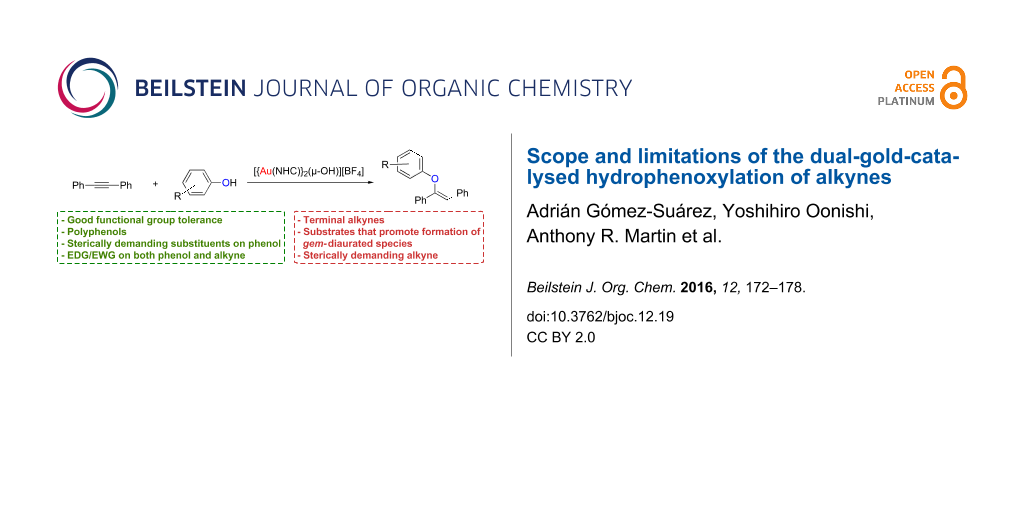
Abstract
Due to the synthetic advantages presented by the dual-gold-catalysed hydrophenoxylation of alkynes, a thorough study of this reaction was carried out in order to fully define the scope and limitations of the methodology. The protocol tolerates a wide range of functional groups, such as nitriles, ketones, esters, aldehydes, ketals, naphthyls, allyls or polyphenols, in a milder and more efficient manner than the previously reported methodologies. We have also identified that while we are able to use highly steric hindered phenols, small changes on the steric bulk of the alkynes have a dramatic effect on the reactivity. More importantly, we have observed that the use of substrates that facilitate the formation of diaurated species such as gem-diaurated or σ,π-digold–acetylide species, hinder the catalytic activity. Moreover, we have identified that the use of directing groups in unsymmetrical alkynes can help to achieve high regioselectivity in the hydrophenoxylation.

Graphical Abstract
Introduction
During the last 30 years, N-heterocyclic carbenes (NHCs) have evolved from mere curiosities to one of the most powerful tools within the synthetic chemist’s arsenal [1]. Due to their unique steric and electronic properties [2-6], NHCs are nowadays used in a myriad of chemical processes, for example as catalysts in organocatalysed reactions [7-9], or as ligands in transition metal and main-group chemistry [10-13]. We have been interested in exploring the use of NHC ligands in transition metal complexes for the development of highly active and well-defined catalysts. One of our main interests during the last decade has been to study the use of gold–NHC species as powerful catalysts for the construction of valuable organic molecules via the construction of C–C or C–O bonds [14-23]. In this regard, we have recently reported the use of [{Au(NHC)}2(µ-OH)][BF4] species as dual-activation catalysts for the hydrophenoxylation of alkynes [24,25]. This transformation proceeds through the interaction of the gold centres with both the phenol and the alkyne motifs, thus generating a synergistic effect that produces unique catalytic activity (Scheme 1) [24,25]. In addition, this reaction is highly stereoselective and only the Z-isomer was observed, regardless of the substrate used.
![[1860-5397-12-19-i1]](/bjoc/content/inline/1860-5397-12-19-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Dual-gold-catalysed hydrophenoxylation of alkynes.
Scheme 1: Dual-gold-catalysed hydrophenoxylation of alkynes.
