-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Fluorination alters the reactivity of aziridines.
Figure 1: Fluorination alters the reactivity of aziridines.
Fluorination has also been shown to influence reactivity in four-membered N-heterocycles (Scheme 1). Kanerva and co-workers [18] investigated a series of β-lactam derivatives (4a–c) in a lipase-catalysed methanolysis process. While the non-fluorinated derivative 4a was found to be unreactive under the reaction conditions specified, successive introduction of one or two fluorine atoms (4b and 4c) led to a marked increase in reactivity. The enantioselectivity of this approach is also worthy of note, and will be discussed further in a later section of this review.
![[1860-5397-9-306-i1]](/bjoc/content/inline/1860-5397-9-306-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Fluorination makes β-lactam derivatives more reactive towards lipase-catalysed methanolysis.
Scheme 1: Fluorination makes β-lactam derivatives more reactive towards lipase-catalysed methanolysis.
Now that our survey of N-heterocycles has reached ring sizes of four atoms or larger, another important consideration emerges: fluorine can affect the molecules’ conformational behaviour [19]. To illustrate this point a series of examples are presented below, drawing from heterocycles with ring sizes of up to eight atoms.
3. Fluorination can influence the conformations of N-heterocycles
3.1 Four-membered rings
O’Hagan and co-workers observed an interesting conformational effect in a computational study of fluorinated azetidine derivatives (Figure 2) [20]. The neutral molecule 6 was calculated to prefer a ring pucker which placed the fluorine atom far away from the neutral nitrogen atom (N–C–C–F dihedral angle = 137.2°). However, the story changed markedly with the charged derivative 7: in this case, the ring pucker inverted and the fluorine atom more closely approached the charged nitrogen atom (N+–C–C–F dihedral angle = 100.0°). This contrast was explained by invoking a favourable interaction between the C–F dipole and the charged N+ atom, and the magnitude of this charge–dipole effect is revealed by comparison with the non-fluorinated control molecule 8 in which the ring pucker is less pronounced (N–C–C–H dihedral angle = 102.3°). It transpires that this C–F…N+ interaction is a general effect which has also been observed in larger N-heterocycles, as discussed below.
![[1860-5397-9-306-2]](/bjoc/content/figures/1860-5397-9-306-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: The ring pucker in azetidine derivatives can be influenced by a C–F…N+ charge–dipole interaction.
Figure 2: The ring pucker in azetidine derivatives can be influenced by a C–F…N+ charge–dipole interaction.
3.2 Five-membered rings
The C–F…N+ interaction can have a more dramatic impact on the conformations of pyrrolidines, since they are inherently more flexible than azetidines [21]. For example, O’Hagan and co-workers investigated the pyrrolidine-containing molecules 9 and 10 (Figure 3) as ligands of G-quadruplex DNA [22]. The non-fluorinated ligand 9 had some conformational disorder because the pyrrolidine rings were able to interconvert between exo and endo puckers. In contrast, the pyrrolidine rings of fluorinated ligand 10 were more rigid, with the fluorine atoms preferring to occupy an axial position consistent with a favourable C–F…N+ interaction (worth approximately 5.0 kcal/mol). This led to a number of changes to the DNA binding mode of 10, including a rotation of the entire pyrrolidine ring by 180° relative to that of 9, and several different H-bonding contacts with the DNA as a result.
![[1860-5397-9-306-3]](/bjoc/content/figures/1860-5397-9-306-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Fluorination ridifies the pyrrolidine rings of ligand 10, with several consequences for its G-quadruplex DNA binding properties.
Figure 3: Fluorination ridifies the pyrrolidine rings of ligand 10, with several consequences for its G-quadr...
In contrast with the strong charge–dipole effect evident in pyrrolidine 10 (Figure 3), another more subtle interaction is observed in neutral fluorinated pyrrolidines. For example, Raines and co-workers found that (4R)-fluoroproline 12 adopts a Cγ-exo ring pucker (Figure 4), in contrast with natural proline 11 which has a more flexible pyrrolidine ring [23]. The increased rigidity of 12 was explained by a stabilising hyperconjugation phenomenon (Figure 4), in which an appropriately-aligned σCH orbital is able to donate electron density into the vacant σ*CF antibonding orbital. This stabilising interaction is only possible if the C–F and C–N bonds are aligned gauche to one another, and is analogous to the well-known fluorine gauche effect [24]. The importance of the rigid Cγ-exo ring pucker of 12 was demonstrated in spectacular fashion: Raines and co-workers showed that the thermal stability of collagen was increased when 12 was incorporated in place of collagen’s naturally-present (4R)-hydroxyproline residues [25].
![[1860-5397-9-306-4]](/bjoc/content/figures/1860-5397-9-306-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Proline 11 readily undergoes a ring-flip process, but (4R)-fluoroproline 12 is more rigid because of hyperconjugation (σCH → σ*CF).
Figure 4: Proline 11 readily undergoes a ring-flip process, but (4R)-fluoroproline 12 is more rigid because o...
This hyperconjugation effect has also been exploited in the context of organocatalysis. Fluorination of proline itself, as well as related N-heterocycles, has been shown to increase enantioselectivity in certain organocatalytic processes [13]. For example, Alexakis and co-workers found that the non-fluorinated catalyst 13 (Scheme 2) catalysed an alkylation reaction (15→17) with only moderate enantioselectivity [26]. This was ascribed to the flexibility of the pyrrolidine moiety in the enamine intermediate 16. In contrast, the fluorinated catalyst 14 has a relatively strong (1.5 kcal/mol) preference for an endo pucker, stabilised by hyperconjugation, and this increased ridigity was credited with a dramatic improvement in the enantioselectivity.
![[1860-5397-9-306-i2]](/bjoc/content/inline/1860-5397-9-306-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Hyperconjugation rigidifies the ring pucker of a fluorinated organocatalyst 14, leading to higher enantioselectivity.
Scheme 2: Hyperconjugation rigidifies the ring pucker of a fluorinated organocatalyst 14, leading to higher e...
3.3 Six-membered rings
The conformational analysis of six-membered rings is a cornerstone in physical chemistry. Substituted saturated six-membered compounds usually adopt a chair conformation with substituents preferring the equatorial positions. However, in 1993 Lankin and Snyder [27] observed that fluoropiperidine 18 preferentially adopted a conformation in which the fluorine substituent resides in the axial position (Figure 5). This study was then extended to include piperidines 19 and 20, and in each case the axial conformers are preferred by a substantial ~5.0 kcal/mol over the equatorial conformers (not shown) [28,29]. This pioneering work constituted the original discovery of the C–F…N+ interaction which has already been discussed above in the context of azetidines and pyrrolidines. Interestingly, Lankin and Snyder were also able to rule out hydrogen bonding as the source of the axial preference, since the N,N-dimethyl analogue 20 exhibited a similar effect.
![[1860-5397-9-306-5]](/bjoc/content/figures/1860-5397-9-306-5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Fluorinated piperidines prefer the axial conformation, due to stabilising C–F…N+ interactions.
Figure 5: Fluorinated piperidines prefer the axial conformation, due to stabilising C–F…N+ interactions.
3.4 Seven-membered rings
Seven-membered rings exhibit much more complex conformational behaviour than six-membered rings. Hence, it is perhaps unsurprising that a twenty year gap separated the pioneering work of Lankin and Snyder (Figure 5) from the first analysis of fluorinated seven-membered N-heterocycles. Liu and co-workers [30] have recently explored the conformational behaviour of the substituted azepanes 21–23 (Figure 6), and observed that the rigidifying power of a fluorine substituent is strongly dependent on the other groups present. The non-fluorinated azepane 21 was found to exhibit extensive conformational disorder, and this was attributed to competing preferences for the OBn/N3 substituents to adopt pseudoequatorial positions and for the azide group to align gauche to the ring nitrogen. The situation was not greatly changed upon introduction of a (6S)-fluorine atom (compound 22): in this case, no single conformation of 22 was able to satisfy a C–F…N+ gauche alignment as well as the two conformational preferences described for 21. In contrast however, introduction of a (6R)-fluorine atom (compound 23) greatly rigidified the ring system, to the extent that a single conformer of 23 dominated in solution. This work highlights the subtleties that can arise when fluorine atoms are incorporated into highly flexible molecules with pre-existing substituents.
