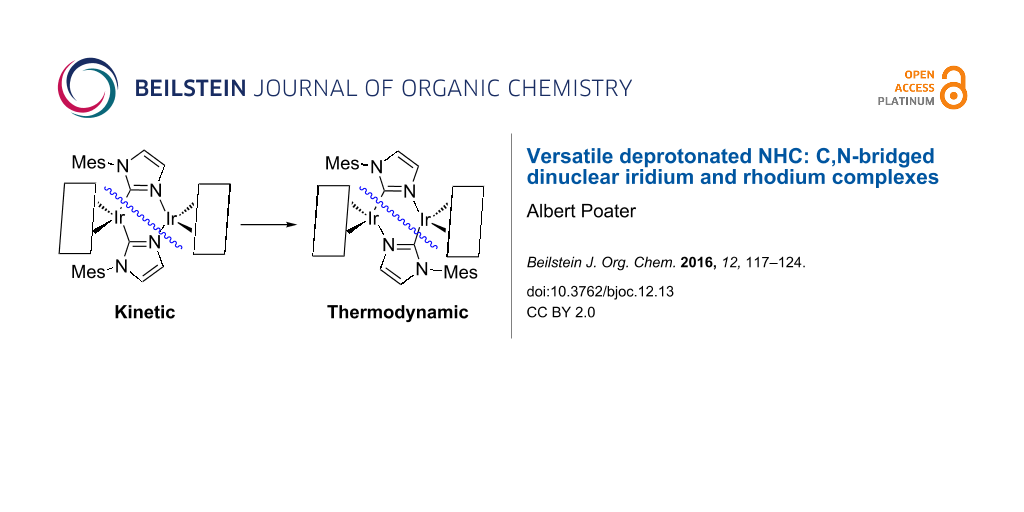
Abstract
Bearing the versatility of N-heterocyclic carbene (NHC) ligands, here density functional theory (DFT) calculations unravel the capacity of coordination of a deprotonated NHC ligand (pNHC) to generate a doubly C2,N3-bridged dinuclear complex. Here, in particular the discussion is based on the combination of the deprotonated 1-arylimidazol (aryl = mesityl (Mes)) with [M(cod)(μ-Cl)] (M = Ir, Rh) generated two geometrical isomers of complex [M(cod){µ-C3H2N2(Mes)-κC2,κN3}]2). The latter two isomers display conformations head-to-head (H-H) and head-to-tail (H-T) of CS and C2 symmetry, respectively. The isomerization from the H-H to the H-T conformation is feasible, whereas next substitutions of the cod ligand by CO first, and PMe3 later confirm the H-T coordination as the thermodynamically preferred. It is envisaged the exchange of the metal, from iridium to rhodium, confirming here the innocence of the nature of the metal for such arrangements of the bridging ligands.

Graphical Abstract
Introduction
In the framework of organometallic chemistry, N-heterocyclic carbenes (NHC) centre a well stablished class of relatively new ligands since in 1991 Arduengo and collaborators isolated the first stable NHC of the imidazole type with bulky N-substituents [1]. However the existence of stable NHCs was before postulated by Wanzlick et al. during the 1960s [2-4] and supported later by Öfele [5,6]. NHCs have become useful ligands in many transition metal-catalyzed reactions, stimulating the study of the unique features of the M–NHC bond [7,8], which favored the synthesis of new NHCs and to their use as ligands in transition metal complexes. The latter complexes were usually obtained by an easy replacement of a phosphine by the new NHC ligand, displaying a very high stability under many catalytic conditions. Furthermore, NHCs exhibit better activity, despite bearing its carbene functionality. Of course, these good results in basic research supposed and explosion of industrial efforts to design the right metal NHC-based catalyst for any kind of reaction. Anyway, neither a unique nor a few list of catalysts turned out to be effective for any catalytic reaction, but some successful applications were achieved in the field of Ru-catalyzed metathesis of olefins [9-12], Ir-catalyzed hydrogenation [13,14], Pd-catalyzed C=C coupling reactions [15,16], Ir-catalyzed CO2 fixation [17,18], and/or functionalization of alkenes and alkynes by Au-catalyzed reactions [19-21].
It is not feasible to exclude the asymmetry thanks to the modification of any of the two groups on the imidazolin-2-ylidene ring for two reasons. First, H atoms on the backbone of the either saturated or unsaturated imidazolin-2-ylidene ring suppose a key structural feature for the introduction of asymmetry in the NHC ring. Second, these H atoms might transform the corresponding NHCs in potentially efficient chiral NHCs in asymmetric synthesis [22]. Following with the latter recipe, protic NHCs (pNHCs) consist of the presence of a N-bound H atom. Although most of the studies on NHCs together with metal moieties are based on the interaction of the carbene carbon of the NHC with the metal [23-26], the high instability due the NH group of the pNHCs can induce secondary interactions leading to bifunctional catalysis [27,28], substrate recognition [29] and/or biological systems [30,31]. Among the synthetic methodologies to access to pNHC metal complexes [32-40], recently N-arylimine functionalized pNHC iridium complexes were obtained using excess of [Ir(cod)(µ-Cl)]2 [41], and next deprotonation of the pNHC leads to an equilibrium between a mononuclear complex containing a C-bound anionic imidazolide [42-44] and its dimer [45-58], where the NHC moiety binds in a µ-C,N bridging mode (see Scheme 1) [44].
![[1860-5397-12-13-i1]](/bjoc/content/inline/1860-5397-12-13-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Equilibrium between the monoiridum complex bearing a C-bound anionic imidazolide and its corresponding dimer, once deprotonated a pNHC.
Scheme 1: Equilibrium between the monoiridum complex bearing a C-bound anionic imidazolide and its correspond...
Even though during the last two decades thousands of papers have presented and described the NHC based catalysis, bearing a carbene–metal coordination [7], catalytically few efforts have been dedicated to other types of coordination of the NHC with the metal. Braunstein and collaborators have smartly faced the challenge to mix the reactivity of both coordinative atoms of pNHCs [59], either the carbene carbon or the non-substituted nitrogen, i.e., the N from the former N–H group, bearing 1-arylimidazolide ligands. Scheme 2 contains the general scheme that leads to a particular case of the “equilibrium” between two dinuclear complexes, labelled 3H-H and 3H-T (where H-H = head-to-head and H-T = head-to-tail). By DFT calculations here we contribute in the understanding of the thermodynamics of the subsequent C,N-bridged dinuclear iridium and rhodium complexes [59], and the facility for the interconversion between these latter dimeric species.
![[1860-5397-12-13-i2]](/bjoc/content/inline/1860-5397-12-13-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Experimental routes to the “equilibrium” between 3H-H and 3H-T.
Scheme 2: Experimental routes to the “equilibrium” between 3H-H and 3H-T.
Results
To shed light about both isomers of the dinuclear complex 3, 3H-H and 3H-T, we envisaged DFT calculations (see Figure 1). The optimized geometry of 3H-H is in perfect agreement with the X-ray structure [60] (rmsd = 0.065 Å and 1.1° for the selected main distances and angles) [61,62]. In agreement with experiments that indicated 3H-T is 5.2 kcal/mol more stable than 3H-H, which means that with free tautomerism only the thermodynamic isomer 3H-T would not coexist with 3H-H, but as the unique isomer [59].
![[1860-5397-12-13-1]](/bjoc/content/figures/1860-5397-12-13-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: View of the molecular structure of a) 3H-H and b) 3H-T. Hydrogen atoms have been omitted for the sake of clarity (main distances in Å).
Figure 1: View of the molecular structure of a) 3H-H and b) 3H-T. Hydrogen atoms have been omitted for the sa...
