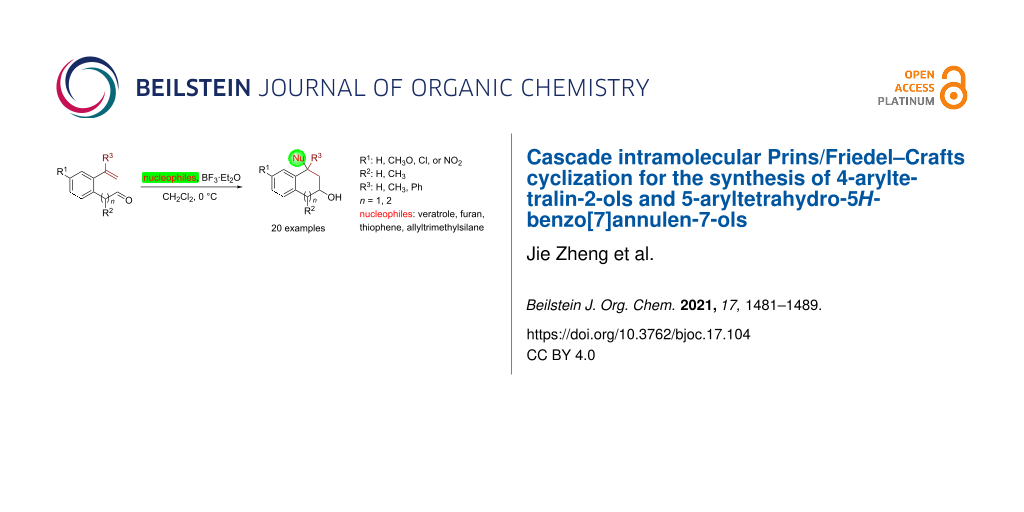
Abstract
The treatment of 2-(2-vinylphenyl)acetaldehydes or 3-(2-vinylphenyl)propanals with BF3·Et2O results in an intramolecular Prins reaction affording intermediary benzyl carbenium ions, which are then trapped by a variety of electron-rich aromatics via Friedel–Crafts alkylation. This cascade Prins/Friedel–Crafts cyclization protocol paves an expedient path to medicinally useful 4-aryltetralin-2-ol and 5-aryltetrahydro-5H-benzo[7]annulen-7-ol derivatives.

Graphical Abstract
Introduction
2,4-Disubstituted tetralins (Figure 1, 1), especially 2-functionalized tetralins are privileged building blocks for medicinal chemistry applications which are known to exhibit a wide spectrum of biological activities [1-3]. Some representative compounds comprising this skeleton are illustrated in Figure 1. Cycloolivil (Figure 1, 2) [4], which is isolated from the stem bark of Olea europaea, has been recognized as inhibitor of cyclic AMP dependent phosphodiesterase, can act as a Ca2+ antagonist, and exhibits promising anti-oxidant properties. 4-Phenyl-2-propionamidotetralin (4-P-PDOT, 3, Figure 1) [5] is a melatonin MT2 selective antagonist that can be used to map melatonin receptor subtypes in tissue and serves as a chemical biology tool to identify sub-type selective analogues with therapeutic potential. In addition, trans-4-phenyl-N,N-dimethyl-2-aminotetralin (trans-H2-PAT, 4, Figure 1) [6] has been determined to modulate tyrosine hydroxylase activity and dopamine synthesis in rodent forebrain and is also a ligand binding to histamine H1 receptors, and thus is a potentially useful therapeutic for psychoses, addiction, and other neuropsychiatric disorders.
![[1860-5397-17-104-1]](/bjoc/content/figures/1860-5397-17-104-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Parent structure of 2,4-disubstituted tetralins (1) and selected medicinally useful derivatives 2–4.
Figure 1: Parent structure of 2,4-disubstituted tetralins (1) and selected medicinally useful derivatives 2–4....
Although 4-substitituted tetralin-2-ols and derivatives with significant pharmaceutical activities have been identified, only a limited number of synthetic methods is documented in the literature (Scheme 1) [7-9]. Moreover, these methods generally require multiple steps, proceed in low overall yields, and have a limited ability for structural modifications to prepare analogues with new substitution patterns for enhancing activities. Consequently, it is highly desirable to develop new synthetic methods that provide efficient access to 2,4-disubstituted tetralin compounds and thus facilitate their biological investigations.
![[1860-5397-17-104-i1]](/bjoc/content/inline/1860-5397-17-104-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Reported strategies for the synthesis of tetralin-2-ol ring systems.
Scheme 1: Reported strategies for the synthesis of tetralin-2-ol ring systems.
