Abstract
N-Alkyl-N-chlorosulfonamides add to alkenes under copper(I) catalysis. In reactions of styrene derivatives with terminal double bonds the addition products were obtained in excellent yield and high regioselectivity. Lower yields are obtained in addition reactions to non-aromatic alkenes. The reaction most likely proceeds via a redox catalysis and amidyl radicals, a concerted mechanism has been ruled out and a polar mechanism via chloronium ions would lead to the opposite regiochemistry.

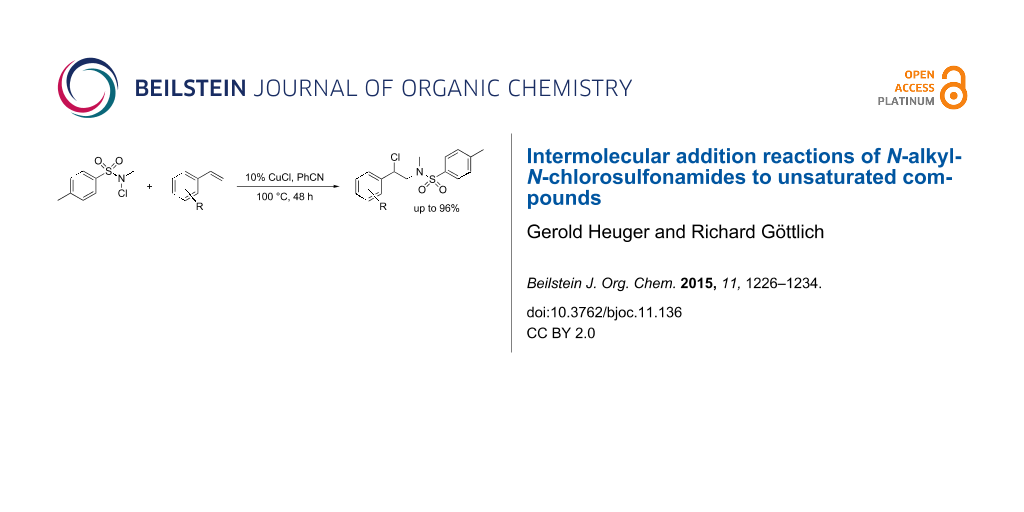
Graphical Abstract
Introduction
In earlier publications we described the cyclisation of various unsaturated N-hetero-substituted amines and amides via radicals [1-3] and other mechanistic pathways [4-8]. Although reported by other groups [9-11] in our hands an efficient intermolecular addition reaction of N-hetero substituted amines via radicals was not possible in appreciable yields. The reactivity of the intermediate aminyl radicals towards alkenes was simply not high enough and various side reactions became predominant. We therefore turned our attention to more electrophilic, thus more reactive, nitrogen-centered radicals and chose sulfonamidyl radicals as such electrophilic intermediates for an efficient intermolecular addition reaction.
Addition reactions with N-alkyl-N-halosulfonamides to unsaturated compounds have not been examined in detail so far. In earlier works Komori added a secondary N-chloro-sulfonamide to 1-hexene under photoirridation [12,13] and Priestly [14], Seden [15] and Daniher [16] published addition reactions of secondary N-halosulfonamides and N,N-dihalosulfonamides to alkenes. Neale [17] discussed a radical mechanism via nitrogen radicals as intermediates. In a more recent series of publications Li developed a new aminohalogenation of cinnamic esters using N,N-dichloro-p-toluenesulfonamide and ZnCl2 or Cu(OTf)2 [18] as catalysts and transferred these conditions to reactions with alkynes [19-21] and α,β-unsaturated ketones [19-21]. An ionic mechanism via halonium ions was proposed. The amidofluorination of alkenes has been achieved by Zhang [22] using copper or palladium catalysts and proceeds via radicals and fluoropalladation, respectively. Cyclisation reactions of unsaturated sulfonamides which proceed via amidyl radicals have been described by Li [23] and by Oshima [24]. Chemler [25,26] discusses radical and polar pathways as competing mechanisms and has developed a nice copper-catalyzed oxidative amidation of alkenes whilst Muñiz [27] in a recent publication proposes a polar sulfonamido-chlorination mechanism of alkenes.
Similar, intramolecular and intermolecular additions of N-chlorosulfonamides and derivatives like Chloramine-T to alkenes have been described by Sharpless [28,29], Komatsu [30-32] and Dodd [33]. In these examples the nitrogen center carries no substituents, which limits the scope of the reactions. In our own studies we wanted to develop a radical addition, using amidyl radicals with an N-alkyl substituent, which we anticipated to generate readily from the corresponding N-chloroamides by electron transfer from copper(I) catalysts.
Results and Discussion
The N-chlorosulfonamides can be easily prepared by reaction of the sulfonamide with calcium hypochlorite and moist alumina [34], which produced the corresponding N-chloro compounds 2a and 2b in quantitative yield (Scheme 1).
![[1860-5397-11-136-i1]](/bjoc/content/inline/1860-5397-11-136-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Preparation of the chloroamides.
Scheme 1: Preparation of the chloroamides.
Other procedures for the synthesis of these compounds including N-chlorination with an fivefold excess of Oxone® in the presence of NaCl/Al2O3 [35] or deprotonation and reaction with NCS [36,37] did not lead to higher or led in distinctly lower yields to the N-chloro compounds.
We chose styrene as the model compound for the first addition reactions of N-chlorosulfonamide 2a and used the complex [(MeCN)4Cu]PF6 as catalyst (Scheme 2). However even after a prolonged reaction time of 24 hours no addition product could be detected by TLC (Table 1, entry 1). Increasing the reaction temperature to 50 °C led to the formation of the addition product 3 in low yield. The yield was further raised to 30% by a longer reaction time of 48 h (Table 1, entries 2 and 3).
![[1860-5397-11-136-i2]](/bjoc/content/inline/1860-5397-11-136-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: First experiments for the intermolecular radical addition.
Scheme 2: First experiments for the intermolecular radical addition.
Table 1: Optimized addition reactions of 2a with styrene.
| Entry | 2a (equiv) |
Styrene
(equiv) |
Catalyst | Solvent |
Reaction
time (h) |
Temperature
(°C) |
Yield of 3
(%) |
|---|---|---|---|---|---|---|---|
| 1 | 1.2 | 1 | 10% [(MeCN)4Cu]PF6 | MeCN | 24 | rt | – |
| 2 | 1.2 | 1 | 10% [(MeCN)4Cu]PF6 | MeCN | 24 | 50 | 16 |
| 3 | 1.2 | 1 | 10% [(MeCN)4Cu]PF6 | MeCN | 48 | 50 | 30 |
| 4 | 1.2 | 1 | 50% [(MeCN)4Cu]PF6 | MeCN | 48 | 50 | 25 |
| 5 | 1 | 3 | 10% [(MeCN)4Cu]PF6 | MeCN | 48 | 75 | 43 |
| 6 | 1 | 3 | 10% CuCl | MeCN | 48 | 75 | 60 |
| 7 | 1 | 3 | 10% CuCl | PhCN | 48 | 100 | 92 |
| 8 | 1 | 1 | 10% CuCl | PhCN | 48 | 100 | 48 |
| 9 | 2 | 1 | 10% CuCl | PhCN | 48 | 100 | 34 |
Increasing the amount of catalyst led to no better results whilst rising the temperature to 75 °C and adding an excess of styrene increased the yield to 43%. Under these conditions the N-chlorosulfonamide 2a was completely consumed, undesired products were the sulfonamide 1a as well as oligostyrenes. The oligomerisation should be slowed down by using copper(I) chloride as the catalyst, which (after oxidation to copper(II) chloride) captures carbon radicals at a diffusion controlled rate [38].
