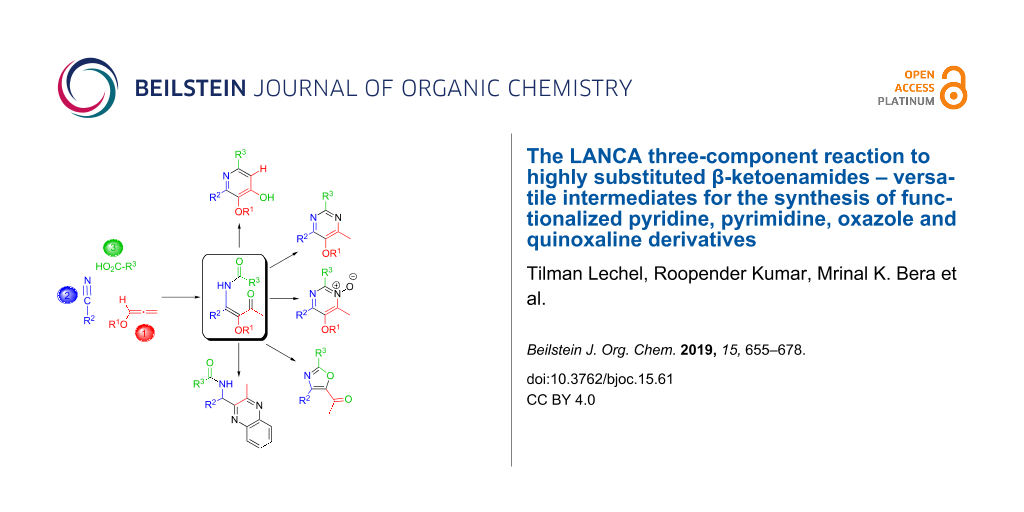
The LANCA three-component reaction to highly substituted β-ketoenamides – versatile intermediates for the synthesis of functionalized pyridine, pyrimidine, oxazole and quinoxaline derivatives
1,
1,2,
1,3,
1 and
1
Tilman Lechel
Institut für Chemie und Biochemie, Freie Universität Berlin, Takustr. 3, D-14195 Berlin, Germany
Guest Editor: T. J. J. Müller Beilstein J. Org. Chem.2019,15, 655–678.https://doi.org/10.3762/bjoc.15.61 Received 20 Dec 2018,
Accepted 20 Feb 2019,
Published 13 Mar 2019
The LANCA three-component reaction of lithiated alkoxyallenes LA, nitriles N and carboxylic acids CA leads to β-ketoenamides KE in good to excellent yields. The scope of this reaction is very broad and almost all types of nitriles and carboxylic acids have successfully been used. The alkoxy group introduced via the allene component is also variable and hence the subsequent transformation of this substituent into a hydroxy group can be performed under different conditions. Enantiopure nitriles or carboxylic acids can also be employed leading to chiral KE with high enantiopurity and dinitriles or dicarboxylic acids also lead to the expected bis-β-ketoenamides. β-Ketoenamides incorporate a unique combination of functional groups and hence a manifold of subsequent reactions to highly substituted heterocyclic compounds is possible. An intramolecular aldol-type condensation reaction efficiently furnishes pyridin-4-ols PY that can be further modified by palladium-catalyzed reactions, e.g., to specifically substituted furopyridine derivatives. Condensations of β-ketoenamides with ammonium salts or with hydroxylamine hydrochloride afford pyrimidines PM or pyrimidine N-oxides PO with a highly flexible substitution pattern in good yields. The functional groups of these heterocycles also allow a variety of subsequent reactions to various pyrimidine derivatives. On the other hand, acid-labile alkoxy substituents such as a 2-(trimethylsilyl)ethoxy group are required for the conversion of β-ketoenamides into 5-acetyl-substituted oxazoles OX, again compounds with high potential for subsequent functional group transformations. For acid labile β-ketoenamides bearing bulky substituents the acid treatment leads to acylamido-substituted 1,2-diketones DK that could be converted into quinoxalines QU. All classes of heterocycles accessed through the key β-ketoenamides show a unique substitution pattern – not easily accomplishable by alternative methods – and therefore many subsequent reactions are possible.
Multicomponent reactions are known to create unique product skeletons in an atom economic, efficient and time saving fashion. In many cases, compounds bearing functional groups of relatively high energy level with the potential of multiple reactivity are employed, for instance nitriles, isonitriles or alkynes [1-9]. Not surprisingly, simple or functionalized allenes have also been used in multicomponent processes and – dependent on the substitution pattern of the allene – a remarkable variety of reactions and product types are known using the three-carbon backbone of these reactive compounds [10]. During the exploration of alkoxyallene chemistry [11-20] we accidently discovered a new three-component reaction leading to β-ketoenamides that are uniquely functionalized alkenes and suitable for a variety of subsequent reactions, in particular in heterocyclic synthesis.
This LANCA three-component reaction (LA = lithiated alkoxyallene, N = nitrile, CA = carboxylic acid) was observed for the first time by Oliver Flögel, who treated pivalonitrile (1) with lithiated methoxyallene 2 and isolated the expected primary addition product 3[21]. This intermediate was subjected to different cyclization conditions (Scheme 1) and the desired pyrrole derivative 4 was produced under specific conditions employing silver nitrate as catalyst. However, the treatment of 3 with an excess of trifluoroacetic acid led to a mixture of β-ketoenamide 5 and pyridin-4-ol derivative 6. Thus, the carboxylic acid did not act as a catalyst in this reaction as expected, it was incorporated into the products!
Scheme 1:
Discovery of the LANCA three-component reaction. The reaction of pivalonitrile (1) with lithiated methoxyallene 2 leads to allenyl-substituted imine 3 which upon reaction with trifluoroacetic acid affords β-ketoenamide 5 and pyridin-4-ol derivative 6.
Scheme 1:
Discovery of the LANCA three-component reaction. The reaction of pivalonitrile (1) with lithiated m...
The unique mechanism leading to the β-ketoenamide and the pyridin-4-ol has been discussed earlier [21-23], but the essentials of the involved cascade reactions have to be presented again in order to understand the formation of the crucial β-ketoenamide intermediates (Scheme 2). The protonation of the primarily formed allenylimine A by the added carboxylic acid at the nitrogen gives an allenyl iminium/aminobutadienyl cation intermediate B that accepts the present carboxylate at the electrophilic carbon to provide an acyloxy-substituted aminobutadiene derivative C. The acyl group is subsequently transferred to the close amino group giving D thus accomplishing the final connectivity of the three components. Enol/carbonyl tautomerization gives the isolated β-ketoenamide KE with E-configuration being the result of the intramolecular acyl transfer. Even after storage of β-ketoenamides there is no evidence that an isomerization to the corresponding Z-isomers occurs.
Scheme 2:
Proposed mechanism of the LANCA three-component reaction to β-ketoenamides KE and pyridin-4-ol derivatives PY.
Scheme 2:
Proposed mechanism of the LANCA three-component reaction to β-ketoenamides KE and pyridin-4-ol deri...
It should be noted here that the protonation to the allenyl iminium species B implies an “umpolung of reactivity” of the alkoxyallene subunit converting the central allene carbon to an electrophilic center whereas this carbon is a nucleophilic center in the neutral compound. The obtained β-ketoenamides are alkenes with a remarkable assembly of functional groups: they are enamides, enol ethers and α,β-unsaturated carbonyl compounds at the same time. In addition, their methyl ketone subunit is required for some of the subsequent transformations, e.g., the synthesis of pyridin-4-ols PY.
The first reaction shown in Scheme 1 gave a mixture of β-ketoenamide 5 and its subsequent cyclization product pyridin-4-ol 6 in low yields. This new route to highly substituted pyridin-4-ol derivatives could be streamlined as a one-pot procedure and after completion of the condensation reaction with trimethylsilyl trifluoromethanesulfonate (TMSOTf) and a tertiary amine as base a broad range of pyridine derivatives was accessible (Scheme 3). According to its discoverer, we named this reaction Flögel pyridine synthesis [21]. The reaction is very flexible with respect to the employed alkoxyallenes, nitriles and carboxylic acids and due to the two differently protected oxygen functions of the pyridin-4-ols these intermediates could be further substituted, e.g., through palladium-catalyzed reactions, to give highly substituted pyridine derivatives in a great variety. The scope and limitations of this approach as well as many subsequent reactions to a broad range of pyridine or furopyridine derivatives has recently been summarized in a comprehensive review [23]. It should be mentioned here, that alkoxyallenes are no exotic compounds but easily available in two steps from simple starting materials [24,25]. They can smoothly be prepared in multigram scale and recently a flow chemistry approach on the use of lithiated methoxyallene was published [26].
