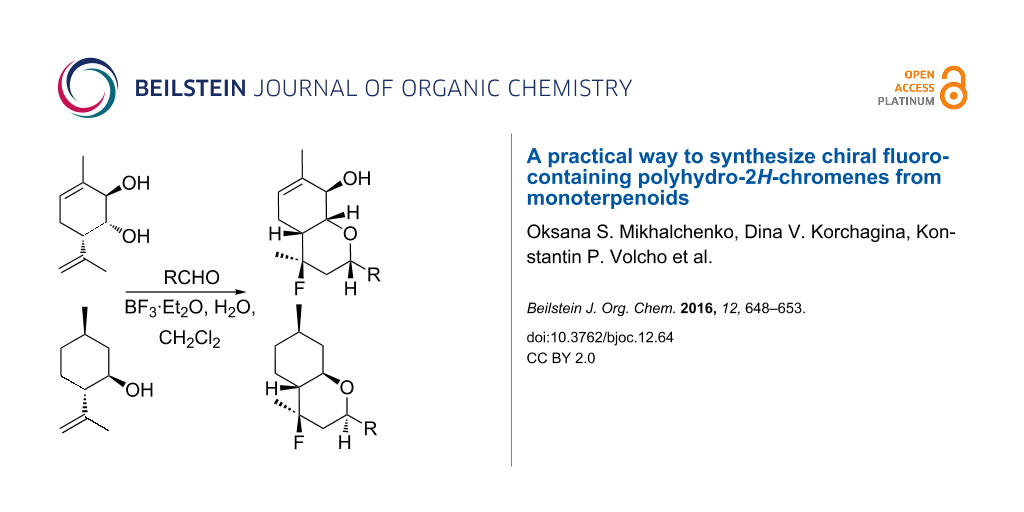
Abstract
Conditions enabling the single-step preparative synthesis of chiral 4-fluoropolyhydro-2H-chromenes in good yields through a reaction between monoterpenoid alcohols with para-menthane skeleton and aldehydes were developed for the first time. The BF3·Et2O/H2O system is used both as a catalyst and as a fluorine source. The reaction can involve aliphatic aldehydes as well as aromatic aldehydes containing various acceptor and donor substituents. 4-Hydroxyhexahydro-2H-chromenes were demonstrated to be capable of converting to 4-fluorohexahydro-2H-chromenes under the developed conditions, the reaction occurs with inversion of configuration.

Graphical Abstract
Introduction
Recently, we have found that a reaction between para-mentha-6,8-dien-2,3-diol (1) and aromatic aldehydes in the presence of K10 montmorillonite clay forms chiral heterocyclic compounds with the hexahydro-2H-chromene scaffold 2 (Scheme 1) [1-4]. Products of these reactions are of interest as many of them exhibit a significant analgesic activity in vivo [2-4].
![[1860-5397-12-64-i1]](/bjoc/content/inline/1860-5397-12-64-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Reaction between monoterpenoid 1 and aromatic aldehydes in the presence of K10 montmorillonite clay.
Scheme 1: Reaction between monoterpenoid 1 and aromatic aldehydes in the presence of K10 montmorillonite clay....
In terms of structure–activity relationship studies of hexahydro-2H-chromenes and similar compounds it is important to replace the hydroxy group at the C(5) position by another functional group. Thus, the approaches for synthesis of thio- [5] and nitrogen [6] containing analogous with reasonable yields (50−80%) by using a third component (corresponding thiols or acetonitrile) were elaborated. Of particular interest is the introduction of a fluorine atom into the molecule of the biologically active compound. The introduction of a highly electronegative centre can lead to an increase in stability and changes in lipophilicity. Furthermore, it alters the patterns of reactivity of the C–F versus the C–H or the C–OH bond [7-9]. As a result it can have a significant impact on the biological activity of a compound.
