Abstract
In the search for high-energy materials, novel 3D-fluorophosphates, Li2Co1−xFexPO4F and Li2Co1−xMnxPO4F, have been synthesized. X-ray diffraction and scanning electron microscopy have been applied to analyze the structural and morphological features of the prepared materials. Both systems, Li2Co1−xFexPO4F and Li2Co1−xMnxPO4F, exhibited narrow ranges of solid solutions: x ≤ 0.3 and x ≤ 0.1, respectively. The Li2Co0.9Mn0.1PO4F material demonstrated a reversible electrochemical performance with an initial discharge capacity of 75 mA·h·g−1 (current rate of C/5) upon cycling between 2.5 and 5.5 V in 1 M LiBF4/TMS electrolyte. Galvanostatic measurements along with cyclic voltammetry supported a single-phase de/intercalation mechanism in the Li2Co0.9Mn0.1PO4F material.
Introduction
In recent years the range of application of Li-ion batteries has been expanded from small-sized portable electronics to large-scale electric vehicles and stationary energy storage systems. Large-scale energy applications require batteries that are economically efficient, highly safe and that provide a high energy and power density. Today most of the cells in use have almost reached their intrinsic limits, and no significant improvements are expected. Therefore, current research in this field is directed towards the development of new high-performance materials. The specific energy of Li-ion batteries can be enhanced by applying cathode materials that operate at high voltages, and/or by increasing the specific capacity with materials that could cycle more than one Li atom per active transition metal atom. In this respect, fluorophosphates of the general formula A2MPO4F seem to be very attractive since they are expected to exhibit a high operating potential because of the increased ionicity of the M–F bond. Furthermore, A2MPO4F cathode materials may reach capacity values larger than 200 mA·h·g−1, if more than one lithium atom would participate in the reversible de/intercalation process.
Li2CoPO4F, which exhibits an electrochemical activity above 5 V vs Li/Li+, is one of the attractive candidates in the fluorophosphate family [1]. This fluorophosphate possesses a three-dimensional (3D) tunnel structure and, by analogy to the olivine phase, is expected to demonstrate a good stability and reversibility upon cycling. It is built of edge-shared CoO4F2-octahedra interconnected with PO4-tetrahedra, which generate a framework with channels through which alkali-ion diffusion can take place [2-4] (Figure 1). The reversible electrochemical activity of Li2CoPO4F has been studied by several groups [4-9]. Our previous investigation of this cathode material has revealed the de/intercalation of lithium occurs through a single-phase reaction mechanism. Moreover, according to the capacity–voltage dependence the extraction of more than one Li+ ion should take place at potentials larger than 5.5 V [4], which is beyond the stability range of conventional electrolytes. An initial discharge capacity of 132 mA·h·g−1 that is delivered by Li2CoPO4F in a high-voltage electrolyte with fluorinated alkyl carbonates has been reported by S. Amaresh et al., however noticeable capacity fading has been observed upon prolonged cycling [8]. Therefore, the evaluation of the electrochemical performance of Li2CoPO4F and the other representative of this family such as Li2NiPO4F [2,10], is limited to conventional electrolytes. Hence, the development of new organic electrolytes with a wide range of application voltages and the investigation of high-voltage fluorophosphates using these new electrolyte systems are strongly required.
![[2190-4286-4-97-1]](/bjnano/content/figures/2190-4286-4-97-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Crystal structure of 3D-Li2MPO4F, positions of Li atoms are denoted.
Figure 1: Crystal structure of 3D-Li2MPO4F, positions of Li atoms are denoted.
Another way to explore this fluorophopshate family is to adjust the operating voltage of these compounds to values that are sustained by conventional electrolytes. This might be achieved through a complete or a partial substitution of Co2+ by Fe2+ or Mn2+ with lower values of the of M2+/M3+ redox potential. Here, we report on the synthesis and the investigation of Li2Co1−xMxPO4F (M = Fe, Mn) fluorophophates, which have not been yet identified. Furthermore, different high-voltage electrolytes systems were tested and utilized to evaluate the electrochemical performance of the new synthesized compounds.
Results and Discussion
Testing of electrolytes
An electrochemical window that extends above 5.5 V (vs Li/Li+) has been reported for several electrolytes systems based on sulfone or dinitrile solvents [10-14]. For instance, tetramethylene sulfone (TMS) in the presence of an imide salt (LiTFSI) demonstrated a resistance to electrochemical oxidation up to 6 V vs Li/Li+ [11], while 1 M LiBF4/(EC)/DMC/sebaconitrile was used to examine the high-voltage performance of the fluorophosphate Li2NiPO4F [10]. We chose 1 M LiBF4/TMS to investigate the electrochemical activity of the fluorophosphate materials. LiBF4 salt was chosen instead of LiTFSI, because the last one corrodes the aluminum current collector at high potentials.
Preliminarily, the stability of both electrolytes was investigated by cyclic voltammetry to further establish their compatibility with high-voltage cathode materials. Two types of working electrodes were used to evaluate the electrochemical window of the electrolytes: 1) Al-foil (since it is used as a current collector for the positive electrode); 2) an “idle electrode”, which consisted of Al2O3/C/PVdF in a ratio of 80/10/10, in order to imitate the effect of the carbon- and binding electrode components at high potentials. Because the loading mass and the effective surface area of the active material on the electrodes that were used for electrolyte testing were similar in all experiments, the obtained current values were compared without normalization.