Indeed using copper(I) chloride as the catalyst we obtained a yield of 60% of the addition product with sulfonamide 1a being the remaining side product. We supposed that this sulfonamide was generated by H-abstraction from the solvent and therefore we used benzonitrile, a solvent from which hydrogen cannot easily be abstracted and which allowed us a higher reaction temperature (100 °C). Using these conditions we obtained a nearly quantitative yield of 92% of 3 (Table 1, entry 7), whilst changing the chloroamide/styrene ratio led to reduced yields.
We next wanted to check the scope of this addition reaction and used, with the chloroamide 2b, a sulfonamide with an extreme sterical hinderance (tert-butyl group). This led to the addition product 4 in a distinct lower yield (Scheme 3).
![[1860-5397-11-136-i3]](/bjoc/content/inline/1860-5397-11-136-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Reaction of sterically hindered N-chlorosulfonamides.
Scheme 3: Reaction of sterically hindered N-chlorosulfonamides.
In a next step we added chloroamide 2a to a variety of styrene derivatives (Table 2). Whilst the addition proceeds well with electron-poor styrenes such as 4-nitro and 4-fluorostyrene, an electron rich substrate with a methoxy substituent (Table 2, entry 4) leads to a complex mixture of products. Most likely the electron-rich aromatic ring is oxidized under these conditions, leading to a variety of products which could not be separated.
![[Graphic 1]](/bjoc/content/inline/1860-5397-11-136-i7.svg?max-width=637&scale=1.0)
![[Graphic 2]](/bjoc/content/inline/1860-5397-11-136-i8.svg?max-width=637&scale=1.0)
![[Graphic 3]](/bjoc/content/inline/1860-5397-11-136-i9.svg?max-width=637&scale=1.0)
![[Graphic 4]](/bjoc/content/inline/1860-5397-11-136-i10.svg?max-width=637&scale=1.0)
![[Graphic 5]](/bjoc/content/inline/1860-5397-11-136-i11.svg?max-width=637&scale=1.0)
![[Graphic 6]](/bjoc/content/inline/1860-5397-11-136-i12.svg?max-width=637&scale=1.0)
![[Graphic 7]](/bjoc/content/inline/1860-5397-11-136-i13.svg?max-width=637&scale=1.0)
With styrene derivatives giving high yields of addition products, we next turned our attention to the less reactive non-aromatic alkenes.
Addition to non-aromatic alkenes
As expected non-aromatic alkenes are less good substrates for the radical addition of amidyl radicals and the yields of addition products of 2a decreased significantly (Table 3). A conjugated diene like cyclooctadiene (Table 3, entry 4) and a terminal, sterically not hindered alkene like 1-decene (Table 3, entry 1) still gave reasonable yields of the addition product. The yield is lower in addition to norbornene and cyclooctene, both of which are more sterically hindered and do not allow any mesomeric stabilization of the intermediate radical (Table 3, entries 2 and 3). Addition to an alkyne (Table 3, entry 5) produced a complex mixture of products, which could not be separated. Electron poor alkenes like an unsaturated ketone (Table 3, entry 6) are not good substrates for the addition of amidyl radicals either, as amidyl radicals are expected to be electrophilic themselves. Therefore we expected the addition to electron-rich enol ethers to proceed smoothly, however, with such electron-rich alkenes polar reaction pathways seem to become predominant (Table 3, entries 8 and 9).
Table 3: Addition reactions of 2a with electron-deficient olefines.
| Entry | Olefine | Producta | Yield (%) |
|---|---|---|---|
| 1 | 1-decene |
![[Graphic 8]](/bjoc/content/inline/1860-5397-11-136-i14.svg?max-width=637&scale=1.0)
7 |
34 |
| 2 | cyclooctene |
![[Graphic 9]](/bjoc/content/inline/1860-5397-11-136-i15.svg?max-width=637&scale=1.0)
8 |
13 |
| 3 | norbornene |
![[Graphic 10]](/bjoc/content/inline/1860-5397-11-136-i16.svg?max-width=637&scale=1.0)
9 |
26
dr 2:1 |
| 4 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-11-136-i17.svg?max-width=637&scale=1.0)
|
![[Graphic 12]](/bjoc/content/inline/1860-5397-11-136-i18.svg?max-width=637&scale=1.0)
|
33
(10a) 10 (10b) |
| 5 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-11-136-i19.svg?max-width=637&scale=1.0)
|
unidentified
reaction mixture |
– |
| 6 |
![[Graphic 14]](/bjoc/content/inline/1860-5397-11-136-i20.svg?max-width=637&scale=1.0)
|
no addition | – |
| 7 | cyclohexene |
![[Graphic 15]](/bjoc/content/inline/1860-5397-11-136-i21.svg?max-width=637&scale=1.0)
11 |
17 |
| 8 |
![[Graphic 16]](/bjoc/content/inline/1860-5397-11-136-i22.svg?max-width=637&scale=1.0)
|
![[Graphic 17]](/bjoc/content/inline/1860-5397-11-136-i23.svg?max-width=637&scale=1.0)
12 |
26 |
| 9 |
![[Graphic 18]](/bjoc/content/inline/1860-5397-11-136-i24.svg?max-width=637&scale=1.0)
|
![[Graphic 19]](/bjoc/content/inline/1860-5397-11-136-i25.svg?max-width=637&scale=1.0)
13 |
34 |
aAll products are obtained as racemates.
Whilst the formation of 11 from cyclohexene could result from an elimination of HCl after the radical addition due to the high reaction temperature, the formation of regioisomers of 12 and especially the formation of 13 can be explained by a halonium-ion transfer to the alkene. This could produce an chloro-substituted alkene 14, which in a second step would undergo radical addition of 2b (Scheme 4).
![[1860-5397-11-136-i4]](/bjoc/content/inline/1860-5397-11-136-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Proposed mechanism of the chlorination.
Scheme 4: Proposed mechanism of the chlorination.
This result surprised us, as we did not regard the chloroamide as a chloronium ion source. For the addition reactions to styrenes a polar mechanism can be ruled out due to the observed regiochemistry, however, in all other cases a polar mechanism is possible too and we therefore wanted to verify the radical-type mechanism which we initially anticipated.