To create halogenated tetrahydropyranyl rings the Prins reactions between homoallylic alcohols and aldehydes catalyzed by appropriate halogen-containing Lewis acids or ionic liquids are usually used (Scheme 2) [10-16]. However, only a few examples of reactions for introducing a fluorine atom via the halo-Prins cyclization have been reported to date [15-21]. In these reactions, BF3·Et2O acts both as a catalyst and as a fluorine source. At the same time, as a rule, reactions in the presence of BF3·Et2O involved relatively simple homoallylic alcohols, such as but-3-en-1-ol (3) and its analogues, as substrates (Scheme 2). If more complex substrates, such as isopulegol (4) or geraniol (5), were involved in transformations, the formation of non-fluorinated heterocyclic products was observed (Scheme 2) [22,23]. The aim of the present study was to find conditions for the synthesis of fluorinated chiral hexahydrochromenes based on monoterpenoids.
![[1860-5397-12-64-i2]](/bjoc/content/inline/1860-5397-12-64-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: The Prins reaction between homoallylic alcohols and aldehydes.
Scheme 2: The Prins reaction between homoallylic alcohols and aldehydes.
Results and Discussion
The interaction of para-mentha-6,8-dien-2,3-diol (1) with 3,4,5-trimethoxybenzaldehyde (6a) in the presence of BF3·Et2O was chosen as a model reaction.
The study was started with the investigation of the effect of the reaction conditions and the reactant ratio on the yield of a fluorinated product (Table 1). The addition of an equimolar amount of BF3·Et2O to a mixture of monoterpenoid 1 and aldehyde 6a at room temperature (Table 1, entry 1) resulted, after 1 h, in the formation of a reaction mixture containing compounds 2a and 7a as the major low molecular weight products and only minor amounts of target compound 8a. It is important that the reaction is accompanied by a significant resinification. The addition of water to the initial reactants enabled the reduction of the amount of undesirable products 2a and 7a and had a minor effect on the amount of compound 8a. This indicates a significant contribution of side processes that are likely related to the formation of high molecular weight products. Lowering the reaction temperature to 2 °C made it possible to reduce a contribution of resinification processes that led to an increase in the amount of product 8a to 31%, but with incomplete conversion of monoterpenoid 1. When the reaction time was increased to 8 h, the conversion increased, but was still not quantitative. The complete conversion of compound 1 was achieved by using a 1.5-fold excess of BF3·Et2O, and the use of a slight excess of aldehyde led to a marked increase in the amount of fluorinated product 8a (61%). These conditions (Table 1, entry 6) were chosen as suitable for further research.
Table 1: The variation of reaction conditions.
![[Graphic 1]](/bjoc/content/inline/1860-5397-12-64-i6.svg?max-width=637&scale=1.0)
|
|||||||
| Entry | Reagent ratio 1:6:BF3·Et2O:H2O | Temp (°C) | Time (h) | Conv. 1 (%) | Yield (%)a | ||
|---|---|---|---|---|---|---|---|
| 2a | 7ab | 8a | |||||
| 1 | 1:1:1:0 | rt | 1 | 100 | 12 | 13 | 5 |
| 2 | 1:1:1:7.4 | rt | 1 | 85 | 0 | 4 | 6 |
| 3 | 1:1:1:7.4 | 2 | 1 | 78 | 8 | 16 | 31 |
| 4 | 1:1:1:7.4 | 2 | 8 | 88 | 8 | 26 | 50 |
| 5 | 1:1:1.5:7.4 | 2 | 8 | 100 | 10 | 20 | 48 |
| 6 | 1:1.2:1.5:7.4 | 2 | 8 | 100 | 11 | 12 | 61 |
aThe yields of products obtained from the GC–MS chromatograms using the standard (2,5-hexanediol) and the correction factors. bOlefin 7a is a mixture of double bond position isomers, and amount of 7a shown in the table corresponds to the total yield of the olefin isomers. The structure of 7a shown on the scheme is the major olefin isomer.
Preparative production under the chosen conditions provided 72% of 8a by GC–MS and after separation by column chromatography the target fluorinated product 8a was isolated in 69% yield; the yield of compound 2a was 7% (Table 2). Thus, we found for the first time the conditions enabling production of chiral fluoro-containing heterocyclic compounds from a monoterpenoid using the halo-Prins cyclisation.
