Abstract
An extension of the substrate scope of the Flögel-three-component reaction of lithiated alkoxyallenes, nitriles and carboxylic acids is presented. The use of dicarboxylic acids allowed the preparation of symmetrical bis(β-ketoenamides) from simple starting materials in moderate yields. Cyclocondensations of these enamides to 4-hydroxypyridine derivatives or to functionalized pyrimidines efficiently provided symmetrically and unsymmetrically substituted fairly complex (hetero)aromatic compounds containing up to six conjugated aryl and hetaryl groups. In addition, subsequent functionalizations of the obtained heterocycles by palladium-catalyzed couplings or by oxidations are reported. We also describe the simple synthesis of a structurally interesting macrocyclic bispyrimidine derivative incorporating a 17-membered ring, whose configuration was elucidated by DFT calculations and by subsequent reactions.

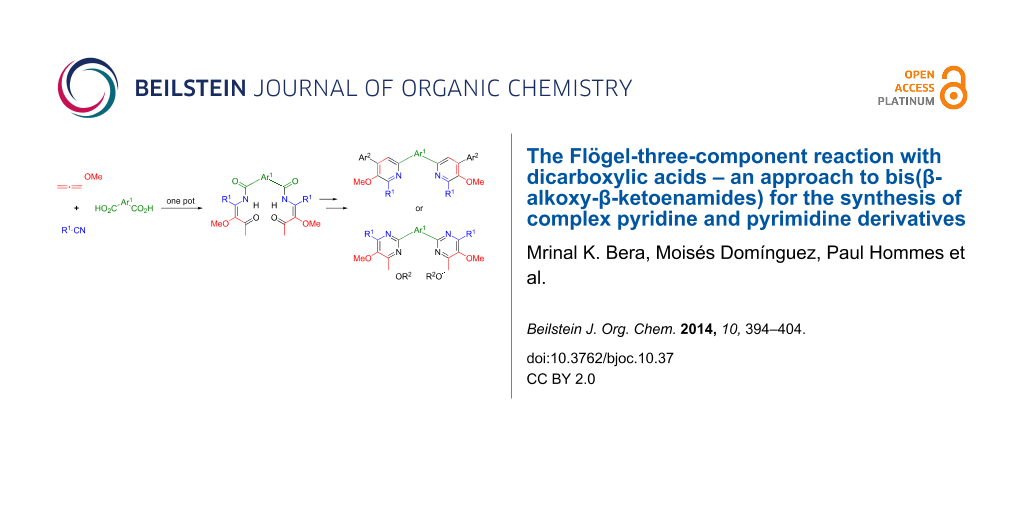
Graphical Abstract
Introduction
Multicomponent reactions (MCRs) generally allow a diversity-oriented fast and efficient access to complex synthetic intermediates and are thus powerful tools for the assembly of small-molecule libraries [1,2]. MCRs leading to functionalized N-heterocycles [3-7] have long been known before the general concept of MCRs was introduced, e.g. the Hantzsch dihydropyridine synthesis [8] or the Biginelli reaction [9] leading to dihydropyrimidinones or the corresponding dihydropyrimidinethiones. Due to their general importance (e.g. as biologically active compounds) the development of efficient protocols for the preparation of functionalized pyridine [10-20] and pyrimidine derivatives [21-33], in particular by MCRs, is of permanent high interest. In the course of exploring the reactivity of alkoxyallenes and their utilization as C-3 building blocks [34-37] our group developed a highly flexible method to synthesize β-alkoxy-β-ketoenamides of type 1 that are remarkably versatile cyclization precursors for the synthesis of functionalized heterocycles such as 4-hydroxypyridines [38-44], furopyridines [45], 5-acetyloxazoles [46,47], pyrimidines [43,48,49] and their corresponding N-oxides [50] (Scheme 1). This approach – discovered and mechanistically elucidated by Oliver Flögel – features a three-component reaction that employs alkoxyallenes, nitriles and carboxylic acids: upon treatment with n-butyllithium the allene is lithiated in α-position to the alkoxy moiety; the addition of a nitrile as electrophile to this highly reactive nucleophile results in the formation of an iminoallene adduct [38] that is protonated and subsequently acylated by the addition of a carboxylic acid furnishing a β-alkoxy-β-ketoenamide 1. A detailed mechanistic proposal for this reaction has been disclosed in previous reports [38,39].
![[1860-5397-10-37-i1]](/bjoc/content/inline/1860-5397-10-37-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Flögel-three-component reaction of lithiated alkoxyallenes, nitriles and carboxylic acids providing β-alkoxy-β-ketoenamides 1 – versatile precursors for the synthesis of functionalized N-heteroaromatics 2–6.
Scheme 1: Flögel-three-component reaction of lithiated alkoxyallenes, nitriles and carboxylic acids providing...
Our earlier investigations revealed that this method tolerates a broad variety of differently substituted starting materials – inter alia (het-)aromatic and (branched) aliphatic nitriles and carboxylic acids. It is also noteworthy to mention that the configurational integrity of enantiopure α-chiral carboxylic acids and/or nitriles is retained during this reaction [40]. In the present report we describe our efforts to further broaden the substrate scope of this multicomponent reaction and the subsequent cyclizations by employing aromatic dicarboxylic acids. This extension should allow a rapid access to fairly complex heteroaromatic systems containing up to six conjugated aryl and hetaryl groups. Complementary examples employing aromatic dinitriles in this Flögel-three-component reaction have previously been presented [39].
Results and Discussion
As typical model substrates we chose to employ isophthalic acid (11) and diphenic acid (12) in combination with methoxyallene (7), pivalonitrile (9) and thiophene-2-carbonitrile (10) in the three-component reaction (Scheme 2). Gratifyingly we were able to isolate the expected bis(β-ketoenamides) 13–15 in reasonable yields of 15–28%. Taking the number of individual steps into account (six new bonds are formed for each product) and considering possible (unknown) side reactions these yields are quite satisfactory. In analogy to our previously published results [38,51,52] the double bond geometry of the enamide moiety is likely to be E-configured as shown in Scheme 2, allowing an intramolecular hydrogen bridge between the amide NH and the β-carbonyl group. However, we did not further investigate the nature of the double bond geometry, since it was irrelevant for the planed subsequent cyclization reactions where the (Lewis-)acidic conditions allow a facile isomerization of E- and Z-configured enamide moieties [51,52], finally leading to identical products.
![[1860-5397-10-37-i2]](/bjoc/content/inline/1860-5397-10-37-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of bis(β-ketoenamides) 13–15 by three-component reactions of lithiated methoxyallene 8 with nitriles 9 or 10 and isophthalic acid (11) or diphenic acid (12).
Scheme 2: Synthesis of bis(β-ketoenamides) 13–15 by three-component reactions of lithiated methoxyallene 8 wi...
After these successful multicomponent reactions we investigated the intramolecular condensations of the bis(β-ketoenamides) 13–15 to pyridine and pyrimidine derivatives. Enamides 13 and 14 were treated with trimethylsilyl trifluoromethanesulfonate (TMSOTf) and triethylamine to provide the bis(4-hydoxypyridines) 16 in 50% yield and 18a in 60% yield, respectively (Scheme 3). A mechanistic proposal for this aldol type condensation has been presented in a previous report [53]. For precursor 14 partial monocyclization was observed under the applied conditions, affording in 18% yield 4-hydroxypyridine 18b with a retained β-ketoenamide moiety. Treatment of compounds 16, 18a and 18b with sodium hydride followed by nonafluorobutanesulfonyl fluoride (NfF) provided the corresponding sulfonates 17, 19 and 20 in yields in the range of 60–72%. Pyrid-4-yl nonaflates are excellent precursors for transition metal-catalyzed cross-coupling reactions [42,54-58], which was demonstrated here by the successful Suzuki coupling of bisnonaflate 19 with (E)-styrylboronic acid and the Stille coupling of 19 with 2-(tributylstannyl)thiophene. Albeit the expected twofold coupling products 21 and 22 were obtained in only moderate yields, the presented approach nevertheless features a quite rapid access to these fairly complex heteroaromatic systems containing six conjugated aryl and hetaryl groups. Upon excitation with UV light (253 nm) compound 22 shows fluorescence with a maximum intensity at 378 nm (see Supporting Information File 1 for details). The photophysical properties of structurally related pyridine–thiophene conjugates were recently investigated in detail [55,57,58].