The cascade Prins/Friedel–Crafts reaction to form multiple chemical bonds in one operation has emerged as an atom-economic and straightforward strategy for the construction of oxygen-containing heterocycles [10-14]. For example, Nagumo and coworkers have developed a Prins/Friedel–Crafts cyclization of homocinnamyl alcohols with aromatic aldehydes under the action of BF3·Et2O affording 2H-indeno[1,2-b]furan derivatives [15]. Likewise, Hinkle and coworkers reported in 2017 a three-step domino alkynyl-Prins cyclization, Friedel–Crafts alkenylation, and dehydration/aromatization reaction between 1-aryl-3-hexyne-2,6-diol derivatives and aldehydes, that led to the formation of 1,4-dihydro-2H-benzo[f]isochromenes [16].
The Prins reaction-induced cyclization, inter alia, became a versatile tool for the assembly of complex molecules from relatively simple and inexpensive materials/reagents in a single operation. The reaction continues to be an interesting and profitable field of research with high impact on synthetic organic chemistry [17,18]. Many of the existing protocols rely on an acid-promoted condensation of a homoallylic alcohol and an aldehyde to give an oxocarbenium ion, which is then reacted with an olefinic/alkynic bond generating a carbocation that undergoes a Friedel–Crafts reaction. Given the potential value of tetralin-2-ol scaffolds to drug research programs, we decided to develop a novel Prins/Friedel–Crafts cyclization strategy for the synthesis of 4-aryl-2-hydroxytetralins starting from 2-(2-vinylphenyl)acetaldehydes (Scheme 2). In this protocol, we envisioned that the aldehyde 5 would give rise to an oxocarbenium ion species 6 upon treatment with a Lewis acid. The intermediate 6 then would undergo a Prins-type intramolecular cyclization with the olefinic bond to produce a stable benzyl carbocation 7, that may be trapped through a Friedel–Crafts alkylation with an aromatic substrate or through the reaction with an external nucleophile to afford the target product 8.
![[1860-5397-17-104-i2]](/bjoc/content/inline/1860-5397-17-104-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Designed cascade reactions to 4-substituted tetralin-2-ols.
Scheme 2: Designed cascade reactions to 4-substituted tetralin-2-ols.
Results and Discussion
Our research began with the preparation of 2-(2-vinylphenyl)acetaldehydes (13) required as substrates for the Prins/Friedel–Crafts cyclization reactions. Commonly, these aromatic alkenyl aldehydes were previously prepared via a three step process as exemplified by 13a shown in Scheme 3 consisting of the following steps: (i) Wittig reaction of 2-bromobenzaldehyde with methyltriphenylphosphonium iodide ylide, (ii) lithiation of the resultant o-bromostyrene with n-BuLi and reaction of the aryl lithium species with ethylene oxide, and (iii) oxidation of the resultant primary alcohol using Dess–Martin periodinane [19,20].
![[1860-5397-17-104-i3]](/bjoc/content/inline/1860-5397-17-104-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: The documented synthesis of 2-(2-vinylphenyl)acetaldehyde (13a).
Scheme 3: The documented synthesis of 2-(2-vinylphenyl)acetaldehyde (13a).
The reported methods involved the use of ethylene oxide, a hazardous and carcinogenic gas. This prompted us to work out a more practical and flexible method to access the aromatic enal compounds 13. At the offset, we examined the synthesis of 2-(2-vinylphenyl)acetaldehyde (13a) using the route as outlined in Scheme 4. The synthesis started with the Wittig reaction of 2-bromobenzaldehyde (9a) with (methoxymethyl)triphenylphosphonium chloride (MTPPC) upon action with n-butyllithium in THF at 0 °C to give the vinyl ether 10a that was subjected to acidic hydrolysis using 18% aq HCl furnishing the corresponding aldehyde [21]. Without purification, the resultant aldehyde intermediate was then directly reduced using potassium borohydride to the corresponding primary alcohol 11a in 74% yield starting from 9a. Pd-catalyzed cross-coupling of 11a with pinacol vinylboronate afforded the o-hydroxyethyl-styrene 12a in 78% yield [22,23]. Next, Dess–Martin oxidation of the alcohol 12a was carried out, and the desired 2-(2-vinylphenyl)acetaldehyde (13a) was successfully obtained in 85% yield. Obviously, this modified method has the advantages of mild reaction conditions, operational simplicity, and using cheap and non-toxic reagents.
![[1860-5397-17-104-i4]](/bjoc/content/inline/1860-5397-17-104-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Modified synthesis of 2-(2-vinylphenyl)acetaldehydes 13a–g and 1-vinyl-2-naphthaldehyde (13h).