Ionic or radical reaction mechanism
For these studies we first chose vinylcyclopropane as the substrate to rule out a concerted mechanism, as this alkene upon addition of a radical or a cation should react fast under ring-opening [39], producing an acyclic product. Upon copper(I) catalyzed addition of 2a to vinylcyclopropane only the ring-opend product 15 was obtained, which rules out a concerted mechanism and is, due to the observed regiochemistry, a strong indication for a radical pathway of the addition reaction (Scheme 5).
![[1860-5397-11-136-i5]](/bjoc/content/inline/1860-5397-11-136-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Ring opening in the case of cationic or radical intermediates.
Scheme 5: Ring opening in the case of cationic or radical intermediates.
Another strong argument for a mechanism via radicals is the complete surpression of the reaction of 2a with decene in the presence of TEMPO (2,2,6,6-tetramethyl-1-piperidinyloxy) as a radical trap, which we observed.
In a last experiment we added 2a to 5-hexen-1-ol, a substrate that easily reacts with halonium ions in a halocyclization [40] (Scheme 6).
![[1860-5397-11-136-i6]](/bjoc/content/inline/1860-5397-11-136-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Addition to unsaturated alcohols prone to halocyclization.
Scheme 6: Addition to unsaturated alcohols prone to halocyclization.
We only obtained the simple addition product 16 with no traces of halocyclization, again ruling out a polar pathway.
Conclusion
In summary we have shown that N-chlorosulfonamides can be added to styrenes efficiently under copper(I) catalysis via amidyl radicals. Addition to non-aromatic alkenes proceeds less readily in lower yield and electron-rich alkenes react via a competing polar reaction pathway.
Experimental
All solvents were purified by distillation and dried, if necessary, prior to use. Products were purified by flash chromatography on silica gel (40–63 μm). 1H and 13C NMR spectra were recorded on Bruker WM 300 and ARX400 spectrometers or on a Varian Unity plus 600 spectrometer in CDCl3 using TMS as internal standard.
Preparation of the N-chlorosulfonamides 2a, 2b; general procedure: 5.72 g Ca(OCl)2 (40 mmol, 2 equiv), moist alumina (10 g) and chloroform (50 mL) were efficiently stirred at 40 °C for 10 min. The sulfonamide (20 mmol) was then added, and the mixture was stirred at 40 °C until the sulfonamide had disappeared (monitored by TLC (pentane/ether 1:1), 1.5–3 h). The crude product was separated from solids and purified by chromatography (silica gel, pentane/ether 3:1).
N-Chloro-N,4-dimethylbenzenesulfonamide (2a) as white crystals. Rf 0.48 (pentane/ether 3:1); 1H NMR (400 MHz, CDCl3) δ 2.49 (s, 3H), 3.11 (s, 3H), 7.42 (d with fine-splitting, J = 8.0 Hz, 2H), 7.84 (d with fine-splitting, J = 8.4 Hz); 13C NMR (100 MHz, CDCl3) δ 21.6, 45.3, 128.5, 129.6, 129.8, 145.5.
N-tert-Butyl-N-chlorobenzenesulfonamide (2b) as white crystals. Rf 0.52 (pentane/ether 3:1); 1H NMR (400 MHz, CDCl3) δ 1.50 (s, 9H), 7.52–7.57 (m, 2H), 7.62–7.66 (m, 1H), 7.99 (dd, J = 8.4, 1.2 Hz, 2H) ppm; 13C NMR (100 MHz, CDCl3) δ 29.1, 68.1, 128.80, 128.84, 133.5, 138.5.
Metal-catalyzed addition reactions; general procedure: In a heat-dried Schlenk vessel 0.1 equiv (related to the N-chlorosulfonamide) of the metal salt were dissolved in anhydrous benzonitrile under an Ar atmosphere and heated to 100 °C. To this solution the N-chlorosulfonamide and the olefine were added and stirred for 48 h at this temperature. The solvent was removed by bulb-to-bulb distillation and the residue was taken up in dichloromethane. Approx. 10 g of silica gel were added and the solvent was removed in vacuo. Purifying by flash chromatography (pentane/ether 10:1 to 1:1) gave the addition products as described below.
N-(2-Chloro-2-phenylethyl)-N-methylbenzenesulfonamide (3) as a colourless oil [27]. Rf 0.47 (pentane/ether 3:1); 1H NMR (600 MHz, CDCl3) δ 2.41, (s, 3H), 2.62 (s, 3H), 3.40 (dd, J = 14.4, 7.2 Hz, 1H), 3.58 (dd, J = 14.4, 7.8, 1H), 5.10 (t, 7.2 Hz, 1H), 7.29–7.38 (m, 5H), 7.41 (m, 2H), 7.63 (d, J = 8.4 Hz, 2H); 13C NMR (150 MHz, CDCl3) δ 21.4, 36.8, 57.9, 61.2, 127.2, 127.4, 128.71, 128.78, 129.7, 134.6, 138.6, 143.5; MS (ESI, 1.57 kV, MeOH) m/z (%): 324 (38) [M+], 310 (20), 288 (70) [M − Cl+], 262 (8); HRMS (ESI) m/z: [M+] calcd for C16H18N35ClO2S, 324.0796; found, 324.0820. C16H18N37ClO2S [M+]: 326.0770; found: 326.0793. C16H18N35ClO2S + Na [M + Na]+, 346.0608; found, 346.0639; C16H18N37ClO2S + Na [M + Na]+, 348.0584; found, 348.0613; Anal. calcd for C15H16NClO2S (323.843): C, 59.34; H, 5.60; N, 4.33; found: C, 59.51; H, 5.69; N, 4.06.
N-tert-Butyl-N-(2-chloro-2-phenylethyl)benzenesulfonamide (4) as a colourless oil. Rf 0.61 (pentane/ether 3:1); 1H NMR (400 MHz, CDCl3) δ 1.25 (s, 9H), 3.71 (dd, J = 15.6, 8.8 Hz, 1H), 3.91 (dd, J = 15.8, 4.6 Hz, 1H), 5.55 (dd, J = 8.8, 4.4 Hz, 1H), 7.32–7.41 (m, 5H), 7.46–7.56 (m, 3H), 7.93 (dd, J = 7.2, 1.2 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 29.5, 53.3, 59.7, 62.8, 127.1, 127.5, 128.62, 128.66, 128.9, 132.2, 139.2, 143.9; MS (ESI, 1.57 kV, MeOH) m/z (Intens. ×106): 374 (1.15) [M + Na+], 338 (0.64) [M − Cl+], 316 (0.43), 282 (0.5), 260 (0.8); HRMS (ESI) m/z: [M + Na]+ calcd for C18H22N35ClO2S + Na, 374.0952; found, 374.0960; C18H22N37ClO2S + Na [M + Na]+, 376.0927; found, 376.0933.