![[1860-5397-10-37-i3]](/bjoc/content/inline/1860-5397-10-37-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Cyclocondensations of β-ketoenamides 13 and 14 to 4-hydroxypyridines 16, 18a and 18b, their subsequent nonaflations and palladium-catalyzed coupling reactions of 19 leading to compounds 21 and 22. NfF = C4F9SO2F
Scheme 3: Cyclocondensations of β-ketoenamides 13 and 14 to 4-hydroxypyridines 16, 18a and 18b, their subsequ...
Next, we investigated the cyclocondensation of bis(β-ketoenamides) 13–15 to pyrimidines (Scheme 4) using ammonium acetate as ammonia source. Initially we subjected enamide 13 to conditions that had been optimized for mono-β-ketoenamides [48,49], in this case resulting in incomplete conversion: after heating 13 with 8 equiv of ammonium acetate in a sealed tube we obtained a 1:1 mixture of bis(pyrimidine) derivative 23a and pyrimidine 23b still containing one β-ketoenamide unit with an overall yield of 68%. However, full conversion of 13 into 23a was achieved by increasing the amount of ammonium acetate to 16 equiv and using a higher reaction temperature, raising the yield of 23a from 34% to 55% yield. When enamide 14 was cyclized under these optimized conditions the conversion was nevertheless incomplete giving the desired bis(pyrimidine) derivative 24a in 56% yield and the corresponding mono-pyrimidine 24b in 23% yield. For enamide 15 however, the cyclization was complete under these conditions furnishing bis(pyrimidine) derivative 25 as a single product in 60% yield.
![[1860-5397-10-37-i4]](/bjoc/content/inline/1860-5397-10-37-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Cyclocondensations of β-ketoenamides 13–15 with ammonium acetate to bis(pyrimidine) derivatives 23a, 24a and 25 and mono-pyrimidines 23b and 24b.
Scheme 4: Cyclocondensations of β-ketoenamides 13–15 with ammonium acetate to bis(pyrimidine) derivatives 23a...
Although initially not desired the incomplete conversions of the bis(β-ketoenamides) leading to mono-pyridine derivatives such as 18b or to mono-pyrimidine derivatives like 23b and 24b provided new synthetic options to construct unsymmetrically substituted mixed heteroaromatic systems. As an example we used mono-pyrimidine derivative 24b and cyclized its β-ketoenamide moiety by treatment with TMSOTf and triethylamine. Pyrimidine/pyridinol derivative 26 was isolated in 79% yield (Scheme 5) and subsequently converted into the corresponding nonaflate 27 in 70% yield.
![[1860-5397-10-37-i5]](/bjoc/content/inline/1860-5397-10-37-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Conversion of mono-pyrimidine derivative 24b into unsymmetrically substituted biphenylen-bridged pyrimidine/nonafloxypyridine conjugate 27. NfF = C4F9SO2F
Scheme 5: Conversion of mono-pyrimidine derivative 24b into unsymmetrically substituted biphenylen-bridged py...
As recently described, β-alkoxy-β-ketoenamides may also be directly cyclized to pyrimidine-N-oxides under mild conditions if hydroxylamine hydrochloride is used as reagent [50]. Accordingly, the reactions of β-ketoenamides 14 and 20 with hydroxylamine hydrochloride provided the symmetric bis(pyrimidine-N-oxide) 28 in 39% yield or the mono-pyrimidine-N-oxide 30 in 54% yield (Scheme 6). The acetoxylation of 2- and 4-alkyl substituted pyridine-N-oxides by treatment with acetic anhydride is known as the Boekelheide rearrangement [59,60]. For pyrimidine-N-oxides however, only few examples of this type of transformation have been reported [50,61-65]. Therefore we were pleased to find that upon treatment with acetic anhydride the obtained pyrimidine-N-oxides 28 and 30 smoothly underwent the expected rearrangement to give the acetoxymethyl-substituted pyrimidine derivatives 29 and 31 in 61% and 55% yield, respectively. This approach thus allows the simple functionalization of the 4-methyl group of the pyrimidine derivatives and is a very useful tool for the preparation of other compounds.
![[1860-5397-10-37-i6]](/bjoc/content/inline/1860-5397-10-37-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Condensation of β-ketoenamides 14 and 20 with hydroxylamine hydrochloride to pyridine-N-oxides 28 and 30 and their subsequent Boekelheide rearrangements furnishing functionalized bis(pyrimidine) derivative 29 and pyrimidine/pyridine conjugate 31.
Scheme 6: Condensation of β-ketoenamides 14 and 20 with hydroxylamine hydrochloride to pyridine-N-oxides 28 a...
An alternative option for the side chain functionalization of 4- or 6-methyl substituted pyrimidines involves an oxidation with selenium dioxide (Riley oxidation [66-68]). To explore the synthetic potential of the newly prepared compounds we exemplarily oxidized bis(pyrimidine) 23a by this method in order to finally prepare a macrocyclic compound such as 34 (Scheme 7). Treatment of 23a with an excess of selenium dioxide at 90 °C resulted in the formation of an inseparable mixture of two different aldehydes (probably the dialdehyde and the monoaldehyde). After reduction of the mixture with sodium borohydride the obtained products could be separated by column chromatography providing the dialcohol 32a in 51% yield over two steps and the monoalcohol 32b in 25% yield, respectively. The subsequent O-allylation of 32a furnished bisallyl ether 33 with 77% yield that was subjected to a ring closing metathesis (RCM) [69] with Grubbs-II-catalyst smoothly leading to the structurally interesting macrocyclic compound 34 in 73% yield. Compounds of this type – incorporating a 17-membered ring – have the potential to serve as structurally quite unique ligands for a variety of applications, e.g. in catalysis.
![[1860-5397-10-37-i7]](/bjoc/content/inline/1860-5397-10-37-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Riley oxidation of bis(pyrimidine) derivative 23a and conversion of diol 32a into macrocycle 34.
Scheme 7: Riley oxidation of bis(pyrimidine) derivative 23a and conversion of diol 32a into macrocycle 34.
With ruthenium-based catalysts bearing N-heterocyclic carbene (NHC) ligands, RCM usually delivers macrocyclic olefins as mixtures of E- and Z-isomers, in most cases in favor of the E-isomer [70-73]. The E/Z-ratio is often under thermodynamic control, reflecting the energy difference between the two isomers. According to TLC and NMR spectroscopy, macrocycle 34 was isolated as a single compound. Due to the symmetry of 34 no couplings of the olefinic protons in its 1H NMR spectrum can be observed. Thus at this stage, we were unable to assign the configuration of the double bond. In lack of suitable crystals for an X-ray analysis, we calculated the energy for the two possible isomers of 34, suggesting that the E-isomer should be considerably more stable than the corresponding Z-isomer (Table 1). Using the semi-empirical AM1 method an energy difference of ΔEZ-E of 28.7 kJ/mol was determined. DFT calculations using the B3LYP method with the basis sets 6-31(d) or 6-31G(d,p) both gave a ΔEZ-E value of 16.4 kJ/mol. This energy difference may be attributed to the strain of the macrocycle and higher torsion angles between the central benzene unit and the pyrimidine rings for the Z-isomer of 34, resulting in less efficient conjugation of the aromatic π-systems. The optimized molecular geometries of E-34 and Z-34 as well as the calculated torsion angles are depicted in Figure 1.
![[1860-5397-10-37-1]](/bjoc/content/figures/1860-5397-10-37-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Optimized geometries of (a) E-configured and (b) Z-configured macrocycle 34 at B3LYP/6-31G(d,p) level. The numbers represent the calculated torsion angles between the aromatic rings.
Figure 1: Optimized geometries of (a) E-configured and (b) Z-configured macrocycle 34 at B3LYP/6-31G(d,p) lev...