Scheme 3:
One-pot preparation of pyridin-4-ols PY and their subsequent transformations to highly substituted pyridine derivatives and furopyridines.
Scheme 3:
One-pot preparation of pyridin-4-ols PY and their subsequent transformations to highly substituted ...
For the case, that substituents R3 are strongly electron withdrawing the relatively electrophilic amido carbonyl group of the β-ketoenamides partially undergoes a subsequent intramolecular aldol-type condensation reaction to furnish the pyridin-4-ols. Therefore, trifluoroacetic acid or related fluorinated carboxylic acids [22] lead to mixtures of the two products as shown in Scheme 1. For other carboxylic acids the multistep reaction of the three components stops at the stage of the β-ketoenamides that were usually isolated in good yields. These intermediates also provide pyridin-4-ols under slightly more rigorous condensation conditions, but they can also be used in alternative synthetic operations providing other compound classes, in particular heterocyclic compounds. The synthesis of pyrimidines PM, pyrimidine N-oxides PO, oxazoles OX, 1,2-diketones DK and quinoxalines QU starting from β-ketoenamides KE is the topic of the present review.
Review
Scope of the LANCA three-component synthesis of β-ketoenamides
The scope of the LANCA three-component synthesis of β-ketoenamides KE through the reaction of alkoxyallenes, nitriles and carboxylic acids is very broad and only a few clear limitations were found (Scheme 4). With lithiated methoxyallene (R1 = Me) – the standard alkoxyallene generally used to study new reactions – many conceivable combinations of nitriles as second and carboxylic acids as third component were examined.
Scheme 4:
Synthesis of β-ketoenamides KE by the LANCA three-component reaction of alkoxyallenes, nitriles and carboxylic acids.
Scheme 4:
Synthesis of β-ketoenamides KE by the LANCA three-component reaction of alkoxyallenes, nitriles and...
In Table 1 the resulting products KE1–35 are collected, showing that simple alkyl, branched alkyl, cycloalkyl, functionalized alkyl, alkenyl, aryl or heteroaryl substituents (R2 or R3) can be introduced into the resulting β-ketoenamides KE. The use of α,β-unsaturated nitriles as second component did not provide reasonable results, possibly due to a competing 1,4-addition of the lithiated methoxyallene to the double bonds. On the other hand, α,β-unsaturated carboxylic acids were excellent third components as shown in various examples (Table 1, entries 15–19, 26–28, and 34). Even with acrylic acid the expected product KE15 was isolated in 91% yield. Although we did not systematically study nitriles with heterocyclic substituents, we showed that thiophene-2-carbonitrile is an excellent substrate leading to KE32–35 in good yields (Table 1, entries 32–35). Unfortunately, pyridine-2-carbonitrile could not be used as electrophilic component; the reason for this failure is unclear. By use of heterocyclic carboxylic acids we could smoothly introduce 2-thienyl and 2-pyridyl substituents into the β-ketoenamides KE2, KE23, KE30, KE31 and KE35 (Table 1, entries 2, 23, 30, 31, and 35).
Table 1:
Synthesis of β-ketoenamides KE1–35 through the LANCA three-component reaction of lithiated methoxyallene, nitriles (R2-CN) and carboxylic acids (R3-CO2H) according to Scheme 4.a
aAbbreviations: Ad = 1-adamantyl, Fu = furyl, Py = pyridyl, Th = thienyl; all alkenyl substituents are E-configured.
The present approach does not allow the synthesis of β-ketoenamides with substituents R2 = H or R3 = H. The reaction of lithiated methoxyallene with hydrogen cyanide as second component was not examined due to the assumed Brønsted acid property of the latter. As a substitute, cyano trimethylsilane was examined, however, this experiment did not afford the corresponding β-ketoenamide. Unexpectedly, using formic acid as the third component afforded only mixtures of compounds whose structures could not be elucidated. The role of formic acid in the three-component reaction should be investigated by finding the proper substrates and conditions. The β-ketoenamides with N-formyl substituents should be valuable precursors for subsequent transformations.
As expected, the reactions involving trifluoroacetic acid gave only low yields of the β-ketoenamides KE due to the competing in situ cyclization to the corresponding pyridin-4-ols (PY, Table 1, entries 12 and 24). The related reactions with trichloroacetic acid provided the β-ketoenamides in slightly better yields with lower amounts of the corresponding pyridin-4-ols (Table 1, entries 3, 14, and 25) showing that the electrophilicity of the amide carbonyl group is lower in these substrates.
Stereogenic centers could also successfully be introduced into the β-ketoenamides as shown by the examples collected in Scheme 5. All three possibilities to use enantiopure starting materials were examined in the three-component reaction. The products KE36–39 are derived from the O-protected nitrile obtained from (S)-lactic acid [35,36]. This chiral acid itself was incorporated as third component resulting in β-ketoenamides KE39–41. There is no indication of an erosion of the enantiopurity and the chiral compounds were converted into the corresponding pyridin-4-ol derivatives and tested as chiral ligands in asymmetric catalysis [37]. Using the N-trityl-substituted proline as carboxylic acid provided the expected β-ketoenamide KE42 in low yield and as major product we isolated compound 7 in 49% yield (1:1 mixture of the two possible diastereomers). Probably due to the bulkiness of the acyl group, the migration to the nitrogen is strongly hampered and hence the three-component cascade almost completely stops at the stage of aminobutadiene C (see Scheme 2) that is hydrolyzed during work-up to give α-methoxy carbonyl compound 7. The isolation of this compound supports our mechanistic proposal as shown in Scheme 2 and it shows that sterically very hindered carboxylic acids are probably poor components in the route to β-ketoenamides. A systematic study of this possible limitation was not carried out, but a component such as Mosher acid with a tertiary carbon next to the carboxylic acid function was successfully used in the three-component reaction and the corresponding β-ketoenamide was converted into the corresponding pyridin-4-ol in good overall yield [36]. This demonstrates that carboxylic acids with tertiary centers are possible candidates for the route to β-ketoenamides.
Scheme 5:
β-Ketoenamides KE36–43 derived from enantiopure components.
Scheme 5:
β-Ketoenamides KE36–43 derived from enantiopure components.
The last example shown in Scheme 5 demonstrates that allenes with chiral alkoxy substituents are also suitable starting materials in the three-component reaction. We did not study lithiated carbohydrate-derived alkoxyallenes that were good precursors for other applications [38,39], but prepared the allene derived from N-protected alaninol that was converted into β-ketoenamide KE43 in 26% yield [30].
It should be mentioned here that most of the reactions to β-ketoenamides were performed only once under standard conditions without detailed optimization reaction conditions such as stoichiometry of components, applied temperatures and reaction times. It is therefore very likely that in the cases where low or moderate yields were recorded improvements are easily possible. For a few examples, we also performed the reactions in larger scale, e.g., the synthesis of β-ketoenamides KE35 that was prepared in 3.5 g quantity [34]. The scalability of the three-component reactions seems therefore no problem which is important for the multistep preparation of subsequent products (see below).
Aromatic dinitriles such as 1,3- and 1,4-dicyanobenzene were also examined as second component in the three-component reaction. The obtained bis-β-ketoenamides were not isolated and purified, but directly converted into the corresponding bis-pyridin-4-ol derivatives by cyclocondensation [40]. The overall yields were only in the range of 20% probably due to solubility problems with employed aromatic dinitriles. Nevertheless, these examples showed the feasibility of this approach to highly substituted β-ketoenamides. Similar results were obtained by the use of aromatic dicarboxylic acids (Scheme 6). Again, the moderate efficacy may be due to their low solubility in ethereal solvents at low temperatures – a problem that could only partially be solved by use of DMF as cosolvent. The yields of β-ketoenamides KE44–46 are only in the range of 25%, but the three-component approach to unique multifunctional products is nevertheless remarkable [41].
Scheme 6:
Bis-β-ketoenamides KE44–46 derived from aromatic dicarboxylic acids.
Scheme 6:
Bis-β-ketoenamides KE44–46 derived from aromatic dicarboxylic acids.