This new, dual-catalysed methodology presents several advantages over previously reported protocols [26,27], such as milder reaction conditions or the use of lower catalyst loading. Therefore, encouraged by our preliminary studies [24], and the potential applications of the vinyl ether derivatives 3 as synthetic building blocks [28], we decided to further explore the scope and limitations of the dual-gold-catalysed hydrophenoxylation of alkynes. With this study, we sought to answer the following questions: what is the functional group tolerance of our transformation? How do steric and electronic factors affect both reaction partners? Can regioselectivity be achieved for the addition of phenols to unsymmetrical alkynes?
Results and Discussion
Functional group tolerance
We commenced our studies by assessing the functional group tolerance of the methodology (Scheme 2). With that aim we reacted diphenylacetylene (1a) with several phenols, 2a–o, in toluene, using 0.5 mol % of [{Au(IPr)}2(µ-OH)][BF4] (IPr = 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene) as the catalyst. Functional groups such as nitriles (3aa), ketones (3ab), esters (3ac), aldehydes (3ad), acetals (3ae) and naphthyls (3af) were well tolerated and the corresponding vinyl ethers were obtained in moderate to high yields (50–98%). On the contrary, the use of azide (3ag) or boronic ester (3ah) functionalities did not afford the desired products [29]. The latter case can be explained due to formation of the corresponding gem-diaurated aryl species, by reaction of catalyst [{Au(IPr)}2(µ-OH)][BF4] with the boronic ester [30], thus inhibiting the catalytic activity. The electronic properties of the phenol were also examined. If the electron density on the phenol is decreased, its nucleophilicity decreases and an increase in catalyst loading is needed in order to maintain relatively short reaction times (3ai and 3al) [31]. The steric hindrance on the phenol was studied next. Increasing it, with either allyl (3am) or tert-butyl (3an) groups on the ortho-position also required 1 mol % of catalyst, but the reaction proceeded smoothly. Encouraged by these and past results with highly hindered substrates, we attempted the addition of 2,6-di-tert-butylphenol (2ao). Unfortunately, this reaction did not proceed at all and only starting materials were observed by GC–FID. These results, in combination with our previous studies [24], where para-MeO, -CF3, -NO2, -Cl or -F substituents were also tolerated, support the robustness of our protocol, as it is highly efficient in the presence of a wide range of functional groups.
![[1860-5397-12-19-i2]](/bjoc/content/inline/1860-5397-12-19-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Exploring the functional group tolerance. Reaction conditions: 1a (0.50 mmol, 1 equiv), 2a–o (0.55 mmol, 1.1 equiv), [Au] (2.5 µmol, 0.5 mol %), toluene (1 mL), 80 °C. Isolated yields, average of two runs. a[Au] (5 µmol, 1 mol %), 110 °C. bGC analysis showed a complex mixture.
Scheme 2: Exploring the functional group tolerance. Reaction conditions: 1a (0.50 mmol, 1 equiv), 2a–o (0.55 ...
Polyphenols as nucleophiles
Intrigued by the versatility of our protocol, we explored the reaction using polyphenols as substrates (Scheme 3). Interestingly, we were able to selectively functionalize one (3ap) or both (4ap) hydroxy groups of catechol (2p) by using one or two equiv of 1a, respectively. Unfortunately, for the other polyphenol derivatives only the difunctionalised products could be observed (4aq–4as). We hypothesize that this difference in reactivity may be due to the steric hindrance around the monofunctionalised product 3ap, allowing for a significant rate difference in the reactions of 3ap and 4ap. In other cases, the greater separation between the hydroxy functional groups likely reduces this difference. To put these results in context, the previously reported methodology required 10 mol % of AuCl3 and 96 h in order to obtain similar yields for 4aq [26]. Moreover, only 7% of 4ap could be isolated after 168 h [26].
![[1860-5397-12-19-i3]](/bjoc/content/inline/1860-5397-12-19-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Hydrophenoxylation using polyphenols. Reaction conditions: 1a (1 mmol, 2 equiv), 2p–s (0.50 mmol, 1 equiv), [Au] (2.5 µmol, 0.5 mol %), toluene (1 mL), 80 °C. Isolated yields, average of two runs. a1 equiv 1a. b[Au] (5 µmol, 1 mol %), 110 °C.
Scheme 3: Hydrophenoxylation using polyphenols. Reaction conditions: 1a (1 mmol, 2 equiv), 2p–s (0.50 mmol, 1...