Both electrolytes exhibited an electrochemical stability up to 5.5 V (vs Li/Li+) with aluminum as the working electrode (Figure 2a). For the first cycle the current detected at the highest potential did not exceed 0.4 μА, and it decreased (to 0.001 μА) upon subsequent cycling. It is clearly seen that the effect of the oxidation processes occurred at the Al electrode is negligible for both electrolytes when compared to a scanning with the idle electrode (Figure 2b). In the anodic sweep the commercial electrolyte showed a small increase in oxidation current at 4.8 V followed by drastic growth (up to 40 μA) around 5.2 V, while for the TMS electrolyte irreversible oxidation current peaks of 5 μA were detected. These results confirmed the reasonable stability of 1 M LiBF4/TMS electrolyte up to 5.5 V, which agrees with data reported previously [12,13].
![[2190-4286-4-97-2]](/bjnano/content/figures/2190-4286-4-97-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Cyclovoltammetry curves (first cycle) of 1 M LiPF6 in EC/DMC (black) and 1 M LiBF4 in TMS (red) at scan rate of 0.1 mV·s−1. (a) Aluminum electrode, (b) idle electrode consisting of Al2O3/C/PVdF in an 80/10/10 ratio.
Figure 2: Cyclovoltammetry curves (first cycle) of 1 M LiPF6 in EC/DMC (black) and 1 M LiBF4 in TMS (red) at ...
Investigation of Li2(Co,M)PO4F (M = Mn, Fe)
Applied synthesis approaches were directed not only towards the investigation of Li2Co1−xMxPO4F solid solutions, but also to the preparation of the corresponding electrode materials. Because of poor electronic and ionic conductivity that is inherent to polyanionic compounds, a carbon coating (for improving the electronic surface conductivity) and a downsizing of the particles (in order to shorten the Li-ion transfer paths) were applied to enhance the electrochemical performance of the investigated materials. In order to reduce the particle size and to prevent grain coalescence the lowest temperatures usable for the formation of the pure olivine precursors and the fluorophosphates were always chosen.
The Li2CoPO4F/C composite for electrochemical measurements was synthesized according to a procedure that was optimized previously [4]. A mixture of LiCoPO4/C with 1.05 equiv of LiF was annealed at 670 °C for 1 h under Ar-flow and subsequently quenched to room temperature. The XRD pattern confirmed the formation of Li2CoPO4F, though a small amount of WC (about 1%, from the ball-milling media) was also detected (Figure 3). The refined unit cell parameters of Li2CoPO4F/C (a = 10.444(3) Å, b = 6.374(2) Å, c = 10.868(3) Å, V = 723.6(5) Å3) were in agreement with previously reported data [1,4]. The residual carbon in Li2CoPO4F/C was found to be 1.7%. According to the SEM images the synthesized material consisted of almost uniform particles with an average size of 0.7–0.9 μm (Figure 4).
![[2190-4286-4-97-3]](/bjnano/content/figures/2190-4286-4-97-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Powder XRD patterns of Li2CoPO4F/C (a) and Li2Co0.9Mn0.1PO4F (b). A theoretical pattern of Li2CoPO4F calculated by using PDF 56-149 is shown on the bottom. Reflections corresponding to WC are marked by an asterisk.
Figure 3: Powder XRD patterns of Li2CoPO4F/C (a) and Li2Co0.9Mn0.1PO4F (b). A theoretical pattern of Li2CoPO4...
![[2190-4286-4-97-4]](/bjnano/content/figures/2190-4286-4-97-4.jpg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: SEM images of fluorophosphate materials a) Li2CoPO4F/C, b) Li2Co0.9Mn0.1PO4F/C, c) Li2Co0.7Fe0.3PO4F.
Figure 4: SEM images of fluorophosphate materials a) Li2CoPO4F/C, b) Li2Co0.9Mn0.1PO4F/C, c) Li2Co0.7Fe0.3PO4...
In order to investigate the Li2Co1−xMnxPO4F solid solutions a combination of freeze-drying and solid-state techniques was applied. A mixture of LiCo0.9Mn0.1PO4 obtained from cryogranulate was annealed with 1.05 equiv of LiF in the temperature range of 650–700 °C for 1–2 h. Annealing at 680 °C for 1 h was found to be optimal for the preparation of the Li2Co0.9Mn0.1PO4F phase. The XRD pattern of this sample (Figure 3) was indexed as an orthorhombic unit cell with parameters a = 10.465(2) Å, b = 6.3998(9) Å, c = 10.898(2) Å and V = 729.9(2) Å3. No peaks of the olivine phase were observed, though a small amount of WC (about 1%, from the ball-milling media) was detected in the XRD pattern. Further attempts to increase the Mn content in Li2Co1−xMnxPO4F (x = 0.2, 0.3) by varying the annealing temperature and the heating duration ended up with multiphase samples that contained impurities of olivine and Li3PO4. Moreover, the unit cell parameters of the formed fluorophosphates were found to be close to those of Li2Co0.9Mn0.1PO4F. These results clearly indicated that Li2Co1−xMnxPO4F exhibited a very limited range for the solid solution (x ≤ 0.10). For electrochemical testing Li2Co0.9Mn0.1PO4F/C was synthesized by adding carbon black (5 wt %) to the olivine precursor at an intermediate step of preparation. The XRD pattern of the obtained sample confirmed the formation of pure fluorophosphate with cell parameters similar to those given above. EDX analysis of the prepared material found the Co/Mn ratio to be 0.89(1)/0.11(1), which agreed with the expected values from the chemical formula. The morphology of this sample was investigated by SEM and showed particles of submicron size (Figure 4). The residual carbon in the prepared composite was determined to be as 3.1% by TG analysis. This value was taken into account during the preparation of the electrode.