N-[2-Chloro-2-(4-fluorphenyl)ethyl]-N-methylbenzenesulfonamide (5) as a yellow oil. Rf 0.34 (pentane/ether 3:1); 1H NMR (300 MHz, CDCl3) δ 2.42 (s, 3H), 2.63 (s, 3H), 3.39 (dd, J = 14.5, 7.8 Hz, 1H), 3.55 (dd, J = 14.5, 6.9 Hz, 1H), 5.09 (t, J = 7.5 Hz, 1H), 7.05 (t, J = 8.5 Hz, 2H), 7.30 (d, J = 8.4 Hz, 2H), 7.39 (dd, J = 8.7, 5.1 Hz, 2H), 7.64 (d, J = 8.4 Hz, 2H); 13C NMR (75 MHz, CDCl3) δ 21.4, 37.0, 58.0, 60.4, 115.8 (d, J = 26.8 Hz), 127.3, 129.3 (d, J = 8.3 Hz), 129.8, 134.5, 134.7, 143.6, 162.8 (d, J = 246.8 Hz); 19F NMR (282 MHz, CDCl3) δ −112.79; MS (EI, 70 eV) m/z (%): 341 (3) [M+], 305 (7) [M − HCl+], 198 (97) [M − C7H5ClF+], 168 (2), 155 (71) [C7H7O2S+], 113 (19), 91 (100) [C7H7+], 65 (17) [C5H5+]; HRMS (ESI) m/z: [M + Na]+ calcd for C16H17N35ClFO2S + Na, 364.0545; found, 364.0574; C16H17N37ClFO2S + Na [M + Na]+, 366.0518; found, 366.0546; Anal. calcd for C15H16NClFO2S (341.834): C, 56.22; H, 5.01; N, 4.10; found, C, 56.43; H, 5.01; N, 4.05.
N-[2-Chloro-2-(3-nitrophenyl)ethyl]-N-methylbenzenesulfonamide (6) as a yellow resin. Rf 0.14 (pentane/ether 3:1); 1H NMR (600 MHz, CDCl3) δ 2.43 (s, 3H), 2.69 (s, 3H), 3.50 (dd, J = 14.0, 8.0 Hz, 1H), 3.55 (dd, J = 14.4, 6.8 Hz, 1H), 5.21 (t, J = 7.6 Hz, 1H), 7.32 (d, J = 8.0 Hz, 2H), 7.59 (t, J = 8.0 Hz, 1H), 7.64 (d, J = 8.4 Hz, 2H), 7.80 (d, 7.6 Hz, 1H), 8.22 (m, 1H), 8.28 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 21.4, 37.1, 57.8, 59.5, 122.5, 123.7, 127.3, 129.82, 129.88, 133.8, 134.3, 140.7, 143.9, 148.4; MS (EI, 70 eV) m/z (%): 368 (0.1) [M+], 198 (99) [M − C7H5ClNO2+], 170 (5) [C7H5ClNO2+], 155 (100) [C7H7O2S+], 127 (7), 91 (97) [C7H7+], 65 (16) [C5H5+]; Anal. calcd for C16H17N2ClO4S (368.841): C, 52.10; H, 4.65; N, 7.59; found, C, 52.35; H, 4.81; N, 7.40.
N-(2-Chlorodecyl)-4,N-dimethylbenzenesulfonamide (7) as a colourless oil. Rf 0.60 (pentane/ether 3:1); 1H NMR (400 MHz, CDCl3) δ 0.86 (t, J = 6.9 Hz, 3H), 1.25–1.39 (m, 10H), 1.52–1.63 (m, 3H), 1.85–1.91 (m, 1H), 2.41 (s, 3H), 2.80 (s, 3H), 3.06 (dd, J = 14.1, 6.6 Hz, 1H) oder (10’), 3.34 (dd, J = 14.4, 6.6 Hz, 1H), 4.01–4.05 (m, 1H), 7.30 (d with fine-splitting, J = 7.8 Hz, 2H), 7.65 (d with fine-splitting, J = 8.4 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 14.0, 21.4, 22.6, 26.1, 29.0, 29.1, 29.3, 31.8, 35.3, 36.9, 56.8, 60.8, 127.3, 129.7, 134.4, 143.5; MS (EI, 70 eV) m/z (%): 359 (11) [M+], 310 (29) [M − CH2Cl+], 282 (3) [M − C6H5+], 246 (13), 198 (97), 186 (10), 155 (98) [C7H7O2S+], 91 (100) [C7H7+], 65 (38) [C5H5+]; HRMS (EI) m/z: [M+] calcd for C18H30N35ClO2S, 359.16778; found, 359.16858; Anal. calcd for C18H30NClO2S (359.961): C, 60.06; H, 8.40; N, 3.89; found, C, 60.29; H, 8.41; N, 3.90.
N-(2-Chlorocyclooctyl)-N-methylbenzenesulfonamide (8) as a colourless oil. Rf 0.32 (pentane/ether 3:1); 1H NMR (600 MHz, CDCl3) δ 1.38–1.44 (m, 1H), 1.49–1.71 (m, 7H), 1.72–1.79 (m, 1H), 1.89–1.95 (m, 1H), 2.01–2.06 (m, 1H), 2.10–2.16 (m, 1H), 2.41 (s, 3H), 2.70 (s, 3H), 4.11 (m, 1H), 4.30 (m, 1H), 7.28 (d with fine-splitting, J = 8.4 Hz, 2H), 7.74 (d with fine-splitting, J = 8.4 Hz, 2H); 13C NMR (150 MHz, CDCl3) δ 21.4, 22.2, 24.7, 26.1, 27.8, 28.1 (broad), 30.85, 30.88, 62.2 (broad), 62.5, 127.4, 129.3, 137.1, 142.9; MS (EI, 70 eV) m/z (%): 329 (15) [M+], 294 (24) [M − Cl+], 272 (3) [M − C4H9+], 225 (8), 224 (47), 198 (5), 155 (21) [C7H7O2S+], 127 (42), 110 (42) [C8H14+], 84 (100), 57 (37) [C4H9+]; HRMS (EI) m/z: [M+] calcd for C16H24N35ClO2S, 329.12164; found 329.12040; Anal. calcd for C16H24NClO2S (329.891): C, 58.25; H, 7.33; N, 4.25; found: C, 58.47; H, 7.18; N, 4.03.