![[1860-5397-9-306-6]](/bjoc/content/figures/1860-5397-9-306-6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Fluorination can rigidify a substituted azepane, but only if it acts in synergy with the other substituents: azepanes 21 and 22 are disordered, while azepane 23 has one dominant geometry in solution.
Figure 6: Fluorination can rigidify a substituted azepane, but only if it acts in synergy with the other subs...
3.5 Eight-membered rings
The eight-membered ring is the largest stereoselectively fluorinated N-heterocycle that has been investigated to date [20]. O’Hagan and co-workers investigated the structure 24 (Figure 7), and calculated that the axial conformation of 24 should be strongly preferred over the equatorial conformation (9.2 kcal/mol) because of two stabilising C–F…N+ interactions. An X-ray structure of 24 was also obtained (Figure 7), and it revealed a geometry consistent with the calculated minimum-energy structure, with no evidence of disorder.
![[1860-5397-9-306-7]](/bjoc/content/figures/1860-5397-9-306-7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: The eight-membered N-heterocycle 24 prefers an axial orientation of the fluorine substituent, giving two C–F…N+ interactions.
Figure 7: The eight-membered N-heterocycle 24 prefers an axial orientation of the fluorine substituent, givin...
So far in this review, we have primarily been considering fluorine as a replacement for hydrogen in N-heterocycles. However a new vista opens up if we consider fluorine as a replacement for the hydroxy group in bioactive molecules.
4. Fluorine can serve as a tool to probe the importance of hydrogen bonding
The replacement of a hydroxy group in a bioactive molecule with a fluorine atom can cause the loss of hydrogen bond donor ability, which may have profound effects on the ligand–receptor interaction. The study of fluorinated iminosugars serves as a good platform to discuss this issue.
Naturally occurring iminosugars, also referred as polyhydroxylated alkaloids or azasugars, are sugar mimics in which a nitrogen atom replaces the ring oxygen of the corresponding monosaccharide (Figure 8) [31-36]. Iminosugars can competitively bind to glycosidase enzymes because of their structural resemblance to the terminal sugar moiety of natural substrates, or to the activated intermediate of hydrolysis (i.e. the oxocarbenium ion). As a consequence, iminosugars show great promise for the treatment of a variety of diseases including diabetes, viral infection, bacterial infection, and lysosomal storage disorders [37].
![[1860-5397-9-306-8]](/bjoc/content/figures/1860-5397-9-306-8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: Some iminosugars are “privileged structures” that serve as valuable drug leads.
Figure 8: Some iminosugars are “privileged structures” that serve as valuable drug leads.
Fluorinated analogues of several of these privileged structures have been prepared, in order to probe the importance of hydrogen bonding in these systems [38-43]. For example, 1-deoxynojirimycin (28) is the C1-deoxy product of nojirimycin, the first iminosugar isolated from Nature. Iminosugar 28 is a potent inhibitor of yeast α-glycosidase (Figure 9), and the fluorinated analogues 32–34 suggest that the C2 and C4 hydroxy groups of 28 act as H-bond donors when binding to the enzyme, while the C6 hydroxy of 28 does not [44,45].
![[1860-5397-9-306-9]](/bjoc/content/figures/1860-5397-9-306-9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 9: Fluorinated iminosugar analogues 32–34 illuminate the binding interactions of the α-glycosidase inhibitor 28.
Figure 9: Fluorinated iminosugar analogues 32–34 illuminate the binding interactions of the α-glycosidase inh...
Miglitol (30, Figure 10) is an orally-available drug used for the treatment of type II diabetes. It was first marketed by Merck in 1996. The biological activity of the fluorinated analogues 35–37 (Figure 10) suggest that the C6 hydroxy group of 30 acts as a hydrogen bond donor in its binding to yeast α-glycosidase, while the C2' and C2 hydroxy groups of 30 do not [46,47]. The fluorinated analogue 37 is particularly worthy of note, since this compound is five times more potent than the existing drug 30, and exhibits no toxicity in human cells.
![[1860-5397-9-306-10]](/bjoc/content/figures/1860-5397-9-306-10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 10: Fluorinated miglitol analogues, and their inhibitory activity towards yeast α-glycosidase.
Figure 10: Fluorinated miglitol analogues, and their inhibitory activity towards yeast α-glycosidase.
However, a word of warning: in the fluorinated iminosugar examples discussed above (Figure 9 and Figure 10) the inhibition data must be interpreted with some caution, because another effect could be in operation. As well as changing the molecules’ hydrogen bonding properties, fluorination can also affect the basicity of the amine group. This latter effect can be rationally exploited, for example to improve the bioavailability of a drug molecule; this concept is explored in the next section.
5. Fluorination alters the basicity of N-heterocycles
The 3-piperidinylindole derivative 38 (Table 1) binds to the human 5-HT2A serotonin receptor, and was identified as a promising antipsychotic drug lead [48]. However, the bioavailability of 38 was poor, and this was attributed to the basicity of the secondary amine group which made the molecule positively charged at physiological pH and hence unable to traverse biological membranes. This problem was overcome by introducing a fluorine atom onto the piperidine ring (39): the basicity of the secondary amine was thereby reduced by nearly two orders of magnitude, and this led to a marked improvement in bioavailability. Incidentally, it is also worthy of note that the bioavailability (and 5-HT2A binding affinity) could be further improved by the introduction of a second fluorine atom, this time onto the indole moiety (40); this further improvement in bioavailability was attributed to blockage of the metabolic degradation of 38 and 39 which commenced with hydroxylation of the indole moiety.
Table 1: Fluorination improves the bioavailability of 3-piperidinylindole derivatives 38–40 by reducing the basicity of the secondary amine.
![[Graphic 1]](/bjoc/content/inline/1860-5397-9-306-i12.svg?max-width=637&scale=1.0)
38 |
![[Graphic 2]](/bjoc/content/inline/1860-5397-9-306-i13.svg?max-width=637&scale=1.0)
39 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-9-306-i14.svg?max-width=637&scale=1.0)
40 |
|
| pKaH | 10.4 | 8.5 | ~8.5a |
| 5-HT2A affinity | 0.99 nM | 0.43 nM | 0.06 nM |
| Bioavailability | “Poor” | 18% | 80% |
aNot measured, but assumed to be similar to 39.
In the next example, we return to the world of iminosugars. Isofagomine (31, Figure 11) is an inhibitor of the β-glucosidase from sweet almond, and it is thought to exert its inhibitory activity by mimicking the oxocarbenium intermediate of glycoside cleavage [49]. Several analogues of 31 have been investigated (41–44, Figure 11) [50-52], and on first inspection it is difficult to rationalise the observed trends in biological activity. One possible explanation for the dramatically improved activity of e.g. 43 over 42 is to invoke the “polar hydrophobic” nature of the fluorine substituent [53,54]. But another important factor is the basicity of the amine group [55]. To best mimic the oxocarbenium ion, the iminosugars 31 and 41–44 (Figure 11) must bear a positive charge, and since the pKaH values vary considerably amongst the different derivatives, it follows that each derivative has a different “optimal” pH for maximal inhibitory potency. This explains why the Ki values in Figure 11 do not seem to follow a clear trend: the quoted Ki values were all measured at the same pH, whereas it would be more revealing to consider the Ki of each molecule at its “optimal” pH. This is a very interesting situation, because it opens up the possibility of developing drugs that are selective for particular pH environments.