Furthermore, the strength of the two H-H and H-T arrangements of the bridging pNHC ligand with the iridium was examined with the Mayer bond order (MBO) [63], which is valuable for evaluating bonding in main group compounds, but has been also used as a tool for the characterization of transition metal systems [64,65]. For 3H-H the NHC coordination to the metal through the carbene carbon, the MBO is 0.874, whereas through the N atom is only 0.438. The type of coordination of pNHCs around the iridium atoms, either H-H or H-T, does not modify significantly the strength of the Ir–C and Ir–N bonds. For 3H-T the MBO values are 0.841 and 0.410. Further, the structure of the dinuclear complexes may be determined by steric effects of both monomeric moieties. To evaluate only the sterics, topographic steric maps were used, which are calculated through the SambVca package developed by Cavallo et al. [66]. This analysis allows the rationalization of the first coordination sphere around metal centres where catalytic processes take place. Basically the method calculates the buried volume of a given ligand [67] based on the quantification of the proportion of the first coordination sphere of the metal occupied by this ligand. The encumbered zones (color-coded in brown) belong to the part of the ligand that protrudes in the direction of the reacting groups, thus restricting the space they can fill, whereas empty zones (color coded in blue) correspond to the part where the ligand retracts from the reacting groups [68,69].
For 3H-H the percentage of buried volume (%VBur) is 26.2 bearing a metal–carbene coordination, whereas it decreases to only 19.1 when the NHC bonds to the metal through the deprotonated nitrogen atom. However, the steric maps in Figure 2 confirm completely that the system prefers the latter N-bound coordination. Splitting the map into four quadrants the carbene coordination reveals a quadrant highly sterically hindered (37.2) due to the rotation of the aromatic ring on the NHC which facilitates the allocation of such a NHC ligand in the dinuclear complexes. The other quadrant where this aromatic ring participates displays a value of 29.2, whereas the other two are innocuous for the metal sphere with low values of 19.3 and 19.1. On the other hand, the coordination of the NHC through the nitrogen is sterically innocent towards the metal sphere because the four quadrants show low sterical occupations (19.1, 18.7, 19.4 and 19.1). Thus, the N-bound coordination of the NHC can be regarded as the perfect coordination to facilitate a free “first coordination sphere”, with enough space for allowing the formation of a dinuclear complex. For 3H-T the %VBur is nearly identical, displaying values of 26.4 and 19.0, for carbene and nitrogen coordination to the iridium centre, respectively.
![[1860-5397-12-13-2]](/bjoc/content/figures/1860-5397-12-13-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Steric maps for the NHC ligand of 3H-H, coordinated to iridium by a) carbene or b) nitrogen. The isocontour curves are given in Å. The systems are oriented along the z-axis defined by the metal and the coordinating atom bonded to the metal. The two maps are computed with a radius of the sphere equal to 3.5 Å.
Figure 2: Steric maps for the NHC ligand of 3H-H, coordinated to iridium by a) carbene or b) nitrogen. The is...
Natural bond order (NBO) analysis on the iridium reveal that the two equivalent iridium centres in 3H-T display a charge on the metal of 0.009e, whereas −0.143 and 0.189e for 3H-H for the iridium bonded to two carbene carbons and two nitrogen atoms of the NHC, respectively. This confirms that the metal–carbene coordination allocates more electron density on the metal than through the nitrogen.
Despite the dimeric nature of complex 3, hypothetically they might be discussed as aggregates of two monomeric moieties. However the coupling between the two metal centres seems demonstrated by removing an electron of the system, thus the expected mixed valent Ir(I)/Ir(II) species turns out to display two identical metal centres that distribute equally the cost of the electrolysis of 3. Geometrically no asymmetry is observed, and together with the positive charge increase of 0.306e on each former Ir(I) centre, shows that the effect on system 3 of the released electron is mainly paid by the metal centres, but also partially spread over the ligands [59].
To follow up the experimental results the tautomerism/metallotropism between pNHC and imidazole ligands in these iridium complexes bearing a doubly C,N-bridged dinuclear core was also computationally studied after the displacement of cod ligands of 3H-H and 3H-T by CO in Scheme 3 [59], affording the tetracarbonyl complexes [M(CO)2{µ-C3H2N2(Mes)-κC2,κN3}]2, 4H-H and 4H-T, respectively. Next we evaluated the substitution of CO ligands by PMe3 affording complex [M(CO)(PMe3){µ-C3H2N2(Mes)-κC2,κN3}]2 (5) and finally, oxidative addition of MeI to the latter complex 5 afforded the dinuclear complex [M(CO)2(PMe3)2(Me)I{µ-C3H2N2(Mes)-κC2,κN3}]2.
![[1860-5397-12-13-i3]](/bjoc/content/inline/1860-5397-12-13-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Equilibrium between complexes 3–6, in the presence of CO, PMe3, and MeI.
Scheme 3: Equilibrium between complexes 3–6, in the presence of CO, PMe3, and MeI.
In agreement with experiments [59], without reflux conditions, the substitution of cod ligands by CO is extremely favored, being isomer 4H-T 48.5 kcal/mol more stable than 3H-T, and again the equilibrium between 4H-H and 4H-T is displaced towards the latter species, by a difference of 3.0 kcal/mol. Furthermore, the third CO ligand coordination on each iridium atom in 4 was faced but discarded due to a destabilization of 6.7 and 14.8 kcal/mol with respect to 4H-H and 4H-T, respectively. Basically this lower stability is not only due to the sterical hindrance, but to the preferred quasi perfect square planar type of coordination on each iridium centre (see Figure 3). Going into electronic details, this 3→4 transformation also follows the principle of maximum hardness [70,71], i.e., the chemical hardness evolves from 39.6 to 46.7 kcal/mol bearing a H-T type of coordination. This increase of chemical hardness is a consequence of the increased stability of the HOMO, which results in a larger HOMO–LUMO gap [61,65]. To point out that the two types of coordination, H-H and H-T, do not suppose a significant change of chemical hardness, just an increase for H-T of only 0.6 kcal/mol for species 4, whereas a decrease of 0.1 for species 3. Thus the electronics do not affect the equilibrium between both arrangements of the bridging ligands, but sterics as stated above. Further, the NBO charges show a decrease of the charge on both metals of 0.777e in the 3→4 transformation. Despite the π-backdonation of CO the donation to the iridium centres is larger than the corresponding electron density transferred by the cod ligands in species 3.
![[1860-5397-12-13-3]](/bjoc/content/figures/1860-5397-12-13-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: View of the molecular structure of a) 4H-H and b) 4H-T (main distances in Å).
Figure 3: View of the molecular structure of a) 4H-H and b) 4H-T (main distances in Å).
4H-H and 4H-T evolves to complex 5, exchanging one CO by a PMe3 ligand on each iridium centre, releasing 5.0 and 2.0 kcal/mol, respectively. Bearing an energy difference of 11.7 kcal/mol the H-T isomer is favored with respect to the H-H, in agreement with experiments [59], because the H-H coordination was not locate for 5, and further thermodynamics do not support the H-H coordination for 5, with an energy endergonicity of 6.7 kcal/mol with respect the previous corresponding complex 4. Oxidative addition of MeI to 5 affords complex 6, in an exergonic release of 16.5 kcal/mol, pointing out that there are several other isomers of complex 6 that differ from the Me and I coordination to each corresponding iridium centre. However all these alternative isomers of complex 6 are placed higher in energy by at least 12.9 kcal/mol (see Supporting Information File 1). The Ir–Ir distance in 6 is only 2.857 Å (see Figure 4), which is a clear proof of concept of a formally metal–metal bonded d7–d7 complex, to be compared with complex 5, where this Ir–Ir distance elongates till 3.497 Å. Furthermore, the MBO for the metal–metal bond reveals a significant value of 0.626 for complex 6 [72], being null for 5 and the previous complexes. Electronically, the charge on metals for complex 6 is 0.334 less charged, which is explained by, among other reasons, the weaker back-bonding from the metal to the CO ligands compared to 5, which affords the metal–metal interaction in 6.