It should also be noted that preparative isolation of compounds of type 7a is complicated by the presence of their double bond position isomers in the reaction mixtures. In this study, we did not seek to isolate the individual byproducts.
Compounds 2a and 8a produced by a reaction of diol 1 with aldehyde 6a are formed as a mixture of diastereomers differing in the position of substituents at the C(5) carbon. The (S)-isomer predominates in the case of compound 2a, while the (R)-isomer is the major one in the case of compound 8a (Table 2). The ratio of the major and minor diastereomeric products of types 2 and 8 was determined from 1H NMR spectra by integration of the Ha-3 signal (Table 2). The methyl group at the C(5) carbon atom is axial in the (S)-isomers and equatorial in the (R)-isomers. Consequently, in the (R)-isomers the axial OH or F group causes a paramagnetic shift to a weak field of the Ha-3 signal due to the 1,3-diaxial interaction in comparison with the corresponding signal in the (S)-isomers.
Table 2: Yields of products 8a–m and 2a–m obtained in the reaction of diol 1 with aldehydes 6a–m.
![[Graphic 2]](/bjoc/content/inline/1860-5397-12-64-i7.svg?max-width=637&scale=1.0)
|
||||
| 6 | R | Reaction time (h) | Yielda (R:S) | |
|---|---|---|---|---|
| 2 | 8 | |||
| 6a | (3,4,5-MeO)C6H2 | 8 | 7% (2:3) | 69% (4:1) |
| 6b | C6H5 | 8 | 24% (1:3) | 55% (7:1) |
| 6c | 4-MeOC6H4 | 8 | 24% (1:4) | 34% (4:1) |
| 6d | (3,4-MeO)C6H3 | 8 | 20% (1:5) | 35% (3:1) |
| 6e | (2,4,6-MeO)C6H2 | 8 | 35% (1:1) | 42% (10:1) |
| 6fb | (2,4,5-MeO)C6H2 | 8 | 8% (1:3) | 20% (7:1) |
| 6g | 4-NO2C6H4 | 72 | 17% (1:1) | 53% (12:1) |
| 6h | 4-FC6H4 | 72 | 17% (1:2) | 47% (6:1) |
| 6i | 4-ClC6H4 | 72 | 17% (1:3) | 58% (6:1) |
| 6j | 4-BrC6H4 | 72 | 13% (1:2) | 60% (10:1) |
| 6k | cyclo-C6H11 | 8 | 27% (1:3) | 61% (3:1) |
| 6l | CH3CH=CH | 8 | 20% (1:2) | 57% (3:1) |
| 6m | 4-OH-3-MeO-C6H3 | 8 | 35% (1:3) | 60% (3:1) |
aReaction conditions: diol 1 (2.4 mmol), aldehyde (2.9 mmol), BF3·Et2O (3.6 mmol) and H2O (17.8 mmol). bProduct 9f with epoxychromene framework was also isolated in 14% yield.
The next stage of our research was to study the effect of substituents at the aldehyde aromatic ring on the yield and the ratio of the reaction products. Thus, a reaction of monoterpenoid 1 with benzaldehyde (6b) under previously chosen conditions provided fluorinated product 8b in 55% yield. In addition, compound 2b was isolated from the reaction mixture in 24% yield (Table 2).
Introduction of one or two methoxy groups into the aldehyde aromatic ring reduced the yield of fluorinated products 8c and 8d to ca. 35%, without affecting the yields of compounds 2.
To find out how much this reaction is sensitive to steric hindrances, we studied a reaction of monoterpenoid 1 with 2,4,6-trimethoxybenzaldehyde (6e), in which both ortho-positions are occupied. Previously, using the aldehyde as a reactant in a reaction with diol 1 catalyzed by K10 montmorillonite clay led to a sharp decrease in the yield of product 2e [2]. In our case, the yields of products containing a fluorine atom (8e) and a hydroxy group (2e) were 42% and 35%, respectively (Table 2).