![[1860-5397-9-306-11]](/bjoc/content/figures/1860-5397-9-306-11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 11: Analogues of isofagomine (31) have different pKaH values, and therefore exhibit maximal β-glucosidase inhibition at different pH values.
Figure 11: Analogues of isofagomine (31) have different pKaH values, and therefore exhibit maximal β-glucosida...
We have now seen that fluorination can affect N-heterocycles’ stability, their conformational behaviour, their hydrogen bonding ability, and their basicity. It is hopefully clear to the reader that these effects have already led to several benefits in fields such as medicinal chemistry and organocatalysis. If these concepts are to be continued to be exploited in the future, then robust methods must be available for the synthesis of new fluorinated N-heterocycles. Hence, in the final section of this review we will examine some of the stereoselective synthetic methods that have been developed in recent years.
6. There are many ways to synthesise stereoselectively fluorinated N-heterocycles
6.1 Deoxyfluorination
Because of the ease of synthesis of enantiomerically pure alcohols, and the ever-increasing availability of deoxyfluorination reagents [10], the deoxyfluorination of N-protected alcohols is the most obvious strategy for synthesising fluorinated N-heterocycles (Scheme 3). Deoxyfluorination methods have already been extensively reviewed [8], and of course they are not limited in scope to N-heterocyclic systems, so we will not attempt to comprehensively cover this topic here. Instead, we will focus on two recent developments in deoxyfluorination methods that are particularly relevant to N-heterocyclic targets.
![[1860-5397-9-306-i3]](/bjoc/content/inline/1860-5397-9-306-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: General strategy for the synthesis of fluorinated N-heterocycles via deoxyfluorination.
Scheme 3: General strategy for the synthesis of fluorinated N-heterocycles via deoxyfluorination.
Late stage deoxyfluorination is an attractive method for synthesising multifunctional fluorinated N-heterocycles, but mild and selective reagents are required if this is to be successfully achieved. One such reagent, PhenoFluor (45, Figure 12), was originally developed by Ritter and co-workers for the direct fluorination of phenols [56]. Recent work showed that 45 can also be used to effect late-stage fluorination of hydroxy groups within complex molecular architectures. For example, 45 can react selectively with primary and allylic alcohols in the presence of secondary and tertiary alcohols, and the reaction will also tolerate the presence of carbonyl groups [57]. Some N-heterocyclic targets that have been synthesised in one step using 45 as the deoxyfluorination reagent are highlighted in Figure 12.
![[1860-5397-9-306-12]](/bjoc/content/figures/1860-5397-9-306-12.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 12: Late stage deoxyfluorination in the synthesis of multifunctional N-heterocycles.
Figure 12: Late stage deoxyfluorination in the synthesis of multifunctional N-heterocycles.
A unique complication sometimes arises when deoxyfluorination is attempted in N-heterocyclic systems: side reactions can occur, bought about by neighbouring group participation (Scheme 4) [58]. Such processes can lead to rearrangement, and this outcome has been rationally exploited to synthesise fluorinated five- [59], six- [60] and seven-membered [61] N-heterocycles that may have been otherwise difficult to access (e.g. 48→49, Scheme 4). Alternatively, neighbouring group participation sometimes results in an unexpected pattern of substitution with retention (e.g. 50→51, Scheme 4); in the latter example, note that the ring nitrogen does not directly engage in the anchimeric process [62].
![[1860-5397-9-306-i4]](/bjoc/content/inline/1860-5397-9-306-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: During the deoxyfluorination of N-heterocycles, neighbouring group participation can sometimes lead to rearrangement (48→49) or substitution with retention (50→51).
Scheme 4: During the deoxyfluorination of N-heterocycles, neighbouring group participation can sometimes lead...
6.2 The fluorinated building block approach
An alternative to the strategy of deoxyfluorination (section 6.1) is to synthesise fluorinated N-heterocycles starting from fluorine-containing organic building blocks. Such an approach benefits from the wide variety, and frequently the low cost, of today’s commercially available organofluorine molecules [63,64].
For example, the fluorinated aziridines 2 and 3 presented earlier (Figure 1) were synthesised through a building block approach. De Kimpe and co-workers [15,16] developed a strategy to synthesise such targets via cyclization of β-fluoro-β-chloroamines (54, Scheme 5), which in turn are derived from the readily-available fluoroacetate derivatives 52.
![[1860-5397-9-306-i5]](/bjoc/content/inline/1860-5397-9-306-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: A building block approach for the synthesis of fluorinated aziridines 2 and 3.
Scheme 5: A building block approach for the synthesis of fluorinated aziridines 2 and 3.
Percy and co-workers’ synthesis of a difluorinated analogue of calystegine B (63, Scheme 6) is a more elaborate example of the strategy of using a readily available fluorinated starting material for the synthesis of a complex target [65]. Percy’s approach commenced with protected trifluoroethanol 55 (Scheme 6), and the multistep route to 63 featured a [2,3]-Wittig rearrangement, a diastereoselective epoxidation, and a microwave assisted transannular epoxide opening reaction. It is also noteworthy that the starting material 55 contains an extraneous fluorine atom which is deleted during the synthetic sequence; this approach takes advantage of the often low cost and ready availability of perfluorinated building blocks.
![[1860-5397-9-306-i6]](/bjoc/content/inline/1860-5397-9-306-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Building block approach for the synthesis of a difluorinated analogue of calystegine B (63).
Scheme 6: Building block approach for the synthesis of a difluorinated analogue of calystegine B (63).
It should be noted, however, that access to enantiopure targets is not straightforward via the building block approach. Such targets may be better obtained through diastereoselective or enantioselective fluorination methods, and examples of these types of approaches are examined in the following sections.
6.3 Diastereoselective fluorocyclisation
The use of fluorocyclisation processes for the production of heterocycles and carbocycles has attracted considerable attention in recent years. Such processes have the advantage of forming multiple bonds in one pot [66]. Electrophilic fluorocyclisation involving the intrinsic nucleophilicity of nitrogen can be a powerful tool to synthesise stereoselectively fluorinated N-heterocycles. This concept was exemplified by Shibata and co-workers [67], who in 2001 reported an elegant and efficient method for synthesising fluorinated analogues of the natural product brevianamide E [68] (65, Scheme 7). This synthesis was remarkable for its rapid generation of molecular complexity, which is a defining feature of the fluorocyclisation approach. Even more spectacular was the extension of this methodology to create analogues of the natural product gypsetin [69,70] (68) via a double fluorocyclisation sequence (Scheme 7). The one drawback of this approach was its disappointing lack of diastereoselectivity, which presumably arose because nonselective fluoroquaternisation of the indole moiety preceded the cyclisation event.
![[1860-5397-9-306-i7]](/bjoc/content/inline/1860-5397-9-306-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Synthesis of fluorinated analogues of brevianamide E (65) and gypsetin (68) via electrophilic fluorocyclisation.
Scheme 7: Synthesis of fluorinated analogues of brevianamide E (65) and gypsetin (68) via electrophilic fluor...
6.4 Enantioselective fluorocyclisation
The lack of diastereoselectivity seen in Scheme 7 is attributable to the fluorination event preceding the cyclisation event, and this is a significant issue which inhibits the further development of diastereoselective processes. However, this issue does not preclude the development of enantioselective variants, provided the initial fluorination event can be controlled [11]. Gouverneur and co-workers recently reported the first enantioselective electrophilic fluorocyclisation (Scheme 8) [71]. Their substrates (e.g. 69) were indole derivatives bearing a pendant nitrogen nucleophile, and the source of chirality was a substoichiometric quantity of the cinchona alkaloid derivative (DHQ)2PHAL (70). This method was shown to work very well with several different pendant nucleophiles, but the N-acetamido nucleophile was found to be optimal, giving the corresponding product 71 in an impressive 92% ee. Elucidating the mechanism of chiral induction in this type of process is not straightforward, but preliminary experiments showed that associative complexation between the substrate 69 and the alkaloid catalyst 70 may account for the observed enantioselectivity.