In order to unambiguously identify the double bond configuration of 34, we oxidized this compound with potassium osmate/NMO to obtain the vicinal diol 35 in 76% yield (Scheme 8). In the case of a Z-configured olefin 34 this dihydroxylation should give a cis-configured diol (meso compound), whereas an E-configured olefin 34 would lead to a racemic mixture of the corresponding trans-configured diol. However, due to the symmetry of both vicinal diols a distinction between cis- and trans-35 (σv- or C2-symmetry respectively) by NMR is still not possible. The resulting diol 35 was therefore treated with an excess of (S)-Mosher's acid chloride to obtain the bis-(R)-Mosher ester 36 [74]. TLC analysis and NMR-spectroscopy revealed, that compound 36 was obtained as a pair of C2-symmetric diastereomers and that the obtained diol 35 was in fact a racemic mixture. This observation allowed the conclusion that the RCM reaction of 33 produced the expected thermodynamically more stable E-configured macrocyclic olefin 34. Hence this experimental result is in perfect agreement with the DFT calculations.
![[1860-5397-10-37-i8]](/bjoc/content/inline/1860-5397-10-37-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Dihydroxylation of the macrocyclic olefin 34 to diol 35 and subsequent esterification to the bis-(R)-Mosher ester 36; (S)-MTPA-Cl = (S)-3,3,3-trifluoro-2-methoxy-2-phenylpropanoyl chloride.
Scheme 8: Dihydroxylation of the macrocyclic olefin 34 to diol 35 and subsequent esterification to the bis-(R...
Conclusion
We were able to extend the substrate scope of the Flögel-three-component reaction of alkoxyallenes, nitriles and carboxylic acids by successfully utilizing aromatic dicarboxylic acids to prepare three new bis(β-methoxy-β-ketoenamides). With these products of a multicomponent reaction we performed cyclizations to rapidly construct symmetrically and unsymmetrically substituted pyridine and pyrimidine derivatives. Hence a very short approach to fairly complex functionalized oligoaromatic systems was established. In addition we exemplarily investigated subsequent transformations of these compounds either by palladium-catalyzed cross-couplings or by oxidations of the 4-methyl groups of the pyrimidine subunits. Although the yields for the crucial initial multicomponent reactions leading to the bis(β-methoxy-β-ketoenamides) are only moderate when dicarboxylic acids are used the simplicity of the processes and the diversity of the products accessible is impressive. The described methods allow the preparation of oligo(hetero)aromatic compounds not available by alternative procedures.
Experimental
General methods
Reactions were performed under an atmosphere of argon in flame-dried flasks. Solvents and liquid reagents were added by syringe. Et2O, CH2Cl2 and THF were transferred from a MB SPS-800-dry solvent system into the reaction vessels. Dry DMF was purchased from Acros Organics and stored in the presence of molecular sieve under an atmosphere of argon. NEt3 was distilled from CaH2 and stored over KOH under argon. Methoxyallene was prepared from propargylic alcohol in two steps according to literature procedures [34,75]. All other solvents and reagents were purchased from commercial suppliers and were used without further purification. Thin-layer chromatography (TLC) analyses were performed on TLC plates purchased from Merck (silica gel 60, fluorescence indicator F254, 0.25 mm layer thickness). Products were purified by flash column chromatography on silica gel 60 (230–400 mesh, Macherey-Nagel). NMR spectra were recorded with Bruker (AC 500, AVIII 700) and JEOL (ECX 400, Eclipse 500) instruments. Chemical shifts are reported relative to solvent residual peaks or TMS. Integrals are in accordance with assignments, and coupling constants are given in Hz. All 13C NMR spectra are proton-decoupled. 13C NMR signals of Nf-groups [CF3(CF2)3] are not given since unambiguous assignment is not possible due to strong splitting by coupling with the 19F nuclei. IR spectra were measured with a Jasco FT/IR-4100 spectrometer. HRMS analyses were performed with a Varian Ionspec QFT-7 (ESI–FT ICRMS) or an Agilent 6210 (ESI–TOF) instrument. Melting points were measured with a Reichert apparatus (Thermovar) and are uncorrected.
Three-component-reaction of methoxyallene, nitriles and dicarboxylic acids (typical procedure 1)
To a solution of methoxyallene (7, 2.07 g, 29.6 mmol) in dry Et2O (25 mL) was added n-BuLi (10.8 mL, 27.0 mmol, 2.5 M in hexanes) at −50 °C. After 30 min stirring at −50 °C, the reaction mixture was cooled to −78 °C and pivalonitrile (9, 0.752 g, 9.06 mmol) in dry Et2O (10 mL) was added to the mixture. After stirring for 4 h a suspension of diphenic acid (12, 6.54 g, 27.0 mmol) in dry Et2O (50 mL) was added. The temperature was allowed to rise to rt and the mixture was stirred overnight. The reaction was quenched with sat. aq NaHCO3 solution (25 mL) and the layers were separated. The aqueous layer was extracted with Et2O (3 × 50 mL) and the combined organic layers were washed with brine (25 mL), dried with Na2SO4 and filtered. The solvent was removed under reduced pressure and the obtained crude product was purified by column chromatography (silica gel, hexanes/EtOAc = 1:2) to provide bis(β-ketoenamide) 14 (1.39 g, 28%) as a pale yellow solid.
N2,N2'-Bis(4-methoxy-2,2-dimethyl-5-oxohex-3-en-3-yl)biphenyl-2,2'-dicarboxamide (14): mp 140–143 °C; IR (ATR) ν: 3145 (NH), 3040–2835 (=C-H, C-H), 1695 (C=O), 1525–1390 (C=C) cm−1; 1H NMR (CDCl3, 500 MHz) δ 0.96 (s, 18H, t-Bu), 2.09 (s, 6H, Me), 3.42 (s, 6H, OMe), 7.07–7.09, 7.31–7.37, 7.49–7.51 (3 m, 2H, 4H, 2H, Ar), 8.13 (br s, 2H, NH) ppm; 13C NMR (CDCl3, 126 MHz) δ 27.6 (q, Me), 28.4, 36.5 (q, s, t-Bu), 58.8 (q, OMe), 127.0, 127.9, 129.6, 130.4 (4 d, Ar), 131.9, 136.4, 138.4, 151.0 (4 s, C=C, Ar), 169.5 (s, CONH), 200.1 (s, C=O) ppm; ESI–TOF (m/z): [M + Na]+ calcd for C32H40N2NaO6, 571.2779; found, 571.2783.
Cyclization of β-ketoenamides to 4-hydroxypyridines (typical procedure 2)
Bis(β-ketoenamide) 14 (0.310 g, 0.57 mmol) was placed in an ACE-sealed tube and dissolved in DCE (10 mL). NEt3 (0.40 mL, 2.89 mmol) and TMSOTf (0.50 mL, 2.76 mmol) were added and the resulting mixture was stirred at 90 °C for 3 d. After cooling to rt the reaction was quenched with sat. aq NH4Cl solution (10 mL) and the layers were separated. The aqueous layer was extracted with CH2Cl2 (3 × 25 mL) and the combined organic layers were dried with Na2SO4 and filtered. The solvent was removed under reduced pressure and the obtained crude product was purified by column chromatography (silica gel, EtOAc) to provide bis(4-hydroxypyridine) 18a (0.174 g, 60%) as a brown liquid and 18b (54 mg, 18%) as pale yellow oil. The products were directly converted into the corresponding nonaflates 19 and 20.
Nonaflation of 4-hydroxypyridines (typical procedure 3)
Bis(4-hydroxypyridine) 18a (0.805 g, 1.57 mmol) was dissolved in THF (25 mL) and NaH (0.313 g, 7.86 mmol, 60% in mineral oil) was added under argon atmosphere. Nonafluorobutanesulfonyl fluoride (2.35 g, 7.79 mmol) was added drop-wise and the mixture was stirred at rt for 12 h. After dilution with Et2O (25 mL), the reaction was slowly quenched with ice and water (25 mL). The layers were separated and the aqueous layer was extracted with Et2O (3 × 25 mL). The combined organic layers were dried with Na2SO4, filtered and concentrated to dryness under reduced pressure. The residue was purified by column chromatography (silica gel, hexanes/EtOAc = 9:1 to 4:1) to provide pyridyl nonaflate 19 (1.20 g, 71%) as a pale yellow oil.