Interestingly, in the case of 2,4,5-trimethoxybenzaldehyde (6f), containing one of the three methoxy groups at the ortho-position, the overall yield of reaction products unexpectedly decreased, and the yield of fluorinated product 8f was only 20%. In addition to the expected products 2f and 8f, tricyclic compound 9f with an epoxychromene scaffold was isolated from the reaction mixture (Scheme 3), whose formation was previously observed only when using K10 clay [24].
![[1860-5397-12-64-i3]](/bjoc/content/inline/1860-5397-12-64-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Reaction of compound 1 with aldehyde 6f.
Scheme 3: Reaction of compound 1 with aldehyde 6f.
The use of 4-nitrobenzaldehyde (6g) required the increase of the reaction time to 72 h for achieving complete conversion of monoterpenoid 1, which is obviously due to the electron-withdrawing effect of the nitro group; the yield of compound 8g was 53%. Reactions with 4-halogen-substituted aldehydes 6h–j proceeded similarly and provided target products 8h–j with yields of 47−60%.
Aliphatic aldehydes 6k and 6l were comparable in terms of reactivity with benzaldehyde (6b) and its methoxyderivatives; complete conversion was achieved after 8 h. It should be noted that the presence of a double bond in aldehyde 6l had no significant effect on the reaction; yields of fluorinated products 8k and 8l were about 60%.
A reaction of diol 1 with 3-methoxy-4-hydroxybenzaldehyde (6m) in the presence of BF3·Et2O and water for 8 h also produces fluoro-containing hexahydrochromenes 8m in 60% yield despite the presence of a phenolic group in the aldehyde aromatic ring.
It is known that the presence of water in the reaction medium containing BF3·Et2O can result in the formation of BF3·H2O, since the interaction of BF3 with H2O is relatively stronger than that with Et2O [25,26]. BF3·H2O, in turn, is a strong Brønsted acid and may be presented as H+(BF3·OH)− [27,28]. Based on these data, it may be supposed that in the case of a 5-fold excess of water relative to BF3·Et2O, the reaction medium may contain both BF3·H2O and products of the partial hydrolysis of BF3·Et2O that may act both as catalysts and as fluorine sources.
Presumably, the reaction starts with the Prins cyclisation resulting in the formation of cation 10. There are then several mechanistic pathways, some or all of them may be in operation: a) cation 10 may undergo stereoselective trap by [F−] to form fluoride epimers 8; b) cation 10 may undergo stereoselective trap by H2O to form alcohol epimers 2; these may then undergo a stereospecific SN2 reaction to form fluoride epimers 8; c) the fluorination and/or hydroxylation of cation 10, forming fluoride epimers 8 and alcohol epimers 2, respectively, may be reversible. This has several effects: firstly, reversible hydroxylation means that alcohol epimers 2 may convert to fluoride epimers 8 via cation 10 (pathway (a)); secondly, reversible fluorination and/or hydroxylation means that the diastereoselectivity of formation and/or 2 may be governed by product stability and not inherent stereoselectivity of the trapping of cation 10 (Scheme 4).
![[1860-5397-12-64-i4]](/bjoc/content/inline/1860-5397-12-64-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: A possible mechanism of the compound 2 and 8 formation.
Scheme 4: A possible mechanism of the compound 2 and 8 formation.
Re-subjection of alcohol 2b epimers ((R):(S) = 1:4) to the reaction conditions gave fluoride epimers 8b ((R):(S) = 8:1), albeit along with a number of side products and resinification processes. This result establishes that the alcohol epimers 2b can convert to fluoride epimers 8b under the reaction conditions but the apparent inversion of epimeric ratio (2b (R):(S) = 1:4, 8b (R):(S) = 8:1) is not necessarily a specific evidence for the operation of pathway (b): the alcohol epimers 2b may rect via different pathways and the amplification of the epimeric ratio would be consistent with a stereoselective aspect to the conversion. Further experimentation is therefore needed to elucidate the precise mechanistic details of this reaction.