For subsequent reactions, alkoxy groups other than the methoxy group were desirable because the latter substituent can only be converted into free hydroxy groups by treatment with strong (Lewis) acids. We therefore examined benzyloxyallene as starting material in the three-component reaction to β-ketoenamides. The resulting products were deprotectable under milder conditions as shown below. The examples, including one with a p-methoxybenzyloxy group (PMB, Table 2, entry 10) are collected in Table 2. As expected there were no fundamental differences to the observations with methoxyallene. The β-ketoenamides derived from trifluoroacetic acid are only available in low yield due to the fast formation of the corresponding pyridin-4-ols (PY, Table 2, entries 1, 6, and 10). For the other combinations of substituents the unoptimized yields of β-ketoenamides are satisfying.
Table 2:
Synthesis of β-ketoenamides KE47–56 through the LANCA three-component reaction of lithiated benzyloxyallene, nitriles (R2-CN) and carboxylic acids (R3-CO2H) according to Scheme 4.a
aAbbreviations: Fu = furyl, Py = pyridyl, Th = thienyl, PMB = CH2C6H4-4-OMe; all alkenyl substituents are E-configured.
Another good alternative to methoxyallene is the 2-(trimethylsilyl)ethoxy-substituted allene. The 2-(trimethylsilyl)ethyl substituent can be removed from the products either by fluoride or acid treatment under mild conditions. Again, there were no great differences in the performance of this component compared to methoxyallene or benzyloxyallene. In Table 3 the result with tetrahydropyranyl-substituted allene is also included (Table 3, entry 1) that gave a moderate yield of β-ketoenamide KE57.
Table 3:
Synthesis of β-ketoenamides KE57–77 by the LANCA three-component reaction of lithiated 2-(trimethylsilyl)ethoxyallene, nitriles (R2-CN) and carboxylic acids (R3-CO2H) according to Scheme 4.a
aAbbreviations: THP = tetrahydropyranyl, Ad = 1-adamantyl, Fu = furyl, Py = pyridyl, Th = thienyl; all alkenyl substituents are E-configured. bAs second product, 15% of the imine tautomer of β-ketoenamide KE67 was isolated.
The alkoxyallenes so far listed are unsubstituted at the C-3 terminus and related allenes bearing alkyl groups at this carbon are not directly accessible [44]. In contrast, products that are formally derived from 3-aryl-substituted alkoxyallenes can smoothly be prepared from the corresponding alkyl propargyl ethers E (Scheme 7). Their deprotonation with n-butyllithium proceeds with a proton shift delivering the intermediate F that reacts with electrophiles at C-1. The three-component reaction with nitriles and carboxylic acids then leads to the corresponding β-ketoenamides KEAr in moderate yields. The reaction sequence is illustrated in Scheme 7 also showing the three products KE78[22], KE79[45] and KE80[46] that were prepared by this largely unexplored, but very promising method. It opens a route to highly substituted heterocycles as shown by the cyclocondensation of KE78 that gave the corresponding pentasubstituted pyridin-4-ol derivative in 91% yield [22].
Scheme 7:
Conversion of alkyl propargyl ethers E into aryl-substituted β-ketoenamides KEAr and selected products KE78–80 obtained by this route.
Scheme 7:
Conversion of alkyl propargyl ethers E into aryl-substituted β-ketoenamides KEAr and selected produ...
As mentioned above, a large number of the prepared β-ketoenamides KE was converted into the corresponding pyridin-4-ol derivatives PY and subsequent products of these versatile heterocyclic intermediates. Our published review on this topic [23] presents many examples of β-ketoenamides KE that were not purified but directly transferred into these pyridine derivatives. Hence, the scope of available β-ketoenamides KE is broader than the eighty examples presented here. This fact should be kept in mind when the reactions of β-ketoenamides to alternative subsequent products are discussed in the following chapters.
Synthesis of pyrimidine derivatives
The β-ketoenamides KE also serve as excellent starting materials for the preparation of highly substituted pyrimidine derivatives PM[47-49]. Cyclocondensation reactions with ammonium salts in methanol afford these versatile heterocycles in good to excellent yields (Scheme 8). In most cases, ammonium acetate gave the best results (method A) and in a few examples ammonium bicarbonate was tested as alternative (method B) [29,33]. The plausible mechanism of this transformation involves the formation of an α,β-unsaturated imine G and its cyclization to H followed by water elimination. As characteristic substitution pattern, the available pyrimidines PM contain a methyl group at C-4 and an alkoxy group OR1 at C-5.
Scheme 8:
Condensation of LANCA-derived β-ketoenamides KE with ammonium salts to give 5-alkoxy-substituted pyrimidine derivatives PM.
Scheme 8:
Condensation of LANCA-derived β-ketoenamides KE with ammonium salts to give 5-alkoxy-substituted py...
The remarkably wide scope of this pyrimidine synthesis is demonstrated by the thirty examples collected in Table 4. All tested β-ketoenamides were successfully converted into the pyrimidines PM and the examples show that the method is fully compatible with methoxy, benzyloxy and 2-(trimethylsilyl)ethoxy substituents. The groups R2 and R3 can be unbranched, branched or functionalized alkyl, aryl or heteroaryl groups. In addition, there are many examples with alkenyl substituents R3.
Table 4:
Condensation of β-ketoenamides KE with ammonium salts to give pyrimidine derivatives PM1–30 according to Scheme 8.a
aAbbreviations: Fu = furyl, Py = pyridyl, Th = thienyl; all alkenyl substituents are E-configured. bMethod A: NH4OAc, MeOH, 60 °C; method B: NH4HCO3, MeOH, 65 °C.
In Scheme 9 additional examples PM31–34 having stereogenic centers are presented, that were obtained from β-ketoenamides KE37, KE38, KE40 and KE41 (see Scheme 5) [43]. The pyrimidine PM35 is derived from β-ketoenamide KE78 (see Scheme 7) and bears a benzyl group at C-4 instead of the standard methyl group [33].
Scheme 9:
Synthesis of PM31–35 from β-ketoenamides KE37, KE38, KE40, KE41 and KE78 obtained by method A (NH4OAc, MeOH, 60 °C).
Scheme 9:
Synthesis of PM31–35 from β-ketoenamides KE37, KE38, KE40, KE41 and KE78 obtained by method A (NH4O...
The bis-β-ketoenamides KE44–46 also provide the expected bis-pyrimidine derivatives (Scheme 10) [41]. With precursor KE44 a full conversion into the expected bis-pyrimidine product PM36 was achieved in 55% yield after 48 h reaction time, when a large excess of ammonium acetate (16 equiv) is employed, whereas with only eight equivalents (reaction time: 36 h) a 1:1 mixture of PM36 and the intermediate mono-pyrimidine PM37 – containing still one β-ketoenamide moiety – was isolated. The use of sixteen equivalents ammonium acetate afforded good yields of bis-pyrimidine derivative PM39 and PM40, whereas in the case of KE45 as starting material, considerable amounts of the mono-pyrimidine derivative PM38 were isolated as side product. Subsequently, both mono-pyrimidine derivatives PM37 and PM38 were subjected to alternative cyclization reactions involving the remaining β-ketoenamide moiety [41]. It is worth mentioning that the relatively complex heterocyclic compounds depicted in Scheme 10 are accessible through the three-component reaction and subsequent condensation reaction in only two steps.
Scheme 10:
Synthesis of bis-pyrimidine derivatives PM36, PM39 and PM40 from β-ketoenamides KE44–46 by method A (NH4OAc, MeOH, 60 °C).
Scheme 10:
Synthesis of bis-pyrimidine derivatives PM36, PM39 and PM40 from β-ketoenamides KE44–46 by method A...
The substitution pattern of the prepared pyrimidine derivatives PM allows a variety of subsequent transformations to new derivatives. The C-4 methyl (in one case benzyl) group is an inevitable structural feature of these pyrimidines, but it can smoothly be used for oxidation reactions to introduce new functional groups. As typical examples selenium dioxide oxidations of PM5, PM9, PM15 and PM19 furnishing aldehydes PM41, PM42, PM44 and PM48 are shown in Scheme 11[33]. In case of the benzyl-substituted substrate PM5, the (probably faster) oxidation of the C-2 benzyl group could not be avoided and hence the dicarbonyl compound PM41 was isolated [50]. The formyl group of the prepared intermediates allows further conversion into other functional groups as depicted in the scheme. Wittig reactions provided 4-alkenyl-substituted pyrimidine derivatives such as PM43 or PM47, whereas further oxidation of PM44 afforded the carboxylic acid PM45 in good yield [33]. Alternatively, the conversion of PM44 into oxime PM46 or a van Leusen oxazole synthesis [51] of PM48 with tosylmethyl isocyanide giving PM49 were possible. The synthesis of pyrimidine derivative PM49 with three heterocyclic substituents is remarkable and stresses the flexibility of the methods presented here.