Scheme 4: Modified synthesis of 2-(2-vinylphenyl)acetaldehydes 13a–g and 1-vinyl-2-naphthaldehyde (13h).
The modified procedure was then expanded to the synthesis of a set of 2-(2-vinylphenyl)acetaldehydes 13b–f starting from differently substituted 2-bromobenzaldehydes 9 or 1-(2-bromophenyl)ethan-1-one (9e) in comparable yields. Likewise, 2-(1-vinylnaphthalen-2-yl)acetaldehyde (13h) was prepared from 1-bromo-2-naphthaldehyde in 48% yield over the three steps. It should be noted that the nitro-substituted intermediate 11d was prepared by nitration of 11a with nitric acid under the promotion of acetic anhydride.
With the accessibility of the aromatic vinyl aldehydes 13, next the cascade Prins/Friedel–Crafts reaction was examined. We started our investigations by applying aldehyde 13a as the model substrate (Scheme 5). A Lewis acid screening was carried out to identify the best catalyst for the tandem intramolecular Prins/Friedel–Crafts reaction (Table 1). Thus, the portion-wise addition of AlCl3 (1.1 equiv) to a stirred mixture of 13a (1.0 equiv) and veratrole (1.05 equiv) in CH2Cl2 at 0 °C resulted in the intramolecular Prins reaction to generate a benzyl carbenium ion that concurrently underwent Friedel–Crafts reaction with veratrole, leading to the formation of the expected 4-(3,4-dimethoxyphenyl)-1,2,3,4-tetrahydronaphthalen-2-ol (14aa), 51:49 mixture of cis/trans-diastereomers) as a colorless oil in 50% yield (Table 1, entry 1). The use of Et2AlCl as the Lewis acid gave tetralin 14aa in a slightly improved 55% yield (Table 1, entry 2). However, the reaction with AlMe3 as the promotor resulted in a competing reduction of 13a to 2-bromophenylethanol (12a) that was obtained as the major product (Table 1, entry 3). Switching to the weaker Lewis acid In(OTf)3 failed to induce any intramolecular Prins cyclization (Table 1, entry 4), whilst the use of FeCl3 produced 14aa in a similar 52% yield as observed for AlCl3 (Table 1, entry 5). To our delight, 1.1 equivalents of BF3·Et2O were found to promote the transformation efficiently, and a 70% isolated yield of 14aa was obtained (Table 1, entry 6). However, experiments with BF3·Et2O at substoichiometric amounts afforded significantly decreased yields of 14aa (Table 1, entries 7 and 8).
![[1860-5397-17-104-i5]](/bjoc/content/inline/1860-5397-17-104-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Lewis acid-catalyzed Prins/Friedel–Crafts reaction of 13a with veratrole.
Scheme 5: Lewis acid-catalyzed Prins/Friedel–Crafts reaction of 13a with veratrole.
Table 1: Screening of Lewis acid catalysts.a
| entry | Lewis acid | amount of LA [mol %] | cis/trans ratiob | yield [%]c |
| 1 | AlCl3 | 110 | 51:49 | 50 |
| 2 | Et2AlCl | 110 | 50:50 | 55 |
| 3 | AlMe3 | 110 | NA | 0d |
| 4 | In(OTf)3 | 110 | NA | 0 |
| 5 | FeCl3 | 110 | 50:50 | 52 |
| 6 | BF3·Et2O | 110 | 49:51 | 70 |
| 7 | BF3·Et2O | 80 | 50:50 | 50 |
| 8 | BF3·Et2O | 50 | 50:50 | 35 |
aReaction conditions: a mixture of 13a (1.40 mmol), veratrole (1.47 mmol) and Lewis acid (1.54 mmol) in CH2Cl2 (6 mL) was stirred at 0 °C for 2 h; bcis/trans ratios were determined by 1H NMR spectroscopy; cisolated yield after chromatography; dreduction product 12a instead of the desired 14aa was identified.
The relative cis- and trans-configuration of the C-2 hydroxy group and the C-4 aryl substituent (Figure 2) were assigned on the basis of 1H-1H COSY analysis. Firstly, the HSQC analysis was used to determine H3. The 13C NMR chemical shift for C2 is expected to be in the range of 60 to 70 ppm and the assignment of H3 was based on the HSQC correlation between H3 and C2. Then, H1 and H2 could be assigned by COSY and HSQC experiments. Following that, NOE analysis was applied to analyze the relative cis- and trans-configuration. If there is an NOE correlation between H1 and H3, and meanwhile H1 and H3 also have a strong NOE correlation with H2a, the compound is assigned to be cis-configured. Otherwise, it was assigned to be the trans-isomer (see Supporting Information File 1 for details).