Isopulegol (4), like diol 1, has a hydroxy group and an isopropenyl group at the 1- and 2-positions; these two groups are trans-located in compound 4 (while cis-located in monoterpenoid 1).
When a reaction of isopulegol (4) with 3,4,5-trimethoxybenzaldehyde (6a) was conducted under the conditions previously optimised for diol 1, fluorinated product 11 with the chromene scaffold and its analogue 12 with a hydroxy group were obtained in 76% and 15% yield, respectively (Scheme 5).
![[1860-5397-12-64-i5]](/bjoc/content/inline/1860-5397-12-64-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Reaction of isopulegol (4) with aldehyde 6a in the presence BF3·Et2O.
Scheme 5: Reaction of isopulegol (4) with aldehyde 6a in the presence BF3·Et2O.
Conclusion
We developed for the first time conditions enabling the single-step preparative production of chiral 4-fluoropolyhydro-2H-chromenes via a reaction between monoterpenoid alcohols with the para-menthane skeleton and aldehydes. The BF3·Et2O/H2O system is used both as a catalyst and as a fluorine source. The reaction can involve saturated and unsaturated aliphatic aldehydes as well as aromatic aldehydes containing various acceptor and donor substituents, including a phenolic hydroxy group. The yield of target fluorinated products usually ranges from 53% to 76%; byproducts having a hydroxy group instead of the fluorine atom are formed in smaller quantities. The possibility of a transformation of 4-hydroxyhexahydro-2H-chromenes to 4-fluorohexahydro-2H-chromenes with inversion of the configuration was demonstrated.
Supporting Information
| Supporting Information File 1: Detailed experimental procedures, compound characterization data, and copies of NMR spectra. | ||
| Format: PDF | Size: 4.0 MB | Download |
References
-
Il'ina, I. V.; Volcho, K. P.; Mikhalchenko, O. S.; Korchagina, D. V.; Salakhutdinov, N. F. Helv. Chim. Acta 2011, 94, 502–513. doi:10.1002/hlca.201000269
Return to citation in text: [1] -
Mikhalchenko, O.; Il’ina, I.; Pavlova, A.; Morozova, E.; Korchagina, D.; Tolstikova, T.; Pokushalov, E.; Volcho, K.; Salahutdinov, N. Med. Chem. Res. 2013, 22, 3026–3034. doi:10.1007/s00044-012-0310-9
Return to citation in text: [1] [2] [3] -
Il’ina, I.; Mikhalchenko, O.; Pavlova, A.; Korchagina, D.; Tolstikova, T.; Volcho, K.; Salakhutdinov, N.; Pokushalov, E. Med. Chem. Res. 2014, 23, 5063–5073. doi:10.1007/s00044-014-1071-4
Return to citation in text: [1] [2] -
Pavlova, A.; Mikhalchenko, O.; Rogachev, A.; Il’ina, I.; Korchagina, D.; Gatilov, Y.; Tolstikova, T.; Volcho, K.; Salakhutdinov, N. Med. Chem. Res. 2015, 24, 3821–3830. doi:10.1007/s00044-015-1426-5
Return to citation in text: [1] [2] -
Sarmah, B.; Baishya, G.; Baruah, R. K. Eur. J. Org. Chem. 2014, 7561–7565. doi:10.1002/ejoc.201403080