N-(3-Chlorobicyclo[2.2.1]hept-2-yl)-N-methylbenzenesulfonamide (9) as a colourless oil. 9a (endo-Product): Rf 0.26 (pentane/ether 3:1); 1H NMR (400 MHz, CDCl3) δ 1.29–1.37 (m, 2H), 1.40–1.60 (m, 3H), 1.86–1.93 (m, 2H), 2.39–2.41 (m, 1H), 2.42 (s, 3H), 2.78 (s, 3H), 3.71 (dd, J = 5.4, 1.8Hz, 1H), 4.06 (m, 1H), 7.30 (d with fine-splitting, J = 8.0 Hz, 2H), 7.71 (d with fine-splitting, J = 8.4 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 20.9, 21.4, 29.3, 30.5, 35.8, 39.7, 43.1, 64.9, 69.1, 127.3, 129.5, 135.9, 143.2; MS (EI, 70 eV) m/z (%): 313 (36) [M+], 278 (6) [M − Cl+], 250 (9) [M − C2H4Cl+], 238 (21), 224 (5), 198 (51), 158 (100) [M − C7H7O2S+], 131 (13), 121 (31), 91 (88) [C7H7+], 65 (20) [C5H5+]; HRMS (EI) m/z: [M+] calcd for C15H20N35ClO2S, 313.09033; found, 313.08982.
9b (exo-Product): Rf 0.42 (pentane/ether 3:1); 1H NMR (400 MHz, CDCl3) δ 1.19–1.22 (m, 3H), 1.44–1.54 (m, 1H), 1.59–1.72 (m, 1H), 1.93 (m, 1H), 2.06 (m, 1H), 2.42–2.44 (m, 1H), 2.42 (s, 3H), 2.93 (s, 3H), 4.06 (m, 1H), 4.14 (m, 1H), 7.29 (d with fine-splitting, J = 8.0 Hz, 2H), 7.86, d with fine-splitting, J = 8.0 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 21.4, 25.6, 29.2, 31.7, 34.7, 38.6, 45.0, 63.2, 68.5, 127.0, 129.5, 136.8, 143.0; MS (EI, 70 eV) m/z (%): 313 (50) [M+]; 278 (10) [M − Cl+]; 250 (6) [M − C2H4Cl+], 238 (31), 224 (7), 198 (67), 158 (100) [M − C7H7O2S+], 139 (11), 122 (34), 91 (88) [C7H7+], 65 (15) [C5H5+].
N-(4-Chlorocyclooct-2-enyl)-N-methylbenzenesulfonamide (10b) as a colourless oil. Rf 0.32 (pentane/ether 3:1); 1H NMR (600 MHz, CDCl3) δ 1.49–1.60 (m, 4H), 1.63–1.71 (m, 2H), 1.73–1.79 (m, 1H), 2.12–2.17 (m, 1H), 2.40 (s, 3H), 2.74 (s, 3H), 4.72–4.80 (m, 2H), 5.19 (ddd, J = 11.1, 8.4, 1.2 Hz, 1H), 5.50 (ddd, J = 10.8, 7.8, 1.2 Hz, 1H), 7.27 (d, J = 8.4 Hz, 2H), 7.63 (d, J = 8.4 Hz); 13C NMR (150 MHz, CDCl3) δ 21.4, 23.6, 25.0, 28.8, 34.4, 40.1, 54.5, 56.7, 125.2, 127.3, 129.5, 132.8, 135.7, 143.2; MS (EI, 70 eV) m/z (%): 327 (36) [M+], 292 (14) [M − Cl+], 250 (26) [M − C6H5+], 231 (4), 224 (12) [M − C8H7+], 198 (8), 172 (43) [M − C7H7O2S+], 127 (45), 106 (16), 91 (100) [C7H7+], 57 (37) [C4H9+]; HRMS (Schubstange) m/z [M+]: calcd for C16H22N35ClO2S, 327.10599; found, 327.10532.
Byproduct: N-(2-Chlorocyclooct-3-enyl)-N-methylbenzenesulfonamide (10a) as a colourless oil. Rf 0.39 (pentan/ether 3:1); 1H NMR (300 MHz, CDCl3) δ 1.26–1.35 (m, 1H), 1.57–1.76 and 1.85–1.92 (m, 6H), 2.21–2.28 (m, 1H), 2.42 (s, 3H), 2.76 (s, 3H), 4.44–4.51 (m, 1H), 4.90–5.03 (m, 1H), 5.66 (ddd, J = 12.3, 6.5, 1.6 Hz, 1H), 5.77–5.86 (m, 1H), 7.29 (d, J = 8.1 Hz, 2H), 7.73 (d, J = 7.8 Hz, 2H); 13C NMR (150 MHz, CDCl3) δ 21.9, 23.5, 29.1, 30.3, 37.5, 57.3, 62.1, 127.2, 128.3, 129.4, 131.8 (selected peaks); MS (ESI, 1.30 kV, MeOH) m/z (Intens. ×106): 677 (2.2) [2*M + Na+], 350 (3.1) [M + Na+], 314 (1.8) [MNa − Cl+].
N-Cyclohex-2-enyl-N-methylbenzenesulfonamide (11) as a colourless oil [41]. Rf 0.42 (pentane/ether 3:1); 1H NMR (600 MHz, CDCl3) δ 1.46–1.52 (m, 1H), 1.57–1.61 (m, 1H), 1.74 (m, 2H), 1.93 (m, 2H), 2.43 (s, 3H), 2.70 (s, 3H), 5.11 (m, 1H), 5.57 (m, 1H), 5.81 (m, 1H), 7.30 (d, J = 8.4 Hz, 2H), 7.71 (d with fine-splitting, J = 8.4 Hz, 2H); 13C NMR (150 MHz, CDCl3) δ 21.3, 21.4, 24.3, 26.7, 29.1, 54.2, 127.0, 127.1, 129.6, 132.3, 137.2, 142.9; MS (EI, 70 eV) m/z (%): 265 (15) [M+], 237 (100) [M − C2H4+], 213 (24) [M − C4H4+], 186 (6), 155 (27) [C7H7O2S+], 126 (25) [M − C7H7OS+], 110 (66) [C7H12N+], 91 (75) [C7H7+], 55 (25); HRMS (ESI) m/z: [M + Na+] calcd for C14H19NO2SNa, 288.1029; found, 288.0984.
N-(2-Chlorotetrahydropyran-3-yl)-N-methylbenzenesulfonamide (12) as a yellow oil. Rf 0.21 (pentane/ether 3:1); 1H NMR (400 MHz, CDCl3) δ 1.63–1.73 (m, 2H), 1.87–1.97 (m, 1H), 2.42 (s, 3H), 2.42–2.46 (m, 1H), 2.75 (s, 3H), 3.56–3.62 (m, 1H), 3.77 (ddd, J = 11.6, 11.6, 4.6 Hz, 1H), 3.90–3.96 (m, 1H), 5.10 (d, J = 9.2 Hz, 1H), 7.29 (d, J = 8.0 Hz, 2H), 7.77 (d with fine-splitting, J = 8.4 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 21.4, 26.4, 28.0, 34.6, 54.3, 67.5, 89.0, 127.8, 129.3, 136.2, 143.3; MS (EI, 70 eV) m/z (%): 303 (80) [M+], 259 (4) [M − C2H4O+], 241 (11), 214 (14), 196 (12), 155 (31) [C7H7O2S+], 127 (41), 108 (100), 91 (66) [C7H7+], 55 (42) [C3H3O+]; HRMS (EI) m/z: [M+] calcd for C13H18N35ClO3S, 303.06958; found, 303.06931.