6,6'-(Biphenyl-2,2'-diyl)bis(2-tert-butyl-3-methoxypyridine-6,4-diyl) bisnonaflate (19): IR (ATR) ν: 3065–2870 (=C-H, C-H), 1555–1410 (C=C) cm−1; 1H NMR (CDCl3, 500 MHz) δ 1.19 (s, 18H, t-Bu), 3.89 (s, 6H, OMe), 6.92 (s, 2H, Py), 7.10 (dd, J = 7.5, 1.2 Hz, 2H, Ar), 7.30 (td, J = 7.5, 1.4 Hz, 2H, Ar), 7.36 (dd, J = 7.5, 1.4 Hz, 2H, Ar), 7.59 (dd, J = 7.5, 1.2 Hz, 2H, Ar) ppm; 13C NMR (CDCl3, 126 MHz) δ 29.1, 38.7 (q, s, t-Bu), 61.7 (q, OMe), 115.2 (d, Py), 127.4, 128.6, 130.1, 131.6 (4 d, Ar), 138.2, 140.6 (2 s, Ar), 145.3, 149.3, 153.2, 163.7 (4 s, Py) ppm; 19F NMR (CDCl3, 470 MHz) δ −80.6 (t, J = 9.6 Hz, 6F, CF3), −109.5 (t, J = 13.7 Hz, 4F, CF2), −120.7, −125.8 (2 mc, 4F each, CF2) ppm; ESI–TOF (m/z): [M + Na]+ calcd for C40H34F18N2NaO8S2, 1099.1361; found, 1099.1394.
Cyclization of β-ketoenamides to pyrimidines (typical procedure 4)
Bis(β-ketoenamide) 14 (0.162 g, 0.296 mmol) and NH4OAc (0.365 g, 4.73 mmol) were placed in an ACE-sealed tube. The mixture was dissolved in MeOH (5 mL) and stirred for 2 d at 90 °C. After addition of H2O (10 mL) and Et2O (20 mL) the layers were separated and the aqueous layer was extracted with Et2O (2 × 25 mL). The combined organic layers were dried with Na2SO4, filtered and the solvent was evaporated under reduced pressure. The residue was purified by column chromatography (silica gel, hexanes/EtOAc = 5:1) to provide pyrimidines 24a (88 mg, 56%) and 24b (35 mg, 23%), both as colorless oils.
2,2'-Bis(4-tert-butyl-5-methoxy-6-methylpyrimidin-2-yl)biphenyl (24a): IR (ATR) ν: 3070–2855 (=C-H, C-H), 1550–1440 (C=C) cm−1; 1H NMR (CDCl3, 500 MHz) δ 0.99 (s, 18H, t-Bu), 2.28 (s, 6H, Me), 3.70 (s, 6H, OMe), 7.30 (dt, J = 7.7, 1.9 Hz, 2H, Ar), 7.34–7.39 (m, 4H, Ar), 7.70 (dd, J = 7.7, 1.0 Hz, 2H, Ar) ppm; 13C NMR (CDCl3, 126 MHz) δ 19.7 (q, Me), 28.7, 37.6 (q, s, t-Bu), 60.9 (q, OMe), 126.4, 128.7, 130.2, 131.4 (4 d, Ar), 138.4, 142.6 (2 s, Ar), 149.8, 159.3, 159.4, 166.9 (4 s, Py) ppm; ESI–TOF (m/z): [M + H]+ calcd for C32H39N4O2, 511.3068; found, 511.3085.
2'-(4-tert-Butyl-5-methoxy-6-methylpyrimidin-2-yl)-N-(4-methoxy-2,2-dimethyl-5-oxohex-3-en-3-yl)biphenyl-2-carboxamide (24b): IR (ATR) ν: 3325 (N-H), 3065–2865 (=C-H, C-H), 1700, 1665 (C=O), 1550–1445 (C=C) cm−1; 1H NMR (CDCl3, 500 MHz) δ 0.71 (s, 9H, t-Bu), 1.26 (s, 9H, t-Bu), 2.31, 2.33 (2 s, 3H each, Me), 3.45, 3.70 (2 s, 3H each, OMe), 6.64 (dd, J = 7.5, 1.0 Hz, 1H, Ar), 7.07, 7.25 (2 dt, J = 7.5, 1.2 Hz, 1H each, Ar), 7.32 (dd, J = 7.5, 1.2 Hz, 1H, Ar), 7.39 (dt, J = 7.5, 1.8 Hz, 1H, Ar), 7.43 (dt, J = 7.5, 1.0 Hz, 1H, Ar), 7.50 (dd, J = 7.8, 1.2 Hz, 1H, Ar), 7.91 (dd, J = 7.8, 1.8 Hz, 1H, Ar), 8.40 (br s, 1H, NH) ppm; 13C NMR (CDCl3, 126 MHz) δ 19.2 (q, Me), 27.2 (q, Me), 28.1, 29.2, 35.9, 37.9 (2 q, 2 s, t-Bu), 58.9, 61.0 (2 q, OMe), 126.8, 127.9, 128.0, 128.5, 129.0, 129.4, 130.3, 130.7, 131.0 (8 d, s, Ar, =C), 137.5, 138.4, 138.9, 140.5, 150.1 (5 s, Ar, =C), 150.4, 159.6, 160.0, 168.4 (4 s, Py), 169.3 (s, CONH), 199.8 (s, C=O) ppm; ESI–TOF (m/z): [M + Na]+ calcd for C32H34N3NaO4, 552.2833; found, 552.2844.
Supporting Information
| Supporting Information File 1: Additional experimental procedures and analytical data, as well as copies of NMR spectra of representative examples. | ||
| Format: PDF | Size: 3.3 MB | Download |
Acknowledgements
Generous support of this work by the Alexander von Humboldt Foundation by a postdoctoral research fellowship for M. K. B. and by Bayer HealthCare is most gratefully acknowledged. A postdoctoral research fellowship for M. D. by BECAS CHILE and his support by the Dierks-von-Zweck-Stiftung is gratefully acknowledged. We also thank Dr. R. Zimmer for discussions and assistance during preparation of this manuscript.
References
-
Zhu, J.; Bienaymé, H., Eds. Multicomponent Reactions; Wiley-VCH: Weinheim, Germany, 2005.
Return to citation in text: [1] -
Tietze, L. F.; Brasche, G.; Gericke, K. M. Domino Reactions in Organic Synthesis; Wiley-VCH: Weinheim, Germany, 2006.
Return to citation in text: [1] -
D'Souza, D. M.; Müller, T. J. J. Chem. Soc. Rev. 2007, 36, 1095–1108. doi:10.1039/b608235c
Return to citation in text: [1] -
Brauch, S.; van Berkel, S. S.; Westermann, B. Chem. Soc. Rev. 2013, 42, 4948–4962. doi:10.1039/c3cs35505e
Return to citation in text: [1] -
Simon, C.; Constantieux, T.; Rodriguez, J. Eur. J. Org. Chem. 2004, 4957–4980. doi:10.1002/ejoc.200400511
Return to citation in text: [1] -
Eckert, H. Molecules 2012, 17, 1074–1102. doi:10.3390/molecules17011074
Return to citation in text: [1] -
Ruijter, E.; Scheffelaar, R.; Orru, R. V. A. Angew. Chem., Int. Ed. 2011, 50, 6234–6246. doi:10.1002/anie.201006515
Angew Chem. 2011, 123, 6358–6371. doi:10.1002/ange.201006515
Return to citation in text: [1] -
Hantzsch, A. Justus Liebigs Ann. Chem. 1882, 215, 1–82. doi:10.1002/jlac.18822150102
Return to citation in text: [1] -
Biginelli, P. Gazz. Chim. Ital. 1983, 23, 360–416.