Return to citation in text: [1] -
Sarmah, B.; Baishya, G.; Baruah, R. K. RSC Adv. 2014, 4, 22387–22397. doi:10.1039/c4ra02124j
Return to citation in text: [1] -
Wang, J.; Sánchez-Roselló, M.; Aceña, J. L.; del Pozo, C.; Sorochinsky, A. E.; Fustero, S.; Soloshonok, V. A.; Liu, H. Chem. Rev. 2014, 114, 2432–2506. doi:10.1021/cr4002879
Return to citation in text: [1] -
Shibata, N.; Ishimaru, T.; Nakamura, S.; Toru, T. J. Fluorine Chem. 2007, 128, 469–483. doi:10.1016/j.jfluchem.2006.12.014
Return to citation in text: [1] -
Wender, P. A.; Billingsley, K. L. Synthesis 2013, 45, 1815–1824. doi:10.1055/s-0033-1338860
Return to citation in text: [1] -
Coppi, L.; Ricci, A.; Taddei, M. J. Org. Chem. 1988, 53, 911–913. doi:10.1021/jo00239a053
Return to citation in text: [1] -
Yang, J.; Viswanathan, G. S.; Li, C.-J. Tetrahedron Lett. 1999, 40, 1627–1630. doi:10.1016/S0040-4039(99)00027-1
Return to citation in text: [1] -
Biermann, U.; Lützen, A.; Metzger, J. O. Eur. J. Org. Chem. 2006, 2631–2637. doi:10.1002/ejoc.200500701
Return to citation in text: [1] -
Yang, X.-F.; Mague, J. T.; Li, C.-J. J. Org. Chem. 2001, 66, 739–747. doi:10.1021/jo001136i
Return to citation in text: [1] -
Dobbs, A. P.; Pivnevi, L.; Penny, M. J.; Martinović, S.; Iley, J. N.; Stephenson, P. T. Chem. Commun. 2006, 3134–3136. doi:10.1039/b606121d
Return to citation in text: [1] -
Kishi, Y.; Nagura, H.; Inagi, S.; Fuchigami, T. Chem. Commun. 2008, 3876–3878. doi:10.1039/b806389c
Return to citation in text: [1] [2] -
Kishi, Y.; Inagi, S.; Fuchigami, T. Eur. J. Org. Chem. 2009, 103–109. doi:10.1002/ejoc.200800872
Return to citation in text: [1] [2] -
Cresswell, A. J.; Davies, S. G.; Roberts, P. M.; Thomson, J. E. Chem. Rev. 2015, 2, 566–611. doi:10.1021/cr5001805
Return to citation in text: [1] -
Bondalapati, S.; Reddy, U. C.; Kundu, D. S.; Saikia, A. K. J. Fluorine Chem. 2010, 131, 320–324. doi:10.1016/j.jfluchem.2009.11.002
Return to citation in text: [1] -
Kataoka, K.; Ode, Y.; Matsumoto, M.; Nokami, J. Tetrahedron 2006, 62, 2471–2483. doi:10.1016/j.tet.2005.12.054
Return to citation in text: [1] -
Launay, G. G.; Slawin, A. M. Z.; O’Hagan, D. Beilstein J. Org. Chem. 2010, 6, No. 41. doi:10.3762/bjoc.6.41
Return to citation in text: [1] -
Bahnck, K. B.; Rychnovsky, S. D. J. Am. Chem. Soc. 2008, 130, 13177–13181. doi:10.1021/ja805187p
Return to citation in text: [1] -
Bondalapati, S.; Reddy, U. C.; Saha, P.; Saikia, A. K. Org. Biomol. Chem. 2011, 9, 3428–3438. doi:10.1039/c1ob00033k
Return to citation in text: [1] -
Saha, P.; Gogoi, P.; Saikia, A. K. Org. Biomol. Chem. 2011, 9, 4626–4634. doi:10.1039/c1ob05172e
Return to citation in text: [1] -