N-(1,2-Dichloro-2-isobutoxyethyl)-N-methylbenzenesulfonamide (13) as a yellow oil. Rf 0.56 (pentane/ether, 3:1); 1H NMR (400 MHz, CDCl3) δ 0.88 (d, J = 1.6 Hz, 3H), 0.89 (d, J = 1.6 Hz, 3H), 1.85 (sept, J = 6.6 Hz, 1H), 2.44 (s, 3H), 2.76 (s, 3H), 3.20 (d, J = 6.8 Hz, 2H), 5.30 (d, J = 6.4 Hz, 1H), 5.58 (d, J = 6.4 Hz, 2H), 7.32 (d, J = 8.4 Hz, 2H), 7.77 (d with fine-splitting, J = 8.4 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 19.0, 19.1, 21.4, 27.6, 28.0, 71.2, 76.5, 89.6, 127.5, 129.5, 136.1, 143.8; MS (ESI, 1.30 kV, MeOH) m/z (%): 376 (60) [M + Na+], 318 (11), 304 (3) [M − C4H8O+], 285 (6) [M − C7H7+], 262 (6), 226 (60), 208 (4), 197 (11), 187 (4), 163 (9), 155 (56) [C7H7O2S+], 139 (11) [C7H7OS+], 128 (4), 106 (9), 91 (6) [C7H7+], 72 (58) [C4H8O+], 57 (17) [C4H9+]; HRMS (ESI) m/z: [M + Na]+ calcd for C14H21N35Cl2O3S + Na, 376.0511; found, 376.0506; C14H21N37Cl2O3S + Na [M + Na]+, 378.0483; found, 378.0480.
N-(5-Chloropent-2-enyl)-N-methylbenzenesulfonamide (15) as a yellow liquid and a non separable 4:1 mixture of isomers. Rf 0.23 (pentane/ether 3:1). Main isomer 15a (probably the E-isomer): 1H NMR (400 MHz, CDCl3) δ 2.43 (s, 3H), 2.45–2.51 (m, 2H), 2.66 (s, 3H), 3.50 (t, J = 6.6 Hz, 2H), 3.60 (d, J = 6.4 Hz, 2H), 5.43–5.53 (m, 1H), 5.56–5.65 (m, 1H), 7.32 (d, J = 8.0 Hz, 2H), 7.67 (d with fine-splitting, J = 8.4 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 21.4, 34.1, 35.1, 43.7, 52.0, 127.45, 127.48, 129.61, 131.0, 134.5, 143.3. Minor isomer 15b (probably the Z-isomer)(selected Peaks): 1H NMR (400 MHz, CDCl3) δ 2.44 (s, 3H), 2.67 (s, 3H), 3.51 (t, J = 6.6 Hz, 2H), 3.68 (d, J = 6.4 Hz, 2H), 7.33 (d, J = 8.0 Hz, 2H), 7.67 (d, J = 8.0 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 30.3, 34.2, 46.8, 126.7, 129.68, 130.2, 143.4; MS (EI, 70 eV) m/z (%): 287 (2) [M+], 272 (5) [M − CH3+], 238 (8) [M − CH2Cl+], 224 (16) [M − C2H4Cl+], 198 (14), 186 (100), 155 (58) [C7H7O2S+], 132 (53), 91 (93) [C7H7+], 57 (46) [C4H9+]; HRMS (ESI) m/z: [M + Na]+ calcd for C13H18N35ClO2S + Na, 310.0639; found, 310.0620; C13H18N37ClO2S + Na [M + Na]+, 312.0611; found, 312.0594; Anal. calcd for C13H18NClO2S (287.806): C, 54.25; H, 6.30; N, 4.87; found, C, 54.18; H, 6.40; N, 5.02.
N-(2-Chloro-6-hydroxyhexyl)-N-methylbenzenesulfonamide (16) as a colourless oil. Rf 0,06 (pentane/ether 3:1); 1H NMR (400 MHz, CDCl3) δ 1.49–1.79 (m, 6H), 1.92–2.01 (m, 1H), 2.44 (s, 3H), 2.83 (s, 3H), 3.08 (dd, J = 14.0, 6.4 Hz, 1H), 3.39 (dd, J = 14.2, 7.0 Hz, 1H), 3.67 (t, J = 6.2 Hz, 2H), 4.05–4.11 (m, 1H), 7.33 (d, J = 8.0 Hz, 2H), 7.67 (d with fine-splitting, J = 8.8 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 21.4, 22.2, 32.0, 34.8, 36.9, 56.6, 60.4, 62.4, 127.3, 129.7, 134.4, 143.6; MS (EI, 70 eV) m/z (%): 318 (1) [M+], 283 (16) [M − Cl+], 270 (12), 246 (3), 198 (88), 186 (8), 155 (100) [C7H7O2S+], 127 (15), 91 (93) [C7H7+], 65 (43) [C5H5+]; HRMS (ESI) m/z: [M + Na]+ calcd for C14H22N35ClO3S + Na 342.0901; found, 342.0890; C14H22N37ClO3S + Na [M + Na]+: 344.0874; found, 344.0864.
References
-
Noack, M.; Göttlich, R. Chem. Commun. 2002, 536. doi:10.1039/b111656h
Return to citation in text: [1] -
Heuger, G.; Kalsow, S.; Göttlich, R. Eur. J. Org. Chem. 2002, 1848. doi:10.1002/1099-0690(200206)2002:11<1848::AID-EJOC1848>3.0.CO;2-V
Return to citation in text: [1] -
Schulte-Wülwer, I. A.; Helaja, J.; Göttlich, R. Synthesis 2003, 1886. doi:10.1055/s-2003-40997
Return to citation in text: [1] -
Helaja, J.; Göttlich, R. Chem. Commun. 2002, 720. doi:10.1039/b201209j
Return to citation in text: [1] -
Noack, M.; Göttlich, R. Eur. J. Org. Chem. 2002, 3171. doi:10.1002/1099-0690(200209)2002:18<3171::AID-EJOC3171>3.0.CO;2-L
Return to citation in text: [1] -
Noack, M.; Kalsow, S.; Göttlich, R. Synlett 2004, 1110. doi:10.1055/s-2004-822907
Return to citation in text: [1] -
Göttlich, R. Sci. Synth. 2006, 35, 189–250.
See for a review on the aminochlorination of alkenes.
Return to citation in text: [1] -
Chemler, S. R.; Bovino, M. T. ACS Catal. 2013, 3, 1076. doi:10.1021/cs400138b
See for a review on the aminochlorination of alkenes.
Return to citation in text: [1] -
Neale, R. S. Synthesis 1971, 1. doi:10.1055/s-1971-21662
See for a review in this field.