The synthesis of the iron-substituted fluorophosphates, Li2Co1−xFexPO4F, was performed by a two-step solid-state process. The optimization of the preparation conditions was done for the composition of x = 0.3. Figure 5a represents XRD patterns of the samples obtained by annealing mixtures of LiCo0.7Fe0.3PO4 and LiF (with 10 wt % excess) at different temperatures. According to the XRD data, the fluorophosphate phase started to form above 700 °С, and further enhancement of the annealing temperature resulted in a decrease of the olivine impurities and in an increase of the fluorophosphate constituent. The formation of the almost pure Li2Co0.7Fe0.3PO4F was observed upon heating at 740–750 °С. Above these temperatures (>760 °С) samples melted and were heavily contaminated by cobalt oxide. Thus, the annealing at 750 °С for 1 h in Ar was found to be optimum to yield Li2Co0.7Fe0.3PO4F. A tuning of the annealing temperature allowed us to synthesize pure fluorophosphates with different levels of substitution, Li2Co1−xFexPO4F (x = 0.1–0.3) (Figure 5b). The XRD patterns of obtained samples were indexed on the base of an orthorhombic structure with a Pnma space group and the unit cell parameters that are listed in Table 1. Careful inspection of the XRD data revealed negligible amounts of Li3PO4 and Co admixtures. It is evident from the obtained results that the synthesis of Fe-substituted compounds requires increased annealing temperatures that depend on the Fe-content in Li2Co1−xFexPO4F. For a higher Fe-substitution higher annealing temperatures are needed. The solid-state synthesis at elevated temperatures resulted in large micrometer-sized particles (2–4 μm) as observed by SEM (Figure 4). It should be noted that the presence of LiF, which is used as the reagent, promoted the coalescence of small particles and induced crystallite growth because of fluxing at elevated temperatures. In spite of varying the preparation conditions all attempts to increase the substitution level of Fe in Li2Co1−xFexPO4F (x = 0.4, 0.5) led to multi-phase samples, with the fluorophosphate phases having cell parameters close to those of Li2Co0.7Fe0.3PO4F. Thus, it was concluded that the solid-solution range of Li2Co1−xFexPO4F was limited to x ≤ 0.3. Efforts to prepare a Li2Co0.7Fe0.3PO4F/C composite by adding carbon black or glucose to the initial mixtures of reagents resulted in multiphase samples that contained large amounts of metallic Co (>10%), which can be explained by the strongly reductive conditions that appeared at elevated temperatures (>700 °С) because of the presence of C-containing additives. Therefore, for the electrochemical evaluation of the Fe-substituted fluorophosphates the electrodes were prepared from the carbon-free product Li2Co0.7Fe0.3PO4F by thoroughly mixing it with Super-C carbon (10 wt %).
![[2190-4286-4-97-5]](/bjnano/content/figures/2190-4286-4-97-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: a) XRD patterns of a mixture of LiCo0.7Fe0.3PO4 and LiF, annealed at different temperatures, starting from 670 °С. XRD peaks that correspond to impurities are marked. b) XRD patterns of Li2Co1−xFexPO4F (x = 0.1, 0.2, 0.3), synthesized at the denoted temperatures.
Figure 5: a) XRD patterns of a mixture of LiCo0.7Fe0.3PO4 and LiF, annealed at different temperatures, starti...
Table 1: Unit cell parameters of fluorophosphates Li2Co1−xMxPO4F (M = Mn, Fe).