![[1860-5397-17-104-2]](/bjoc/content/figures/1860-5397-17-104-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: The speculated stereostructures of compound cis-14aa and trans-14aa.
Figure 2: The speculated stereostructures of compound cis-14aa and trans-14aa.
Having determined the suitable reaction conditions, we surveyed the scope and limitation of the cascade protocol. Initially, we explored the range of nucleophiles that were used to intercept the benzyl carbenium ion and the results are summarized in Scheme 6. All reactions with electron-rich aromatics containing a p- and/or o-methoxy substituent as the nucleophile proceeded well to give the desired 2-hydroxy-4-aryltetralins 14aa–ae as 49:51 to 60:40 mixtures of cis/trans diastereomers in moderate to good yields.
![[1860-5397-17-104-i6]](/bjoc/content/inline/1860-5397-17-104-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Use of different nucleophiles for the cascade reaction with 13a. Reaction conditions: a mixture of 13a (1.40 mmol), nucleophile (1.47 mmol), BF3·Et2O (1.54 mmol) in anhydr. CH2Cl2 (6 mL) was stirred at 0 °C for 2 h. aIsolated yield by chromatography; bisolated by preparative HPLC.
Scheme 6: Use of different nucleophiles for the cascade reaction with 13a. Reaction conditions: a mixture of ...
The electron-rich 5-membered heterocycles like furans and thiophene participated also smoothly in the reaction sequence, leading to the clean formation of the respective 2-hydroxy-4-heteroaryltetralins 14af–ah, although the yields were somewhat lower than that with substituted anisole derivatives. As an attempt to enlarge the generality, tetraallysilane was also examined. To our delight, this substrate also participated in the reaction leading to the 4-allyl-substituted tetrahydronaphthalen-2-ol 14ai in 65% yield.
On comparing the results from the anisole-type nucleophiles or thiophene with that from furans, it was observed that the reactions with furans furnished predominantly trans-14af and trans-14ag with a high degree of diastereoselectivity (cis/trans ratio = 1:99). The preferential formation of the trans-configured products for furan nucleophiles may be due to the fact that the addition of furan is reversible leading to equilibration to the more stable trans product. To test this hypothesis, we monitored the reaction by HPLC (Table 2). As expected, we observed that the initially formed cis-isomer of 14af turned gradually to trans-14af and finally reached 1:99 after 2 hours (for further details, see Supporting Information File 1).
Table 2: Dependency of cis/trans ratio of product 14af on conditions and time.
| entry | conditions/reaction time | cis/trans ratioa |
| 1 | addition 20% of BF3·Et2O | 29:71 |
| 2 | addition 100% of BF3·Et2O | 23:77 |
| 3 | further stirred for 30 min | 21:79 |
| 4 | further stirred for 60 min | 12:88 |
| 5 | further stirred for 120 min | 1:99 |
acis/trans ratio was examined by HPLC.
To further expand the substitution pattern, we then tried the reaction of 13a with allysilane as a carbon-nucleophile. As expected, the 4-allyl-substituted tetrahydronaphthalen-2-ol 14ai was obtained, again, as a mixture of cis/trans-isomers in a ratio of 44:56. This example demonstrates the general synthetic utility of this cascade protocol.
Encouraged by the success of using 13a as the substrate, the reactions with other 2-(2-vinylphenyl)ethanals 13b–g carrying different substituents on the benzene ring or on the side chain with veratrole and furan as the nucleophiles were investigated. As can be seen from Scheme 7, under comparable conditions, most reactions proceeded smoothly with the attempted alkenyl-aldehydes 13 to furnish the corresponding 2,4-disubstituted tetralins 14ba–hb in acceptable to good isolated yields. For instance, the reaction with aldehydes 13 containing π-donating substituents like methoxy and chloro substituents afforded the 2-hydroxy-4-aryltetralin products 14ba–cb in 38–72% yield. To our gladness, aldehyde 13d, with an electron-deficient nitro group residing on the benzene ring reacted with veratrole under the standard conditions, delivering tetralin 14da in 55% yield. However, using furan as the nucleophile component, the reaction sequence with 13d failed to give the tetralin product. Instead, we only isolated 30% yield of the difuranyl-substituted compound 15 as the major product.
![[1860-5397-17-104-i7]](/bjoc/content/inline/1860-5397-17-104-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Reaction of aldehydes 13b–h with veratrole or furan. Reaction conditions: a mixture of 13b–h (1.40 mmol), nucleophile (veratrole or furan, 1.47 mmol), BF3·Et2O (1.54 mmol) in anhydr. CH2Cl2 (6 mL) was stirred at 0 °C for 2 h. aIsolated yield by chromatography; bcis-14ga refers to the structure with furyl and hydroxy substituents residing at the same side.