![[1860-5397-9-306-i8]](/bjoc/content/inline/1860-5397-9-306-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Organocatalysed enantioselective fluorocyclisation.
Scheme 8: Organocatalysed enantioselective fluorocyclisation.
6.5 Radical reactions
Examples of direct fluorination of C–H bonds with fluorine-containing radicals are rare in the literature, especially if stereoselective versions of such reactions are sought. However, this transformation can be a very effective and concise method for synthesising fluorinated N-heterocycles. For example, L-cis-3-fluoroazetidine-2-carboxylic acid (73) was synthesised in one step from the corresponding amino acid 72 by photofluorination with fluoroxytrifluoromethane as the source of the fluorine radical, in 53% yield [72] (Scheme 9).
![[1860-5397-9-306-i9]](/bjoc/content/inline/1860-5397-9-306-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Synthesis of 3-fluoroazetidine 73 via radical fluorination.
Scheme 9: Synthesis of 3-fluoroazetidine 73 via radical fluorination.
Radical reactions can also be used to generate gem-difluorinated N-heterocycles. For example, Hu and Li [73] employed the versatile reagent 74 (Scheme 10) in their synthesis of the chiral 3,3-difluoropyrrolidine derivative 78. Reagent 74 can act as either a CF2 anion equivalent or a CF2 radical equivalent (Scheme 10, inset), and in Hu’s synthesis this reagent fulfils both functions at different stages: thus, the target 78 is achieved from N-(tert-butylsulfinyl)imine 75 through a nucleophilic addition/radical cyclisation sequence. The selectivity during the radical cyclisation (77→78) can be explained by the Beckwith–Houk transition-state model [74,75]. The 3,3-difluoropyrrolidine moiety (e.g. 78) is found in a variety of enzyme inhibitors such as thrombin inhibitors and cathepsin inhibitors, and so this synthetic methodology (Scheme 10) is likely to have valuable future applications in medicinal chemistry.
![[1860-5397-9-306-i10]](/bjoc/content/inline/1860-5397-9-306-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Synthesis of 3,3-difluoropyrrolidine 78 via a radical cyclisation.
Scheme 10: Synthesis of 3,3-difluoropyrrolidine 78 via a radical cyclisation.
6.6 Chemoenzymatic synthesis
Enzyme catalysis was presented earlier (Scheme 1) as a strategy for synthesising fluorinated β-lactams (4) [18]. At that time, we were interested in the effect that the fluorine substituents had on the reactivity of the β-lactam derivatives. However this work now merits further attention, because it also illustrates a strategy for achieving stereoselectivity in C–F bond formation. The racemic β-lactam 4b was synthesised as a single diastereoisomer from the Schiff base 79 (Scheme 11), by a Reformatsky addition followed by spontaneous cyclisation; removal of the amine protecting group under oxidative conditions then furnished rac-4b, the substrate for enzymatic resolution. Using an immobilized lipase enzyme as the catalyst (and under slightly different conditions from those described in Scheme 1), one enantiomer of the racemic β-lactam 4b was completely transformed into the ester 5b, while the other enantiomer of β-lactam 4b remained intact. The net result was that a fluorinated stereocentre was rapidly constructed, with defined absolute configuration, within a nitrogen heterocycle.
![[1860-5397-9-306-i11]](/bjoc/content/inline/1860-5397-9-306-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 11: Chemoenzymatic synthesis of fluorinated β-lactam 4b.
Scheme 11: Chemoenzymatic synthesis of fluorinated β-lactam 4b.
Conclusion
When the concept of selective fluorination is applied in the context of N-heterocycles, the resulting molecules have a variety of unique properties. In this brief review, we have examined some of these features, including the effects on stability, conformation, hydrogen bonding ability and basicity, and we have also surveyed some of the synthetic methods that are currently available for the production of such molecules. Throughout, we have seen that the unique properties of stereoselectively fluorinated N-heterocycles have led to a variety of valuable applications of these molecules, particularly in the field of medicinal chemistry.
What does the future hold? It is interesting to note that the molecules described in this review all comprise ring sizes of three to eight atoms; in contrast, macrocyclic structures have been little explored to date. It will be fascinating to learn whether similar effects operate in much larger ring sizes, for example in fluorinated analogues of cyclic peptides [76-78]. More generally however, it seems safe to predict that the unique properties of stereoselectively fluorinated N-heterocycles will ensure that their importance and utility continue to grow in the future.
References
-
Ismail, F. M. D. J. Fluorine Chem. 2002, 118, 27–33. doi:10.1016/S0022-1139(02)00201-4
Return to citation in text: [1] -
O’Hagan, D. J. Fluorine Chem. 2010, 131, 1071–1081. doi:10.1016/j.jfluchem.2010.03.003
Return to citation in text: [1] -
Grushin, V. V. Acc. Chem. Res. 2010, 43, 160–171. doi:10.1021/ar9001763
Return to citation in text: [1] -
Furuya, T.; Kamlet, A. S.; Ritter, T. Nature 2011, 473, 470–477. doi:10.1038/nature10108
Return to citation in text: [1] -
Shimizu, M.; Hiyama, T. Angew. Chem., Int. Ed. 2004, 44, 214–231. doi:10.1002/anie.200460441
Return to citation in text: [1] -
Tomashenko, O. A.; Grushin, V. V. Chem. Rev. 2011, 111, 4475–4521. doi:10.1021/cr1004293
Return to citation in text: [1] -
Fujiwara, Y.; Dixon, J. A.; O’Hara, F.; Daa Funder, E.; Dixon, D. D.; Rodriguez, R. A.; Baxter, R. D.; Herlé, B.; Sach, N.; Collins, M. R.; Ishihara, Y.; Baran, P. S. Nature 2012, 492, 95–99. doi:10.1038/nature11680
Return to citation in text: [1] -
Liang, T.; Neumann, C. N.; Ritter, T. Angew. Chem., Int. Ed. 2013, 52, 8214–8264. doi:10.1002/anie.201206566
Return to citation in text: [1] [2] -
Sandford, G. Top. Heterocycl. Chem. 2012, 27, 1–32. doi:10.1007/7081_2011_63
Return to citation in text: [1] -
Al-Maharik, N.; O’Hagan, D. Aldrichimica Acta 2011, 44, 65–75.
Return to citation in text: [1] [2] -
Ma, J.-A.; Cahard, D. Chem. Rev. 2004, 104, 6119–6146. doi:10.1021/cr030143e
Return to citation in text: [1] [2] -
Petrov, C. A., Ed. Fluorinated Heterocyclic Compounds: Synthesis, Chemistry and Applications; John Wiley & Sons: Hoboken, NJ, USA, 2009.