Scheme 11:
Functionalization of pyrimidine derivatives PM through selenium dioxide oxidations of PM5, PM9, PM15 and PM19 leading to 4-formyl-substituted pyrimidines PM41, PM42, PM44 and PM48 and selected subsequent transformations (TosMIC = tosylmethyl isocyanide).
Scheme 11:
Functionalization of pyrimidine derivatives PM through selenium dioxide oxidations of PM5, PM9, PM15...
The easily introduced C-2 alkenyl groups may also be oxidized. Thus, dihydroxylation of the vinyl group in PM7 followed by oxidative cleavage afforded pyrimidine derivative PM50 having a formyl group at C-2 (Scheme 12) [31].
Scheme 12:
Conversion of 2-vinyl-substituted pyrimidine PM7 into aldehyde PM50; (NMO = N-methylmorpholine N-oxide).
Scheme 12:
Conversion of 2-vinyl-substituted pyrimidine PM7 into aldehyde PM50; (NMO = N-methylmorpholine N-ox...
Next, the conversion of the 5-alkoxy groups of the pyrimidine derivatives PM into a 1-nonafluorobutanesulfonate group is presented, that – like the closely related triflate group – allows transition metal-catalyzed coupling reactions or nucleophilic substitutions [52]. The selected three examples presented in Scheme 13 show striking differences in the deprotection step [33]. The methoxy-substituted compound PM2 requires harsh conditions employing trimethylsilyl iodide at 80 °C to provide the intermediate hydroxy derivative PM51. In contrast, the removal of the benzyl group in PM20 can be achieved by palladium-catalyzed hydrogenolysis at room temperature to give hydroxy compound PM53. This method is certainly not applicable to pyrimidines with alkenyl substituents, but in this case 2-(trimethylsilyl)ethoxy-substituted compounds such as PM29 can be used, whose deprotection with trifluoroacetic acid proceeds at room temperature. The obtained 5-hydroxy-pyrimidines can be purified and characterized or, for further transformations, the crude products are directly converted into the corresponding nonaflates by deprotonation with sodium hydride and treatment with 1-nonafluorobutanesulfonyl fluoride (NfF). Scheme 13 shows two examples, PM52 and PM54 that are ready for palladium-catalyzed reactions.
Scheme 13:
Deprotection of 5-alkoxy-substituted pyrimidines PM2, PM20 and PM29 and conversion into nonaflates PM52 and PM54; (Nf =1-nonafluorobutanesulfonyl).
Scheme 13:
Deprotection of 5-alkoxy-substituted pyrimidines PM2, PM20 and PM29 and conversion into nonaflates ...
As mentioned above, pyridyl nonaflates derived from the β-ketoenamides KE are excellent substrates for palladium-catalyzed coupling reactions as briefly discussed in our review [23]. Pyrimidyl nonaflates can analogously be used to achieve higher substitution degrees as illustrated by the examples shown in Scheme 14[33]. Nonaflate PM54 underwent a Suzuki–Miyaura reaction to PM55 or a Sonogashira coupling to PM56 under standard conditions. The ethynyl-substituted pyrimidine derivative PM12 could also be employed in C–C coupling reactions as shown by its connection to pyridyl nonaflate 8 – readily available from β-ketoenamide KE61[33] – efficiently furnishing the disubstituted alkyne PM57. This intermediate was directly converted into pyrimidyl-substituted furopyridine derivative PM58 in very good overall yield. The example of compound PM58 nicely demonstrates the combination of different heterocycles that were generated from the two β-ketoenamides KE20 and KE61 and shows the potential of these methods in heterocyclic chemistry.
Scheme 14:
Palladium-catalyzed coupling reactions of PM54 and PM12 giving rise to new pyrimidine derivatives PM55–58.
Scheme 14:
Palladium-catalyzed coupling reactions of PM54 and PM12 giving rise to new pyrimidine derivatives P...
Options for palladium-catalyzed reactions are also offered by compound PM60 that was prepared from mono-pyrimidyl-substituted β-ketoenamide PM38 (see Scheme 10). This compound was converted into PM59 by the standard cyclocondensation reaction (Scheme 15) leading to a pyridin-4-ol moiety that was converted to the nonaflate. Compound PM60 bears a pyrimidyl and a pyridinyl substituent at the 2,2’-position of the biphenyl part [41].
Scheme 15:
Synthesis of pyrimidyl-substituted pyridyl nonaflate PM60.
Scheme 15:
Synthesis of pyrimidyl-substituted pyridyl nonaflate PM60.
The condensation of β-ketoenamides KE with hydroxylamine hydrochloride can either deliver pyrimidine N-oxides PO or oxazepine derivatives J. However, only the six-membered heterocycles were isolated under the conditions employed (Scheme 16) [32]. Remarkably, the condensations occurred under milder conditions compared with those involving ammonium salts and smoothly provided the pyrimidine N-oxides at room temperature. An additional advantage of this approach to the pyrimidine skeleton is the fact that the N-oxide moiety could be exploited for the functionalization of the adjacent 4-alkyl group.
Scheme 16:
Condensation of LANCA-derived β-ketoenamides KE with hydroxylamine hydrochloride leading to pyrimidine N-oxides PO.
Scheme 16:
Condensation of LANCA-derived β-ketoenamides KE with hydroxylamine hydrochloride leading to pyrimid...
The scope of this method is again very broad as demonstrated by the 25 examples compiled in Table 5 and those of Scheme 17 and Scheme 18. This condensation method is compatible with all substituents that are available by the three-component reactions to β-ketoenamides KE, however, due to the slightly acidic reaction conditions the tert-butyldimethylsilyl protection group of KE40 is removed during the formation of PO7 (Table 5, entry 7).
Table 5:
Preparation of pyrimidine N-oxides PO1–25 through condensation of β-ketoenamides KE with hydroxylamine hydrochloride.a
Under the conditions applied the vinyl-substituted β-ketoenamide KE15 furnished the expected pyrimidine N-oxide PO26. However, the addition of the solvent methanol to the double bond provided compound PO27 as major product (Scheme 17) [32]. It was not studied whether the use of other solvents can suppress this addition reaction. The allyl-substituted β-ketoenamide KE7 was converted under the standard conditions into condensation product PO28, but in this case a second compound, PO29 bearing a shifted double bond, was isolated as main product [32].
A few pyrimidine N-oxides with special substituents are depicted in Scheme 18. They are generated from the enantiopure β-ketoenamide KE43[30], the biphenyl derivative KE45[41] and the aryl-substituted β-ketoenamides KE78 and KE80[46]. In all cases, the corresponding heterocycles PO30–33 were isolated in moderate to good yields.
Typical subsequent reactions of pyrimidine N-oxides
The N-oxide moiety of pyrimidine N-oxides can easily be reduced by various methods, as shown by the reduction of PO4 with hydrogen/palladium to give pyrimidine PM5 (Scheme 19) [30]. Although compounds such as PM5 are also directly available by condensation with ammonium salts (see above), the detour via pyrimidine N-oxides may have advantages in certain cases due to the milder reaction conditions of the condensation step. However, a more important transformation of pyrimidine N-oxides PO represents the Boekelheide rearrangement [55] to afford 4-acetoxymethyl-substituted pyrimidines and some typical examples of this side-chain functionalization are depicted in Scheme 19. Treatment of pyrimidine N-oxide PO13 with acetic anhydride at 90 °C furnished the expected pyrimidine derivative PM61 in 69% yield [32] showing that this transformation involves an internal redox reaction. However, the mechanism of this rearrangement is still under discussion [56,57] and side-products such as PM62 having a 4-ethyl group (3% yield) and other compounds evidence the participation of radicals [30]. After the efficient conversion of pyrimidine N-oxide PO14 into pyrimidine PM63 no products of this type were isolated. The regioselectivity is another important feature of the Boekelheide rearrangement if alkyl groups are present at C-2 or C-4 next to the N-oxide moiety. The pyrimidine N-oxide PO4 offers a benzyl substituent and a methyl group whereas PO30 bears two methyl groups. In both cases, 1:1 mixtures of the two possible rearranged products, PM64 and PM65[50] or PM66 and PM67[30] were isolated, respectively.
Scheme 19:
Reduction of PO4 to PM5 and Boekelheide rearrangements of PO13, PO14, PO4 and PO30 to 4-acetoxymethyl-substituted pyrimidine derivatives; Ad = 1-adamantyl.