| x (M) | unit cell parameters of Li2Co1−xMxPO4F | |||
|---|---|---|---|---|
| a, Å | b, Å | c, Å | V, Å3 | |
| 0 | 10.439(2) | 6.3731(12) | 10.864(2) | 722.8(2) |
| 0.1 (Mn) | 10.465(2) | 6.3998(9) | 10.898(2) | 729.9(2) |
| 0.1 (Fe) | 10.440(2) | 6.3862(13) | 10.867(3) | 724.5(4) |
| 0.2 (Fe) | 10.442(2) | 6.4103(14) | 10.884(2) | 728.6(3) |
| 0.3 (Fe) | 10.453(1) | 6.4096(8) | 10.888(12) | 729.5(2) |
According to the obtained results Li2Co1−xFexPO4F and Li2Co1−xMnxPO4F systems exhibit limited ranges of solid solution. This finding might be explained by differences in the sizes of transition metal ions: Apparently, the structure framework becomes unstable upon higher substitution of Co2+ (0.735 Å) by larger Fe2+ (0.780 Å) and Mn2+ (0.820 Å) [15]. Indeed, while Li2MPO4F (M = Co, Ni) can be obtained by direct synthesis, the preparation of 3D-Li2FePO4F requires the electrochemical ion-exchange of the Na-counterpart, and the corresponding Mn-based fluorophosphate has not been yet identified [16]. It is reasonable, that a substitution of Co2+ by Mn2+, which has the largest ionic radius, only takes place in a smaller range (x ≤ 0.10) than in the case of Fe2+ (x ≤ 0.30). In both cases the substitution results in considerable expansion of the unit cell (ca. 7 Å3) for the highest level of substitution (Table 1).
Electrochemical performance of Li2(Co,M)PO4F (M = Mn, Fe)
According to galvanostatic measurements performed at a rate of C/5 (Figure 6) Li2CoPO4F starts to discharge at approx. 5 V, which agrees well with previous results. The Li/Li2CoPO4F cells delivered initial discharge capacities of ca. 90 and 85 mA·h·g−1 with the commercial and the sulfone-based electrolyte, respectively, and these values corresponded to a reversible de/intercalation of about 0.65 Li. During the initial cycles the charge capacity values were remarkably higher than the corresponding discharge capacities. This discrepancy in the capacities may result from a decomposition of the electrolyte on the conductive carbon and on the flurophosphate material at high potentials. For the TMS electrolyte this discrepancy disappeared upon subsequent cycling. During the 10th cycle the corresponding values became almost equal, with a coulombic efficiency of 98% (Figure 6). Moreover, there is less capacity fading when using the TMS electrolyte. During the 10th cycle the discharge capacity decreased to about 83% of the initial value in contrast to a decrease to about 45% found with the commercial electrolyte. The obtained results indicated a rather stable electrochemical performance of the Li2CoPO4F material at high voltages in the 1 M LiBF4/TMS electrolyte. The decrease of the irreversible capacity, which leads to the high columbic efficiency, implies that this electrolyte forms a stable solid-electrolyte interface on the electrode surface, but this suggestion should be further investigated and confirmed.
![[2190-4286-4-97-6]](/bjnano/content/figures/2190-4286-4-97-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Charge-discharge curves of Li2CoPO4F in the commercial (a) and in the sulfone-based electrolytes (b) measured at C/5.
Figure 6: Charge-discharge curves of Li2CoPO4F in the commercial (a) and in the sulfone-based electrolytes (b...
A preliminary investigation of the electrochemical behavior of Li2Co0.7Fe0.3PO4F was carried out with electrodes prepared from the well crystallized sample. Potentiodynamic measurements in both electrolytes resulted in broad peaks on the anodic and cathodic branches with the discharge capacity values being lower than 10 mA·h·g−1. Because of the poor electrochemical activity, which is ascribed to the non-optimized morphology of the electrode material (particle size of 2–4 μm), any comparisons of Li2Co0.7Fe0.3PO4F with the unsubstituted material were unreasonable.
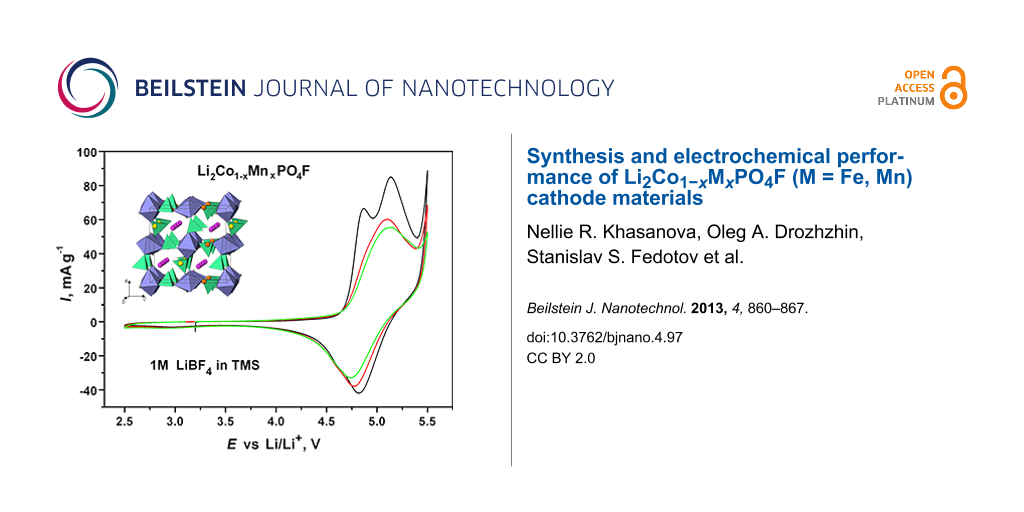
Figure 7 shows the cyclovoltammetry (CV) curves of the Li/Li2Co0.9Mn0.1PO4F cells cycled in both electrolytes. For the TMS electrolyte two oxidative peaks (at 4.9 V and 5.2 V) and a broad reductive peak (at 4.8 V) were observed in the first anodic and cathodic scans, respectively. During the second cycle the two oxidative peaks merged, and the broad peaks on the anodic (≈5.1 V) and cathodic (4.8 V) branches showed charge and discharge capacities of 135 and 70 mA·h·g−1, respectively. The CV curves that were recorded in the commercial electrolyte were quite similar. The presence of two oxidative peaks in the first anodic scan (Figure 7) hints at the occurrence of at least two redox processes. We related them to the structure transformation upon deintercalation of Li, followed by a further removal of Li from the transformed structure. This irreversible structure transformation, which occurs upon first charging, was investigated by ex-situ XRD studies and described in detail in our previous paper. This transformation resulted in an expansion of the framework and a probable redistribution of Li ions within the framework [4]. Similar features were observed in CV curves of Li2CoPO4F by D. Wang et al. [5] and S. Amaresh et al. [8]. This indicates the intrinsic nature of this transformation.