Return to citation in text: [1] -
Zimmer, L. E.; Sparr, C.; Gilmour, R. Angew. Chem., Int. Ed. 2011, 50, 11860–11871. doi:10.1002/anie.201102027
Return to citation in text: [1] [2] -
O’Hagan, D. Chem. Soc. Rev. 2008, 37, 308–319. doi:10.1039/b711844a
Return to citation in text: [1] -
van Hende, E.; Verniest, G.; Surmont, R.; De Kimpe, N. Org. Lett. 2007, 9, 2935–2937. doi:10.1021/ol071127x
Return to citation in text: [1] [2] -
Verniest, G.; Colpaert, F.; Van Hende, E.; De Kimpe, N. J. Org. Chem. 2007, 72, 8569–8572. doi:10.1021/jo071253e
Return to citation in text: [1] [2] -
Banks, H. D. J. Org. Chem. 2006, 71, 8089–8097. doi:10.1021/jo061255j
Return to citation in text: [1] -
Li, X.-G.; Lähitie, M.; Päiviö, M.; Kanerva, L. T. Tetrahedron: Asymmetry 2007, 18, 1567–1573. doi:10.1016/j.tetasy.2007.06.033
Return to citation in text: [1] [2] -
Hunter, L. Beilstein J. Org. Chem. 2010, 6, No. 38. doi:10.3762/bjoc.6.38
Return to citation in text: [1] -
Gooseman, N. E. J.; O’Hagan, D.; Slawin, A. M. Z.; Teale, A. M.; Tozer, D. J.; Young, R. J. Chem. Commun. 2006, 3190–3192. doi:10.1039/b606334a
Return to citation in text: [1] [2] -
Gerig, J. T.; McLeod, R. S. J. Am. Chem. Soc. 1973, 95, 5725–5729. doi:10.1021/ja00798a046
Return to citation in text: [1] -
Campbell, N. H.; Smith, D. L.; Reszka, A. P.; Neidle, S.; O’Hagan, D. Org. Biomol. Chem. 2011, 9, 1328–1331. doi:10.1039/c0ob00886a
Return to citation in text: [1] -
Holmgren, S. K.; Bretscher, L. E.; Taylor, K. M.; Raines, R. T. Chem. Biol. 1999, 6, 63–70. doi:10.1016/S1074-5521(99)80003-9
Return to citation in text: [1] -
Kirsch, P. Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications; Wiley-VCH: Weinheim, Germany, 2004. doi:10.1002/352760393X
Return to citation in text: [1] -
Holmgren, S. K.; Taylor, K. M.; Bretscher, L. E.; Raines, R. T. Nature 1998, 392, 666–667. doi:10.1038/33573
Return to citation in text: [1] -
Quintard, A.; Langlois, J.-B.; Emery, D.; Mareda, J.; Guénée, L.; Alexakis, A. Chem.–Eur. J. 2011, 17, 13433–13437. doi:10.1002/chem.201102757
Return to citation in text: [1] -
Lankin, D. C.; Chandrakumar, N. S.; Rao, S. N.; Spangler, D. P.; Snyder, J. P. J. Am. Chem. Soc. 1993, 115, 3356–3357. doi:10.1021/ja00061a055
Return to citation in text: [1] -
Snyder, J. P.; Chandrakumar, N. S.; Sato, H.; Lankin, D. C. J. Am. Chem. Soc. 2000, 122, 544–545. doi:10.1021/ja9934504
Return to citation in text: [1] -
Sum, A. M.; Lankin, D. C.; Hardcastle, K.; Snyder, J. P. Chem.–Eur. J. 2005, 11, 1579–1591. doi:10.1002/chem.200400835
Return to citation in text: [1] -
Patel, A. R.; Ball, G.; Hunter, L.; Liu, F. Org. Biomol. Chem. 2013, 11, 3781–3785. doi:10.1039/c3ob40391b
Return to citation in text: [1] -
Compain, P.; Martin, O. R., Eds. Iminosugars: From Synthesis to Therapeutic Applications; Wiley-VCH: Chichester, UK, 2007.
Return to citation in text: [1] -
Stütz, A. E., Ed. Iminosugars as Glycosidase Inhibitors: Nojirimycin and Beyond; Wiley-VCH: Weinheim, Germany, 1999.
Return to citation in text: [1] -
Asano, N.; Nash, R. J.; Molyneux, R. J.; Fleet, G. W. J. Tetrahedron: Asymmetry 2000, 11, 1645–1680. doi:10.1016/S0957-4166(00)00113-0
Return to citation in text: [1] -
Watson, A. A.; Fleet, G. W. J.; Asano, N.; Molyneux, R. J.; Nash, R. J. Phytochemistry 2001, 56, 265–295. doi:10.1016/S0031-9422(00)00451-9
Return to citation in text: [1] -
Lillelund, V. H.; Jensen, H. H.; Liang, X.; Bols, M. Chem. Rev. 2002, 102, 515–554. doi:10.1021/cr000433k
Return to citation in text: [1] -
Whalen, L. J.; Wong, C.-H. Aldrichimica Acta 2006, 39, 63–71.
Return to citation in text: [1] -
Nash, R. J.; Kato, A.; Yu, C.-Y.; Fleet, G. W. J. Future Med. Chem. 2011, 3, 1513–1521. doi:10.4155/fmc.11.117
Return to citation in text: [1] -
Thaharn, W.; Bootwicha, T.; Soorukram, D.; Kuhakarn, C.; Prabpai, S.; Kongsaeree, P.; Tuchinda, P.; Reutrakul, V.; Pohmakotr, M. J. Org. Chem. 2012, 77, 8465–8479. doi:10.1021/jo301327s
Return to citation in text: [1] -
Reymond, J.-L.; Pinkerton, A. A.; Vogel, P. J. Org. Chem. 1991, 56, 2128–2135. doi:10.1021/jo00006a031
Return to citation in text: [1] -
Fumeaux, R. H.; Mason, J. M.; Tyler, P. C. Tetrahedron Lett. 1994, 35, 3143–3146. doi:10.1016/S0040-4039(00)76852-3
Return to citation in text: [1] -
Furneaux, R. H.; Gainsford, G. J.; Mason, J. M.; Tyler, P. C. Tetrahedron 1994, 50, 2131–2160. doi:10.1016/S0040-4020(01)85075-4
Return to citation in text: [1] -
Furneaux, R. H.; Gainsford, G. J.; Mason, J. M.; Tyler, P. C.; Hartley, O.; Winchester, B. G. Tetrahedron 1995, 51, 12611–12630. doi:10.1016/0040-4020(95)00803-G
Return to citation in text: [1] -
Li, Y.-X.; Huang, M.-H.; Yamashita, Y.; Kato, A.; Jia, Y.-M.; Wang, W.-B.; Fleet, G. W. J.; Nash, R. J.; Yu, C.-Y. Org. Biomol. Chem. 2011, 9, 3405–3414. doi:10.1039/c0ob01063d
Return to citation in text: [1] -
Kajimoto, T.; Liu, K. K. C.; Derson, R. L.; Zhong, Z. Y.; Ichikawa, Y.; Porco, J. A.; Wong, C. H. J. Am. Chem. Soc. 1991, 113, 6187–6196. doi:10.1021/ja00016a039
Return to citation in text: [1] -
Anderson, S. M.; Ebner, M.; Ekhart, C. W.; Gradnig, G.; Legler, G.; Lundt, I.; Stütz, A. E.; Withers, S. G.; Wrodnigg, T. Carbohydr. Res. 1997, 301, 155–166. doi:10.1016/S0008-6215(97)00099-2