Scheme 19:
Reduction of PO4 to PM5 and Boekelheide rearrangements of PO13, PO14, PO4 and PO30 to 4-acetoxymeth...
4-Acetoxymethyl-substituted pyrimidine derivatives offer many options for the introduction of new substituents. For example the removal of the acetyl group by treatment with potassium carbonate in methanol and oxidation with Dess–Martin periodinane (DMP) converted PM61 and PM63 into aldehydes PM69 and PM71 in reasonable overall yields (Scheme 20) [30]. This pathway via the pyrimidine N-oxides represents a good alternative to the direct oxidation of the 4-methyl group by selenium dioxide (see Scheme 11). The subsequent transformation of the aldehyde PM71 to the oxime followed by dehydration afforded nitrile PM72 in good yield [30]. The latter should be a suitable precursor for three-component reactions with alkoxyallenes and carboxylic acids to furnish new β-ketoenamides KE bearing a 6-adamantyl-2-cyclopropyl-5-methoxypyrimidin-4-yl substituent. This again stresses the flexibility and versatility of our approach to complex heterocycles.
Scheme 20:
Deprotection of 4-acetoxymethyl-substituted pyrimidine derivatives PM61 and PM63, oxidations to formyl-substituted pyrimidines PM69 and PM71 and synthesis of nitrile PM72 (DMP = Dess–Martin periodinane).
Scheme 20:
Deprotection of 4-acetoxymethyl-substituted pyrimidine derivatives PM61 and PM63, oxidations to for...
The aldehyde PM69 was further converted into the terminal alkyne PM73 by employing the Bestmann–Ohira protocol (Scheme 21). After its Sonogashira reaction with iodobenzene to the intermediate disubstituted alkyne PM74 this compound was converted into furopyrimidine derivative PM75[30]. Finally, the bromoaryl group in PO3 was engaged in a coupling with ethynylbenzene to give PO34. This latter reaction proves that the N-oxide moiety is compatible with palladium/copper-catalyzed reactions [30]. The conversion of the 5-alkoxy substituent into a nonafloxy group (as shown above with the pyrimidine derivatives, see Scheme 13) was not examined so far, however, it should be possible. Hence, the pyrimidine N-oxides may also be used in other palladium-catalyzed processes in order to introduce new substituents at C-5 of the heterocycles.
Scheme 21:
Synthesis of pyrimidinyl-substituted alkyne PM74 and conversion into furopyrimidine PM75 and Sonogashira reaction of PO3 with ethynylbenzene to pyrimidine N-oxide PO34.
Scheme 21:
Synthesis of pyrimidinyl-substituted alkyne PM74 and conversion into furopyrimidine PM75 and Sonoga...
By brief heating with trifluoroacetic acid β-ketoenamides KE with acid-labile alkoxy substituents OR1 underwent an unexpected formation of 5-acetyl-substituted oxazole derivatives OX (Scheme 22) [42,45]. This useful transformation proceeds with benzyloxy-, p-methoxybenzyl-, 2-tetrahydropyranyl- and 2-(trimethylsilyl)ethoxy-substituted β-ketoenamides KE as precursors and - mainly depending on the size of substituent R2 - oxazoles OX and/or the simple hydrolysis products 1,2-diketones DK were isolated in moderate to excellent yields (Table 6). With substituents R2 of moderate bulkiness the oxazoles OX are formed exclusively (Table 6, entries 1, 3–6, 17, and 20–23), whereas for the two compounds KE57 and KE58 (R2 = R3 = cyclopropyl) the corresponding oxazoles OX6 and OX7 were isolated as highly predominating products (Table 6, entries 7 and 8), but traces of the corresponding 1,2-diketones DK2 and DK3 were detected in the crude product. The reactions of β-ketoenamides KE60, KE61 and KE68 (R2 = tert-butyl or adamantanyl, R3 = methyl, trifluoromethyl or cyclopropyl) provided mixtures of oxazoles OX7, OX8 and OX10, respectively, and of 1,2-diketones DK4, DK5 and DK11 (Table 6, entries 9, 10, and 16). For examples with very bulky substituents R2 and R3 the exclusive formation of the 1,2-diketones DK1 and DK6–10 was observed (Table 6, entries 2, and 11–15). Trifluoroacetic acid treatment of β-ketoenamides KE70 and KE71 not only furnished DK12 and DK13 in moderate yield, but also dimeric products whose structure has still to be established [43].
Scheme 22:
Trifluoroacetic acid-promoted conversion of LANCA-derived β-ketoenamides KE into oxazoles OX and 1,2-diketones DK.
Scheme 22:
Trifluoroacetic acid-promoted conversion of LANCA-derived β-ketoenamides KE into oxazoles OX and 1,...
aAbbreviations: Ad = 1-adamantyl, Py = pyridyl, Fu = furyl, Th = thienyl, Ac = acetyl; all alkenyl substituents are E-configured. bIn addition, ca. 30% of a dimeric compound were isolated.
The method could also be extended to aryl-substituted β-ketoenamide KE79 that delivered oxazole derivative OX16 in 59% yield (Scheme 23) [45]. A subsequent reduction of the carbonyl group followed by iodine-induced elimination gave the 5-styryl-substituted oxazole OX17. This sequence demonstrates the potential of the C-5 functionalized oxazoles to be used for further transformations (see below).
Scheme 23:
Conversion of β-ketoenamide KE79 into oxazole OX16 and transformation into 5-styryl-substituted oxazole OX17.
Scheme 23:
Conversion of β-ketoenamide KE79 into oxazole OX16 and transformation into 5-styryl-substituted oxa...
The examples collected in Table 6 show a dichotomy of oxazole and 1,2-diketone formation that is not fully understood so far. As mentioned above, the presence of bulky substituents R2 (and R3) seems to be a prerequisite of the 1,2-diketone formation, however, for the series with R2 = phenyl the observed product distributions are not easy to explain (Table 6, entries 18–23). Nevertheless, a plausible mechanism is presented in Scheme 24 showing the analogy to the Gabriel–Robinson oxazole synthesis [58]. For β-ketoenamides KE with OR1 groups that are not easily cleaved by acids the cyclization to pyridin-4-ol derivatives PY occurs without touching of the alkoxy group. If this group is reacting with trifluoroacetic acid the E-configured enol E-EN is generated first and its prototropy directly delivers the isolated 1,2-diketones DK. Experiments with labelled oxygen showed that the oxazole oxygen originates from the alkoxy group and not from the amide moiety [45]. The oxazole formation therefore requires a configurational switch from enol E-EN to Z-EN. Very likely, this step is acid-catalyzed as the subsequent cyclization to form the five-membered intermediate L and the final water elimination to oxazole OX. The formation of Z-EN is possibly disfavored by bulky groups R2 due to repulsion with the acetyl group. The cyclization step leading to L may also be hampered if R3 is too bulky. In these cases, no sufficient concentrations of Z-EN or of L are formed and hence the 1,2-diketones DK are obtained as the products. It should also be mentioned that isolated 1,2-diketones DK do not undergo cyclizations to OX even after extended treatment with trifluoroacetic acid.
Scheme 24:
Mechanisms of the formation of 1,2-diketones DK and of acetyl-substituted oxazole derivatives OX.
Scheme 24:
Mechanisms of the formation of 1,2-diketones DK and of acetyl-substituted oxazole derivatives OX.
As an alternative to the strongly acidic conditions, palladium-catalyzed hydrogenolysis of the benzyloxy-substituted derivatives is possible, thus avoiding the condensation to oxazoles. Scheme 25 shows the conversion of KE52 into 1,2-diketone DK14 (compare entry 3 of Table 6). Longer reaction times lead to a subsequent reduction of the internal carbonyl group as shown by the conversion of KE54 into the two diastereomeric α-hydroxy-β-amino ketones 9[45]. Due to the moderate mass balance of this transformation we cannot exclude that the second carbonyl group was also partially reduced.
Scheme 25:
Hydrogenolyses of benzyloxy-substituted β-ketoenamides KE52 and KE54 to 1,2-diketone DK14 and to diastereomeric α-hydroxy-β-amino ketones 9.
Scheme 25:
Hydrogenolyses of benzyloxy-substituted β-ketoenamides KE52 and KE54 to 1,2-diketone DK14 and to di...
As already shown in Scheme 23, the carbonyl group at C-5 of oxazole derivatives OX offers possibilities for subsequent reactions to other functionalized oxazoles. Typical examples are depicted in Scheme 26 and Scheme 27 employing 2,4-dicyclopropyl-substituted oxazole OX7 as the starting material. The efficient conversion of the acetyl group into the corresponding silyl enol ether moiety delivered OX18 that may be used for further transformations. Alternatively, OX7 and phenyl hydrazine afforded the corresponding hydrazone OX19 in excellent yield that was further treated with polyphosphoric acid to undergo a Fischer indole reaction to 5-indolyl-substituted oxazole OX20.