Mikhalchenko, O. S.; Korchagina, D. V.; Volcho, K. P.; Salakhutdinov, N. F. Helv. Chim. Acta 2014, 97, 1406–1421. doi:10.1002/hlca.201300461
Return to citation in text: [1] -
Huang, J.-W.; Shi, M. Tetrahedron Lett. 2003, 44, 9343–9347. doi:10.1016/j.tetlet.2003.10.086
Return to citation in text: [1] -
Li, Y.; Xiong, Y.; Li, X.; Ling, X.; Huang, R.; Zhang, X.; Yang, J. Green Chem. 2014, 16, 2976–2981. doi:10.1039/c4gc00005f
Return to citation in text: [1] -
Fǎrcaşiu, D.; Ghenciu, A. J. Catal. 1992, 134, 126–133. doi:10.1016/0021-9517(92)90216-5
Return to citation in text: [1] -
Prakash, G. K. S.; Panja, C.; Shakhmin, A.; Shah, E.; Mathew, T.; Olah, G. A. J. Org. Chem. 2009, 74, 8659–8668. doi:10.1021/jo901668j
Return to citation in text: [1]
| 1. | Il'ina, I. V.; Volcho, K. P.; Mikhalchenko, O. S.; Korchagina, D. V.; Salakhutdinov, N. F. Helv. Chim. Acta 2011, 94, 502–513. doi:10.1002/hlca.201000269 |
| 2. | Mikhalchenko, O.; Il’ina, I.; Pavlova, A.; Morozova, E.; Korchagina, D.; Tolstikova, T.; Pokushalov, E.; Volcho, K.; Salahutdinov, N. Med. Chem. Res. 2013, 22, 3026–3034. doi:10.1007/s00044-012-0310-9 |
| 3. | Il’ina, I.; Mikhalchenko, O.; Pavlova, A.; Korchagina, D.; Tolstikova, T.; Volcho, K.; Salakhutdinov, N.; Pokushalov, E. Med. Chem. Res. 2014, 23, 5063–5073. doi:10.1007/s00044-014-1071-4 |
| 4. | Pavlova, A.; Mikhalchenko, O.; Rogachev, A.; Il’ina, I.; Korchagina, D.; Gatilov, Y.; Tolstikova, T.; Volcho, K.; Salakhutdinov, N. Med. Chem. Res. 2015, 24, 3821–3830. doi:10.1007/s00044-015-1426-5 |
| 7. | Wang, J.; Sánchez-Roselló, M.; Aceña, J. L.; del Pozo, C.; Sorochinsky, A. E.; Fustero, S.; Soloshonok, V. A.; Liu, H. Chem. Rev. 2014, 114, 2432–2506. doi:10.1021/cr4002879 |
| 8. | Shibata, N.; Ishimaru, T.; Nakamura, S.; Toru, T. J. Fluorine Chem. 2007, 128, 469–483. doi:10.1016/j.jfluchem.2006.12.014 |
| 9. | Wender, P. A.; Billingsley, K. L. Synthesis 2013, 45, 1815–1824. doi:10.1055/s-0033-1338860 |
| 6. | Sarmah, B.; Baishya, G.; Baruah, R. K. RSC Adv. 2014, 4, 22387–22397. doi:10.1039/c4ra02124j |
| 5. | Sarmah, B.; Baishya, G.; Baruah, R. K. Eur. J. Org. Chem. 2014, 7561–7565. doi:10.1002/ejoc.201403080 |
| 2. | Mikhalchenko, O.; Il’ina, I.; Pavlova, A.; Morozova, E.; Korchagina, D.; Tolstikova, T.; Pokushalov, E.; Volcho, K.; Salahutdinov, N. Med. Chem. Res. 2013, 22, 3026–3034. doi:10.1007/s00044-012-0310-9 |
| 3. | Il’ina, I.; Mikhalchenko, O.; Pavlova, A.; Korchagina, D.; Tolstikova, T.; Volcho, K.; Salakhutdinov, N.; Pokushalov, E. Med. Chem. Res. 2014, 23, 5063–5073. doi:10.1007/s00044-014-1071-4 |
| 4. | Pavlova, A.; Mikhalchenko, O.; Rogachev, A.; Il’ina, I.; Korchagina, D.; Gatilov, Y.; Tolstikova, T.; Volcho, K.; Salakhutdinov, N. Med. Chem. Res. 2015, 24, 3821–3830. doi:10.1007/s00044-015-1426-5 |