![[2190-4286-4-97-7]](/bjnano/content/figures/2190-4286-4-97-7.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: Cyclovoltammetry curves of the Li2Co0.9Mn0.1PO4F electrodes in the commercial (a) and the sulfone-based (b) electrolytes recorded at 0.1 mV s−1.
Figure 7: Cyclovoltammetry curves of the Li2Co0.9Mn0.1PO4F electrodes in the commercial (a) and the sulfone-b...
Galvanostatic measurements on Li2Co0.9Mn0.1PO4F (Figure 8) revealed the highest discharge capacities of 75 and 85 mA·h·g−1 in TMS and the commercial electrolytes, respectively. As in the case of Li2CoPO4F, the capacity fading of the Mn-substituted fluorophosphate was slower in the TMS electrolyte. Sloping charge–discharge profiles and broad CV peaks suggest a single-phase (solid-solution) reaction mechanism, similar to Li2CoPO4F [4,8]. There is no visible change in the operating potential of Li2Co0.9Mn0.1PO4F. Therefore it was difficult to draw a decisive conclusion on the effect of Mn-substitution on the electrochemical activity of the Li2CoPO4F system. A further optimization in synthesis and formulation of the cathode material (particle investigation and carbon coating) of Mn- and Fe-substituted fluorophosphates will improve their electrochemical performance and, thereby, answer the question about a possible fine tuning of the operating voltage of this fluorophosphate family through substitutions on the transition metal site.
![[2190-4286-4-97-8]](/bjnano/content/figures/2190-4286-4-97-8.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: Charge–discharge curves of Li2Co0.9Mn0.1PO4/C in the commercial (a) and the sulfone-based (b) electrolytes measured at C/5.
Figure 8: Charge–discharge curves of Li2Co0.9Mn0.1PO4/C in the commercial (a) and the sulfone-based (b) elect...
Conclusion
New fluorophosphates, Li2Co1−xMnxPO4F and Li2Co1−xFexPO4F, were successfully synthesized and investigated. Both systems exhibited narrow ranges of solid solution that agreed well with the ionic sizes of the transition metals. Good cycling and capacity behavior was attained with the 1 M LiBF4/TMS electrolyte. Galvanostatic measurements revealed a reversible electrochemical activity with discharge capacities as high as 90 and 75 mA·h·g−1 for Li2CoPO4F and Li2Co0.9Mn0.1PO4F respectively. A further investigation that includes the optimization of the electrode materials and the development of a high-voltage electrolyte is required to evaluate all potentials of this Li2Co1−xMxPO4F (M = Mn, Fe) fluorophosphate family.
Experimental
The fluorophosphates, Li2CoPO4F and Li2Co1−xMxPO4F (M = Fe, Mn) were synthesized in a two-steps process. In the first step LiCo1−xMxPO4 olivine precursors were prepared through freeze-drying or ceramic techniques, depending on the transition metal. In the second step the obtained olivine samples were ball-milled with 1.05 equiv of LiF (5% excess), pelletized, annealed at 650–720 °C for 1–2 h under Ar flow and, subsequently, quenched to room temperature. As noticed above the technique applied to synthesize olivine precursors depended on the transition metal. Thus, LiCoPO4 was prepared by a solid-state reaction from a stoichiometric mixture of Li2CO3 (99.1%), (NH4)H2PO4 (99%), Co(NO3)2·6H2O (99.9%). It should be noted that the purity of initial reagents was checked by X-ray diffraction, the weight form of the crystallohydrates that were used for sample preparation was verified by thermogravimetric analysis. The initial reagents were mixed by planetary ball-milling, pelletized and then annealed in a tubular furnace (under steady Ar flow) at 380 °C for 10 h and at 600 °C for 15 h with intermediate regrinding. The Fe-substituted olivine precursors, LiCo1−xFexPO4, were obtained from stoichiometric mixtures of Li2CO3, NH4H2PO4, FeC2O4·2H2O (99%) and CoC2O4·2H2O (99%). The annealing profile was similar to that described for LiCoPO4. The oxalates, FeC2O4·2H2O and CoC2O4·2H2O, were chosen as initial reagents because the mixture of CO and CO2 released upon their decomposition suppressed the Fe2+ oxidation (in contrast to NO2 evolved by nitrates) and, at the same time, did not reduce Co2+ to metallic Co.