Return to citation in text: [1] -
Evdokimov, N. M.; Kireev, A. S.; Yakovenko, A. A.; Antipin, M. Yu.; Magedov, I. V.; Kornienko, A. J. Org. Chem. 2007, 72, 3443–3453. doi:10.1021/jo070114u
Return to citation in text: [1] -
Sasada, T.; Sakai, N.; Konakahara, T. J. Org. Chem. 2008, 73, 6905–6908. doi:10.1021/jo801090h
Return to citation in text: [1] -
Barluenga, J.; Jiménez-Aquino, A.; Fernández, M. A.; Aznar, F.; Valdés, C. Tetrahedron 2008, 64, 778–786. doi:10.1016/j.tet.2007.10.112
Return to citation in text: [1] -
Da-Qing, S.; Hao, Y. Synth. Commun. 2009, 39, 2481–2491. doi:10.1080/00397910802656034
Return to citation in text: [1] -
Guo, K.; Thompson, M. J.; Chen, B. J. Org. Chem. 2009, 74, 6999–7006. doi:10.1021/jo901232b
Return to citation in text: [1] -
Sha, F.; Huang, X. Angew. Chem., Int. Ed. 2009, 48, 3458–3461. doi:10.1002/anie.200900212
Angew. Chem. 2009, 121, 3510–3513. doi:10.1002/ange.200900212
Return to citation in text: [1] -
Chen, M. Z.; Micalizio, G. C. J. Am. Chem. Soc. 2012, 134, 1352–1356. doi:10.1021/ja2105703
Return to citation in text: [1] -
Al-Awadi, N. A.; Ibrahim, M. R.; Elnagdi, M. H.; John, E.; Ibrahim, Y. A. Beilstein J. Org. Chem. 2012, 8, 441–447. doi:10.3762/bjoc.8.50
Return to citation in text: [1] -
Sha, F.; Wu, L.; Huang, X. J. Org. Chem. 2012, 77, 3754–3765. doi:10.1021/jo300072x
Return to citation in text: [1] -
Zheng, L.; Ju, J.; Bin, Y.; Hua, R. J. Org. Chem. 2012, 77, 5794–5800. doi:10.1021/jo3010414
Return to citation in text: [1] -
He, Z.; Dobrovolsky, D.; Trinchera, P.; Yudin, A. K. Org. Lett. 2013, 15, 334–337. doi:10.1021/ol303246b
Return to citation in text: [1] -
Stonehouse, J. P.; Chekmarev, D. S.; Ivanova, N. V.; Lang, S.; Pairaudeau, G.; Smith, N.; Stocks, M. J.; Sviridov, S. I.; Utkina, L. M. Synlett 2008, 100–104. doi:10.1055/s-2007-1000839
Return to citation in text: [1] -
Sasada, T.; Moriuchi, M.; Sakai, N.; Konakahara, T. Eur. J. Org. Chem. 2009, 5738–5743. doi:10.1002/ejoc.200900639
Return to citation in text: [1] -
Sasada, T.; Kobayashi, F.; Sakai, N.; Konakahara, T. Org. Lett. 2009, 11, 2161–2164. doi:10.1021/ol900382j
Return to citation in text: [1] -
Hekmatshoar, R.; Kenary, G. N.; Sadjadi, S.; Beheshtiha, Y. S. Synth. Commun. 2010, 40, 2007–2013. doi:10.1080/00397910903219385
Return to citation in text: [1] -
Wan, J.-P.; Liu, Y. Synthesis 2010, 3943–3953. doi:10.1055/s-0030-1258290
Return to citation in text: [1] -
Zonouzi, A.; Biniaz, M.; Mirzazadeh, R.; Talebi, M.; Ng, S. W. Heterocycles 2010, 81, 1271–1278. doi:10.3987/com-10-11929
Return to citation in text: [1] -
Majumder, S.; Odom, A. L. Tetrahedron 2010, 66, 3152–3158. doi:10.1016/j.tet.2010.02.066
Return to citation in text: [1] -
Sedenkova, K. N.; Averina, E. B.; Grishin, Y. K.; Kutateladze, A. G.; Rybakov, V. B.; Kuznetsova, T. S.; Zefirov, N. S. J. Org. Chem. 2012, 77, 9893–9899. doi:10.1021/jo301880m
Return to citation in text: [1] -
You, X.; Yu, S.; Liu, Y. Organometallics 2013, 32, 5273–5276. doi:10.1021/om400880r
Return to citation in text: [1] -
Reddy, L. S.; Reddy, T. R.; Reddy, N. C. G.; Mohan, R. B.; Lingappa, Y. Synthesis 2013, 45, 75–84. doi:10.1055/s-0032-1316814
Return to citation in text: [1] -
Yavari, I.; Nematpour, M. Synlett 2013, 24, 165–168. doi:10.1055/s-0032-1317951
Return to citation in text: [1] -
Yang, K.; Xiang, J.; Bao, G.; Dang, Q.; Bai, X. ACS Comb. Sci. 2013, 15, 519–524. doi:10.1021/co400086u
Return to citation in text: [1] -
Gers, C. F.; Rosellen, J.; Merkul, E.; Müller, T. J. J. Beilstein J. Org. Chem. 2011, 7, 1173–1181. doi:10.3762/bjoc.7.136
Return to citation in text: [1] -
Zimmer, R. Synthesis 1993, 165–178. doi:10.1055/s-1993-25823
And references cited therein.
Return to citation in text: [1] [2] -
Krause, N.; Hashmi, A. S. K., Eds. Modern Allene Chemistry; Wiley-VCH: Weinheim, Germany, 2004.
Return to citation in text: [1] -
Brasholz, M.; Reissig, H.-U.; Zimmer, R. Acc. Chem. Res. 2009, 42, 45–56. doi:10.1021/ar800011h
Return to citation in text: [1] -
Lechel, T.; Reissig, H.-U. Pure Appl. Chem. 2010, 82, 1835–1844. doi:10.1351/pac-con-09-09-06
Return to citation in text: [1] -
Flögel, O.; Dash, J.; Brüdgam, I.; Hartl, H.; Reissig, H.-U. Chem.–Eur. J. 2004, 10, 4283–4290. doi:10.1002/chem.200400322
Return to citation in text: [1] [2] [3] [4] -
Dash, J.; Lechel, T.; Reissig, H.-U. Org. Lett. 2007, 9, 5541–5544. doi:10.1021/ol702468s
Return to citation in text: [1] [2] [3] -
Eidamshaus, C.; Reissig, H.-U. Adv. Synth. Catal. 2009, 351, 1162–1166. doi:10.1002/adsc.200800789
Return to citation in text: [1] [2] -
Lechel, T.; Dash, J.; Eidamshaus, C.; Brüdgam, I.; Lentz, D.; Reissig, H.-U. Org. Biomol. Chem. 2010, 8, 3007–3014. doi:10.1039/B925468D
Return to citation in text: [1] -
Lechel, T.; Dash, J.; Hommes, P.; Lentz, D.; Reissig, H.-U. J. Org. Chem. 2010, 75, 726–732. doi:10.1021/jo9022183
Return to citation in text: [1] [2] -
Bera, M. K.; Reissig, H.-U. Synthesis 2010, 2129–2138. doi:10.1055/s-0029-1218787
Return to citation in text: [1] [2] -
Eidamshaus, C.; Kumar, R.; Bera, M. K.; Reissig, H.-U. Beilstein J. Org. Chem. 2011, 7, 962–975. doi:10.3762/bjoc.7.108
Return to citation in text: [1] -
Lechel, T.; Dash, J.; Brüdgam, I.; Reissig, H.-U. Eur. J. Org. Chem. 2008, 3647–3655. doi:10.1002/ejoc.200800398
Return to citation in text: [1] -