![[1860-5397-12-13-4]](/bjoc/content/figures/1860-5397-12-13-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: View of the molecular structure of 6. Hydrogen atoms have been omitted for the sake of clarity (main distances in Å).
Figure 4: View of the molecular structure of 6. Hydrogen atoms have been omitted for the sake of clarity (mai...
To extent the scope of the reactivity of the NHC here, the cod ligands were first exchanged by ethylene, revealing a preferred H-T type of coordination of the NHC (5.1 kcal/mol). On the other hand, the iridium was replaced by rhodium to verify the innocent nature of the electronic properties of the metal, maintaining nearly completely the geometry features for complexes 3–6. Bearing rhodium complex 3H-T is again more stable than 3H-H, even 0.2 kcal/mol more stable. To sum up, no change of behaviour between iridium and homologous rhodium complexes was observed. On the other hand, the evolution from H-T to H-H arrangement was evaluated and mechanistically this process is predicted to be dissociative since neither intramolecular transition state was located nor the linear transits suggested energy barriers affordable at the experimental conditions.
Conclusion
The two possible H-T and H-H arrangements of a bridging NHC here have been studied in detail by DFT calculations. This is a contribution in the understanding of the thermodynamics of the subsequent C,N-bridged dinuclear iridium and rhodium complexes defined by Braunstein et al. [59] and the facility for the interconversion between these latter dimeric species. Steric maps confirm that the H-T is preferred since the metal centres are less sterically hindered, thus revealing the relatively unstable kinetic H-H isomers with respect to the H-T ones. However, screening the evolution from complex 3 to 6, going through complexes 4 and 5, the relative thermodynamics show a constant and sharp decay of energy, not due to steric factors, but mainly electronics.
Computational Details
The density functional calculations were performed on all the systems with the Gaussian 09 set of programs [73], Revision D.01, at the BP86 GGA level [74-76], adding the Grimme D3 dispersion term [77]. For iridium and rhodium we used the small-core, quasi-relativistic Stuttgart/Dresden (SDD) effective core potential with an associated valence contracted basis set (standard SDD keywords in Gaussian 09) [78-80]. The electronic configuration of the molecular systems was described with the triple-ζ valence plus polarization (TZVP keyword in Gaussian) basis set on all main group atoms during geometry optimizations [81]. The reported energies have been obtained via single point calculations on the BP86 geometries with triple-ζ valence plus polarization using the M06 functional [82]. Solvent effects, using either tetrahydrofuran, dichloromethane or toluene, were calculated with the polarizable continuous solvation model polarizable continuum model (PCM) model [83,84], and non-electrostatic terms were also included. The cavity is created via a series of overlapping spheres. However, the numbers reported throughout the text here are based on THF because this is the solvent employed in the experiments bearing the H-H to H-T rearrangement [59]. The geometry optimizations were performed without symmetry constraints, and the nature of the extrema was checked by analytical frequency calculations. Furthermore, all the extrema were confirmed by calculation of the intrinsic reaction paths.
The reported free energies in this work include energies obtained at the M06/TZVP level of theory in solvent corrected with zero-point energies at 298.15 K, together with the model of Martin et al. [85] which consists of thermal corrections and entropy effects evaluated at 1354 atm [86-89], with the BP86-d3/TZVP method in the gas phase [90,91].
%VBur calculations: The buried volume calculations were performed with the SambVca package developed by Cavallo et al. [90]. The radius of the sphere around the metal centre was set to 3.5 Å, while for the atoms we adopted the Bondi radii scaled by 1.17, and a mesh of 0.1 Å was used to scan the sphere for buried voxels. The steric maps were evaluated with a development version of the SambVca package.
Supporting Information
| Supporting Information File 1: Energies, cartesian coordinates, and 3D view for all DFT optimized species. | ||
| Format: PDF | Size: 404.5 KB | Download |
References
-
Arduengo III, A. J.; Harlow, R. L.; Kline, M. J. Am. Chem. Soc. 1991, 113, 361–363. doi:10.1021/ja00001a054
Return to citation in text: [1] -
Wanzlick, H.-W.; Kleiner, H.-J. Angew. Chem. 1961, 73, 493. doi:10.1002/ange.19610731408
Return to citation in text: [1] -
Wanzlick, H.-W. Angew. Chem., Int. Ed. Engl. 1962, 1, 75–80. doi:10.1002/anie.196200751
Return to citation in text: [1] -
Wanzlick, H.-W.; Schönherr, H.-J. Angew. Chem., Int. Ed. Engl. 1968, 7, 141–142. doi:10.1002/anie.196801412
Return to citation in text: [1] -
Öfele, K. J. Organomet. Chem. 1968, 12, P42–P43. doi:10.1016/S0022-328X(00)88691-X
Return to citation in text: [1] -
Öfele, K.; Herberhold, M. Angew. Chem., Int. Ed. Engl. 1970, 9, 739–740. doi:10.1002/anie.197007391
Return to citation in text: [1] -
Jacobsen, H.; Correa, A.; Poater, A.; Costabile, C.; Cavallo, L. Coord. Chem. Rev. 2009, 253, 687–703. doi:10.1016/j.ccr.2008.06.006
Return to citation in text: [1] [2] -
Jacobsen, H.; Correa, A.; Poater, A.; Costabile, C.; Cavallo, L. Coord. Chem. Rev. 2009, 253, 2784. doi:10.1016/j.ccr.2009.04.007
Return to citation in text: [1] -
Vougioukalakis, G.; Grubbs, R. H. Chem. Rev. 2010, 110, 1746–1787. doi:10.1021/cr9002424
Return to citation in text: [1] -
Poater, A.; Cavallo, L. Beilstein J. Org. Chem. 2015, 11, 1767–1780. doi:10.3762/bjoc.11.192
Return to citation in text: [1] -
Yuan, W.; Wei, Y.; Shi, M. ChemistryOpen 2013, 2, 63–68. doi:10.1002/open.201300002
Return to citation in text: [1] -
Credendino, R.; Poater, A.; Ragone, F.; Cavallo, L. Catal. Sci. Technol. 2011, 1, 1287–1297. doi:10.1039/c1cy00052g
Return to citation in text: [1] -
Vázquez-Serrano, L. D.; Owens, B. T.; Buriak, J. M. Chem. Commun. 2002, 2518–2519. doi:10.1039/b208403a
Return to citation in text: [1] -
Lee, H. M.; Jiang, T.; Stevens, E. D.; Nolan, S. P. Organometallics 2001, 20, 1255–1258. doi:10.1021/om000990x
Return to citation in text: [1] -
Grasa, G. A.; Viciu, M. S.; Huang, J.; Nolan, S. P. J. Org. Chem. 2001, 66, 7729–7737. doi:10.1021/jo010613+
Return to citation in text: [1] -
Marion, N.; Navarro, O.; Mei, J.; Stevens, E. D.; Scott, N. M.; Nolan, S. P. J. Am. Chem. Soc. 2006, 128, 4101–4111. doi:10.1021/ja057704z
Return to citation in text: [1] -
Vummaleti, S. V. C.; Talarico, G.; Nolan, S. P.; Cavallo, L.; Poater, A. Eur. J. Inorg. Chem. 2015, 4653–4657. doi:10.1002/ejic.201500905
Return to citation in text: [1] -