Scheme 7: Reaction of aldehydes 13b–h with veratrole or furan. Reaction conditions: a mixture of 13b–h (1.40 ...
In addition, aldehydes 13e or 13f bearing a methyl group at the acetaldehyde side or the benzylic position of the alkene side were also suitable substrates for this cascade strategy: the 1,2,4-trisubsitituted tetralins 14ea and 14eb as well as the 2,4,4-trisubsitituted tetralins 14fa and 14fb were obtained in moderate to reasonable yields. The aldehyde 13g bearing a phenyl group at the benzylic position of the alkene side was also tried. Under the standard conditions, the 1,2,4-trisubsitituted tetralin 14ga was isolated as a 17:83 mixture of cis/trans isomers, but with 20% yield. The poor yield may be attributed to the enhanced steric hindrance. This cyclization methodology was also applicable to 2-(1-vinylnaphthalen-2-yl)acetaldehyde (13h), for which the reaction with veratrole or furan led to the formation of the respective tricyclic 4-aryl-1,2,3,4-tetrahydrophenanthren-2-ols 14ha and 14hb in 73% and 43% yields, respectively.
In order to further explore the generality of this cascade Prins/Friedel–Crafts cyclization, the established methodology was also applied to the formation of tetrahydro-5H-benzocyclohepten-7-ol ring systems. As shown in Scheme 8, the required homo-aldehyde substrate 19 was prepared starting from methyl 3-(2-bromophenyl)propionate (16) analogously as for 13. Reduction of the ester 16 with LiAlH4 in THF at 0 °C afforded the alcohol 17 that was subjected to a Suzuki reaction with pinacol vinylboronate using Pd(dppf)Cl2 as catalyst to produce 3-(2-vinylphenyl)propan-1-ol (18). The oxidation of alcohol 18 with Dess–Martin oxidizing reagent furnished the requisite aldehyde 19 in 43% yield over the three steps.
![[1860-5397-17-104-i8]](/bjoc/content/inline/1860-5397-17-104-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Synthesis of 5-aryltetrahydro-5H-benzo[7]annulen-7-ols 20a, b.
Scheme 8: Synthesis of 5-aryltetrahydro-5H-benzo[7]annulen-7-ols 20a, b.
Under the standard conditions, aldehyde 19 underwent satisfactorily the cascade Prins/Friedel–Crafts cyclization with veratrole or furan as the nucleophile furnishing the tetrahydro-5H-benzo[7]annulen-7-ol 20a (cis/trans ratio = 54:46) and 20b (cis/trans ratio = 26:74) in 60% and 31% yield, respectively. The predominance of the trans-product for the reaction with furan further verified the oxophilic character of the employed BF3, although the stereoselectivity considerably decreased in comparison with the formation of tetralin ring system as the distance between the reaction sites is increased. It is worth mentioning that the tetrahydro-5H-benzo[7]annulen-7-ol skeleton is also of considerable medicinal significance and has attracted much synthetic efforts [24,25].
Finally, the ability to structurally diversify the 2-hydroxy-4-substituted tetralin skeletons into medicinally useful derivatives was demonstrated by converting 2-hydroxy-4-furyl-tetralin 14af into the PAT analogue 22 (see Figure 1) [26]. The reaction of 14af with p-toluenesulfonyl chloride in pyridine afforded the tosylate 21 in 90% yield, which was then treated with 40% aqueous dimethylamine to produce the tertiary amine containing PAT analogue 22 (cis/trans ratio = 79:21) in 70% yield (Scheme 9). With regard to the partial epimerization of product 21, it may be due to the action of pyridine. In the preparation of compound 21, pyridine is used both as solvent and the acid acceptor. Because pyridine itself can show nucleophilic reactivity in addition to basicity, the long reaction time of 20 hours may lead to an ion-pair species with 21 and hence erode the stereochemistry. To prove this idea, we performed the reaction with CH2Cl2 as the solvent in the presence of 5.0 equivalents of pyridine and 2.0 equivalents of TsCl. Under these conditions, the tosylate 21 was obtained with full retention of the expected stereochemistry (cis/trans =1:99) (see Supporting Information File 1 for details). The conversion of tosylate 21 to product 22 proceeded in a typical SN2 manner resulting in the expected inversion of the configuration.
![[1860-5397-17-104-i9]](/bjoc/content/inline/1860-5397-17-104-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Conversion of 2-hydroxy-4-(2-furyl)tetralin (14af) into PAT analogue 22.