Lechel, T.; Lentz, D.; Reissig, H.-U. Chem.–Eur. J. 2009, 15, 5432–5435. doi:10.1002/chem.200900386
Return to citation in text: [1] -
Lechel, T.; Gerhard, M.; Trawny, D.; Brusilowskij, B.; Schefzig, L.; Zimmer, R.; Rabe, J. P.; Lentz, D.; Schalley, C. A.; Reissig, H.-U. Chem.–Eur. J. 2011, 17, 7480–7491. doi:10.1002/chem.201100382
Return to citation in text: [1] -
Lechel, T.; Möhl, S.; Reissig, H.-U. Synlett 2009, 1059–1062. doi:10.1055/s-0028-1088220
Return to citation in text: [1] [2] -
Lechel, T.; Reissig, H.-U. Eur. J. Org. Chem. 2010, 2555–2564. doi:10.1002/ejoc.201000056
Return to citation in text: [1] [2] -
Zimmer, R.; Lechel, T.; Rancan, G.; Bera, M. K.; Reissig, H.-U. Synlett 2010, 1793–1796. doi:10.1055/s-0030-1258088
Return to citation in text: [1] [2] [3] -
Dash, J.; Reissig, H.-U. Chem.–Eur. J. 2009, 15, 6811–6814. doi:10.1002/chem.200900939
Return to citation in text: [1] [2] -
Hommes, P.; Jungk, P.; Reissig, H.-U. Synlett 2011, 2311–2314. doi:10.1055/s-0030-1260304
Return to citation in text: [1] [2] -
Eidamshaus, C.; Reissig, H.-U. Eur. J. Org. Chem. 2011, 6056–6069. doi:10.1002/ejoc.201100681
Return to citation in text: [1] -
Högermeier, J.; Reissig, H.-U. Adv. Synth. Catal. 2009, 351, 2747–2763. doi:10.1002/adsc.200900566
Return to citation in text: [1] -
Bera, M. K.; Hommes, P.; Reissig, H.-U. Chem.–Eur. J. 2011, 17, 11838–11843. doi:10.1002/chem.201101739
Return to citation in text: [1] [2] -
Eidamshaus, C.; Hommes, P.; Reissig, H.-U. Synlett 2012, 23, 1670–1674. doi:10.1055/s-0031-1290398
Return to citation in text: [1] -
Gholap, S. L.; Hommes, P.; Neuthe, K.; Reissig, H.-U. Org. Lett. 2013, 15, 318–321. doi:10.1021/ol303231c
Return to citation in text: [1] [2] -
Bera, M. K.; Gholap, S. L.; Hommes, P.; Neuthe, K.; Trawny, D.; Rabe, J. P.; Lentz, D.; Zimmer, R.; Reissig, H.-U. Adv. Synth. Catal. 2013, 355, 3463–3474. doi:10.1002/adsc.201300613
Return to citation in text: [1] [2] -
Boekelheide, V.; Linn, W. J. J. Am. Chem. Soc. 1954, 76, 1286–1291. doi:10.1021/ja01634a026
Return to citation in text: [1] -
Galatsis, P. Boekelheide Reaction. In Name Reactions in Heterocyclic Chemistry; Li, J.-J.; Corey, E. J., Eds.; John Wiley & Sons Inc.: Hoboken, New Jersey, 2005; pp 340–349.
Return to citation in text: [1] -
Fortea, J. J. Prakt. Chem. 1975, 317, 705–711. doi:10.1002/prac.19753170502
Return to citation in text: [1] -
Sakamoto, T.; Yoshizawa, H.; Kaneda, S.; Yamanaka, H. Chem. Pharm. Bull. 1984, 32, 728–732. doi:10.1248/cpb.32.728
Return to citation in text: [1] -
Blake, J. F.; Xu, R.; Bencsik, J. R.; Xiao, D.; Kallan, N. C.; Schlachter, S.; Mitchell, I. S.; Spencer, K. L.; Banka, A. L.; Wallace, E. M.; Gloor, S. L.; Martinson, M.; Woessner, R. D.; Vigers, G. P. A.; Brandhuber, B. J.; Liang, J.; Safina, B. S.; Li, J.; Zhang, B.; Chabot, C.; Do, S.; Lee, L.; Oeh, J.; Sampath, D.; Lee, B. B.; Lin, K.; Liederer, B. M.; Skelton, N. J. J. Med. Chem. 2012, 55, 8110–8127. doi:10.1021/jm301024w
Return to citation in text: [1] -
Lehn, J.-M.; Regnouf de Vains, J.-B. Tetrahedron Lett. 1989, 30, 2209–2212. doi:10.1016/S0040-4039(00)99650-3
Return to citation in text: [1] -
Tikhonov, A. Y.; Volodarskii, L. B.; Vakolova, O. A.; Podgornaya, M. I. Chem. Heterocycl. Compd. 1981, 17, 89–95. doi:10.1007/bf00507100
Return to citation in text: [1] -
Waitkins, G. R.; Clark, C. W. Chem. Rev. 1945, 36, 235–289. doi:10.1021/cr60115a001
Return to citation in text: [1] -
Rabjohn, N. Org. React. 1976, 24, 261–415.
Return to citation in text: [1] -
Tagawa, Y.; Yamashita, K.; Higuchi, Y.; Goto, Y. Heterocycles 2003, 60, 953–957. doi:10.3987/COM-02-9702
Return to citation in text: [1] -
Grubbs, R. H., Ed. Handbook of Metathesis; Wiley-VCH: Weinheim, Germany, 2003.
Return to citation in text: [1] -
Lee, C. W.; Grubbs, R. H. Org. Lett. 2000, 2, 2145–2147. doi:10.1021/ol006059s
Return to citation in text: [1] -
Heckrodt, T. J.; Singh, R. Synth. Commun. 2011, 42, 2854–2865. doi:10.1080/00397911.2011.570891
Return to citation in text: [1] -
Marx, V. M.; Herbert, M. B.; Keitz, B. K.; Grubbs, R. H. J. Am. Chem. Soc. 2013, 135, 94–97. doi:10.1021/ja311241q
Return to citation in text: [1] -
Prunet, J. Angew. Chem., Int. Ed. 2003, 42, 2826–2830. doi:10.1002/anie.200301628
Angew. Chem. 2003, 115, 2932–2936. doi:10.1002/ange.200301628
Return to citation in text: [1] -
Dale, J. A.; Mosher, H. S. J. Am. Chem. Soc. 1973, 95, 512–519. doi:10.1021/ja00783a034
Return to citation in text: [1] -
Hoff, S.; Brandsma, L.; Arens, J. F. Recl. Trav. Chim. Pays-Bas 1968, 87, 916–924. doi:10.1002/recl.19680870807
Return to citation in text: [1]
| 70. | Lee, C. W.; Grubbs, R. H. Org. Lett. 2000, 2, 2145–2147. doi:10.1021/ol006059s |
| 71. | Heckrodt, T. J.; Singh, R. Synth. Commun. 2011, 42, 2854–2865. doi:10.1080/00397911.2011.570891 |
| 72. | Marx, V. M.; Herbert, M. B.; Keitz, B. K.; Grubbs, R. H. J. Am. Chem. Soc. 2013, 135, 94–97. doi:10.1021/ja311241q |
| 73. |
Prunet, J. Angew. Chem., Int. Ed. 2003, 42, 2826–2830. doi:10.1002/anie.200301628
Angew. Chem. 2003, 115, 2932–2936. doi:10.1002/ange.200301628 |
| 74. | Dale, J. A.; Mosher, H. S. J. Am. Chem. Soc. 1973, 95, 512–519. doi:10.1021/ja00783a034 |
| 34. |
Zimmer, R. Synthesis 1993, 165–178. doi:10.1055/s-1993-25823
And references cited therein. |