Return to citation in text: [1] -
Minisci, F. Synthesis 1973, 1. doi:10.1055/s-1973-22123
See for a review in this field.
Return to citation in text: [1] -
Zard, S. Z. Chem. Soc. Rev. 2008, 37, 1603. doi:10.1039/b613443m
See for a review in this field.
Return to citation in text: [1] -
Ohashi, T.; Sugie, M.; Okahara, M.; Komori, S. Tetrahedron Lett. 1968, 9, 4195. doi:10.1016/S0040-4039(00)75406-2
Return to citation in text: [1] -
Ohashi, T.; Sugie, M.; Okahara, M.; Komori, S. Tetrahedron 1969, 25, 5349. doi:10.1016/S0040-4020(01)83035-0
Return to citation in text: [1] -
Kharasch, M. S.; Priestley, H. M. J. Am. Chem. Soc. 1939, 61, 3425. doi:10.1021/ja01267a057
Return to citation in text: [1] -
Seden, T. P.; Turner, R. W. J. Chem. Soc. C 1968, 876. doi:10.1039/j39680000876
Return to citation in text: [1] -
Daniher, F. A.; Butler, P. E. J. Org. Chem. 1968, 33, 4336. doi:10.1021/jo01276a006
Return to citation in text: [1] -
Neale, R. S.; Marcus, N. L. J. Org. Chem. 1969, 34, 1808. doi:10.1021/jo01258a061
Return to citation in text: [1] -
Li, G.; Saibabu Kotti, S. R. S.; Timmons, C. Eur. J. Org. Chem. 2007, 2745. doi:10.1002/ejoc.200600990
And references cited therein.
Return to citation in text: [1] -
Karur, S.; Saibabu Kotti, S. R. S.; Xu, X.; Cannon, J. F.; Headley, A.; Li, G. J. Am. Chem. Soc. 2003, 125, 13340. doi:10.1021/ja0304188
Return to citation in text: [1] [2] -
Chen, D.; Timmons, C.; Guo, L.; Xu, X.; Li, G. Synthesis 2004, 2479. doi:10.1055/s-2004-831203
Return to citation in text: [1] [2] -
Chen, D.; Timmons, C.; Chao, S.; Li, G. Eur. J. Org. Chem. 2004, 3097. doi:10.1002/ejoc.200400050
Return to citation in text: [1] [2] -
Zhang, H.; Song, Y.; Zhao, J.; Zhang, J.; Zhang, Q. Angew. Chem., Int. Ed. 2014, 53, 11079. doi:10.1002/anie.201406797
Return to citation in text: [1] -
Lu, H.; Chen, Q.; Li, C. J. Org. Chem. 2007, 72, 2564. doi:10.1021/jo0625857
Return to citation in text: [1] -
Tsuritani, T.; Shinokubo, H.; Oshima, K. J. Org. Chem. 2003, 68, 3246. doi:10.1021/jo034043k
Return to citation in text: [1] -
Sherman, E. S.; Fuller, P. H.; Kasi, D.; Chemler, S. R. J. Org. Chem. 2007, 72, 3896. doi:10.1021/jo070321u
Return to citation in text: [1] -
Liwosz, T. W.; Chemler, S. R. Chem. – Eur. J. 2013, 19, 12771. doi:10.1002/chem.201301800
Return to citation in text: [1] -
Martinez, C.; Muñiz, K. Adv. Synth. Catal. 2014, 356, 205. doi:10.1002/adsc.201300880
Return to citation in text: [1] [2] -
Gontcharov, A. V.; Liu, H.; Sharpless, K. B. Org. Lett. 1999, 1, 783. doi:10.1021/ol990761a
Return to citation in text: [1] -
Jeong, J. U.; Tao, B.; Sagasser, I.; Henniges, H.; Sharpless, K. B. J. Am. Chem. Soc. 1998, 120, 6844. doi:10.1021/ja981419g
Return to citation in text: [1] -
Ando, T.; Minakata, S.; Ryu, I.; Komatsu, M. Tetrahedron Lett. 1998, 39, 309. doi:10.1016/S0040-4039(97)10553-6
Return to citation in text: [1] -
Minakata, S.; Kano, D.; Oderaotoshi, Y.; Komatsu, M. Org. Lett. 2002, 4, 2097. doi:10.1021/ol026020i
Return to citation in text: [1] -
Minakata, S.; Yoneda, Y.; Oderaotoshi, Y.; Komatsu, M. Org. Lett. 2006, 8, 967. doi:10.1021/ol0600178
Return to citation in text: [1] -
Dauban, P.; Dodd, R. H. Tetrahedron Lett. 2001, 42, 1037. doi:10.1016/S0040-4039(00)02214-0
Return to citation in text: [1] -
Larionov, O. V.; Kozhushkov, S. I.; de Meijere, A. Synthesis 2003, 1916. doi:10.1055/s-2003-41037
Return to citation in text: [1] -
Curini, M.; Epifano, F.; Marcotullio, M. C.; Rosati, O.; Tsadjout, A. Synlett 2000, 813. doi:10.1055/s-2000-6695
Return to citation in text: [1] -
Kuehne, M. E.; Horne, D. A. J. Org. Chem. 1975, 40, 1287. doi:10.1021/jo00897a023
Return to citation in text: [1] -
Tsuritani, T.; Shinokubo, H.; Oshima, K. Org. Lett. 2001, 3, 2709. doi:10.1021/ol016310j
Return to citation in text: [1] -
Kochi, J. K. Acc. Chem. Res. 1974, 7, 351. doi:10.1021/ar50082a006
Return to citation in text: [1] -
Griller, D.; Ingold, K. U. Acc. Chem. Res. 1980, 13, 317. doi:10.1021/ar50153a004
Return to citation in text: [1] -
Bailey, A. D.; Cherney, S. M.; Anzalone, P. W.; Anderson, E. D.; Ernat, J. J.; Mohan, R. S. Synlett 2006, 215. doi:10.1055/s-2005-923586
Return to citation in text: [1] -
Haubenreisser, S.; Niggemann, M. Adv. Synth. Catal. 2011, 353, 469. doi:10.1002/adsc.201000768
Return to citation in text: [1]
| 1. | Noack, M.; Göttlich, R. Chem. Commun. 2002, 536. doi:10.1039/b111656h |
| 2. | Heuger, G.; Kalsow, S.; Göttlich, R. Eur. J. Org. Chem. 2002, 1848. doi:10.1002/1099-0690(200206)2002:11<1848::AID-EJOC1848>3.0.CO;2-V |
| 3. | Schulte-Wülwer, I. A.; Helaja, J.; Göttlich, R. Synthesis 2003, 1886. doi:10.1055/s-2003-40997 |
| 14. | Kharasch, M. S.; Priestley, H. M. J. Am. Chem. Soc. 1939, 61, 3425. doi:10.1021/ja01267a057 |
| 25. | Sherman, E. S.; Fuller, P. H.; Kasi, D.; Chemler, S. R. J. Org. Chem. 2007, 72, 3896. doi:10.1021/jo070321u |
| 26. | Liwosz, T. W.; Chemler, S. R. Chem. – Eur. J. 2013, 19, 12771. doi:10.1002/chem.201301800 |
| 12. | Ohashi, T.; Sugie, M.; Okahara, M.; Komori, S. Tetrahedron Lett. 1968, 9, 4195. doi:10.1016/S0040-4039(00)75406-2 |
| 13. | Ohashi, T.; Sugie, M.; Okahara, M.; Komori, S. Tetrahedron 1969, 25, 5349. doi:10.1016/S0040-4020(01)83035-0 |
| 27. | Martinez, C.; Muñiz, K. Adv. Synth. Catal. 2014, 356, 205. doi:10.1002/adsc.201300880 |
| 9. |
Neale, R. S. Synthesis 1971, 1. doi:10.1055/s-1971-21662
See for a review in this field. |
| 10. |
Minisci, F. Synthesis 1973, 1. doi:10.1055/s-1973-22123
See for a review in this field. |
| 11. |
Zard, S. Z. Chem. Soc. Rev. 2008, 37, 1603. doi:10.1039/b613443m
See for a review in this field. |
| 4. | Helaja, J.; Göttlich, R. Chem. Commun. 2002, 720. doi:10.1039/b201209j |
| 5. | Noack, M.; Göttlich, R. Eur. J. Org. Chem. 2002, 3171. doi:10.1002/1099-0690(200209)2002:18<3171::AID-EJOC3171>3.0.CO;2-L |
| 6. | Noack, M.; Kalsow, S.; Göttlich, R. Synlett 2004, 1110. doi:10.1055/s-2004-822907 |
| 7. |
Göttlich, R. Sci. Synth. 2006, 35, 189–250.