Return to citation in text: [1] -
Prell, E.; Csuk, R. Bioorg. Med. Chem. Lett. 2009, 19, 5673–5674. doi:10.1016/j.bmcl.2009.08.012
Return to citation in text: [1] -
Prell, E.; Korb, C.; Kluge, R.; Ströhl, D.; Csuk, R. Arch. Pharm. 2010, 343, 583–589. doi:10.1002/ardp.200900256
Return to citation in text: [1] -
Rowley, M.; Hallett, D. J.; Goodacre, S.; Moyes, C.; Crawforth, J.; Sparey, T. J.; Patel, S.; Marwood, R.; Patel, S.; Thomas, S.; Hitzel, L.; O’Connor, D.; Szeto, N.; Castro, J. L.; Hutson, P. H.; Macleod, A. M. J. Med. Chem. 2001, 44, 1603–1614. doi:10.1021/jm0004998
Return to citation in text: [1] -
Jespersen, T. M.; Dong, W.; Sierks, M. R.; Skrydstrup, T.; Lundt, I.; Bols, M. Angew. Chem., Int. Ed. Engl. 1994, 33, 1778–1779. doi:10.1002/anie.199417781
Return to citation in text: [1] -
Li, R.-J.; Bols, M.; Rousseau, C.; Zhang, X.-G.; Wang, R.-W.; Qing, F.-L. Tetrahedron 2009, 65, 3717–3727. doi:10.1016/j.tet.2009.02.079
Return to citation in text: [1] -
Yang, Y.; Zheng, F.; Bols, M.; Marinescu, L. G.; Qing, F.-L. J. Fluorine Chem. 2011, 838–845. doi:10.1016/j.jfluchem.2011.05.031
Return to citation in text: [1] -
López, O.; Qing, F.-L.; Pedersen, C. M.; Bols, M. Bioorg. Med. Chem. 2013, 21, 4755–4761. doi:10.1016/j.bmc.2013.03.003
Return to citation in text: [1] -
Biffinger, J. C.; Kim, H. W.; DiMagno, S. G. ChemBioChem 2004, 5, 622–627. doi:10.1002/cbic.200300910
Return to citation in text: [1] -
Ioannou, A.; Cini, E.; Timofte, R. S.; Flitsch, S. L.; Turner, N. J.; Linclau, B. Chem. Commun. 2011, 47, 11228–11230. doi:10.1039/c1cc13956h
Return to citation in text: [1] -
Jensen, H. H.; Lyngbye, L.; Jensen, A.; Bols, M. Chem.–Eur. J. 2002, 8, 1218–1226. doi:10.1002/1521-3765(20020301)8:5<1218::AID-CHEM1218>3.0.CO;2-X
Return to citation in text: [1] -
Tang, P.; Wang, W.; Ritter, T. J. Am. Chem. Soc. 2011, 133, 11482–11484. doi:10.1021/ja2048072
Return to citation in text: [1] -
Sladojevich, F.; Arlow, S. I.; Tang, P.; Ritter, T. J. Am. Chem. Soc. 2013, 135, 2470–2473. doi:10.1021/ja3125405
Return to citation in text: [1] -
Ferret, H.; Déchamps, I.; Gomez Pardo, D.; Van Hijfte, L.; Cossy, J. ARKIVOC 2010, No. 8, 126–159. doi:10.3998/ark.5550190.0011.812
Return to citation in text: [1] -
Drouillat, B.; Couty, F.; David, O.; Evano, G.; Marrot, J. Synlett 2008, 1345–1348. doi:10.1055/s-2008-1072770
Return to citation in text: [1] -
Déchamps, I.; Gomez Pardo, D.; Cossy, J. Synlett 2007, 263–267. doi:10.1055/s-2007-967989
Return to citation in text: [1] -
Déchamps, I.; Gomez Pardo, D.; Cossy, J. Eur. J. Org. Chem. 2007, 4224–4234. doi:10.1002/ejoc.200700237
Return to citation in text: [1] -
Patel, A. R.; Liu, F. Tetrahedron 2012, 69, 744–752. doi:10.1016/j.tet.2012.10.078
Return to citation in text: [1] -
Zhu, S. Z.; Wang, Y. L.; Peng, W. M. Curr. Org. Chem. 2002, 6, 1057–1096. doi:10.2174/1385272023373635
Return to citation in text: [1] -
Percy, J. M. Building Block Approaches to Aliphatic Organofluorine Compounds. In Organofluorine Chemistry: Techniques and Synthons; Chambers, R. D., Ed.; Springer-Verlag: Berlin, Germany, 1997; pp 131–195.
Return to citation in text: [1] -
Kyne, S. H.; Miles, J. A. L.; Percy, J. M.; Singh, K. J. Org. Chem. 2012, 77, 991–998. doi:10.1021/jo2022845
Return to citation in text: [1] -
Wilkinson, S. C.; Salmon, R.; Gouverneur, V. Future Med. Chem. 2009, 1, 847–863. doi:10.4155/fmc.09.47
Return to citation in text: [1] -
Shibata, N.; Tarui, T.; Doi, Y.; Kirk, K. L. Angew. Chem., Int. Ed. 2001, 40, 4461–4463. doi:10.1002/1521-3773(20011203)40:23<4461::AID-ANIE4461>3.0.CO;2-E
Return to citation in text: [1] -
Birch, A. J.; Wright, J. J. J. Chem. Soc. D 1969, 644b–645. doi:10.1039/C2969000644B
Return to citation in text: [1] -
Shinohara, C.; Hasumi, K.; Takei, Y.; Endo, A. J. Antibiot. 1994, 47, 163–167. doi:10.7164/antibiotics.47.163
Return to citation in text: [1] -
Nuber, B.; Hansske, F.; Shinohara, C.; Miura, S.; Hasumi, K.; Endo, A. J. Antibiot. 1994, 47, 168–172. doi:10.7164/antibiotics.47.168
Return to citation in text: [1] -
Lozano, O.; Blessley, G.; del Campo, T. M.; Thompson, A. L.; Giuffredi, G. T.; Bettati, M.; Walker, M.; Borman, R.; Gouverneur, V. Angew. Chem., Int. Ed. 2011, 50, 8105–8109. doi:10.1002/anie.201103151
Return to citation in text: [1] -
Kollonitsch, J.; Barash, L.; Doldouras, G. A. J. Am. Chem. Soc. 1970, 92, 7494–7495. doi:10.1021/ja00728a056
Return to citation in text: [1] -
Li, Y.; Hu, J. Angew. Chem., Int. Ed. 2007, 46, 2489–2492. doi:10.1002/anie.200604783
Return to citation in text: [1] -
Beckwith, A. L. J. Tetrahedron 1981, 37, 3073–3100. doi:10.1016/S0040-4020(01)98839-8
Return to citation in text: [1] -
Spellmeyer, D. C.; Houk, K. N. J. Org. Chem. 1987, 52, 959–974. doi:10.1021/jo00382a001
Return to citation in text: [1] -
Hunter, L.; Chung, J. H. J. Org. Chem. 2011, 76, 5502–5505. doi:10.1021/jo200813a
Return to citation in text: [1] -
Hunter, L.; Jolliffe, K. A. J.; Jordan, M. J. T.; Jensen, P.; Macquart, R. B. Chem.–Eur. J. 2011, 17, 2340–2343. doi:10.1002/chem.201003320
Return to citation in text: [1] -
Hunter, L. Chim. Oggi 2012, 30 (3), 20–22.