| 75. | Hoff, S.; Brandsma, L.; Arens, J. F. Recl. Trav. Chim. Pays-Bas 1968, 87, 916–924. doi:10.1002/recl.19680870807 |
| 1. | Zhu, J.; Bienaymé, H., Eds. Multicomponent Reactions; Wiley-VCH: Weinheim, Germany, 2005. |
| 2. | Tietze, L. F.; Brasche, G.; Gericke, K. M. Domino Reactions in Organic Synthesis; Wiley-VCH: Weinheim, Germany, 2006. |
| 10. | Evdokimov, N. M.; Kireev, A. S.; Yakovenko, A. A.; Antipin, M. Yu.; Magedov, I. V.; Kornienko, A. J. Org. Chem. 2007, 72, 3443–3453. doi:10.1021/jo070114u |
| 11. | Sasada, T.; Sakai, N.; Konakahara, T. J. Org. Chem. 2008, 73, 6905–6908. doi:10.1021/jo801090h |
| 12. | Barluenga, J.; Jiménez-Aquino, A.; Fernández, M. A.; Aznar, F.; Valdés, C. Tetrahedron 2008, 64, 778–786. doi:10.1016/j.tet.2007.10.112 |
| 13. | Da-Qing, S.; Hao, Y. Synth. Commun. 2009, 39, 2481–2491. doi:10.1080/00397910802656034 |
| 14. | Guo, K.; Thompson, M. J.; Chen, B. J. Org. Chem. 2009, 74, 6999–7006. doi:10.1021/jo901232b |
| 15. |
Sha, F.; Huang, X. Angew. Chem., Int. Ed. 2009, 48, 3458–3461. doi:10.1002/anie.200900212
Angew. Chem. 2009, 121, 3510–3513. doi:10.1002/ange.200900212 |
| 16. | Chen, M. Z.; Micalizio, G. C. J. Am. Chem. Soc. 2012, 134, 1352–1356. doi:10.1021/ja2105703 |
| 17. | Al-Awadi, N. A.; Ibrahim, M. R.; Elnagdi, M. H.; John, E.; Ibrahim, Y. A. Beilstein J. Org. Chem. 2012, 8, 441–447. doi:10.3762/bjoc.8.50 |
| 18. | Sha, F.; Wu, L.; Huang, X. J. Org. Chem. 2012, 77, 3754–3765. doi:10.1021/jo300072x |
| 19. | Zheng, L.; Ju, J.; Bin, Y.; Hua, R. J. Org. Chem. 2012, 77, 5794–5800. doi:10.1021/jo3010414 |
| 20. | He, Z.; Dobrovolsky, D.; Trinchera, P.; Yudin, A. K. Org. Lett. 2013, 15, 334–337. doi:10.1021/ol303246b |
| 40. | Eidamshaus, C.; Reissig, H.-U. Adv. Synth. Catal. 2009, 351, 1162–1166. doi:10.1002/adsc.200800789 |
| 39. | Dash, J.; Lechel, T.; Reissig, H.-U. Org. Lett. 2007, 9, 5541–5544. doi:10.1021/ol702468s |
| 8. | Hantzsch, A. Justus Liebigs Ann. Chem. 1882, 215, 1–82. doi:10.1002/jlac.18822150102 |
| 38. | Flögel, O.; Dash, J.; Brüdgam, I.; Hartl, H.; Reissig, H.-U. Chem.–Eur. J. 2004, 10, 4283–4290. doi:10.1002/chem.200400322 |
| 3. | D'Souza, D. M.; Müller, T. J. J. Chem. Soc. Rev. 2007, 36, 1095–1108. doi:10.1039/b608235c |
| 4. | Brauch, S.; van Berkel, S. S.; Westermann, B. Chem. Soc. Rev. 2013, 42, 4948–4962. doi:10.1039/c3cs35505e |
| 5. | Simon, C.; Constantieux, T.; Rodriguez, J. Eur. J. Org. Chem. 2004, 4957–4980. doi:10.1002/ejoc.200400511 |
| 6. | Eckert, H. Molecules 2012, 17, 1074–1102. doi:10.3390/molecules17011074 |
| 7. |
Ruijter, E.; Scheffelaar, R.; Orru, R. V. A. Angew. Chem., Int. Ed. 2011, 50, 6234–6246. doi:10.1002/anie.201006515
Angew Chem. 2011, 123, 6358–6371. doi:10.1002/ange.201006515 |
| 38. | Flögel, O.; Dash, J.; Brüdgam, I.; Hartl, H.; Reissig, H.-U. Chem.–Eur. J. 2004, 10, 4283–4290. doi:10.1002/chem.200400322 |
| 39. | Dash, J.; Lechel, T.; Reissig, H.-U. Org. Lett. 2007, 9, 5541–5544. doi:10.1021/ol702468s |
| 45. | Lechel, T.; Dash, J.; Brüdgam, I.; Reissig, H.-U. Eur. J. Org. Chem. 2008, 3647–3655. doi:10.1002/ejoc.200800398 |
| 43. | Bera, M. K.; Reissig, H.-U. Synthesis 2010, 2129–2138. doi:10.1055/s-0029-1218787 |
| 48. | Lechel, T.; Möhl, S.; Reissig, H.-U. Synlett 2009, 1059–1062. doi:10.1055/s-0028-1088220 |
| 49. | Lechel, T.; Reissig, H.-U. Eur. J. Org. Chem. 2010, 2555–2564. doi:10.1002/ejoc.201000056 |
| 38. | Flögel, O.; Dash, J.; Brüdgam, I.; Hartl, H.; Reissig, H.-U. Chem.–Eur. J. 2004, 10, 4283–4290. doi:10.1002/chem.200400322 |
| 39. | Dash, J.; Lechel, T.; Reissig, H.-U. Org. Lett. 2007, 9, 5541–5544. doi:10.1021/ol702468s |
| 40. | Eidamshaus, C.; Reissig, H.-U. Adv. Synth. Catal. 2009, 351, 1162–1166. doi:10.1002/adsc.200800789 |
| 41. | Lechel, T.; Dash, J.; Eidamshaus, C.; Brüdgam, I.; Lentz, D.; Reissig, H.-U. Org. Biomol. Chem. 2010, 8, 3007–3014. doi:10.1039/B925468D |
| 42. | Lechel, T.; Dash, J.; Hommes, P.; Lentz, D.; Reissig, H.-U. J. Org. Chem. 2010, 75, 726–732. doi:10.1021/jo9022183 |
| 43. | Bera, M. K.; Reissig, H.-U. Synthesis 2010, 2129–2138. doi:10.1055/s-0029-1218787 |
| 44. | Eidamshaus, C.; Kumar, R.; Bera, M. K.; Reissig, H.-U. Beilstein J. Org. Chem. 2011, 7, 962–975. doi:10.3762/bjoc.7.108 |
| 50. | Zimmer, R.; Lechel, T.; Rancan, G.; Bera, M. K.; Reissig, H.-U. Synlett 2010, 1793–1796. doi:10.1055/s-0030-1258088 |
| 34. |
Zimmer, R. Synthesis 1993, 165–178. doi:10.1055/s-1993-25823
And references cited therein. |
| 35. | Krause, N.; Hashmi, A. S. K., Eds. Modern Allene Chemistry; Wiley-VCH: Weinheim, Germany, 2004. |
| 36. | Brasholz, M.; Reissig, H.-U.; Zimmer, R. Acc. Chem. Res. 2009, 42, 45–56. doi:10.1021/ar800011h |
| 37. | Lechel, T.; Reissig, H.-U. Pure Appl. Chem. 2010, 82, 1835–1844. doi:10.1351/pac-con-09-09-06 |
| 21. | Stonehouse, J. P.; Chekmarev, D. S.; Ivanova, N. V.; Lang, S.; Pairaudeau, G.; Smith, N.; Stocks, M. J.; Sviridov, S. I.; Utkina, L. M. Synlett 2008, 100–104. doi:10.1055/s-2007-1000839 |
| 22. | Sasada, T.; Moriuchi, M.; Sakai, N.; Konakahara, T. Eur. J. Org. Chem. 2009, 5738–5743. doi:10.1002/ejoc.200900639 |
| 23. | Sasada, T.; Kobayashi, F.; Sakai, N.; Konakahara, T. Org. Lett. 2009, 11, 2161–2164. doi:10.1021/ol900382j |
| 24. | Hekmatshoar, R.; Kenary, G. N.; Sadjadi, S.; Beheshtiha, Y. S. Synth. Commun. 2010, 40, 2007–2013. doi:10.1080/00397910903219385 |