Vummaleti, S. V. C.; Talarico, G.; Nolan, S. P.; Cavallo, L.; Poater, A. Org. Chem. Front. 2016, 3, 19–23. doi:10.1039/C5QO00281H
Return to citation in text: [1] -
Dorel, R.; Echavarren, A. M. Chem. Rev. 2015, 115, 9028–9072. doi:10.1021/cr500691k
Return to citation in text: [1] -
Nun, P.; Gaillard, S.; Poater, A.; Cavallo, L.; Nolan, S. P. Org. Biomol. Chem. 2011, 9, 101–104. doi:10.1039/C0OB00758G
Return to citation in text: [1] -
Nun, P.; Dupuy, S.; Gaillard, S.; Poater, A.; Cavallo, L.; Nolan, S. P. Catal. Sci. Technol. 2011, 1, 58–61. doi:10.1039/c0cy00055h
Return to citation in text: [1] -
Hoveyda, A. H.; Schrock, R. R. Chem. – Eur. J. 2001, 7, 945–950. doi:10.1002/1521-3765(20010302)7:5<945::AID-CHEM945>3.0.CO;2-3
Return to citation in text: [1] -
Melaimi, M.; Soleilhavoup, M.; Bertrand, G. Angew. Chem., Int. Ed. 2010, 49, 8810–8849. doi:10.1002/anie.201000165
Return to citation in text: [1] -
Bourissou, D.; Guerret, O.; Gabbaï, F. P.; Bertrand, G. Chem. Rev. 2000, 100, 39–92. doi:10.1021/cr940472u
Return to citation in text: [1] -
de Frémont, P.; Marion, N.; Nolan, S. P. Coord. Chem. Rev. 2009, 253, 862–892. doi:10.1016/j.ccr.2008.05.018
Return to citation in text: [1] -
Hahn, F. E.; Jahnke, M. C. Angew. Chem., Int. Ed. 2008, 47, 3122–3172. doi:10.1002/anie.200703883
Return to citation in text: [1] -
Kuwata, S.; Ikariya, T. Chem. – Eur. J. 2011, 17, 3542–3556. doi:10.1002/chem.201003296
Return to citation in text: [1] -
Kuwata, S.; Ikariya, T. Chem. Commun. 2014, 50, 14290–14300. doi:10.1039/C4CC04457F
Return to citation in text: [1] -
Hahn, F. E. ChemCatChem 2013, 5, 419–430. doi:10.1002/cctc.201200567
Return to citation in text: [1] -
Hsieh, C.-H.; Pulukkody, R.; Darensbourg, M. Y. Chem. Commun. 2013, 49, 9326–9328. doi:10.1039/c3cc45091k
Return to citation in text: [1] -
Brackemeyer, D.; Hervé, A.; Schulte to Brinke, C.; Jahnke, M. C.; Hahn, F. E. J. Am. Chem. Soc. 2014, 136, 7841–7844. doi:10.1021/ja5030904
Return to citation in text: [1] -
Hahn, F. E.; Langenhahn, V.; Lügger, T.; Pape, T.; Le Van, D. Angew. Chem., Int. Ed. 2005, 44, 3759–3763. doi:10.1002/anie.200462690
Return to citation in text: [1] -
Kösterke, T.; Pape, T.; Hahn, F. E. J. Am. Chem. Soc. 2011, 133, 2112–2115. doi:10.1021/ja110634h
Return to citation in text: [1] -
Dobereiner, G. E.; Chamberlin, C. A.; Schley, N. D.; Crabtree, R. H. Organometallics 2010, 29, 5728–5731. doi:10.1021/om100452g
Return to citation in text: [1] -
Kösterke, T.; Kösters, J.; Würthwein, E.-U.; Mück-Lichtenfeld, C.; Schulte to Brinke, C.; Lahoz, F.; Hahn, F. E. Chem. – Eur. J. 2012, 18, 14594–14598. doi:10.1002/chem.201202973
Return to citation in text: [1] -
Kunz, P. C.; Wetzel, C.; Kögel, S.; Kassack, M. U.; Spingler, B. Dalton Trans. 2011, 40, 35–37. doi:10.1039/C0DT01089H
Return to citation in text: [1] -
Das, R.; Hepp, A.; Daniliuc, C. G.; Hahn, F. E. Organometallics 2014, 33, 6975–6987. doi:10.1021/om501120u
Return to citation in text: [1] -
Das, R.; Daniliuc, C. G.; Hahn, F. E. Angew. Chem., Int. Ed. 2014, 53, 1163–1166. doi:10.1002/anie.201308269
Return to citation in text: [1] -
Hahn, F. E.; Langenhahn, V.; Pape, T. Chem. Commun. 2005, 5390–5392. doi:10.1039/b510996e
Return to citation in text: [1] -
Ruiz, J.; García, G.; Mosquera, M. E. G.; Perandones, B. F.; Gonzalo, M. P.; Vivanco, M. J. Am. Chem. Soc. 2005, 127, 8584–8585. doi:10.1021/ja042879e
Return to citation in text: [1] -
He, F.; Braunstein, P.; Wesolek, M.; Danopoulos, A. A. Chem. Commun. 2015, 51, 2814–2817. doi:10.1039/C4CC10109J
Return to citation in text: [1] -
Hill, C.; Bosold, F.; Harms, K.; Lohrenz, J. C. W.; Marsch, M.; Schmieczek, M.; Boche, G. Chem. Ber./Recl. 1997, 130, 1201–1212. doi:10.1002/cber.19971300907
Return to citation in text: [1] -
Hilf, C.; Bosold, F.; Harms, K.; Marsch, M.; Boche, G. Chem. Ber./Recl. 1997, 130, 1213–1221. doi:10.1002/cber.19971300908
Return to citation in text: [1] -
Marchenko, A. P.; Koidan, H. N.; Pervak, I. I.; Huryeva, A. N.; Zarudnitskii, E. V.; Tolmachev, A. A.; Kostyuk, A. N. Tetrahedron Lett. 2012, 53, 494–496. doi:10.1016/j.tetlet.2011.11.054
Return to citation in text: [1] [2] -
Beveridge, K. A.; Bushnell, G. W.; Stobart, S. R.; Atwood, J. L.; Zaworotko, M. J. Organometallics 1983, 2, 1447–1451. doi:10.1021/om50004a035
Return to citation in text: [1] -
Bushnell, G. W.; Fjeldsted, D. O. K.; Stobart, S. R.; Zaworotko, M. J.; Knox, S. A. R.; MacPherson, K. A. Organometallics 1985, 4, 1107–1114. doi:10.1021/om00125a029
Return to citation in text: [1] -
Brost, R. D.; Stobart, S. R. Inorg. Chem. 1989, 28, 4307–4308. doi:10.1021/ic00323a001
Return to citation in text: [1] -
Brost, R. D.; Fjeldsted, D. O. K.; Stobart, S. R. J. Chem. Soc., Chem. Commun. 1989, 488–490. doi:10.1039/c39890000488
Return to citation in text: [1] -
Lichtenberger, D. L.; Copenhaver, A. S.; Gray, H. B.; Marshall, J. L.; Hopkins, M. D. Inorg. Chem. 1988, 27, 4488–4493. doi:10.1021/ic00297a030
Return to citation in text: [1] -
Bonati, F.; Oro, L. A.; Pinillos, M. T.; Tejel, C.; Bovio, B. J. Organomet. Chem. 1994, 465, 267–274. doi:10.1016/0022-328X(94)87065-9
Return to citation in text: [1] -
Yuan, Y.; Jiménez, M. V.; Sola, E.; Lahoz, F. J.; Oro, L. A. J. Am. Chem. Soc. 2002, 124, 752–753. doi:10.1021/ja016753i
Return to citation in text: [1] -
Bushnell, G. W.; Fjeldsted, D. O. K.; Stobart, S. R.; Zaworotko, M. J. J. Chem. Soc., Chem. Commun. 1983, 580–581. doi:10.1039/c39830000580
Return to citation in text: [1] -
Atwood, J. L.; Beveridge, K. A.; Bushnell, G. W.; Dixon, K. R.; Eadie, D. T.; Stobart, S. R.; Zaworotko, M. J. Inorg. Chem. 1984, 23, 4050–4057. doi:10.1021/ic00192a042
Return to citation in text: [1] -
Bushnell, G. W.; Fjeldsted, D. O. K.; Stobart, S. R.; Wang, J. Organometallics 1996, 15, 3785–3787. doi:10.1021/om9601864
Return to citation in text: [1] -
Coleman, A. W.; Eadie, D. T.; Stobart, S. R.; Zaworotko, M. J.; Atwood, J. L. J. Am. Chem. Soc. 1982, 104, 922–923. doi:10.1021/ja00367a076
Return to citation in text: [1] -
Bushnell, G. W.; Decker, M. J.; Eadie, D. T.; Stobart, S. R.; Vefghi, R.; Atwood, J. L.; Zaworotko, M. J. Organometallics 1985, 4, 2106–2111. doi:10.1021/om00131a007
Return to citation in text: [1] -
Fjeldsted, D. O. K.; Stobart, S. R.; Zaworotko, M. J. J. Am. Chem. Soc. 1985, 107, 8258–8259. doi:10.1021/ja00312a074
Return to citation in text: [1] -
Arthurs, M. A.; Bickerton, J.; Stobart, S. R.; Wang, J. Organometallics 1998, 17, 2743–2750. doi:10.1021/om970554j
Return to citation in text: [1] -
He, F.; Ruhlmann, L.; Gisselbrecht, J.-P.; Choua, S.; Orio, M.; Wesolek, M.; Danopoulos, A. A.; Braunstein, P. Dalton Trans. 2015, 44, 17030–17044. doi:10.1039/C5DT02403J
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] -
root-mean-square deviation (rmsd) is calculated for distances and angles: sn−1 = [∑i=1→N (CV − EV)2/(N − 1)]1/2, where CV means calculated value, EV means experimental value (X-ray data), and N is the number of distances or angles taken into account.