Return to citation in text: [1]
| 44. | Kajimoto, T.; Liu, K. K. C.; Derson, R. L.; Zhong, Z. Y.; Ichikawa, Y.; Porco, J. A.; Wong, C. H. J. Am. Chem. Soc. 1991, 113, 6187–6196. doi:10.1021/ja00016a039 |
| 45. | Anderson, S. M.; Ebner, M.; Ekhart, C. W.; Gradnig, G.; Legler, G.; Lundt, I.; Stütz, A. E.; Withers, S. G.; Wrodnigg, T. Carbohydr. Res. 1997, 301, 155–166. doi:10.1016/S0008-6215(97)00099-2 |
| 46. | Prell, E.; Csuk, R. Bioorg. Med. Chem. Lett. 2009, 19, 5673–5674. doi:10.1016/j.bmcl.2009.08.012 |
| 47. | Prell, E.; Korb, C.; Kluge, R.; Ströhl, D.; Csuk, R. Arch. Pharm. 2010, 343, 583–589. doi:10.1002/ardp.200900256 |
| 48. | Rowley, M.; Hallett, D. J.; Goodacre, S.; Moyes, C.; Crawforth, J.; Sparey, T. J.; Patel, S.; Marwood, R.; Patel, S.; Thomas, S.; Hitzel, L.; O’Connor, D.; Szeto, N.; Castro, J. L.; Hutson, P. H.; Macleod, A. M. J. Med. Chem. 2001, 44, 1603–1614. doi:10.1021/jm0004998 |
| 56. | Tang, P.; Wang, W.; Ritter, T. J. Am. Chem. Soc. 2011, 133, 11482–11484. doi:10.1021/ja2048072 |
| 57. | Sladojevich, F.; Arlow, S. I.; Tang, P.; Ritter, T. J. Am. Chem. Soc. 2013, 135, 2470–2473. doi:10.1021/ja3125405 |
| 8. | Liang, T.; Neumann, C. N.; Ritter, T. Angew. Chem., Int. Ed. 2013, 52, 8214–8264. doi:10.1002/anie.201206566 |
| 53. | Biffinger, J. C.; Kim, H. W.; DiMagno, S. G. ChemBioChem 2004, 5, 622–627. doi:10.1002/cbic.200300910 |
| 54. | Ioannou, A.; Cini, E.; Timofte, R. S.; Flitsch, S. L.; Turner, N. J.; Linclau, B. Chem. Commun. 2011, 47, 11228–11230. doi:10.1039/c1cc13956h |
| 55. | Jensen, H. H.; Lyngbye, L.; Jensen, A.; Bols, M. Chem.–Eur. J. 2002, 8, 1218–1226. doi:10.1002/1521-3765(20020301)8:5<1218::AID-CHEM1218>3.0.CO;2-X |
| 49. | Jespersen, T. M.; Dong, W.; Sierks, M. R.; Skrydstrup, T.; Lundt, I.; Bols, M. Angew. Chem., Int. Ed. Engl. 1994, 33, 1778–1779. doi:10.1002/anie.199417781 |
| 50. | Li, R.-J.; Bols, M.; Rousseau, C.; Zhang, X.-G.; Wang, R.-W.; Qing, F.-L. Tetrahedron 2009, 65, 3717–3727. doi:10.1016/j.tet.2009.02.079 |
| 51. | Yang, Y.; Zheng, F.; Bols, M.; Marinescu, L. G.; Qing, F.-L. J. Fluorine Chem. 2011, 838–845. doi:10.1016/j.jfluchem.2011.05.031 |
| 52. | López, O.; Qing, F.-L.; Pedersen, C. M.; Bols, M. Bioorg. Med. Chem. 2013, 21, 4755–4761. doi:10.1016/j.bmc.2013.03.003 |
| 58. | Ferret, H.; Déchamps, I.; Gomez Pardo, D.; Van Hijfte, L.; Cossy, J. ARKIVOC 2010, No. 8, 126–159. doi:10.3998/ark.5550190.0011.812 |
| 59. | Drouillat, B.; Couty, F.; David, O.; Evano, G.; Marrot, J. Synlett 2008, 1345–1348. doi:10.1055/s-2008-1072770 |
| 60. | Déchamps, I.; Gomez Pardo, D.; Cossy, J. Synlett 2007, 263–267. doi:10.1055/s-2007-967989 |
| 67. | Shibata, N.; Tarui, T.; Doi, Y.; Kirk, K. L. Angew. Chem., Int. Ed. 2001, 40, 4461–4463. doi:10.1002/1521-3773(20011203)40:23<4461::AID-ANIE4461>3.0.CO;2-E |
| 68. | Birch, A. J.; Wright, J. J. J. Chem. Soc. D 1969, 644b–645. doi:10.1039/C2969000644B |
| 65. | Kyne, S. H.; Miles, J. A. L.; Percy, J. M.; Singh, K. J. Org. Chem. 2012, 77, 991–998. doi:10.1021/jo2022845 |
| 66. | Wilkinson, S. C.; Salmon, R.; Gouverneur, V. Future Med. Chem. 2009, 1, 847–863. doi:10.4155/fmc.09.47 |
| 63. | Zhu, S. Z.; Wang, Y. L.; Peng, W. M. Curr. Org. Chem. 2002, 6, 1057–1096. doi:10.2174/1385272023373635 |
| 64. | Percy, J. M. Building Block Approaches to Aliphatic Organofluorine Compounds. In Organofluorine Chemistry: Techniques and Synthons; Chambers, R. D., Ed.; Springer-Verlag: Berlin, Germany, 1997; pp 131–195. |
| 15. | van Hende, E.; Verniest, G.; Surmont, R.; De Kimpe, N. Org. Lett. 2007, 9, 2935–2937. doi:10.1021/ol071127x |
| 16. | Verniest, G.; Colpaert, F.; Van Hende, E.; De Kimpe, N. J. Org. Chem. 2007, 72, 8569–8572. doi:10.1021/jo071253e |
| 61. | Déchamps, I.; Gomez Pardo, D.; Cossy, J. Eur. J. Org. Chem. 2007, 4224–4234. doi:10.1002/ejoc.200700237 |
| 62. | Patel, A. R.; Liu, F. Tetrahedron 2012, 69, 744–752. doi:10.1016/j.tet.2012.10.078 |
| 71. | Lozano, O.; Blessley, G.; del Campo, T. M.; Thompson, A. L.; Giuffredi, G. T.; Bettati, M.; Walker, M.; Borman, R.; Gouverneur, V. Angew. Chem., Int. Ed. 2011, 50, 8105–8109. doi:10.1002/anie.201103151 |
| 69. | Shinohara, C.; Hasumi, K.; Takei, Y.; Endo, A. J. Antibiot. 1994, 47, 163–167. doi:10.7164/antibiotics.47.163 |
| 70. | Nuber, B.; Hansske, F.; Shinohara, C.; Miura, S.; Hasumi, K.; Endo, A. J. Antibiot. 1994, 47, 168–172. doi:10.7164/antibiotics.47.168 |
| 1. | Ismail, F. M. D. J. Fluorine Chem. 2002, 118, 27–33. doi:10.1016/S0022-1139(02)00201-4 |
| 2. | O’Hagan, D. J. Fluorine Chem. 2010, 131, 1071–1081. doi:10.1016/j.jfluchem.2010.03.003 |
| 10. | Al-Maharik, N.; O’Hagan, D. Aldrichimica Acta 2011, 44, 65–75. |
| 11. | Ma, J.-A.; Cahard, D. Chem. Rev. 2004, 104, 6119–6146. doi:10.1021/cr030143e |
| 22. | Campbell, N. H.; Smith, D. L.; Reszka, A. P.; Neidle, S.; O’Hagan, D. Org. Biomol. Chem. 2011, 9, 1328–1331. doi:10.1039/c0ob00886a |
| 7. | Fujiwara, Y.; Dixon, J. A.; O’Hara, F.; Daa Funder, E.; Dixon, D. D.; Rodriguez, R. A.; Baxter, R. D.; Herlé, B.; Sach, N.; Collins, M. R.; Ishihara, Y.; Baran, P. S. Nature 2012, 492, 95–99. doi:10.1038/nature11680 |
| 8. | Liang, T.; Neumann, C. N.; Ritter, T. Angew. Chem., Int. Ed. 2013, 52, 8214–8264. doi:10.1002/anie.201206566 |
| 9. | Sandford, G. Top. Heterocycl. Chem. 2012, 27, 1–32. doi:10.1007/7081_2011_63 |