Scheme 26:
Conversions of 2,4-dicyclopropyl-substituted oxazole OX7 into oxazole derivatives OX18–20 (PPA = polyphosphoric acid).
Scheme 26:
Conversions of 2,4-dicyclopropyl-substituted oxazole OX7 into oxazole derivatives OX18–20 (PPA = po...
Scheme 27:
Syntheses of vinyl and ethynyl-substituted oxazole derivatives OX21 and OX23 and their palladium-catalyzed reactions to OX22 and OX24–26 (Schwesinger P2 base).
Scheme 27:
Syntheses of vinyl and ethynyl-substituted oxazole derivatives OX21 and OX23 and their palladium-ca...
To demonstrate the versatility of the route to new oxazole derivatives, typical palladium-catalyzed processes are compiled in Scheme 27. First, the acetyl moiety was converted into a vinyl or an ethynyl substituent. The reduction of OX7 followed by elimination to OX21 proceeded smoothly and as subsequent transformation a Heck reaction with alkenyl nonaflate 10 was performed delivering OX22. The conversion of OX7 to alkyne OX23 applied the protocol of Lyapkalo et al. [59] using Schwesinger’s base [60] as crucial reagent. First, the corresponding nonaflate is generated from OX7 that immediately underwent elimination to the alkyne. Ethynyl-substituted oxazole OX23 was isolated in excellent yield and subsequently employed in Sonogashira couplings. Iodobenzene afforded compound OX24 in high yield and (β-ketoenamide-based) pyridinyl nonaflate 8 gave OX25. The removal of the TMSE group by acid treatment and subsequent cyclization furnished the furopyridyl-substituted oxazole derivative OX26 in good overall yield [45]. The examples shown in Scheme 27 and Scheme 28 demonstrate the manifold options to synthesize complex heterocyclic systems by the building block system derived from β-ketoenamides KE.
Scheme 28:
Synthesis of C3-symmetric oxazole derivative OX28 and the STM current image of its 1-phenyloctane solution on highly oriented pyrolytic graphite (HOPG).
Scheme 28:
Synthesis of C3-symmetric oxazole derivative OX28 and the STM current image of its 1-phenyloctane s...
Finally, the synthesis of star-shaped compound OX28 is presented. A threefold Sonogashira reaction of 1,3,5-tribromobenzene with ethynyl-substituted OX23 gave the desired OX28 in 22% yield; the major product (41%) of this experiment was the double-coupling product OX27 and the mono-coupling product (not shown) was isolated in 5% yield [45]. A solution of the C3-symmetric compound OX28 in 1-phenyloctane was investigated by scanning tunneling microscopy (STM) to reveal its ability to form self-assembled monolayers at the interface with highly oriented pyrolytic graphite (HOPG). The STM current image inserted in Scheme 28 shows bright areas that indicate the positions of the π-systems, whereas the dark areas indicate the cyclopropyl groups.
Synthesis of quinoxalines
The acylamido-substituted 1,2-diketones DK obtained by hydrolysis of several β-ketoenamides KE also offer possibilities of further synthetic applications. The reduction to α-hydroxy-β-amino ketones such as compound 9 has been already mentioned (see Scheme 25), but the vicinal carbonyl groups may also be employed for condensation reactions leading to heterocycles, for instance the Radziszewski reaction to imidazoles [61,62]. As an example, the condensation of 1,2-diketones DK with o-phenylenediamine to quinoxalines QU[63] employing cerium ammonium nitrate [64] as catalyst was investigated (Scheme 29). This transformation proceeded smoothly at room temperature in water as solvent and provided the expected acylamido-substituted quinoxalines QU1–7 in moderate to good yields (Table 7) [43,45].
Scheme 29:
Condensation of 1,2-diketones DK with o-phenylenediamine to quinoxalines QU1–7 (CAN = cerium ammonium nitrate).
Scheme 29:
Condensation of 1,2-diketones DK with o-phenylenediamine to quinoxalines QU1–7 (CAN = cerium ammoni...
aAbbreviation: Ad = 1-adamantyl, Fu = furyl, Th = thienyl; all alkenyl substituents are E-configured.
Conclusion
Lithiated alkoxyallenes LA, nitriles N and carboxylic acids CA undergo a three-component reaction (LANCA reaction) that affords β-ketoenamides KE in good to very good yields (Scheme 30). The reaction proceeds through a unique mechanism being driven by the high energy level of the allenes. The eighty examples of β-ketoenamides KE collected in this review impressively demonstrate the broad scope of this three-component reaction that is compatible with all kinds of substituents R2 and R3 and several functional groups within these substituents. Enantiopure components efficiently lead to products with stereogenic centers. Dinitriles or dicarboxylic acids provide the expected bis-β-ketoenamides in moderate yield.
Scheme 30:
The LANCA three-component reaction leading to β-ketoenamides KE and the structure of functionalized pyridines PY, pyrimidines PM, pyrimidine N-oxides PO, oxazoles OX, 1,2-diketones DK and quinoxalines QU derived thereof.
Scheme 30:
The LANCA three-component reaction leading to β-ketoenamides KE and the structure of functionalized...
The prepared β-ketoenamides KE are excellent precursors for the synthesis of specifically substituted heterocycles (Scheme 30). The intramolecular aldol-type condensations leading to a manifold of pyridine derivatives PY was already subject of a review article [23]. In this report, we demonstrate that the β-ketoenamides KE are also excellent precursors for the synthesis of a variety of pyrimidines PM, pyrimidine N-oxides PO, 4-acetyl-substituted oxazoles OX and – via 1,2-diketones DK – of quinoxalines QU. The substitution pattern of all compounds allows specific subsequent reactions, for instance, by substitution of the alkoxy groups with a nonafloxy group all kinds of palladium-catalyzed coupling reactions. Specific oxidation reactions also lead to a variety of new heterocyclic compounds. All the examples collected here show the potential of this approach to highly functionalized heterocycles, furnishing compounds with a very high degree of structural diversity that should be of interest in drug synthesis or material science. The versatility of alkoxyallenes [11-20,65,66] as easily available C3 building blocks is key for this prosperousness.
Acknowledgements
We thank the group members mentioned in the references who contributed to this project and gratefully acknowledge the German Academic Exchange Service, the Alexander von Humboldt Foundation and the German Research Foundation (DFG) for support of this work, in part through the collaborative research center SFB 765.
Reissig, H.-U.; Zimmer, R. Allenes in Multicomponent Synthesis of Heterocycles. Multicomponent Reactions in Organic Synthesis; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2014; pp 301–332. doi:10.1002/9783527678174.ch11
Return to citation in text:
[1]
Reissig, H.-U.; Hormuth, S.; Schade, W.; Okala Amombo, M.; Watanabe, T.; Pulz, R.; Hausherr, A.; Zimmer, R. J. Heterocycl. Chem.2000,37, 597–606. doi:10.1002/jhet.5570370316
Return to citation in text:
[1]
[2]
Reissig, H.-U.; Schade, W.; Okala Amombo, G. M.; Pulz, R.; Hausherr, A. Pure Appl. Chem.2002,74, 175–180. doi:10.1351/pac200274010175
Return to citation in text:
[1]
[2]
Zimmer, R.; Reissig, H.-U. In Donor-Substituted Allenes in Modern Allene Chemistry; Krause, N.; Hashmi, A. S. K., Eds.; Wiley-VCH: Weinheim, Germany, 2004; pp 425–492.