Return to citation in text: [1] -
Costas, M.; Ribas, X.; Poater, A.; López Balvuena, J. M.; Xifra, R.; Company, A.; Duran, M.; Solà, M.; Llobet, A.; Corbella, M.; Usón, M. A.; Mahía, J.; Solans, X.; Shan, X.; Benet-Buchholz, J. Inorg. Chem. 2006, 45, 3569–3581. doi:10.1021/ic051800j
Return to citation in text: [1] [2] -
Mola, J.; Rodríguez, M.; Romero, I.; Llobet, A.; Parella, T.; Poater, A.; Duran, M.; Solà, M.; Benet-Buchholz, J. Inorg. Chem. 2006, 45, 10520–10529. doi:10.1021/ic061126l
Return to citation in text: [1] -
Mayer, I. Int. J. Quantum Chem. 1984, 26, 151–154. doi:10.1002/qua.560260111
Return to citation in text: [1] -
Poater, A.; Ragone, F.; Correa, A.; Cavallo, L. J. Am. Chem. Soc. 2009, 131, 9000–9006. doi:10.1021/ja902552m
Return to citation in text: [1] -
Poater, A. J. Phys. Chem. A 2009, 31, 9030–9040. doi:10.1021/jp9040716
Return to citation in text: [1] [2] -
Poater, A.; Cosenza, B.; Correa, A.; Giudice, S.; Ragone, F.; Scarano, V.; Cavallo, L. Eur. J. Inorg. Chem. 2009, 1759–1766. doi:10.1002/ejic.200801160
Return to citation in text: [1] -
Bosson, J.; Poater, A.; Cavallo, L.; Nolan, S. P. J. Am. Chem. Soc. 2010, 132, 13146–13149. doi:10.1021/ja104961s
Return to citation in text: [1] -
Poater, A.; Ragone, F.; Mariz, R.; Dorta, R.; Cavallo, L. Chem. – Eur. J. 2010, 16, 14348–14353. doi:10.1002/chem.201001938
Return to citation in text: [1] -
Giovanni, C.; Poater, A.; Benet-Buchholz, J.; Cavallo, L.; Solà, M.; Llobet, A. Chem. – Eur. J. 2014, 20, 3898–3902. doi:10.1002/chem.201304699
Return to citation in text: [1] -
Parr, R. G.; Donnelly, R. A.; Levy, M.; Palke, W. E. J. Chem. Phys. 1978, 68, 3801–3807. doi:10.1063/1.436185
Return to citation in text: [1] -
Parr, R. G.; Pearson, R. G. J. Am. Chem. Soc. 1983, 105, 7512–7516. doi:10.1021/ja00364a005
Return to citation in text: [1] -
Poater, A.; Moradell, S.; Pinilla, E.; Poater, J.; Solà, M.; Martínez, M. A.; Llobet, A. Dalton Trans. 2006, 1188–1196. doi:10.1039/B511625M
Return to citation in text: [1] -
Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, 2009.
Return to citation in text: [1] -
Becke, A. D. Phys. Rev. A 1988, 38, 3098–3100. doi:10.1103/PhysRevA.38.3098
Return to citation in text: [1] -
Perdew, J. P. Phys. Rev. B 1986, 33, 8822–8824. doi:10.1103/PhysRevB.33.8822
Return to citation in text: [1] -
Perdew, J. P. Phys. Rev. B 1986, 34, 7406. doi:10.1103/PhysRevB.34.7406
Return to citation in text: [1] -
Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. J. Chem. Phys. 2010, 132, 154104. doi:10.1063/1.3382344
Return to citation in text: [1] -
Häussermann, U.; Dolg, M.; Stoll, H.; Preuss, H.; Schwerdtfeger, P.; Pitzer, R. M. Mol. Phys. 1993, 78, 1211–1224. doi:10.1080/00268979300100801
Return to citation in text: [1] -
Küchle, W.; Dolg, M.; Stoll, H.; Preuss, H. J. Chem. Phys. 1994, 100, 7535–7542. doi:10.1063/1.466847
Return to citation in text: [1] -
Leininger, T.; Nicklass, A.; Stoll, H.; Dolg, M.; Schwerdtfeger, P. J. Chem. Phys. 1996, 105, 1052–1059. doi:10.1063/1.471950
Return to citation in text: [1] -
Schäfer, A.; Horn, H.; Ahlrichs, R. J. Chem. Phys. 1992, 97, 2571–2577. doi:10.1063/1.463096
Return to citation in text: [1] -
Zhao, Y.; Truhlar, D. G. Theor. Chem. Acc. 2008, 120, 215–241. doi:10.1007/s00214-007-0310-x
Return to citation in text: [1] -
Barone, V.; Cossi, M. J. Phys. Chem. A 1998, 102, 1995–2001. doi:10.1021/jp9716997
Return to citation in text: [1] -
Tomasi, J.; Persico, M. Chem. Rev. 1994, 94, 2027–2094. doi:10.1021/cr00031a013
Return to citation in text: [1] -
Martin, R. L.; Hay, P. J.; Pratt, L. R. J. Phys. Chem. A 1998, 102, 3565–3573. doi:10.1021/jp980229p
Return to citation in text: [1] -
Poater, A.; Pump, E.; Vummaleti, S. V. C.; Cavallo, L. J. Chem. Theory Comput. 2014, 10, 4442–4448. doi:10.1021/ct5003863
Return to citation in text: [1] -
Pump, E.; Slugovc, C.; Cavallo, L.; Poater, A. Organometallics 2015, 34, 3107–3111. doi:10.1021/om501246q
Return to citation in text: [1] -
Urbina-Blanco, C. A.; Poater, A.; Lebl, T.; Manzini, S.; Slawin, A. M. Z.; Cavallo, L.; Nolan, S. P. J. Am. Chem. Soc. 2013, 135, 7073–7079. doi:10.1021/ja402700p
Return to citation in text: [1] -
Manzini, S.; Poater, A.; Nelson, D. J.; Cavallo, L.; Slawin, A. M. Z.; Nolan, S. P. Angew. Chem., Int. Ed. 2014, 53, 8995–8999. doi:10.1002/anie.201403770
Return to citation in text: [1] -
Manzini, S.; Poater, A.; Nelson, D. J.; Cavallo, L.; Nolan, S. P. Chem. Sci. 2014, 5, 180–188. doi:10.1039/C3SC52612G
Return to citation in text: [1] [2] -
García-Melchor, M.; Pacheco, M. C.; Nájera, C.; Lledós, A.; Ujaque, G. ACS Catal. 2012, 2, 135–144. doi:10.1021/cs200526x