Scheme 9: Conversion of 2-hydroxy-4-(2-furyl)tetralin (14af) into PAT analogue 22.
To unequivocally support the configuration assignment made by NMR analysis, the sulfonate derivative 21 from 14af was prepared by reaction with tosyl chloride. For compound 21, we were able to obtain single crystals suitable for X-ray analysis and the X-ray diffraction studies on 21 confirmed undoubtedly its trans-configuration. The ORTEP structure is shown in Figure 3 [27].
![[1860-5397-17-104-3]](/bjoc/content/figures/1860-5397-17-104-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Crystal structure of the tosylate 21. The displacement ellipsoids are drawn at the 30% probability level.
Figure 3: Crystal structure of the tosylate 21. The displacement ellipsoids are drawn at the 30% probability ...
Conclusion
In summary, a Prins cyclization and Friedel–Crafts cascade reaction strategy for the synthesis of 4-aryl-tetralin-2-ols and 5-aryl-tetrahydro-5H-benzo[7]annulen-7-ols has been established. The sequence involved the Prins cyclization of 2-(2-vinylphenyl)acetaldehydes or 3-(2-vinylphenyl)propanal by action with BF3 to generate benzyl carbenium ions that are captured by a Friedel–Crafts alkylation reaction with a range of electron-rich benzenes or heteroaromatics. The method has a relatively broad applicability allowing variation in the benzene ring as well as in the side chain. The further manipulation of the hydroxy group affording the PAT analogue demonstrated the synthetic potential for accessing medicinally useful derivatives.
Supporting Information
The Supporting Information contains experimental procedures, characterization data of all isolated products as well as copies of NMR spectra and XRPD data for compound 21.
| Supporting Information File 1: Experimental section. | ||
| Format: PDF | Size: 8.3 MB | Download |
References
-
Ichikawa, K.; Kinoshita, T.; Nishibe, S.; Sankawa, U. Chem. Pharm. Bull. 1986, 34, 3514–3517. doi:10.1248/cpb.34.3514
Return to citation in text: [1] -
Dubocovich, M. L.; Masana, M. I.; Iacob, S.; Sauri, D. M. Naunyn-Schmiedeberg's Arch. Pharmacol. 1997, 355, 365–375. doi:10.1007/pl00004956
Return to citation in text: [1] -
Wyrick, S. D.; Booth, R. G.; Myers, A. M.; Owens, C. E.; Kula, N. S.; Baldessarini, R. J.; McPhail, A. T.; Mailman, R. B. J. Med. Chem. 1993, 36, 2542–2551. doi:10.1021/jm00069a013
Return to citation in text: [1] -
Moritani, Y.; Ukita, T.; Ohmizu, H.; Iwasaki, T. J. Chem. Soc., Chem. Commun. 1995, 671–672. doi:10.1039/c39950000671
Return to citation in text: [1] -
Lucarini, S.; Bedini, A.; Spadoni, G.; Piersanti, G. Org. Biomol. Chem. 2008, 6, 147–150. doi:10.1039/b713904g
Return to citation in text: [1] -
Bucholtz, E. C.; Brown, R. L.; Tropsha, A.; Booth, R. G.; Wyrick, S. D. J. Med. Chem. 1999, 42, 3041–3054. doi:10.1021/jm980428x
Return to citation in text: [1] -
Makowski, P.; Cakan, R. D.; Antonietti, M.; Goettmann, F.; Titirici, M.-M. Chem. Commun. 2008, 999, 999–1001. doi:10.1039/b717928f
Return to citation in text: [1] -