Electronic and steric effects on the alkyne
Next, we sought to assess how the electron density on the alkyne affected the reaction. Therefore, both electron-rich (1b, 1e) and electron-poor (1c) alkynes were tested using phenol 2t as nucleophile. The desired vinyl ethers, 3bt, 3ct and 3et, were obtained in moderate to good yields (Scheme 4). In order to expand the alkyne scope, propargylic ethers were also tested. In this case, 2 equiv of the alkyne were needed to obtain good yields of the desired product, 3dt. Next, we targeted the steric hindrance tolerated around the alkyne. With this aim in mind we decided to use diaryl-substituted alkynes bearing ortho-substituents due to synthetic reasons. The use of the ortho-chlorophenyl derivative 1f afforded the desired vinyl ether 3ft in good yields, while changing the chlorides for methyl groups resulted in no reaction, even when using harsher reaction conditions (3gt, Scheme 4). One more highly hindered substrate, 1h, bearing a sterically demanding naphthyl group on one side, and a phenyl on the other was studied. No reaction was observed even after increasing the catalyst loading and temperature (3ht, Scheme 4). Next, we focused our attention on whether the chain length on unsymmetrical alkyl–aryl alkynes, or the electron density on the phenol could affect the regioselectivity of the reaction. The use of 1-phenylbut-1-yne (1i) afforded the desired vinyl ethers 3it and 3it' in a 1:0.23 ratio and in good yields. This selectivity is slightly lower than our previously reported hydrophenoxylation of phenylpropyne (1j, 1:0.18) [24]. We also studied if the electron density on the phenol could alter the selectivity. Addition of p-methoxyphenol (2u) to 1j afforded a 1:0.22 ratio between the two expected products, 3ju and 3ju'. This selectivity is slightly diminished compared to the addition of phenol to 1j (1:0.18), but better than the addition of p-nitrophenol (2v, 1:0.35), both reported in our previous study (3jt and 3jv, Scheme 4) [24]. Finally, we tested whether terminal alkynes were tolerated by our methodology. Phenylacetylene (1k) was reacted with phenol (2t) under our standard reaction conditions, 0.5 mol % [{Au(IPr)}2(µ-OH)][BF4] as catalyst, in toluene at 80 °C for 1 h. However, GC–FID analysis of the reaction mixture revealed only traces of product. A more careful study of the crude reaction mixture by 1H NMR spectroscopy, revealed 8% conversion to the desired vinyl ether 3kt, as well as 10% of acetophenone, the corresponding hydration product. Furthermore, the formation of a new gold species, which was characterised as the previously reported σ,π-digold–acetylide species could also be observed. This diaurated species can be formed by reaction of [{Au(IPr)}2(µ-OH)][BF4] with phenylacetylene [30]. As in the case of the formation of a gem-diaurated species when boronic ester 3h was used, it appears that in situ formation of highly stable diaurated species, inhibits catalytic activity.
![[1860-5397-12-19-i4]](/bjoc/content/inline/1860-5397-12-19-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Hydrophenoxylation of (un)symmetrical alkynes. Reaction conditions: 1b–k (0.50 mmol, 1 equiv), 2t (0.55 mmol, 1.1 equiv), [Au] (2.5 µmol, 0.5 mol %), toluene (1 mL), 80 °C. Isolated yields, average of two runs. Ratio determined by 1H NMR. a[Au] (5 µmol, 1 mol %), 110 °C. b1d (1 mmol, 2 equiv), 2t (0.50 mmol, 1 equiv).
Scheme 4: Hydrophenoxylation of (un)symmetrical alkynes. Reaction conditions: 1b–k (0.50 mmol, 1 equiv), 2t (...
These results suggest that while both electron-rich/poor alkynes are tolerated by our methodology, the tolerated steric hindrance is very low, in contrast to the results observed for the nucleophile. In addition, in the case of unsymmetrical alkynes, the use of electron-rich alkynes appears to favour a better selectivity than their electron-poor counterparts.