The Mn-substituted olivine precursors, LiCo1−xMnxPO4, were prepared through the freeze spraying technique. The initial reagents LiCH3COO (99%), NH4H2PO4, Co(NO3)2·6H2O and Mn(CH3COO)2·3.2H2O were dissolved in distilled water and combined to form a transparent solution with a pH value of 3.0–3.5, which was adjusted by adding 1 M CH3COOH. This solution was exposed to freeze-spraying in liquid nitrogen, and the obtained product was subjected to vacuum sublimation in a Labconco sublimator (pressure 0.2 mbar, temperature range −40 to +30 °C) for 70 h. The prepared granulate was pressed into pellets and annealed at 350 °C for 10 h and at 550 °C for 15 h under Ar flow with intermediate regrinding.
The carbon-containing composites, Li2Co1−xMxPO4F/C, were prepared by adding carbon black (3–5 wt %) to the initial mixtures or, as in the case of M = Mn, to the products obtained from cryogranulates by annealing at 350 °C. The amount of residual carbon in the obtained composites was determined by thermal analysis and taken into account during the preparation of the electrodes.
Mechanical grindings (180–200 rpm for 2–3 h) were carried out in a Fritsch planetary micro-mill Pulverisette 7 while using a WC bowl, ZrO2 balls and acetone media. Thermal analysis was performed in the temperature range of 20–750 °C (10 °C/min heating rate) by using a thermo-gravimetric differential scanning calorimetry (TG-DSC) apparatus STA-449 (Netzsch, Germany).
The samples were characterized by powder X-ray diffraction (XRD) using a Huber G670 Guinier camera (Cu Kα1 radiation, Ge monochromator, image plate detector) and Bruker D8 Advance with a Lynxeye detector (Cu Kα radiation). The quantitative phase analysis for the selected samples was carried out by Rietveld refinement with the program JANA 2006 [17]. SEM investigation of powdered samples was performed with a JEOL JSM-6490LV scanning electron microscope equipped with an energy dispersive X-ray spectroscopy (EDX) attachment.
The electrochemical evaluation was performed in two-electrode-configuration cells with Li-metal foil acting both as the reference and counter electrodes, borosilicate glass was used as a separator. The positive electrodes were prepared by thoroughly mixing the active material (80 wt %) with carbon Timcal Super C (10 wt %) and PVdF (10 wt %) dissolved in a minimal amount of N-methyl-pyrrolidone. This cathode slurry was cast on an Al-foil collector by using the doctor-blade technique with a typical loading of 1 mg·cm−2. The prepared electrodes were dried, rolled and then dried again at 100 °C under vacuum for several hours. The electrochemical evaluation was carried out by using the following electrolytes: 1) 1 М LiPF6 solution in ethylene carbonate (EC) and dimethylcarbonate (DMC) with a volume ratio of 1:1 (commercial electrolyte, Merck); 2) 1 M solution of LiBF4 in tetramethylene sulfone (TMS). The latter electrolyte was prepared by dissolving an appropriate amount of LiBF4 (99.99%, Aldrich) in TMS that was purified up to 99.8% before. The electrochemical cells were assembled in an Ar-filled glove box. All tested cells were left to relax before the measurements (10–20 h). A potentiostat/galvanostat Biologic VMP-3 was used for data collecting. The cyclic voltammetry scanning was performed in the voltage range of 2.5–5.5 V at a scan rate of 0.1 mV·s−1. The galvanostatic charge–discharge cycling was conducted in the voltage range of 2.5–5.5 V at a rate of C/5 (the current required to deintercalate one Li ion from Li2Co1-xMxPO4F in 5 hours).
Acknowledgements
Financial support from LG Chem, Ltd. is gratefully acknowledged. The work was supported in part by Russian Foundation for Basic Research (RFBR grant 13-03-00495a), contract with the Ministry of Education and Science of Russian Federation (No. 16.526.11.6011) and Moscow State University Program of Development.