Zhang, W.; Tang, W. L.; Wang, Z.; Li, Z. Adv. Synth. Catal. 2010, 352, 3380–3390. doi:10.1002/adsc.201000266
Return to citation in text: [1] -
Brenzovich, W. E., Jr.; Brazeau, J.-F.; Toste, F. D. Org. Lett. 2010, 12, 4728–4731. doi:10.1021/ol102194c
Return to citation in text: [1] -
Indukuri, K.; Unnava, R.; Deka, M. J.; Saikia, A. K. J. Org. Chem. 2013, 78, 10629–10641. doi:10.1021/jo401450j
Return to citation in text: [1] -
Reddy, B. V. S.; Sundar, C. S.; Reddy, M. R.; Reddy, C. S.; Sridhar, B. Synthesis 2015, 47, 1117–1122. doi:10.1055/s-0034-1380177
Return to citation in text: [1] -
Li, R.-Q.; He, Y.; Ding, Y.; Ang, C.-K.; Tian, J.-S.; Loh, T.-P. Chem. Commun. 2018, 54, 3150–3153. doi:10.1039/c7cc09875h
Return to citation in text: [1] -
Barakov, R.; Shcherban, N.; Yaremov, P.; Bezverkhyy, I.; Čejka, J.; Opanasenko, M. Green Chem. 2020, 22, 6992–7002. doi:10.1039/d0gc01787f
Return to citation in text: [1] -
Kotipalli, T.; Hou, D.-R. Org. Lett. 2018, 20, 4787–4790. doi:10.1021/acs.orglett.8b01929
Return to citation in text: [1] -
Sakata, Y.; Yasui, E.; Takatori, K.; Suzuki, Y.; Mizukami, M.; Nagumo, S. J. Org. Chem. 2018, 83, 9103–9118. doi:10.1021/acs.joc.8b01195
Return to citation in text: [1] -
Hinkle, R. J.; Chen, Y.; Nofi, C. P.; Lewis, S. E. Org. Biomol. Chem. 2017, 15, 7584–7593. doi:10.1039/c7ob01412k
Return to citation in text: [1] -
Olier, C.; Kaafarani, M.; Gastaldi, S.; Bertrand, M. P. Tetrahedron 2010, 66, 413–445. doi:10.1016/j.tet.2009.10.069
Return to citation in text: [1] -
Padmaja, P.; Reddy, P. N.; Reddy, B. V. S. Org. Biomol. Chem. 2020, 18, 7514–7532. doi:10.1039/d0ob00960a
Return to citation in text: [1] -
Grigg, R. D.; Van Hoveln, R.; Schomaker, J. M. J. Am. Chem. Soc. 2012, 134, 16131–16134. doi:10.1021/ja306446m
Return to citation in text: [1] -
Fustero, S.; Rodríguez, E.; Lázaro, R.; Herrera, L.; Catalán, S.; Barrio, P. Adv. Synth. Catal. 2013, 355, 1058–1064. doi:10.1002/adsc.201201095
Return to citation in text: [1] -
Chua, P. J.; Tan, B.; Yang, L.; Zeng, X.; Zhu, D.; Zhong, G. Chem. Commun. 2010, 46, 7611–7613. doi:10.1039/c0cc01577f
Return to citation in text: [1] -
Dai, X.-J.; Engl, O. D.; León, T.; Buchwald, S. L. Angew. Chem., Int. Ed. 2019, 58, 3407–3411. doi:10.1002/anie.201814331
Return to citation in text: [1] -
Firmansjah, L.; Fu, G. C. J. Am. Chem. Soc. 2007, 129, 11340–11341. doi:10.1021/ja075245r
Return to citation in text: [1] -
Singh, K. N.; Singh, P.; Sharma, E.; Kaur, M.; Deol, Y. S. Synthesis 2015, 47, 3212–3220. doi:10.1055/s-0034-1380453
Return to citation in text: [1] -
Wyrick, S. D.; Booth, R. G.; Myers, A. M.; Owens, C. E.; Bucholtz, E. C.; Hooper, P. C.; Kula, N. S.; Baldessarini, R. J.; Mailman, R. B. J. Med. Chem. 1995, 38, 3857–3864. doi:10.1021/jm00019a016
Return to citation in text: [1] -
Booth, R. G. Therapeutic compounds. Int. Pat. Appl. WO2010129048, Nov 11, 2010.
Return to citation in text: [1] -
CCDC 2060394 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk./data_request/cif.