Enhancing regioselectivity using directing groups
During our preliminary study on the hydrophenoxylation of unsymmetrical alkynes [24], we observed that the use of alkynes bearing a 2-pyridinyl moiety afforded only one of the possible vinyl ether derivatives (3lt and 3mt, Scheme 5). We hypothesised that this directing effect could be a combination of the charge polarization on the alkyne and an interaction between the pyridine nitrogen and one of the gold centres. To test our hypothesis, we synthesized an alkyne bearing a 3-pyridinyl (1n) substituent. In order to reduce the reaction time the catalyst loading was increased to 3 mol %. Interestingly, the selectivity dropped dramatically and a 1:0.43 ratio of products was observed (3nt, Scheme 5). In order to further test our hypothesis, the polarization of the alkyne was next reduced, but the potential chelating heteroatom was retained. The hydrophenoxylation of the unsymmetrical propargylic ether 1o afforded, with complete regioselectivity, the corresponding vinyl ether 3ot [32]. As expected, when homopropargylic ether 1p was used diminished selectivity was observed. Thus supporting our hypothesis that regioselectivity can be obtained by assistance of a directing group, either by polarization of the alkyne or by a chelating effect [33].
![[1860-5397-12-19-i5]](/bjoc/content/inline/1860-5397-12-19-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Regioselective hydrophenoxylation of unsymmetrical alkynes. Reaction conditions: 1l–p (1 equiv), 2a (1.1 equiv), [Au] (1 mol %), toluene (1 mL), 110 °C. Isolated yields, average of two runs. Ratio determined by 1H NMR. nd = not detected. a[Au] (3 mol %). b[Au] (0.5 mol %), 1o (2 equiv.)
Scheme 5: Regioselective hydrophenoxylation of unsymmetrical alkynes. Reaction conditions: 1l–p (1 equiv), 2a...
Conclusion
We have explored an extended range of reactions in order to fully define the scope and limitations of the digold-mediated hydrophenoxylation of alkynes. With these additions we have performed the gold-catalysed hydroalkoxylation of alkynes on ca. 50 substrates, thus demonstrating the robustness and synthetic versatility of the methodology. The protocol tolerates a wide range of functional groups, such as nitriles, ketones, esters, aldehydes, acetals, naphthyls or allyls. Furthermore, we have successfully used polyphenols in a milder and more efficient manner than the previously reported methodologies. We have also identified that while we are able to use highly steric hindered phenols, small changes on the steric bulk of the alkynes have a dramatic effect on reactivity. More importantly, we have observed that the use of substrates that facilitate the formation of diaurated species such as gem-diaurated or σ,π-digold–acetylide species, hinder the catalytic activity. We have identified that the use of directing groups in unsymmetrical alkynes can help achieve regioselectivity in the hydrophenoxylation reaction. With these studies, we have sought to contribute to a better understanding of the interactions involved in dual-gold-catalysed reactions, and anticipate that this information will help to design more efficient dual-activation protocols. We are currently studying the use of digold–hydroxide species in other dual-gold-catalysed reactions and these results will be published in due course.
Supporting Information
| Supporting Information File 1: Experimental procedures and characterisation data for all the compounds. | ||
| Format: PDF | Size: 2.9 MB | Download |
Acknowledgements
The ERC (Advanced Investigator Award-FUNCAT) and Syngenta are gratefully acknowledged for support. Umicore AG is acknowledged for their generous gift of materials. This publication is based upon work supported by the King Abdullah University of Science and Technology (KAUST), Office of Sponsored Research (OSR) under Award No. OSR-2015-CCF-1974-03. Y.O. thanks the Uehara Memorial Foundation for a Research Fellowship. We also thank Danila Gasperini for early synthetic contributions and Dr. David J. Nelson for helpful discussions.