See for a review on the aminochlorination of alkenes. |
| 8. |
Chemler, S. R.; Bovino, M. T. ACS Catal. 2013, 3, 1076. doi:10.1021/cs400138b
See for a review on the aminochlorination of alkenes. |
| 24. | Tsuritani, T.; Shinokubo, H.; Oshima, K. J. Org. Chem. 2003, 68, 3246. doi:10.1021/jo034043k |
| 18. |
Li, G.; Saibabu Kotti, S. R. S.; Timmons, C. Eur. J. Org. Chem. 2007, 2745. doi:10.1002/ejoc.200600990
And references cited therein. |
| 19. | Karur, S.; Saibabu Kotti, S. R. S.; Xu, X.; Cannon, J. F.; Headley, A.; Li, G. J. Am. Chem. Soc. 2003, 125, 13340. doi:10.1021/ja0304188 |
| 20. | Chen, D.; Timmons, C.; Guo, L.; Xu, X.; Li, G. Synthesis 2004, 2479. doi:10.1055/s-2004-831203 |
| 21. | Chen, D.; Timmons, C.; Chao, S.; Li, G. Eur. J. Org. Chem. 2004, 3097. doi:10.1002/ejoc.200400050 |
| 17. | Neale, R. S.; Marcus, N. L. J. Org. Chem. 1969, 34, 1808. doi:10.1021/jo01258a061 |
| 22. | Zhang, H.; Song, Y.; Zhao, J.; Zhang, J.; Zhang, Q. Angew. Chem., Int. Ed. 2014, 53, 11079. doi:10.1002/anie.201406797 |
| 16. | Daniher, F. A.; Butler, P. E. J. Org. Chem. 1968, 33, 4336. doi:10.1021/jo01276a006 |
| 15. | Seden, T. P.; Turner, R. W. J. Chem. Soc. C 1968, 876. doi:10.1039/j39680000876 |
| 19. | Karur, S.; Saibabu Kotti, S. R. S.; Xu, X.; Cannon, J. F.; Headley, A.; Li, G. J. Am. Chem. Soc. 2003, 125, 13340. doi:10.1021/ja0304188 |
| 20. | Chen, D.; Timmons, C.; Guo, L.; Xu, X.; Li, G. Synthesis 2004, 2479. doi:10.1055/s-2004-831203 |
| 21. | Chen, D.; Timmons, C.; Chao, S.; Li, G. Eur. J. Org. Chem. 2004, 3097. doi:10.1002/ejoc.200400050 |
| 33. | Dauban, P.; Dodd, R. H. Tetrahedron Lett. 2001, 42, 1037. doi:10.1016/S0040-4039(00)02214-0 |
| 28. | Gontcharov, A. V.; Liu, H.; Sharpless, K. B. Org. Lett. 1999, 1, 783. doi:10.1021/ol990761a |
| 29. | Jeong, J. U.; Tao, B.; Sagasser, I.; Henniges, H.; Sharpless, K. B. J. Am. Chem. Soc. 1998, 120, 6844. doi:10.1021/ja981419g |
| 30. | Ando, T.; Minakata, S.; Ryu, I.; Komatsu, M. Tetrahedron Lett. 1998, 39, 309. doi:10.1016/S0040-4039(97)10553-6 |
| 31. | Minakata, S.; Kano, D.; Oderaotoshi, Y.; Komatsu, M. Org. Lett. 2002, 4, 2097. doi:10.1021/ol026020i |
| 32. | Minakata, S.; Yoneda, Y.; Oderaotoshi, Y.; Komatsu, M. Org. Lett. 2006, 8, 967. doi:10.1021/ol0600178 |
| 27. | Martinez, C.; Muñiz, K. Adv. Synth. Catal. 2014, 356, 205. doi:10.1002/adsc.201300880 |
| 41. | Haubenreisser, S.; Niggemann, M. Adv. Synth. Catal. 2011, 353, 469. doi:10.1002/adsc.201000768 |
| 39. | Griller, D.; Ingold, K. U. Acc. Chem. Res. 1980, 13, 317. doi:10.1021/ar50153a004 |
| 40. | Bailey, A. D.; Cherney, S. M.; Anzalone, P. W.; Anderson, E. D.; Ernat, J. J.; Mohan, R. S. Synlett 2006, 215. doi:10.1055/s-2005-923586 |
| 36. | Kuehne, M. E.; Horne, D. A. J. Org. Chem. 1975, 40, 1287. doi:10.1021/jo00897a023 |
| 37. | Tsuritani, T.; Shinokubo, H.; Oshima, K. Org. Lett. 2001, 3, 2709. doi:10.1021/ol016310j |
| 34. | Larionov, O. V.; Kozhushkov, S. I.; de Meijere, A. Synthesis 2003, 1916. doi:10.1055/s-2003-41037 |
| 35. | Curini, M.; Epifano, F.; Marcotullio, M. C.; Rosati, O.; Tsadjout, A. Synlett 2000, 813. doi:10.1055/s-2000-6695 |
© 2015 Heuger and Göttlich; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)