| 25. | Wan, J.-P.; Liu, Y. Synthesis 2010, 3943–3953. doi:10.1055/s-0030-1258290 |
| 26. | Zonouzi, A.; Biniaz, M.; Mirzazadeh, R.; Talebi, M.; Ng, S. W. Heterocycles 2010, 81, 1271–1278. doi:10.3987/com-10-11929 |
| 27. | Majumder, S.; Odom, A. L. Tetrahedron 2010, 66, 3152–3158. doi:10.1016/j.tet.2010.02.066 |
| 28. | Sedenkova, K. N.; Averina, E. B.; Grishin, Y. K.; Kutateladze, A. G.; Rybakov, V. B.; Kuznetsova, T. S.; Zefirov, N. S. J. Org. Chem. 2012, 77, 9893–9899. doi:10.1021/jo301880m |
| 29. | You, X.; Yu, S.; Liu, Y. Organometallics 2013, 32, 5273–5276. doi:10.1021/om400880r |
| 30. | Reddy, L. S.; Reddy, T. R.; Reddy, N. C. G.; Mohan, R. B.; Lingappa, Y. Synthesis 2013, 45, 75–84. doi:10.1055/s-0032-1316814 |
| 31. | Yavari, I.; Nematpour, M. Synlett 2013, 24, 165–168. doi:10.1055/s-0032-1317951 |
| 32. | Yang, K.; Xiang, J.; Bao, G.; Dang, Q.; Bai, X. ACS Comb. Sci. 2013, 15, 519–524. doi:10.1021/co400086u |
| 33. | Gers, C. F.; Rosellen, J.; Merkul, E.; Müller, T. J. J. Beilstein J. Org. Chem. 2011, 7, 1173–1181. doi:10.3762/bjoc.7.136 |
| 46. | Lechel, T.; Lentz, D.; Reissig, H.-U. Chem.–Eur. J. 2009, 15, 5432–5435. doi:10.1002/chem.200900386 |
| 47. | Lechel, T.; Gerhard, M.; Trawny, D.; Brusilowskij, B.; Schefzig, L.; Zimmer, R.; Rabe, J. P.; Lentz, D.; Schalley, C. A.; Reissig, H.-U. Chem.–Eur. J. 2011, 17, 7480–7491. doi:10.1002/chem.201100382 |
| 53. | Eidamshaus, C.; Reissig, H.-U. Eur. J. Org. Chem. 2011, 6056–6069. doi:10.1002/ejoc.201100681 |
| 38. | Flögel, O.; Dash, J.; Brüdgam, I.; Hartl, H.; Reissig, H.-U. Chem.–Eur. J. 2004, 10, 4283–4290. doi:10.1002/chem.200400322 |
| 51. | Dash, J.; Reissig, H.-U. Chem.–Eur. J. 2009, 15, 6811–6814. doi:10.1002/chem.200900939 |
| 52. | Hommes, P.; Jungk, P.; Reissig, H.-U. Synlett 2011, 2311–2314. doi:10.1055/s-0030-1260304 |
| 51. | Dash, J.; Reissig, H.-U. Chem.–Eur. J. 2009, 15, 6811–6814. doi:10.1002/chem.200900939 |
| 52. | Hommes, P.; Jungk, P.; Reissig, H.-U. Synlett 2011, 2311–2314. doi:10.1055/s-0030-1260304 |
| 66. | Waitkins, G. R.; Clark, C. W. Chem. Rev. 1945, 36, 235–289. doi:10.1021/cr60115a001 |
| 67. | Rabjohn, N. Org. React. 1976, 24, 261–415. |
| 68. | Tagawa, Y.; Yamashita, K.; Higuchi, Y.; Goto, Y. Heterocycles 2003, 60, 953–957. doi:10.3987/COM-02-9702 |
| 69. | Grubbs, R. H., Ed. Handbook of Metathesis; Wiley-VCH: Weinheim, Germany, 2003. |
| 59. | Boekelheide, V.; Linn, W. J. J. Am. Chem. Soc. 1954, 76, 1286–1291. doi:10.1021/ja01634a026 |
| 60. | Galatsis, P. Boekelheide Reaction. In Name Reactions in Heterocyclic Chemistry; Li, J.-J.; Corey, E. J., Eds.; John Wiley & Sons Inc.: Hoboken, New Jersey, 2005; pp 340–349. |
| 50. | Zimmer, R.; Lechel, T.; Rancan, G.; Bera, M. K.; Reissig, H.-U. Synlett 2010, 1793–1796. doi:10.1055/s-0030-1258088 |
| 61. | Fortea, J. J. Prakt. Chem. 1975, 317, 705–711. doi:10.1002/prac.19753170502 |
| 62. | Sakamoto, T.; Yoshizawa, H.; Kaneda, S.; Yamanaka, H. Chem. Pharm. Bull. 1984, 32, 728–732. doi:10.1248/cpb.32.728 |
| 63. | Blake, J. F.; Xu, R.; Bencsik, J. R.; Xiao, D.; Kallan, N. C.; Schlachter, S.; Mitchell, I. S.; Spencer, K. L.; Banka, A. L.; Wallace, E. M.; Gloor, S. L.; Martinson, M.; Woessner, R. D.; Vigers, G. P. A.; Brandhuber, B. J.; Liang, J.; Safina, B. S.; Li, J.; Zhang, B.; Chabot, C.; Do, S.; Lee, L.; Oeh, J.; Sampath, D.; Lee, B. B.; Lin, K.; Liederer, B. M.; Skelton, N. J. J. Med. Chem. 2012, 55, 8110–8127. doi:10.1021/jm301024w |
| 64. | Lehn, J.-M.; Regnouf de Vains, J.-B. Tetrahedron Lett. 1989, 30, 2209–2212. doi:10.1016/S0040-4039(00)99650-3 |
| 65. | Tikhonov, A. Y.; Volodarskii, L. B.; Vakolova, O. A.; Podgornaya, M. I. Chem. Heterocycl. Compd. 1981, 17, 89–95. doi:10.1007/bf00507100 |
| 48. | Lechel, T.; Möhl, S.; Reissig, H.-U. Synlett 2009, 1059–1062. doi:10.1055/s-0028-1088220 |
| 49. | Lechel, T.; Reissig, H.-U. Eur. J. Org. Chem. 2010, 2555–2564. doi:10.1002/ejoc.201000056 |
| 50. | Zimmer, R.; Lechel, T.; Rancan, G.; Bera, M. K.; Reissig, H.-U. Synlett 2010, 1793–1796. doi:10.1055/s-0030-1258088 |
| 42. | Lechel, T.; Dash, J.; Hommes, P.; Lentz, D.; Reissig, H.-U. J. Org. Chem. 2010, 75, 726–732. doi:10.1021/jo9022183 |
| 54. | Högermeier, J.; Reissig, H.-U. Adv. Synth. Catal. 2009, 351, 2747–2763. doi:10.1002/adsc.200900566 |
| 55. | Bera, M. K.; Hommes, P.; Reissig, H.-U. Chem.–Eur. J. 2011, 17, 11838–11843. doi:10.1002/chem.201101739 |
| 56. | Eidamshaus, C.; Hommes, P.; Reissig, H.-U. Synlett 2012, 23, 1670–1674. doi:10.1055/s-0031-1290398 |
| 57. | Gholap, S. L.; Hommes, P.; Neuthe, K.; Reissig, H.-U. Org. Lett. 2013, 15, 318–321. doi:10.1021/ol303231c |
| 58. | Bera, M. K.; Gholap, S. L.; Hommes, P.; Neuthe, K.; Trawny, D.; Rabe, J. P.; Lentz, D.; Zimmer, R.; Reissig, H.-U. Adv. Synth. Catal. 2013, 355, 3463–3474. doi:10.1002/adsc.201300613 |
| 55. | Bera, M. K.; Hommes, P.; Reissig, H.-U. Chem.–Eur. J. 2011, 17, 11838–11843. doi:10.1002/chem.201101739 |
| 57. | Gholap, S. L.; Hommes, P.; Neuthe, K.; Reissig, H.-U. Org. Lett. 2013, 15, 318–321. doi:10.1021/ol303231c |
| 58. | Bera, M. K.; Gholap, S. L.; Hommes, P.; Neuthe, K.; Trawny, D.; Rabe, J. P.; Lentz, D.; Zimmer, R.; Reissig, H.-U. Adv. Synth. Catal. 2013, 355, 3463–3474. doi:10.1002/adsc.201300613 |
© 2014 Bera et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)