References
-
Hopkinson, M. N.; Richter, C.; Schedler, M.; Glorius, F. Nature 2014, 510, 485–496. doi:10.1038/nature13384
Return to citation in text: [1] -
Nelson, D. J.; Nolan, S. P. Chem. Soc. Rev. 2013, 42, 6723–6753. doi:10.1039/c3cs60146c
Return to citation in text: [1] -
Díez-González, S.; Nolan, S. P. Coord. Chem. Rev. 2007, 251, 874–883. doi:10.1016/j.ccr.2006.10.004
Return to citation in text: [1] -
Jacobsen, H.; Correa, A.; Poater, A.; Costabile, C.; Cavallo, L. Coord. Chem. Rev. 2009, 253, 687–703. doi:10.1016/j.ccr.2008.06.006
Return to citation in text: [1] -
Clavier, H.; Nolan, S. P. Chem. Commun. 2010, 46, 841–861. doi:10.1039/b922984a
Return to citation in text: [1] -
Dröge, T.; Glorius, F. Angew. Chem., Int. Ed. 2010, 49, 6940–6952. doi:10.1002/anie.201001865
Return to citation in text: [1] -
Enders, D.; Niemeier, O.; Henseler, A. Chem. Rev. 2007, 107, 5606–5655. doi:10.1021/cr068372z
Return to citation in text: [1] -
Marion, N.; Díez-González, S.; Nolan, S. P. Angew. Chem., Int. Ed. 2007, 46, 2988–3000. doi:10.1002/anie.200603380
Return to citation in text: [1] -
Grossmann, A.; Enders, D. Angew. Chem., Int. Ed. 2012, 51, 314–325. doi:10.1002/anie.201105415
Return to citation in text: [1] -
Díez-González, S.; Marion, N.; Nolan, S. P. Chem. Rev. 2009, 109, 3612–3676. doi:10.1021/cr900074m
Return to citation in text: [1] -
Asay, M.; Jones, C.; Driess, M. Chem. Rev. 2011, 111, 354–396. doi:10.1021/cr100216y
Return to citation in text: [1] -
Riener, K.; Haslinger, S.; Raba, A.; Högerl, M. P.; Cokoja, M.; Herrmann, W. A.; Kühn, F. E. Chem. Rev. 2014, 114, 5215–5272. doi:10.1021/cr4006439
Return to citation in text: [1] -
Fortman, G. C.; Nolan, S. P. Chem. Soc. Rev. 2011, 40, 5151–5169. doi:10.1039/c1cs15088j
Return to citation in text: [1] -
Marion, N.; Díez-González, S.; de Frémont, P.; Noble, A. R.; Nolan, S. P. Angew. Chem., Int. Ed. 2006, 45, 3647–3650. doi:10.1002/anie.200600571
Return to citation in text: [1] -
Marion, N.; de Frémont, P.; Lemiere, G.; Stevens, E. D.; Fensterbank, L.; Malacria, M.; Nolan, S. P. Chem. Commun. 2006, 2048–2050. doi:10.1039/b602839j
Return to citation in text: [1] -
Marion, N.; Carlqvist, P.; Gealageas, R.; de Frémont, P.; Maseras, F.; Nolan, S. P. Chem. – Eur. J. 2007, 13, 6437–6451. doi:10.1002/chem.200700134
Return to citation in text: [1] -
Marion, N.; Gealageas, R.; Nolan, S. P. Org. Lett. 2007, 9, 2653–2656. doi:10.1021/ol070843w
Return to citation in text: [1] -
Correa, A.; Marion, N.; Fensterbank, L.; Malacria, M.; Nolan, S. P.; Cavallo, L. Angew. Chem., Int. Ed. 2008, 47, 718–721. doi:10.1002/anie.200703769
Return to citation in text: [1] -
Marion, N.; Ramón, R. S.; Nolan, S. P. J. Am. Chem. Soc. 2008, 131, 448–449. doi:10.1021/ja809403e
Return to citation in text: [1] -
Ramón, R. S.; Marion, N.; Nolan, S. P. Chem. – Eur. J. 2009, 15, 8695–8697. doi:10.1002/chem.200901231
Return to citation in text: [1] -