Return to citation in text: [1]
| 66. | Poater, A.; Cosenza, B.; Correa, A.; Giudice, S.; Ragone, F.; Scarano, V.; Cavallo, L. Eur. J. Inorg. Chem. 2009, 1759–1766. doi:10.1002/ejic.200801160 |
| 67. | Bosson, J.; Poater, A.; Cavallo, L.; Nolan, S. P. J. Am. Chem. Soc. 2010, 132, 13146–13149. doi:10.1021/ja104961s |
| 68. | Poater, A.; Ragone, F.; Mariz, R.; Dorta, R.; Cavallo, L. Chem. – Eur. J. 2010, 16, 14348–14353. doi:10.1002/chem.201001938 |
| 69. | Giovanni, C.; Poater, A.; Benet-Buchholz, J.; Cavallo, L.; Solà, M.; Llobet, A. Chem. – Eur. J. 2014, 20, 3898–3902. doi:10.1002/chem.201304699 |
| 72. | Poater, A.; Moradell, S.; Pinilla, E.; Poater, J.; Solà, M.; Martínez, M. A.; Llobet, A. Dalton Trans. 2006, 1188–1196. doi:10.1039/B511625M |
| 59. | He, F.; Ruhlmann, L.; Gisselbrecht, J.-P.; Choua, S.; Orio, M.; Wesolek, M.; Danopoulos, A. A.; Braunstein, P. Dalton Trans. 2015, 44, 17030–17044. doi:10.1039/C5DT02403J |
| 61. | Costas, M.; Ribas, X.; Poater, A.; López Balvuena, J. M.; Xifra, R.; Company, A.; Duran, M.; Solà, M.; Llobet, A.; Corbella, M.; Usón, M. A.; Mahía, J.; Solans, X.; Shan, X.; Benet-Buchholz, J. Inorg. Chem. 2006, 45, 3569–3581. doi:10.1021/ic051800j |
| 65. | Poater, A. J. Phys. Chem. A 2009, 31, 9030–9040. doi:10.1021/jp9040716 |
| 59. | He, F.; Ruhlmann, L.; Gisselbrecht, J.-P.; Choua, S.; Orio, M.; Wesolek, M.; Danopoulos, A. A.; Braunstein, P. Dalton Trans. 2015, 44, 17030–17044. doi:10.1039/C5DT02403J |
| 59. | He, F.; Ruhlmann, L.; Gisselbrecht, J.-P.; Choua, S.; Orio, M.; Wesolek, M.; Danopoulos, A. A.; Braunstein, P. Dalton Trans. 2015, 44, 17030–17044. doi:10.1039/C5DT02403J |
| 70. | Parr, R. G.; Donnelly, R. A.; Levy, M.; Palke, W. E. J. Chem. Phys. 1978, 68, 3801–3807. doi:10.1063/1.436185 |
| 71. | Parr, R. G.; Pearson, R. G. J. Am. Chem. Soc. 1983, 105, 7512–7516. doi:10.1021/ja00364a005 |
| 59. | He, F.; Ruhlmann, L.; Gisselbrecht, J.-P.; Choua, S.; Orio, M.; Wesolek, M.; Danopoulos, A. A.; Braunstein, P. Dalton Trans. 2015, 44, 17030–17044. doi:10.1039/C5DT02403J |
| 59. | He, F.; Ruhlmann, L.; Gisselbrecht, J.-P.; Choua, S.; Orio, M.; Wesolek, M.; Danopoulos, A. A.; Braunstein, P. Dalton Trans. 2015, 44, 17030–17044. doi:10.1039/C5DT02403J |
| 74. | Becke, A. D. Phys. Rev. A 1988, 38, 3098–3100. doi:10.1103/PhysRevA.38.3098 |
| 75. | Perdew, J. P. Phys. Rev. B 1986, 33, 8822–8824. doi:10.1103/PhysRevB.33.8822 |
| 76. | Perdew, J. P. Phys. Rev. B 1986, 34, 7406. doi:10.1103/PhysRevB.34.7406 |
| 77. | Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. J. Chem. Phys. 2010, 132, 154104. doi:10.1063/1.3382344 |
| 86. | Poater, A.; Pump, E.; Vummaleti, S. V. C.; Cavallo, L. J. Chem. Theory Comput. 2014, 10, 4442–4448. doi:10.1021/ct5003863 |
| 87. | Pump, E.; Slugovc, C.; Cavallo, L.; Poater, A. Organometallics 2015, 34, 3107–3111. doi:10.1021/om501246q |
| 88. | Urbina-Blanco, C. A.; Poater, A.; Lebl, T.; Manzini, S.; Slawin, A. M. Z.; Cavallo, L.; Nolan, S. P. J. Am. Chem. Soc. 2013, 135, 7073–7079. doi:10.1021/ja402700p |
| 89. | Manzini, S.; Poater, A.; Nelson, D. J.; Cavallo, L.; Slawin, A. M. Z.; Nolan, S. P. Angew. Chem., Int. Ed. 2014, 53, 8995–8999. doi:10.1002/anie.201403770 |
| 90. | Manzini, S.; Poater, A.; Nelson, D. J.; Cavallo, L.; Nolan, S. P. Chem. Sci. 2014, 5, 180–188. doi:10.1039/C3SC52612G |
| 91. | García-Melchor, M.; Pacheco, M. C.; Nájera, C.; Lledós, A.; Ujaque, G. ACS Catal. 2012, 2, 135–144. doi:10.1021/cs200526x |
| 59. | He, F.; Ruhlmann, L.; Gisselbrecht, J.-P.; Choua, S.; Orio, M.; Wesolek, M.; Danopoulos, A. A.; Braunstein, P. Dalton Trans. 2015, 44, 17030–17044. doi:10.1039/C5DT02403J |
| 85. | Martin, R. L.; Hay, P. J.; Pratt, L. R. J. Phys. Chem. A 1998, 102, 3565–3573. doi:10.1021/jp980229p |
| 82. | Zhao, Y.; Truhlar, D. G. Theor. Chem. Acc. 2008, 120, 215–241. doi:10.1007/s00214-007-0310-x |
| 83. | Barone, V.; Cossi, M. J. Phys. Chem. A 1998, 102, 1995–2001. doi:10.1021/jp9716997 |
| 84. | Tomasi, J.; Persico, M. Chem. Rev. 1994, 94, 2027–2094. doi:10.1021/cr00031a013 |
| 78. | Häussermann, U.; Dolg, M.; Stoll, H.; Preuss, H.; Schwerdtfeger, P.; Pitzer, R. M. Mol. Phys. 1993, 78, 1211–1224. doi:10.1080/00268979300100801 |
| 79. | Küchle, W.; Dolg, M.; Stoll, H.; Preuss, H. J. Chem. Phys. 1994, 100, 7535–7542. doi:10.1063/1.466847 |
| 80. | Leininger, T.; Nicklass, A.; Stoll, H.; Dolg, M.; Schwerdtfeger, P. J. Chem. Phys. 1996, 105, 1052–1059. doi:10.1063/1.471950 |
| 81. | Schäfer, A.; Horn, H.; Ahlrichs, R. J. Chem. Phys. 1992, 97, 2571–2577. doi:10.1063/1.463096 |
| 90. | Manzini, S.; Poater, A.; Nelson, D. J.; Cavallo, L.; Nolan, S. P. Chem. Sci. 2014, 5, 180–188. doi:10.1039/C3SC52612G |