References
-
Okada, S.; Ueno, M.; Uebou, Y.; Yamaki, J.-i. J. Power Sources 2005, 146, 565–569. doi:10.1016/j.jpowsour.2005.03.149
Return to citation in text: [1] [2] -
Dutreilh, M.; Chevalier, C.; El-Ghozzi, M.; Avignant, D. J. Solid State Chem. 1999, 142, 1–5. doi:10.1006/jssc.1998.7908
Return to citation in text: [1] [2] -
Hadermann, J.; Abakumov, A. M.; Turner, S.; Hafideddine, Z.; Khasanova, N. R.; Antipov, E. V.; Van Tendeloo, G. Chem. Mater. 2011, 23, 3540–3545. doi:10.1021/cm201257b
Return to citation in text: [1] -
Khasanova, N. R.; Gavrilov, A. N.; Antipov, E. V.; Bramnik, K. G.; Hibst, H. J. Power Sources 2011, 196, 355–360. doi:10.1016/j.jpowsour.2010.06.086
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Wang, D.; Xiao, J.; Xua, W.; Nie, Z.; Wang, C.; Graff, G.; Zhang, J.-G. J. Power Sources 2011, 196, 2241–2245. doi:10.1016/j.jpowsour.2010.10.021
Return to citation in text: [1] [2] -
Dumont-Botto, E.; Bourbon, C.; Patoux, S.; Rozier, P.; Dolle, M. J. Power Sources 2011, 196, 2274–2278. doi:10.1016/j.jpowsour.2010.09.037
Return to citation in text: [1] -
Wu, X.; Gong, Z.; Tan, S.; Yang, Y. J. Power Sources 2012, 220, 122–129. doi:10.1016/j.jpowsour.2012.07.099
Return to citation in text: [1] -
Amaresh, S.; Kim, G. J.; Karthikeyan, K.; Aravindan, V.; Chung, K. Y.; Choc, B. W.; Lee, Y. S. Phys. Chem. Chem. Phys. 2012, 14, 11904–11909. doi:10.1039/c2cp41624g
Return to citation in text: [1] [2] [3] [4] -
Kosova, N. V.; Devyatkina, E. T.; Slobodyuk, A. B. Solid State Ionics 2012, 225, 570–574. doi:10.1016/j.ssi.2011.11.007
Return to citation in text: [1] -
Nagahama, M.; Hasegawa, N.; Okada, S. J. Electrochem. Soc. 2010, 157, A748–A752. doi:10.1149/1.3417068
Return to citation in text: [1] [2] [3] -
Xu, K. Chem. Rev. 2004, 104, 4303–4417. doi:10.1021/cr030203g
Return to citation in text: [1] [2] -
Abouimrane, A.; Whitfield, P. S.; Niketic, S.; Davidson, I. J. J. Power Sources 2007, 174, 883–888. doi:10.1016/j.jpowsour.2007.06.103
Return to citation in text: [1] [2] -
Watanabe, Y.; Kinoshita, S.-i.; Wada, S.; Hoshino, K.; Morimoto, H.; Tobishima, S.-i. J. Power Sources 2008, 179, 770–779. doi:10.1016/j.jpowsour.2008.01.006
Return to citation in text: [1] [2] -
Abu-Lebdeh, Y.; Davidson, I. J. Electrochem. Soc. 2009, 156, A60–A65. doi:10.1149/1.3023084
Return to citation in text: [1] -
Shannon, R. D. Acta Crystallogr., Sect. A 1976, 32, 751–767. doi:10.1107/S0567739476001551
Return to citation in text: [1] -
Khasanova, N. R.; Drozhzhin, O. A.; Storozhilova, D. A.; Delmas, C.; Antipov, E. V. Chem. Mater. 2012, 24, 4271–4273. doi:10.1021/cm302724a
Return to citation in text: [1] -
Petricek, V.; Dusek, M.; Palatinus, L. JANA 2006. The crystallographic computing system; Institute of Physics: Praha, Czech Republic, 2006.
Return to citation in text: [1]
| 17. | Petricek, V.; Dusek, M.; Palatinus, L. JANA 2006. The crystallographic computing system; Institute of Physics: Praha, Czech Republic, 2006. |
| 8. | Amaresh, S.; Kim, G. J.; Karthikeyan, K.; Aravindan, V.; Chung, K. Y.; Choc, B. W.; Lee, Y. S. Phys. Chem. Chem. Phys. 2012, 14, 11904–11909. doi:10.1039/c2cp41624g |
| 4. | Khasanova, N. R.; Gavrilov, A. N.; Antipov, E. V.; Bramnik, K. G.; Hibst, H. J. Power Sources 2011, 196, 355–360. doi:10.1016/j.jpowsour.2010.06.086 |
| 8. | Amaresh, S.; Kim, G. J.; Karthikeyan, K.; Aravindan, V.; Chung, K. Y.; Choc, B. W.; Lee, Y. S. Phys. Chem. Chem. Phys. 2012, 14, 11904–11909. doi:10.1039/c2cp41624g |
| 1. | Okada, S.; Ueno, M.; Uebou, Y.; Yamaki, J.-i. J. Power Sources 2005, 146, 565–569. doi:10.1016/j.jpowsour.2005.03.149 |