Gaillard, S.; Bosson, J.; Ramón, R. S.; Nun, P.; Slawin, A. M. Z.; Nolan, S. P. Chem. – Eur. J. 2010, 16, 13729–13740. doi:10.1002/chem.201001688
Return to citation in text: [1] -
Nun, P.; Dupuy, S.; Gaillard, S.; Poater, A.; Cavallo, L.; Nolan, S. P. Catal. Sci. Technol. 2011, 1, 58–61. doi:10.1039/c0cy00055h
Return to citation in text: [1] -
Gómez-Suárez, A.; Gasperini, D.; Vummaleti, S. V. C.; Poater, A.; Cavallo, L.; Nolan, S. P. ACS Catal. 2014, 4, 2701–2705. doi:10.1021/cs500806m
Return to citation in text: [1] -
Oonishi, Y.; Gómez-Suárez, A.; Martin, A. R.; Nolan, S. P. Angew. Chem., Int. Ed. 2013, 52, 9767–9771. doi:10.1002/anie.201304182
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Gómez-Suárez, A.; Oonishi, Y.; Martin, A. R.; Vummaleti, S. V. C.; Nelson, D. J.; Cordes, D. B.; Slawin, A. M. Z.; Cavallo, L.; Nolan, S. P.; Poater, A. Chem. – Eur. J. 2016, 22, 1125–1132. doi:10.1002/chem.201503097
Return to citation in text: [1] [2] -
Kuram, M. R.; Bhanuchandra, M.; Sahoo, A. K. J. Org. Chem. 2010, 75, 2247–2258. doi:10.1021/jo1000143
Return to citation in text: [1] [2] [3] -
Richard, M. E.; Fraccica, D. V.; Garcia, K. J.; Miller, E. J.; Ciccarelli, R. M.; Holahan, E. C.; Resh, V. L.; Shah, A.; Findeis, P. M.; Stockland, R. A., Jr. Beilstein J. Org. Chem. 2013, 9, 2002–2008. doi:10.3762/bjoc.9.235
Return to citation in text: [1] -
Oonishi, Y.; Gómez-Suárez, A.; Martin, A. R.; Makida, Y.; Slawin, A. M. Z.; Nolan, S. P. Chem. – Eur. J. 2014, 20, 13507–13510. doi:10.1002/chem.201404630
Return to citation in text: [1] -
Dash, C.; Yousufuddin, M.; Cundari, T. R.; Dias, H. V. R. J. Am. Chem. Soc. 2013, 135, 15479–15488. doi:10.1021/ja406027x
The lack of reactivity observed for 2h could be due to its reaction with cationic gold, which above 50 °C can liberate N2 and generate a reactive nitrene species.
Return to citation in text: [1] -
Gómez-Suárez, A.; Dupuy, S.; Slawin, A. M. Z.; Nolan, S. P. Angew. Chem., Int. Ed. 2013, 52, 938–942. doi:10.1002/anie.201208234
Return to citation in text: [1] [2] -
If these reactions are run under solvent-free conditions, the catalyst loading can be reduced to 0.5 mol %. For further details see reference [28].
Return to citation in text: [1] -
Arcadi, A.; Blesi, F.; Cacchi, S.; Fabrizi, G.; Goggiamani, A.; Marinelli, F. Org. Biomol. Chem. 2012, 10, 9700–9708. doi:10.1039/c2ob26763b
2 Equiv of the alkyne were necessary. See for reactions of 1n under gold catalysis.
Return to citation in text: [1] -
Gorske, B. C.; Mbofana, C. T.; Miller, S. J. Org. Lett. 2009, 11, 4318–4321. doi:10.1021/ol9016782
Return to citation in text: [1]
| 30. | Gómez-Suárez, A.; Dupuy, S.; Slawin, A. M. Z.; Nolan, S. P. Angew. Chem., Int. Ed. 2013, 52, 938–942. doi:10.1002/anie.201208234 |
| 24. | Oonishi, Y.; Gómez-Suárez, A.; Martin, A. R.; Nolan, S. P. Angew. Chem., Int. Ed. 2013, 52, 9767–9771. doi:10.1002/anie.201304182 |
| 24. | Oonishi, Y.; Gómez-Suárez, A.; Martin, A. R.; Nolan, S. P. Angew. Chem., Int. Ed. 2013, 52, 9767–9771. doi:10.1002/anie.201304182 |