| 1. | Arduengo III, A. J.; Harlow, R. L.; Kline, M. J. Am. Chem. Soc. 1991, 113, 361–363. doi:10.1021/ja00001a054 |
| 9. | Vougioukalakis, G.; Grubbs, R. H. Chem. Rev. 2010, 110, 1746–1787. doi:10.1021/cr9002424 |
| 10. | Poater, A.; Cavallo, L. Beilstein J. Org. Chem. 2015, 11, 1767–1780. doi:10.3762/bjoc.11.192 |
| 11. | Yuan, W.; Wei, Y.; Shi, M. ChemistryOpen 2013, 2, 63–68. doi:10.1002/open.201300002 |
| 12. | Credendino, R.; Poater, A.; Ragone, F.; Cavallo, L. Catal. Sci. Technol. 2011, 1, 1287–1297. doi:10.1039/c1cy00052g |
| 32. | Hahn, F. E.; Langenhahn, V.; Lügger, T.; Pape, T.; Le Van, D. Angew. Chem., Int. Ed. 2005, 44, 3759–3763. doi:10.1002/anie.200462690 |
| 33. | Kösterke, T.; Pape, T.; Hahn, F. E. J. Am. Chem. Soc. 2011, 133, 2112–2115. doi:10.1021/ja110634h |
| 34. | Dobereiner, G. E.; Chamberlin, C. A.; Schley, N. D.; Crabtree, R. H. Organometallics 2010, 29, 5728–5731. doi:10.1021/om100452g |
| 35. | Kösterke, T.; Kösters, J.; Würthwein, E.-U.; Mück-Lichtenfeld, C.; Schulte to Brinke, C.; Lahoz, F.; Hahn, F. E. Chem. – Eur. J. 2012, 18, 14594–14598. doi:10.1002/chem.201202973 |
| 36. | Kunz, P. C.; Wetzel, C.; Kögel, S.; Kassack, M. U.; Spingler, B. Dalton Trans. 2011, 40, 35–37. doi:10.1039/C0DT01089H |
| 37. | Das, R.; Hepp, A.; Daniliuc, C. G.; Hahn, F. E. Organometallics 2014, 33, 6975–6987. doi:10.1021/om501120u |
| 38. | Das, R.; Daniliuc, C. G.; Hahn, F. E. Angew. Chem., Int. Ed. 2014, 53, 1163–1166. doi:10.1002/anie.201308269 |
| 39. | Hahn, F. E.; Langenhahn, V.; Pape, T. Chem. Commun. 2005, 5390–5392. doi:10.1039/b510996e |
| 40. | Ruiz, J.; García, G.; Mosquera, M. E. G.; Perandones, B. F.; Gonzalo, M. P.; Vivanco, M. J. Am. Chem. Soc. 2005, 127, 8584–8585. doi:10.1021/ja042879e |
| 7. | Jacobsen, H.; Correa, A.; Poater, A.; Costabile, C.; Cavallo, L. Coord. Chem. Rev. 2009, 253, 687–703. doi:10.1016/j.ccr.2008.06.006 |
| 8. | Jacobsen, H.; Correa, A.; Poater, A.; Costabile, C.; Cavallo, L. Coord. Chem. Rev. 2009, 253, 2784. doi:10.1016/j.ccr.2009.04.007 |
| 41. | He, F.; Braunstein, P.; Wesolek, M.; Danopoulos, A. A. Chem. Commun. 2015, 51, 2814–2817. doi:10.1039/C4CC10109J |
| 5. | Öfele, K. J. Organomet. Chem. 1968, 12, P42–P43. doi:10.1016/S0022-328X(00)88691-X |
| 6. | Öfele, K.; Herberhold, M. Angew. Chem., Int. Ed. Engl. 1970, 9, 739–740. doi:10.1002/anie.197007391 |
| 2. | Wanzlick, H.-W.; Kleiner, H.-J. Angew. Chem. 1961, 73, 493. doi:10.1002/ange.19610731408 |
| 3. | Wanzlick, H.-W. Angew. Chem., Int. Ed. Engl. 1962, 1, 75–80. doi:10.1002/anie.196200751 |
| 4. | Wanzlick, H.-W.; Schönherr, H.-J. Angew. Chem., Int. Ed. Engl. 1968, 7, 141–142. doi:10.1002/anie.196801412 |
| 30. | Hsieh, C.-H.; Pulukkody, R.; Darensbourg, M. Y. Chem. Commun. 2013, 49, 9326–9328. doi:10.1039/c3cc45091k |
| 31. | Brackemeyer, D.; Hervé, A.; Schulte to Brinke, C.; Jahnke, M. C.; Hahn, F. E. J. Am. Chem. Soc. 2014, 136, 7841–7844. doi:10.1021/ja5030904 |
| 19. | Dorel, R.; Echavarren, A. M. Chem. Rev. 2015, 115, 9028–9072. doi:10.1021/cr500691k |
| 20. | Nun, P.; Gaillard, S.; Poater, A.; Cavallo, L.; Nolan, S. P. Org. Biomol. Chem. 2011, 9, 101–104. doi:10.1039/C0OB00758G |
| 21. | Nun, P.; Dupuy, S.; Gaillard, S.; Poater, A.; Cavallo, L.; Nolan, S. P. Catal. Sci. Technol. 2011, 1, 58–61. doi:10.1039/c0cy00055h |
| 23. | Melaimi, M.; Soleilhavoup, M.; Bertrand, G. Angew. Chem., Int. Ed. 2010, 49, 8810–8849. doi:10.1002/anie.201000165 |
| 24. | Bourissou, D.; Guerret, O.; Gabbaï, F. P.; Bertrand, G. Chem. Rev. 2000, 100, 39–92. doi:10.1021/cr940472u |
| 25. | de Frémont, P.; Marion, N.; Nolan, S. P. Coord. Chem. Rev. 2009, 253, 862–892. doi:10.1016/j.ccr.2008.05.018 |
| 26. | Hahn, F. E.; Jahnke, M. C. Angew. Chem., Int. Ed. 2008, 47, 3122–3172. doi:10.1002/anie.200703883 |
| 17. | Vummaleti, S. V. C.; Talarico, G.; Nolan, S. P.; Cavallo, L.; Poater, A. Eur. J. Inorg. Chem. 2015, 4653–4657. doi:10.1002/ejic.201500905 |
| 18. | Vummaleti, S. V. C.; Talarico, G.; Nolan, S. P.; Cavallo, L.; Poater, A. Org. Chem. Front. 2016, 3, 19–23. doi:10.1039/C5QO00281H |
| 27. | Kuwata, S.; Ikariya, T. Chem. – Eur. J. 2011, 17, 3542–3556. doi:10.1002/chem.201003296 |
| 28. | Kuwata, S.; Ikariya, T. Chem. Commun. 2014, 50, 14290–14300. doi:10.1039/C4CC04457F |
| 15. | Grasa, G. A.; Viciu, M. S.; Huang, J.; Nolan, S. P. J. Org. Chem. 2001, 66, 7729–7737. doi:10.1021/jo010613+ |
| 16. | Marion, N.; Navarro, O.; Mei, J.; Stevens, E. D.; Scott, N. M.; Nolan, S. P. J. Am. Chem. Soc. 2006, 128, 4101–4111. doi:10.1021/ja057704z |
| 13. | Vázquez-Serrano, L. D.; Owens, B. T.; Buriak, J. M. Chem. Commun. 2002, 2518–2519. doi:10.1039/b208403a |