| 23. | Holmgren, S. K.; Bretscher, L. E.; Taylor, K. M.; Raines, R. T. Chem. Biol. 1999, 6, 63–70. doi:10.1016/S1074-5521(99)80003-9 |
| 5. | Shimizu, M.; Hiyama, T. Angew. Chem., Int. Ed. 2004, 44, 214–231. doi:10.1002/anie.200460441 |
| 6. | Tomashenko, O. A.; Grushin, V. V. Chem. Rev. 2011, 111, 4475–4521. doi:10.1021/cr1004293 |
| 20. | Gooseman, N. E. J.; O’Hagan, D.; Slawin, A. M. Z.; Teale, A. M.; Tozer, D. J.; Young, R. J. Chem. Commun. 2006, 3190–3192. doi:10.1039/b606334a |
| 76. | Hunter, L.; Chung, J. H. J. Org. Chem. 2011, 76, 5502–5505. doi:10.1021/jo200813a |
| 77. | Hunter, L.; Jolliffe, K. A. J.; Jordan, M. J. T.; Jensen, P.; Macquart, R. B. Chem.–Eur. J. 2011, 17, 2340–2343. doi:10.1002/chem.201003320 |
| 78. | Hunter, L. Chim. Oggi 2012, 30 (3), 20–22. |
| 3. | Grushin, V. V. Acc. Chem. Res. 2010, 43, 160–171. doi:10.1021/ar9001763 |
| 4. | Furuya, T.; Kamlet, A. S.; Ritter, T. Nature 2011, 473, 470–477. doi:10.1038/nature10108 |
| 21. | Gerig, J. T.; McLeod, R. S. J. Am. Chem. Soc. 1973, 95, 5725–5729. doi:10.1021/ja00798a046 |
| 15. | van Hende, E.; Verniest, G.; Surmont, R.; De Kimpe, N. Org. Lett. 2007, 9, 2935–2937. doi:10.1021/ol071127x |
| 16. | Verniest, G.; Colpaert, F.; Van Hende, E.; De Kimpe, N. J. Org. Chem. 2007, 72, 8569–8572. doi:10.1021/jo071253e |
| 18. | Li, X.-G.; Lähitie, M.; Päiviö, M.; Kanerva, L. T. Tetrahedron: Asymmetry 2007, 18, 1567–1573. doi:10.1016/j.tetasy.2007.06.033 |
| 74. | Beckwith, A. L. J. Tetrahedron 1981, 37, 3073–3100. doi:10.1016/S0040-4020(01)98839-8 |
| 75. | Spellmeyer, D. C.; Houk, K. N. J. Org. Chem. 1987, 52, 959–974. doi:10.1021/jo00382a001 |
| 18. | Li, X.-G.; Lähitie, M.; Päiviö, M.; Kanerva, L. T. Tetrahedron: Asymmetry 2007, 18, 1567–1573. doi:10.1016/j.tetasy.2007.06.033 |
| 13. | Zimmer, L. E.; Sparr, C.; Gilmour, R. Angew. Chem., Int. Ed. 2011, 50, 11860–11871. doi:10.1002/anie.201102027 |
| 72. | Kollonitsch, J.; Barash, L.; Doldouras, G. A. J. Am. Chem. Soc. 1970, 92, 7494–7495. doi:10.1021/ja00728a056 |
| 12. | Petrov, C. A., Ed. Fluorinated Heterocyclic Compounds: Synthesis, Chemistry and Applications; John Wiley & Sons: Hoboken, NJ, USA, 2009. |
| 73. | Li, Y.; Hu, J. Angew. Chem., Int. Ed. 2007, 46, 2489–2492. doi:10.1002/anie.200604783 |
| 13. | Zimmer, L. E.; Sparr, C.; Gilmour, R. Angew. Chem., Int. Ed. 2011, 50, 11860–11871. doi:10.1002/anie.201102027 |
| 24. | Kirsch, P. Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications; Wiley-VCH: Weinheim, Germany, 2004. doi:10.1002/352760393X |
| 25. | Holmgren, S. K.; Taylor, K. M.; Bretscher, L. E.; Raines, R. T. Nature 1998, 392, 666–667. doi:10.1038/33573 |
| 37. | Nash, R. J.; Kato, A.; Yu, C.-Y.; Fleet, G. W. J. Future Med. Chem. 2011, 3, 1513–1521. doi:10.4155/fmc.11.117 |
| 38. | Thaharn, W.; Bootwicha, T.; Soorukram, D.; Kuhakarn, C.; Prabpai, S.; Kongsaeree, P.; Tuchinda, P.; Reutrakul, V.; Pohmakotr, M. J. Org. Chem. 2012, 77, 8465–8479. doi:10.1021/jo301327s |
| 39. | Reymond, J.-L.; Pinkerton, A. A.; Vogel, P. J. Org. Chem. 1991, 56, 2128–2135. doi:10.1021/jo00006a031 |
| 40. | Fumeaux, R. H.; Mason, J. M.; Tyler, P. C. Tetrahedron Lett. 1994, 35, 3143–3146. doi:10.1016/S0040-4039(00)76852-3 |
| 41. | Furneaux, R. H.; Gainsford, G. J.; Mason, J. M.; Tyler, P. C. Tetrahedron 1994, 50, 2131–2160. doi:10.1016/S0040-4020(01)85075-4 |
| 42. | Furneaux, R. H.; Gainsford, G. J.; Mason, J. M.; Tyler, P. C.; Hartley, O.; Winchester, B. G. Tetrahedron 1995, 51, 12611–12630. doi:10.1016/0040-4020(95)00803-G |
| 43. | Li, Y.-X.; Huang, M.-H.; Yamashita, Y.; Kato, A.; Jia, Y.-M.; Wang, W.-B.; Fleet, G. W. J.; Nash, R. J.; Yu, C.-Y. Org. Biomol. Chem. 2011, 9, 3405–3414. doi:10.1039/c0ob01063d |
| 20. | Gooseman, N. E. J.; O’Hagan, D.; Slawin, A. M. Z.; Teale, A. M.; Tozer, D. J.; Young, R. J. Chem. Commun. 2006, 3190–3192. doi:10.1039/b606334a |
| 31. | Compain, P.; Martin, O. R., Eds. Iminosugars: From Synthesis to Therapeutic Applications; Wiley-VCH: Chichester, UK, 2007. |
| 32. | Stütz, A. E., Ed. Iminosugars as Glycosidase Inhibitors: Nojirimycin and Beyond; Wiley-VCH: Weinheim, Germany, 1999. |
| 33. | Asano, N.; Nash, R. J.; Molyneux, R. J.; Fleet, G. W. J. Tetrahedron: Asymmetry 2000, 11, 1645–1680. doi:10.1016/S0957-4166(00)00113-0 |
| 34. | Watson, A. A.; Fleet, G. W. J.; Asano, N.; Molyneux, R. J.; Nash, R. J. Phytochemistry 2001, 56, 265–295. doi:10.1016/S0031-9422(00)00451-9 |
| 35. | Lillelund, V. H.; Jensen, H. H.; Liang, X.; Bols, M. Chem. Rev. 2002, 102, 515–554. doi:10.1021/cr000433k |
| 36. | Whalen, L. J.; Wong, C.-H. Aldrichimica Acta 2006, 39, 63–71. |
| 28. | Snyder, J. P.; Chandrakumar, N. S.; Sato, H.; Lankin, D. C. J. Am. Chem. Soc. 2000, 122, 544–545. doi:10.1021/ja9934504 |
| 29. | Sum, A. M.; Lankin, D. C.; Hardcastle, K.; Snyder, J. P. Chem.–Eur. J. 2005, 11, 1579–1591. doi:10.1002/chem.200400835 |
| 30. | Patel, A. R.; Ball, G.; Hunter, L.; Liu, F. Org. Biomol. Chem. 2013, 11, 3781–3785. doi:10.1039/c3ob40391b |
| 26. | Quintard, A.; Langlois, J.-B.; Emery, D.; Mareda, J.; Guénée, L.; Alexakis, A. Chem.–Eur. J. 2011, 17, 13433–13437. doi:10.1002/chem.201102757 |
| 27. | Lankin, D. C.; Chandrakumar, N. S.; Rao, S. N.; Spangler, D. P.; Snyder, J. P. J. Am. Chem. Soc. 1993, 115, 3356–3357. doi:10.1021/ja00061a055 |
© 2013 Hu and Hunter; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)