| 1. | Hopkinson, M. N.; Richter, C.; Schedler, M.; Glorius, F. Nature 2014, 510, 485–496. doi:10.1038/nature13384 |
| 14. | Marion, N.; Díez-González, S.; de Frémont, P.; Noble, A. R.; Nolan, S. P. Angew. Chem., Int. Ed. 2006, 45, 3647–3650. doi:10.1002/anie.200600571 |
| 15. | Marion, N.; de Frémont, P.; Lemiere, G.; Stevens, E. D.; Fensterbank, L.; Malacria, M.; Nolan, S. P. Chem. Commun. 2006, 2048–2050. doi:10.1039/b602839j |
| 16. | Marion, N.; Carlqvist, P.; Gealageas, R.; de Frémont, P.; Maseras, F.; Nolan, S. P. Chem. – Eur. J. 2007, 13, 6437–6451. doi:10.1002/chem.200700134 |
| 17. | Marion, N.; Gealageas, R.; Nolan, S. P. Org. Lett. 2007, 9, 2653–2656. doi:10.1021/ol070843w |
| 18. | Correa, A.; Marion, N.; Fensterbank, L.; Malacria, M.; Nolan, S. P.; Cavallo, L. Angew. Chem., Int. Ed. 2008, 47, 718–721. doi:10.1002/anie.200703769 |
| 19. | Marion, N.; Ramón, R. S.; Nolan, S. P. J. Am. Chem. Soc. 2008, 131, 448–449. doi:10.1021/ja809403e |
| 20. | Ramón, R. S.; Marion, N.; Nolan, S. P. Chem. – Eur. J. 2009, 15, 8695–8697. doi:10.1002/chem.200901231 |
| 21. | Gaillard, S.; Bosson, J.; Ramón, R. S.; Nun, P.; Slawin, A. M. Z.; Nolan, S. P. Chem. – Eur. J. 2010, 16, 13729–13740. doi:10.1002/chem.201001688 |
| 22. | Nun, P.; Dupuy, S.; Gaillard, S.; Poater, A.; Cavallo, L.; Nolan, S. P. Catal. Sci. Technol. 2011, 1, 58–61. doi:10.1039/c0cy00055h |
| 23. | Gómez-Suárez, A.; Gasperini, D.; Vummaleti, S. V. C.; Poater, A.; Cavallo, L.; Nolan, S. P. ACS Catal. 2014, 4, 2701–2705. doi:10.1021/cs500806m |
| 26. | Kuram, M. R.; Bhanuchandra, M.; Sahoo, A. K. J. Org. Chem. 2010, 75, 2247–2258. doi:10.1021/jo1000143 |
| 10. | Díez-González, S.; Marion, N.; Nolan, S. P. Chem. Rev. 2009, 109, 3612–3676. doi:10.1021/cr900074m |
| 11. | Asay, M.; Jones, C.; Driess, M. Chem. Rev. 2011, 111, 354–396. doi:10.1021/cr100216y |
| 12. | Riener, K.; Haslinger, S.; Raba, A.; Högerl, M. P.; Cokoja, M.; Herrmann, W. A.; Kühn, F. E. Chem. Rev. 2014, 114, 5215–5272. doi:10.1021/cr4006439 |
| 13. | Fortman, G. C.; Nolan, S. P. Chem. Soc. Rev. 2011, 40, 5151–5169. doi:10.1039/c1cs15088j |
| 26. | Kuram, M. R.; Bhanuchandra, M.; Sahoo, A. K. J. Org. Chem. 2010, 75, 2247–2258. doi:10.1021/jo1000143 |
| 7. | Enders, D.; Niemeier, O.; Henseler, A. Chem. Rev. 2007, 107, 5606–5655. doi:10.1021/cr068372z |
| 8. | Marion, N.; Díez-González, S.; Nolan, S. P. Angew. Chem., Int. Ed. 2007, 46, 2988–3000. doi:10.1002/anie.200603380 |
| 9. | Grossmann, A.; Enders, D. Angew. Chem., Int. Ed. 2012, 51, 314–325. doi:10.1002/anie.201105415 |
| 31. | If these reactions are run under solvent-free conditions, the catalyst loading can be reduced to 0.5 mol %. For further details see reference [28]. |
| 2. | Nelson, D. J.; Nolan, S. P. Chem. Soc. Rev. 2013, 42, 6723–6753. doi:10.1039/c3cs60146c |
| 3. | Díez-González, S.; Nolan, S. P. Coord. Chem. Rev. 2007, 251, 874–883. doi:10.1016/j.ccr.2006.10.004 |