| 14. | Lee, H. M.; Jiang, T.; Stevens, E. D.; Nolan, S. P. Organometallics 2001, 20, 1255–1258. doi:10.1021/om000990x |
| 22. | Hoveyda, A. H.; Schrock, R. R. Chem. – Eur. J. 2001, 7, 945–950. doi:10.1002/1521-3765(20010302)7:5<945::AID-CHEM945>3.0.CO;2-3 |
| 44. | Marchenko, A. P.; Koidan, H. N.; Pervak, I. I.; Huryeva, A. N.; Zarudnitskii, E. V.; Tolmachev, A. A.; Kostyuk, A. N. Tetrahedron Lett. 2012, 53, 494–496. doi:10.1016/j.tetlet.2011.11.054 |
| 42. | Hill, C.; Bosold, F.; Harms, K.; Lohrenz, J. C. W.; Marsch, M.; Schmieczek, M.; Boche, G. Chem. Ber./Recl. 1997, 130, 1201–1212. doi:10.1002/cber.19971300907 |
| 43. | Hilf, C.; Bosold, F.; Harms, K.; Marsch, M.; Boche, G. Chem. Ber./Recl. 1997, 130, 1213–1221. doi:10.1002/cber.19971300908 |
| 44. | Marchenko, A. P.; Koidan, H. N.; Pervak, I. I.; Huryeva, A. N.; Zarudnitskii, E. V.; Tolmachev, A. A.; Kostyuk, A. N. Tetrahedron Lett. 2012, 53, 494–496. doi:10.1016/j.tetlet.2011.11.054 |
| 45. | Beveridge, K. A.; Bushnell, G. W.; Stobart, S. R.; Atwood, J. L.; Zaworotko, M. J. Organometallics 1983, 2, 1447–1451. doi:10.1021/om50004a035 |
| 46. | Bushnell, G. W.; Fjeldsted, D. O. K.; Stobart, S. R.; Zaworotko, M. J.; Knox, S. A. R.; MacPherson, K. A. Organometallics 1985, 4, 1107–1114. doi:10.1021/om00125a029 |
| 47. | Brost, R. D.; Stobart, S. R. Inorg. Chem. 1989, 28, 4307–4308. doi:10.1021/ic00323a001 |
| 48. | Brost, R. D.; Fjeldsted, D. O. K.; Stobart, S. R. J. Chem. Soc., Chem. Commun. 1989, 488–490. doi:10.1039/c39890000488 |
| 49. | Lichtenberger, D. L.; Copenhaver, A. S.; Gray, H. B.; Marshall, J. L.; Hopkins, M. D. Inorg. Chem. 1988, 27, 4488–4493. doi:10.1021/ic00297a030 |
| 50. | Bonati, F.; Oro, L. A.; Pinillos, M. T.; Tejel, C.; Bovio, B. J. Organomet. Chem. 1994, 465, 267–274. doi:10.1016/0022-328X(94)87065-9 |
| 51. | Yuan, Y.; Jiménez, M. V.; Sola, E.; Lahoz, F. J.; Oro, L. A. J. Am. Chem. Soc. 2002, 124, 752–753. doi:10.1021/ja016753i |
| 52. | Bushnell, G. W.; Fjeldsted, D. O. K.; Stobart, S. R.; Zaworotko, M. J. J. Chem. Soc., Chem. Commun. 1983, 580–581. doi:10.1039/c39830000580 |
| 53. | Atwood, J. L.; Beveridge, K. A.; Bushnell, G. W.; Dixon, K. R.; Eadie, D. T.; Stobart, S. R.; Zaworotko, M. J. Inorg. Chem. 1984, 23, 4050–4057. doi:10.1021/ic00192a042 |
| 54. | Bushnell, G. W.; Fjeldsted, D. O. K.; Stobart, S. R.; Wang, J. Organometallics 1996, 15, 3785–3787. doi:10.1021/om9601864 |
| 55. | Coleman, A. W.; Eadie, D. T.; Stobart, S. R.; Zaworotko, M. J.; Atwood, J. L. J. Am. Chem. Soc. 1982, 104, 922–923. doi:10.1021/ja00367a076 |
| 56. | Bushnell, G. W.; Decker, M. J.; Eadie, D. T.; Stobart, S. R.; Vefghi, R.; Atwood, J. L.; Zaworotko, M. J. Organometallics 1985, 4, 2106–2111. doi:10.1021/om00131a007 |
| 57. | Fjeldsted, D. O. K.; Stobart, S. R.; Zaworotko, M. J. J. Am. Chem. Soc. 1985, 107, 8258–8259. doi:10.1021/ja00312a074 |
| 58. | Arthurs, M. A.; Bickerton, J.; Stobart, S. R.; Wang, J. Organometallics 1998, 17, 2743–2750. doi:10.1021/om970554j |
| 64. | Poater, A.; Ragone, F.; Correa, A.; Cavallo, L. J. Am. Chem. Soc. 2009, 131, 9000–9006. doi:10.1021/ja902552m |
| 65. | Poater, A. J. Phys. Chem. A 2009, 31, 9030–9040. doi:10.1021/jp9040716 |
| 61. | Costas, M.; Ribas, X.; Poater, A.; López Balvuena, J. M.; Xifra, R.; Company, A.; Duran, M.; Solà, M.; Llobet, A.; Corbella, M.; Usón, M. A.; Mahía, J.; Solans, X.; Shan, X.; Benet-Buchholz, J. Inorg. Chem. 2006, 45, 3569–3581. doi:10.1021/ic051800j |
| 62. | Mola, J.; Rodríguez, M.; Romero, I.; Llobet, A.; Parella, T.; Poater, A.; Duran, M.; Solà, M.; Benet-Buchholz, J. Inorg. Chem. 2006, 45, 10520–10529. doi:10.1021/ic061126l |
| 59. | He, F.; Ruhlmann, L.; Gisselbrecht, J.-P.; Choua, S.; Orio, M.; Wesolek, M.; Danopoulos, A. A.; Braunstein, P. Dalton Trans. 2015, 44, 17030–17044. doi:10.1039/C5DT02403J |
| 59. | He, F.; Ruhlmann, L.; Gisselbrecht, J.-P.; Choua, S.; Orio, M.; Wesolek, M.; Danopoulos, A. A.; Braunstein, P. Dalton Trans. 2015, 44, 17030–17044. doi:10.1039/C5DT02403J |
| 60. | root-mean-square deviation (rmsd) is calculated for distances and angles: sn−1 = [∑i=1→N (CV − EV)2/(N − 1)]1/2, where CV means calculated value, EV means experimental value (X-ray data), and N is the number of distances or angles taken into account. |
| 7. | Jacobsen, H.; Correa, A.; Poater, A.; Costabile, C.; Cavallo, L. Coord. Chem. Rev. 2009, 253, 687–703. doi:10.1016/j.ccr.2008.06.006 |
| 59. | He, F.; Ruhlmann, L.; Gisselbrecht, J.-P.; Choua, S.; Orio, M.; Wesolek, M.; Danopoulos, A. A.; Braunstein, P. Dalton Trans. 2015, 44, 17030–17044. doi:10.1039/C5DT02403J |
© 2016 Poater; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)