Return to citation in text: [1]
| 1. | Ichikawa, K.; Kinoshita, T.; Nishibe, S.; Sankawa, U. Chem. Pharm. Bull. 1986, 34, 3514–3517. doi:10.1248/cpb.34.3514 |
| 2. | Dubocovich, M. L.; Masana, M. I.; Iacob, S.; Sauri, D. M. Naunyn-Schmiedeberg's Arch. Pharmacol. 1997, 355, 365–375. doi:10.1007/pl00004956 |
| 3. | Wyrick, S. D.; Booth, R. G.; Myers, A. M.; Owens, C. E.; Kula, N. S.; Baldessarini, R. J.; McPhail, A. T.; Mailman, R. B. J. Med. Chem. 1993, 36, 2542–2551. doi:10.1021/jm00069a013 |
| 7. | Makowski, P.; Cakan, R. D.; Antonietti, M.; Goettmann, F.; Titirici, M.-M. Chem. Commun. 2008, 999, 999–1001. doi:10.1039/b717928f |
| 8. | Zhang, W.; Tang, W. L.; Wang, Z.; Li, Z. Adv. Synth. Catal. 2010, 352, 3380–3390. doi:10.1002/adsc.201000266 |
| 9. | Brenzovich, W. E., Jr.; Brazeau, J.-F.; Toste, F. D. Org. Lett. 2010, 12, 4728–4731. doi:10.1021/ol102194c |
| 27. | CCDC 2060394 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk./data_request/cif. |
| 6. | Bucholtz, E. C.; Brown, R. L.; Tropsha, A.; Booth, R. G.; Wyrick, S. D. J. Med. Chem. 1999, 42, 3041–3054. doi:10.1021/jm980428x |
| 5. | Lucarini, S.; Bedini, A.; Spadoni, G.; Piersanti, G. Org. Biomol. Chem. 2008, 6, 147–150. doi:10.1039/b713904g |
| 24. | Singh, K. N.; Singh, P.; Sharma, E.; Kaur, M.; Deol, Y. S. Synthesis 2015, 47, 3212–3220. doi:10.1055/s-0034-1380453 |
| 25. | Wyrick, S. D.; Booth, R. G.; Myers, A. M.; Owens, C. E.; Bucholtz, E. C.; Hooper, P. C.; Kula, N. S.; Baldessarini, R. J.; Mailman, R. B. J. Med. Chem. 1995, 38, 3857–3864. doi:10.1021/jm00019a016 |
| 4. | Moritani, Y.; Ukita, T.; Ohmizu, H.; Iwasaki, T. J. Chem. Soc., Chem. Commun. 1995, 671–672. doi:10.1039/c39950000671 |
| 26. | Booth, R. G. Therapeutic compounds. Int. Pat. Appl. WO2010129048, Nov 11, 2010. |
| 17. | Olier, C.; Kaafarani, M.; Gastaldi, S.; Bertrand, M. P. Tetrahedron 2010, 66, 413–445. doi:10.1016/j.tet.2009.10.069 |
| 18. | Padmaja, P.; Reddy, P. N.; Reddy, B. V. S. Org. Biomol. Chem. 2020, 18, 7514–7532. doi:10.1039/d0ob00960a |
| 21. | Chua, P. J.; Tan, B.; Yang, L.; Zeng, X.; Zhu, D.; Zhong, G. Chem. Commun. 2010, 46, 7611–7613. doi:10.1039/c0cc01577f |
| 16. | Hinkle, R. J.; Chen, Y.; Nofi, C. P.; Lewis, S. E. Org. Biomol. Chem. 2017, 15, 7584–7593. doi:10.1039/c7ob01412k |
| 22. | Dai, X.-J.; Engl, O. D.; León, T.; Buchwald, S. L. Angew. Chem., Int. Ed. 2019, 58, 3407–3411. doi:10.1002/anie.201814331 |
| 23. | Firmansjah, L.; Fu, G. C. J. Am. Chem. Soc. 2007, 129, 11340–11341. doi:10.1021/ja075245r |
| 15. | Sakata, Y.; Yasui, E.; Takatori, K.; Suzuki, Y.; Mizukami, M.; Nagumo, S. J. Org. Chem. 2018, 83, 9103–9118. doi:10.1021/acs.joc.8b01195 |
| 10. | Indukuri, K.; Unnava, R.; Deka, M. J.; Saikia, A. K. J. Org. Chem. 2013, 78, 10629–10641. doi:10.1021/jo401450j |
| 11. | Reddy, B. V. S.; Sundar, C. S.; Reddy, M. R.; Reddy, C. S.; Sridhar, B. Synthesis 2015, 47, 1117–1122. doi:10.1055/s-0034-1380177 |
| 12. | Li, R.-Q.; He, Y.; Ding, Y.; Ang, C.-K.; Tian, J.-S.; Loh, T.-P. Chem. Commun. 2018, 54, 3150–3153. doi:10.1039/c7cc09875h |
| 13. | Barakov, R.; Shcherban, N.; Yaremov, P.; Bezverkhyy, I.; Čejka, J.; Opanasenko, M. Green Chem. 2020, 22, 6992–7002. doi:10.1039/d0gc01787f |
| 14. | Kotipalli, T.; Hou, D.-R. Org. Lett. 2018, 20, 4787–4790. doi:10.1021/acs.orglett.8b01929 |
| 19. | Grigg, R. D.; Van Hoveln, R.; Schomaker, J. M. J. Am. Chem. Soc. 2012, 134, 16131–16134. doi:10.1021/ja306446m |
| 20. | Fustero, S.; Rodríguez, E.; Lázaro, R.; Herrera, L.; Catalán, S.; Barrio, P. Adv. Synth. Catal. 2013, 355, 1058–1064. doi:10.1002/adsc.201201095 |
© 2021 Zheng et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the author(s) and source are credited and that individual graphics may be subject to special legal provisions.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc/terms)