| 2. | Mikhalchenko, O.; Il’ina, I.; Pavlova, A.; Morozova, E.; Korchagina, D.; Tolstikova, T.; Pokushalov, E.; Volcho, K.; Salahutdinov, N. Med. Chem. Res. 2013, 22, 3026–3034. doi:10.1007/s00044-012-0310-9 |
| 25. | Huang, J.-W.; Shi, M. Tetrahedron Lett. 2003, 44, 9343–9347. doi:10.1016/j.tetlet.2003.10.086 |
| 26. | Li, Y.; Xiong, Y.; Li, X.; Ling, X.; Huang, R.; Zhang, X.; Yang, J. Green Chem. 2014, 16, 2976–2981. doi:10.1039/c4gc00005f |
| 22. | Bondalapati, S.; Reddy, U. C.; Saha, P.; Saikia, A. K. Org. Biomol. Chem. 2011, 9, 3428–3438. doi:10.1039/c1ob00033k |
| 23. | Saha, P.; Gogoi, P.; Saikia, A. K. Org. Biomol. Chem. 2011, 9, 4626–4634. doi:10.1039/c1ob05172e |
| 27. | Fǎrcaşiu, D.; Ghenciu, A. J. Catal. 1992, 134, 126–133. doi:10.1016/0021-9517(92)90216-5 |
| 28. | Prakash, G. K. S.; Panja, C.; Shakhmin, A.; Shah, E.; Mathew, T.; Olah, G. A. J. Org. Chem. 2009, 74, 8659–8668. doi:10.1021/jo901668j |
| 15. | Kishi, Y.; Nagura, H.; Inagi, S.; Fuchigami, T. Chem. Commun. 2008, 3876–3878. doi:10.1039/b806389c |
| 16. | Kishi, Y.; Inagi, S.; Fuchigami, T. Eur. J. Org. Chem. 2009, 103–109. doi:10.1002/ejoc.200800872 |
| 17. | Cresswell, A. J.; Davies, S. G.; Roberts, P. M.; Thomson, J. E. Chem. Rev. 2015, 2, 566–611. doi:10.1021/cr5001805 |
| 18. | Bondalapati, S.; Reddy, U. C.; Kundu, D. S.; Saikia, A. K. J. Fluorine Chem. 2010, 131, 320–324. doi:10.1016/j.jfluchem.2009.11.002 |
| 19. | Kataoka, K.; Ode, Y.; Matsumoto, M.; Nokami, J. Tetrahedron 2006, 62, 2471–2483. doi:10.1016/j.tet.2005.12.054 |
| 20. | Launay, G. G.; Slawin, A. M. Z.; O’Hagan, D. Beilstein J. Org. Chem. 2010, 6, No. 41. doi:10.3762/bjoc.6.41 |
| 21. | Bahnck, K. B.; Rychnovsky, S. D. J. Am. Chem. Soc. 2008, 130, 13177–13181. doi:10.1021/ja805187p |
| 10. | Coppi, L.; Ricci, A.; Taddei, M. J. Org. Chem. 1988, 53, 911–913. doi:10.1021/jo00239a053 |
| 11. | Yang, J.; Viswanathan, G. S.; Li, C.-J. Tetrahedron Lett. 1999, 40, 1627–1630. doi:10.1016/S0040-4039(99)00027-1 |
| 12. | Biermann, U.; Lützen, A.; Metzger, J. O. Eur. J. Org. Chem. 2006, 2631–2637. doi:10.1002/ejoc.200500701 |
| 13. | Yang, X.-F.; Mague, J. T.; Li, C.-J. J. Org. Chem. 2001, 66, 739–747. doi:10.1021/jo001136i |
| 14. | Dobbs, A. P.; Pivnevi, L.; Penny, M. J.; Martinović, S.; Iley, J. N.; Stephenson, P. T. Chem. Commun. 2006, 3134–3136. doi:10.1039/b606121d |
| 15. | Kishi, Y.; Nagura, H.; Inagi, S.; Fuchigami, T. Chem. Commun. 2008, 3876–3878. doi:10.1039/b806389c |
| 16. | Kishi, Y.; Inagi, S.; Fuchigami, T. Eur. J. Org. Chem. 2009, 103–109. doi:10.1002/ejoc.200800872 |
| 24. | Mikhalchenko, O. S.; Korchagina, D. V.; Volcho, K. P.; Salakhutdinov, N. F. Helv. Chim. Acta 2014, 97, 1406–1421. doi:10.1002/hlca.201300461 |
© 2016 Mikhalchenko et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)