Return to citation in text:
[1]
[2]
Tius, M. A.; Zimmer, R.; Reissig, H.-U. 1-Methoxyallenyllithium. Encyclopedia of Reagents for Organic Synthesis; John Wiley & Sons, Ltd: Chichester, United Kingdom, 2006. doi:10.1002/047084289x.rm077.pub2
Return to citation in text:
[1]
[2]
Brasholz, M.; Reissig, H.-U.; Zimmer, R. Acc. Chem. Res.2009,42, 45–56. doi:10.1021/ar800011h
Return to citation in text:
[1]
[2]
Pfrengle, F.; Reissig, H.-U. Chem. Soc. Rev.2010,39, 549–557. doi:10.1039/b914356d
Return to citation in text:
[1]
[2]
Lechel, T.; Reissig, H.-U. Pure Appl. Chem.2010,82, 1835–1844. doi:10.1351/pac-con-09-09-06
Return to citation in text:
[1]
[2]
Bouché, L.; Reissig, H.-U. Pure Appl. Chem.2012,84, 23–36. doi:10.1351/pac-con-11-09-20
Return to citation in text:
[1]
[2]
Zimmer, R.; Reissig, H.-U. Chem. Soc. Rev.2014,43, 2888–2903. doi:10.1039/c3cs60429b
Return to citation in text:
[1]
[2]
Reissig, H.-U.; Zimmer, R. Synthesis2017,49, 3291–3302. doi:10.1055/s-0036-1588846
Return to citation in text:
[1]
[2]
Lechel, T.; Dash, J.; Hommes, P.; Lentz, D.; Reissig, H.-U. J. Org. Chem.2010,75, 726–732. doi:10.1021/jo9022183
Return to citation in text:
[1]
[2]
[3]
[4]
[5]
Lechel, T.; Reissig, H.-U. Synthesis and Reactivity of Pyridin-4-ols Based on the Three-Component Reaction of Alkoxyallenes, Nitriles and Carboxylic Acids. In Targets in Heterocyclic Systems - Chemistry and Properties; Attanasi, O. A.; Merino, P.; Spinelli, D., Eds.; Italian Society of Chemistry: Rome, Italy, 2016; Vol. 20, pp 1–32. doi:10.17374/targets.2017.20.1
Return to citation in text:
[1]
[2]
[3]
[4]
[5]
Hoff, S.; Brandsma, L.; Arens, J. F. Recl. Trav. Chim. Pays-Bas1968,87, 916–924. doi:10.1002/recl.19680870807
Return to citation in text:
[1]
Weiberth, F. J.; Hall, S. S. J. Org. Chem.1985,50, 5308–5314. doi:10.1021/jo00225a061
Return to citation in text:
[1]
Seghers, S.; Heugebaert, T. S. A.; Moens, M.; Sonck, J.; Thybaut, J. W.; Stevens, C. V. ChemSusChem2018,11, 2248–2254. doi:10.1002/cssc.201800760
Return to citation in text:
[1]
Bera, M. K.; Hommes, P.; Reissig, H.-U. Chem. – Eur. J.2011,17, 11838–11843. doi:10.1002/chem.201101739
Return to citation in text:
[1]
[2]
Eidamshaus, C.; Reissig, H.-U. Adv. Synth. Catal.2009,351, 1162–1166. doi:10.1002/adsc.200800789
Return to citation in text:
[1]
Eidamshaus, C.; Kumar, R.; Bera, M. K.; Reissig, H.-U. Beilstein J. Org. Chem.2011,7, 962–975. doi:10.3762/bjoc.7.108
Return to citation in text:
[1]
[2]
Eidamshaus, C.; Reissig, H.-U. Tetrahedron: Asymmetry2011,22, 1644–1652. doi:10.1016/j.tetasy.2011.08.017
Return to citation in text:
[1]
Zimmer, R.; Orschel, B.; Scherer, S.; Reissig, H.-U. Synthesis2002, 1553–1563. doi:10.1055/s-2002-33328
Return to citation in text:
[1]
Hausherr, A.; Zimmer, R.; Reissig, H.-U. Synthesis2019,51, 486–499. doi:10.1055/s-0037-1609942
Return to citation in text:
[1]
Dash, J.; Lechel, T.; Reissig, H.-U. Org. Lett.2007,9, 5541–5544. doi:10.1021/ol702468s
Return to citation in text:
[1]
Bera, M. K.; Domínguez, M.; Hommes, P.; Reissig, H.-U. Beilstein J. Org. Chem.2014,10, 394–404. doi:10.3762/bjoc.10.37
Return to citation in text:
[1]
[2]
[3]
[4]
[5]
Linder, I. Master Thesis, Freie Universität Berlin 2011.
Return to citation in text:
[1]
[2]
Brown, D. J. Pyrimidines and their Benzo Derivatives. In Comprehensive Heterocyclic Chemistry; Katritzky, A. R.; Rees, C. W., Eds.; Pergamon Press: Oxford, 1984; Vol. 3, pp 57–155. doi:10.1016/b978-008096519-2.00035-7
Return to citation in text:
[1]
Angerer, S. Product Class 12: Pyrimidines. In Science of Synthesis; Yamamoto, Y., Ed.; Category 2, Hetarenes and Related Ring Systems, Vol. 16; Thieme: Stuttgart, 2004; pp 379–572. doi:10.1055/sos-sd-016-00463
Return to citation in text:
[1]
Lambert, C. Heterocycles2006,68, 561–603. doi:10.3987/rev-05-604
Return to citation in text:
[1]
Bera, M. K.; Reissig, H.-U. unpublished results.
Return to citation in text:
[1]
[2]
[3]
[4]
Van Leusen, D.; Van Leusen, A. M. Synthetic Uses of Tosylmethyl Isocyanide (TosMIC). Organic Reactions; John Wiley & Sons, Inc.: Hoboken, NJ, U.S.A., 2001; pp 417–666. doi:10.1002/0471264180.or057.03
Return to citation in text:
[1]
Högermeier, J.; Reissig, H.-U. Adv. Synth. Catal.2009,351, 2747–2763. doi:10.1002/adsc.200900566
Return to citation in text:
[1]
Cimen, A. Bachelor Thesis, Freie Universität Berlin, 2011.
Return to citation in text:
[1]
Dreßler, A. Bachelor Thesis, Freie Universität Berlin, 2014.
Return to citation in text:
[1]
[2]
[3]
Boekelheide, V.; Linn, W. J. J. Am. Chem. Soc.1954,76, 1286–1291. doi:10.1021/ja01634a026
Return to citation in text:
[1]
Massaro, A.; Mordini, A.; Mingardi, A.; Klein, J.; Andreotti, D. Eur. J. Org. Chem.2011, 271–279. doi:10.1002/ejoc.201000936
Return to citation in text:
[1]
Further Reactions of N-Oxides. In Introductory Heterocyclic Chemistry; Jacobi, P. A., Ed.; Wiley-VCH: Weinheim, Germany, 2019; pp 61–69.
Return to citation in text:
[1]
Wasserman, H. H.; Vinick, F. J. J. Org. Chem.1973,38, 2407–2408. doi:10.1021/jo00953a028
Return to citation in text:
[1]
Comprehensive Organic Name Reactions and Reagents; Wang, Z., Ed.; John Wiley & Sons: New Jersey, 2009; Vol. 3, p 2293.
Return to citation in text:
[1]
Breslin, H. J.; Miskowski, T. A.; Rafferty, B. M.; Coutinho, S. V.; Palmer, J. M.; Wallace, N. H.; Schneider, C. R.; Kimball, E. S.; Zhang, S.-P.; Li, J.; Colburn, R. W.; Stone, D. J.; Martinez, R. P.; He, W. J. Med. Chem.2004,47, 5009–5020. doi:10.1021/jm030548r
Return to citation in text:
[1]
Brown, D. J. Quinoxalines, Supplement II. In The Chemistry of Heterocyclic Compounds; Taylor, E. C.; Wipf, P., Eds.; John Wiley & Sons: New Jersey, 2004; Vol. 61.
Return to citation in text:
[1]
More, S. V.; Sastry, M. N. V.; Yao, C.-F. Green Chem.2006,8, 91–95. doi:10.1039/b510677j
Return to citation in text:
[1]
Nedolya, N. A.; Tarasova, O. A.; Volostnykh, O. G.; Albanov, A. L.; Klyba, L. V.; Trofimov, B. A. Synthesis2011, 2192–2204. doi:10.1055/s-0030-1260084
Return to citation in text:
[1]
Tius, M. A. Chem. Soc. Rev.2014,43, 2979–3002. doi:10.1039/c3cs60333d
Return to citation in text:
[1]
Reference 43
43.
Kumar, R. Master Thesis, Freie Universität Berlin 2010.
Lechel, T.; Reissig, H.-U. Synthesis and Reactivity of Pyridin-4-ols Based on the Three-Component Reaction of Alkoxyallenes, Nitriles and Carboxylic Acids. In Targets in Heterocyclic Systems - Chemistry and Properties; Attanasi, O. A.; Merino, P.; Spinelli, D., Eds.; Italian Society of Chemistry: Rome, Italy, 2016; Vol. 20, pp 1–32. doi:10.17374/targets.2017.20.1
Van Leusen, D.; Van Leusen, A. M. Synthetic Uses of Tosylmethyl Isocyanide (TosMIC). Organic Reactions; John Wiley & Sons, Inc.: Hoboken, NJ, U.S.A., 2001; pp 417–666. doi:10.1002/0471264180.or057.03
Brown, D. J. Pyrimidines and their Benzo Derivatives. In Comprehensive Heterocyclic Chemistry; Katritzky, A. R.; Rees, C. W., Eds.; Pergamon Press: Oxford, 1984; Vol. 3, pp 57–155. doi:10.1016/b978-008096519-2.00035-7
48.