| 4. | Jacobsen, H.; Correa, A.; Poater, A.; Costabile, C.; Cavallo, L. Coord. Chem. Rev. 2009, 253, 687–703. doi:10.1016/j.ccr.2008.06.006 |
| 5. | Clavier, H.; Nolan, S. P. Chem. Commun. 2010, 46, 841–861. doi:10.1039/b922984a |
| 6. | Dröge, T.; Glorius, F. Angew. Chem., Int. Ed. 2010, 49, 6940–6952. doi:10.1002/anie.201001865 |
| 24. | Oonishi, Y.; Gómez-Suárez, A.; Martin, A. R.; Nolan, S. P. Angew. Chem., Int. Ed. 2013, 52, 9767–9771. doi:10.1002/anie.201304182 |
| 24. | Oonishi, Y.; Gómez-Suárez, A.; Martin, A. R.; Nolan, S. P. Angew. Chem., Int. Ed. 2013, 52, 9767–9771. doi:10.1002/anie.201304182 |
| 29. |
Dash, C.; Yousufuddin, M.; Cundari, T. R.; Dias, H. V. R. J. Am. Chem. Soc. 2013, 135, 15479–15488. doi:10.1021/ja406027x
The lack of reactivity observed for 2h could be due to its reaction with cationic gold, which above 50 °C can liberate N2 and generate a reactive nitrene species. |
| 33. | Gorske, B. C.; Mbofana, C. T.; Miller, S. J. Org. Lett. 2009, 11, 4318–4321. doi:10.1021/ol9016782 |
| 26. | Kuram, M. R.; Bhanuchandra, M.; Sahoo, A. K. J. Org. Chem. 2010, 75, 2247–2258. doi:10.1021/jo1000143 |
| 27. | Richard, M. E.; Fraccica, D. V.; Garcia, K. J.; Miller, E. J.; Ciccarelli, R. M.; Holahan, E. C.; Resh, V. L.; Shah, A.; Findeis, P. M.; Stockland, R. A., Jr. Beilstein J. Org. Chem. 2013, 9, 2002–2008. doi:10.3762/bjoc.9.235 |
| 30. | Gómez-Suárez, A.; Dupuy, S.; Slawin, A. M. Z.; Nolan, S. P. Angew. Chem., Int. Ed. 2013, 52, 938–942. doi:10.1002/anie.201208234 |
| 28. | Oonishi, Y.; Gómez-Suárez, A.; Martin, A. R.; Makida, Y.; Slawin, A. M. Z.; Nolan, S. P. Chem. – Eur. J. 2014, 20, 13507–13510. doi:10.1002/chem.201404630 |
| 24. | Oonishi, Y.; Gómez-Suárez, A.; Martin, A. R.; Nolan, S. P. Angew. Chem., Int. Ed. 2013, 52, 9767–9771. doi:10.1002/anie.201304182 |
| 25. | Gómez-Suárez, A.; Oonishi, Y.; Martin, A. R.; Vummaleti, S. V. C.; Nelson, D. J.; Cordes, D. B.; Slawin, A. M. Z.; Cavallo, L.; Nolan, S. P.; Poater, A. Chem. – Eur. J. 2016, 22, 1125–1132. doi:10.1002/chem.201503097 |
| 24. | Oonishi, Y.; Gómez-Suárez, A.; Martin, A. R.; Nolan, S. P. Angew. Chem., Int. Ed. 2013, 52, 9767–9771. doi:10.1002/anie.201304182 |
| 24. | Oonishi, Y.; Gómez-Suárez, A.; Martin, A. R.; Nolan, S. P. Angew. Chem., Int. Ed. 2013, 52, 9767–9771. doi:10.1002/anie.201304182 |
| 25. | Gómez-Suárez, A.; Oonishi, Y.; Martin, A. R.; Vummaleti, S. V. C.; Nelson, D. J.; Cordes, D. B.; Slawin, A. M. Z.; Cavallo, L.; Nolan, S. P.; Poater, A. Chem. – Eur. J. 2016, 22, 1125–1132. doi:10.1002/chem.201503097 |
| 28. | Oonishi, Y.; Gómez-Suárez, A.; Martin, A. R.; Makida, Y.; Slawin, A. M. Z.; Nolan, S. P. Chem. – Eur. J. 2014, 20, 13507–13510. doi:10.1002/chem.201404630 |
| 32. |
Arcadi, A.; Blesi, F.; Cacchi, S.; Fabrizi, G.; Goggiamani, A.; Marinelli, F. Org. Biomol. Chem. 2012, 10, 9700–9708. doi:10.1039/c2ob26763b
2 Equiv of the alkyne were necessary. See for reactions of 1n under gold catalysis. |
© 2016 Gómez-Suárez et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)