Angerer, S. Product Class 12: Pyrimidines. In Science of Synthesis; Yamamoto, Y., Ed.; Category 2, Hetarenes and Related Ring Systems, Vol. 16; Thieme: Stuttgart, 2004; pp 379–572. doi:10.1055/sos-sd-016-00463
Lechel, T.; Reissig, H.-U. Synthesis and Reactivity of Pyridin-4-ols Based on the Three-Component Reaction of Alkoxyallenes, Nitriles and Carboxylic Acids. In Targets in Heterocyclic Systems - Chemistry and Properties; Attanasi, O. A.; Merino, P.; Spinelli, D., Eds.; Italian Society of Chemistry: Rome, Italy, 2016; Vol. 20, pp 1–32. doi:10.17374/targets.2017.20.1
Brown, D. J. Quinoxalines, Supplement II. In The Chemistry of Heterocyclic Compounds; Taylor, E. C.; Wipf, P., Eds.; John Wiley & Sons: New Jersey, 2004; Vol. 61.
Comprehensive Organic Name Reactions and Reagents; Wang, Z., Ed.; John Wiley & Sons: New Jersey, 2009; Vol. 3, p 2293.
62.
Breslin, H. J.; Miskowski, T. A.; Rafferty, B. M.; Coutinho, S. V.; Palmer, J. M.; Wallace, N. H.; Schneider, C. R.; Kimball, E. S.; Zhang, S.-P.; Li, J.; Colburn, R. W.; Stone, D. J.; Martinez, R. P.; He, W. J. Med. Chem.2004,47, 5009–5020. doi:10.1021/jm030548r
Reissig, H.-U.; Hormuth, S.; Schade, W.; Okala Amombo, M.; Watanabe, T.; Pulz, R.; Hausherr, A.; Zimmer, R. J. Heterocycl. Chem.2000,37, 597–606. doi:10.1002/jhet.5570370316
12.
Reissig, H.-U.; Schade, W.; Okala Amombo, G. M.; Pulz, R.; Hausherr, A. Pure Appl. Chem.2002,74, 175–180. doi:10.1351/pac200274010175
13.
Zimmer, R.; Reissig, H.-U. In Donor-Substituted Allenes in Modern Allene Chemistry; Krause, N.; Hashmi, A. S. K., Eds.; Wiley-VCH: Weinheim, Germany, 2004; pp 425–492.
14.
Tius, M. A.; Zimmer, R.; Reissig, H.-U. 1-Methoxyallenyllithium. Encyclopedia of Reagents for Organic Synthesis; John Wiley & Sons, Ltd: Chichester, United Kingdom, 2006. doi:10.1002/047084289x.rm077.pub2
15.
Brasholz, M.; Reissig, H.-U.; Zimmer, R. Acc. Chem. Res.2009,42, 45–56. doi:10.1021/ar800011h
Lechel, T.; Reissig, H.-U. Synthesis and Reactivity of Pyridin-4-ols Based on the Three-Component Reaction of Alkoxyallenes, Nitriles and Carboxylic Acids. In Targets in Heterocyclic Systems - Chemistry and Properties; Attanasi, O. A.; Merino, P.; Spinelli, D., Eds.; Italian Society of Chemistry: Rome, Italy, 2016; Vol. 20, pp 1–32. doi:10.17374/targets.2017.20.1
Lechel, T.; Reissig, H.-U. Synthesis and Reactivity of Pyridin-4-ols Based on the Three-Component Reaction of Alkoxyallenes, Nitriles and Carboxylic Acids. In Targets in Heterocyclic Systems - Chemistry and Properties; Attanasi, O. A.; Merino, P.; Spinelli, D., Eds.; Italian Society of Chemistry: Rome, Italy, 2016; Vol. 20, pp 1–32. doi:10.17374/targets.2017.20.1
Reissig, H.-U.; Hormuth, S.; Schade, W.; Okala Amombo, M.; Watanabe, T.; Pulz, R.; Hausherr, A.; Zimmer, R. J. Heterocycl. Chem.2000,37, 597–606. doi:10.1002/jhet.5570370316
12.
Reissig, H.-U.; Schade, W.; Okala Amombo, G. M.; Pulz, R.; Hausherr, A. Pure Appl. Chem.2002,74, 175–180. doi:10.1351/pac200274010175
13.
Zimmer, R.; Reissig, H.-U. In Donor-Substituted Allenes in Modern Allene Chemistry; Krause, N.; Hashmi, A. S. K., Eds.; Wiley-VCH: Weinheim, Germany, 2004; pp 425–492.
14.
Tius, M. A.; Zimmer, R.; Reissig, H.-U. 1-Methoxyallenyllithium. Encyclopedia of Reagents for Organic Synthesis; John Wiley & Sons, Ltd: Chichester, United Kingdom, 2006. doi:10.1002/047084289x.rm077.pub2
15.
Brasholz, M.; Reissig, H.-U.; Zimmer, R. Acc. Chem. Res.2009,42, 45–56. doi:10.1021/ar800011h
Lechel, T.; Reissig, H.-U. Synthesis and Reactivity of Pyridin-4-ols Based on the Three-Component Reaction of Alkoxyallenes, Nitriles and Carboxylic Acids. In Targets in Heterocyclic Systems - Chemistry and Properties; Attanasi, O. A.; Merino, P.; Spinelli, D., Eds.; Italian Society of Chemistry: Rome, Italy, 2016; Vol. 20, pp 1–32. doi:10.17374/targets.2017.20.1



![[1860-5397-15-61-i1]](/bjoc/content/inline/1860-5397-15-61-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-15-61-i2]](/bjoc/content/inline/1860-5397-15-61-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-15-61-i3]](/bjoc/content/inline/1860-5397-15-61-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-15-61-i4]](/bjoc/content/inline/1860-5397-15-61-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-15-61-i5]](/bjoc/content/inline/1860-5397-15-61-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-15-61-i6]](/bjoc/content/inline/1860-5397-15-61-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-15-61-i7]](/bjoc/content/inline/1860-5397-15-61-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-15-61-i8]](/bjoc/content/inline/1860-5397-15-61-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-15-61-i9]](/bjoc/content/inline/1860-5397-15-61-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-15-61-i10]](/bjoc/content/inline/1860-5397-15-61-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-15-61-i11]](/bjoc/content/inline/1860-5397-15-61-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-15-61-i12]](/bjoc/content/inline/1860-5397-15-61-i12.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-15-61-i13]](/bjoc/content/inline/1860-5397-15-61-i13.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-15-61-i14]](/bjoc/content/inline/1860-5397-15-61-i14.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-15-61-i15]](/bjoc/content/inline/1860-5397-15-61-i15.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-15-61-i16]](/bjoc/content/inline/1860-5397-15-61-i16.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-15-61-i17]](/bjoc/content/inline/1860-5397-15-61-i17.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-15-61-i18]](/bjoc/content/inline/1860-5397-15-61-i18.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-15-61-i19]](/bjoc/content/inline/1860-5397-15-61-i19.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-15-61-i20]](/bjoc/content/inline/1860-5397-15-61-i20.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-15-61-i21]](/bjoc/content/inline/1860-5397-15-61-i21.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-15-61-i22]](/bjoc/content/inline/1860-5397-15-61-i22.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-15-61-i23]](/bjoc/content/inline/1860-5397-15-61-i23.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-15-61-i24]](/bjoc/content/inline/1860-5397-15-61-i24.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-15-61-i25]](/bjoc/content/inline/1860-5397-15-61-i25.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-15-61-i26]](/bjoc/content/inline/1860-5397-15-61-i26.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-15-61-i27]](/bjoc/content/inline/1860-5397-15-61-i27.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-15-61-i28]](/bjoc/content/inline/1860-5397-15-61-i28.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-15-61-i29]](/bjoc/content/inline/1860-5397-15-61-i29.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-15-61-i30]](/bjoc/content/inline/1860-5397-15-61-i30.svg?scale=2.0&max-width=1024&background=FFFFFF)