| 8. | Amaresh, S.; Kim, G. J.; Karthikeyan, K.; Aravindan, V.; Chung, K. Y.; Choc, B. W.; Lee, Y. S. Phys. Chem. Chem. Phys. 2012, 14, 11904–11909. doi:10.1039/c2cp41624g |
| 4. | Khasanova, N. R.; Gavrilov, A. N.; Antipov, E. V.; Bramnik, K. G.; Hibst, H. J. Power Sources 2011, 196, 355–360. doi:10.1016/j.jpowsour.2010.06.086 |
| 4. | Khasanova, N. R.; Gavrilov, A. N.; Antipov, E. V.; Bramnik, K. G.; Hibst, H. J. Power Sources 2011, 196, 355–360. doi:10.1016/j.jpowsour.2010.06.086 |
| 5. | Wang, D.; Xiao, J.; Xua, W.; Nie, Z.; Wang, C.; Graff, G.; Zhang, J.-G. J. Power Sources 2011, 196, 2241–2245. doi:10.1016/j.jpowsour.2010.10.021 |
| 4. | Khasanova, N. R.; Gavrilov, A. N.; Antipov, E. V.; Bramnik, K. G.; Hibst, H. J. Power Sources 2011, 196, 355–360. doi:10.1016/j.jpowsour.2010.06.086 |
| 5. | Wang, D.; Xiao, J.; Xua, W.; Nie, Z.; Wang, C.; Graff, G.; Zhang, J.-G. J. Power Sources 2011, 196, 2241–2245. doi:10.1016/j.jpowsour.2010.10.021 |
| 6. | Dumont-Botto, E.; Bourbon, C.; Patoux, S.; Rozier, P.; Dolle, M. J. Power Sources 2011, 196, 2274–2278. doi:10.1016/j.jpowsour.2010.09.037 |
| 7. | Wu, X.; Gong, Z.; Tan, S.; Yang, Y. J. Power Sources 2012, 220, 122–129. doi:10.1016/j.jpowsour.2012.07.099 |
| 8. | Amaresh, S.; Kim, G. J.; Karthikeyan, K.; Aravindan, V.; Chung, K. Y.; Choc, B. W.; Lee, Y. S. Phys. Chem. Chem. Phys. 2012, 14, 11904–11909. doi:10.1039/c2cp41624g |
| 9. | Kosova, N. V.; Devyatkina, E. T.; Slobodyuk, A. B. Solid State Ionics 2012, 225, 570–574. doi:10.1016/j.ssi.2011.11.007 |
| 15. | Shannon, R. D. Acta Crystallogr., Sect. A 1976, 32, 751–767. doi:10.1107/S0567739476001551 |
| 2. | Dutreilh, M.; Chevalier, C.; El-Ghozzi, M.; Avignant, D. J. Solid State Chem. 1999, 142, 1–5. doi:10.1006/jssc.1998.7908 |
| 3. | Hadermann, J.; Abakumov, A. M.; Turner, S.; Hafideddine, Z.; Khasanova, N. R.; Antipov, E. V.; Van Tendeloo, G. Chem. Mater. 2011, 23, 3540–3545. doi:10.1021/cm201257b |
| 4. | Khasanova, N. R.; Gavrilov, A. N.; Antipov, E. V.; Bramnik, K. G.; Hibst, H. J. Power Sources 2011, 196, 355–360. doi:10.1016/j.jpowsour.2010.06.086 |
| 16. | Khasanova, N. R.; Drozhzhin, O. A.; Storozhilova, D. A.; Delmas, C.; Antipov, E. V. Chem. Mater. 2012, 24, 4271–4273. doi:10.1021/cm302724a |
| 10. | Nagahama, M.; Hasegawa, N.; Okada, S. J. Electrochem. Soc. 2010, 157, A748–A752. doi:10.1149/1.3417068 |
| 4. | Khasanova, N. R.; Gavrilov, A. N.; Antipov, E. V.; Bramnik, K. G.; Hibst, H. J. Power Sources 2011, 196, 355–360. doi:10.1016/j.jpowsour.2010.06.086 |
| 1. | Okada, S.; Ueno, M.; Uebou, Y.; Yamaki, J.-i. J. Power Sources 2005, 146, 565–569. doi:10.1016/j.jpowsour.2005.03.149 |
| 4. | Khasanova, N. R.; Gavrilov, A. N.; Antipov, E. V.; Bramnik, K. G.; Hibst, H. J. Power Sources 2011, 196, 355–360. doi:10.1016/j.jpowsour.2010.06.086 |
| 10. | Nagahama, M.; Hasegawa, N.; Okada, S. J. Electrochem. Soc. 2010, 157, A748–A752. doi:10.1149/1.3417068 |
| 11. | Xu, K. Chem. Rev. 2004, 104, 4303–4417. doi:10.1021/cr030203g |
| 12. | Abouimrane, A.; Whitfield, P. S.; Niketic, S.; Davidson, I. J. J. Power Sources 2007, 174, 883–888. doi:10.1016/j.jpowsour.2007.06.103 |
| 13. | Watanabe, Y.; Kinoshita, S.-i.; Wada, S.; Hoshino, K.; Morimoto, H.; Tobishima, S.-i. J. Power Sources 2008, 179, 770–779. doi:10.1016/j.jpowsour.2008.01.006 |
| 14. | Abu-Lebdeh, Y.; Davidson, I. J. Electrochem. Soc. 2009, 156, A60–A65. doi:10.1149/1.3023084 |
| 2. | Dutreilh, M.; Chevalier, C.; El-Ghozzi, M.; Avignant, D. J. Solid State Chem. 1999, 142, 1–5. doi:10.1006/jssc.1998.7908 |
| 10. | Nagahama, M.; Hasegawa, N.; Okada, S. J. Electrochem. Soc. 2010, 157, A748–A752. doi:10.1149/1.3417068 |
| 12. | Abouimrane, A.; Whitfield, P. S.; Niketic, S.; Davidson, I. J. J. Power Sources 2007, 174, 883–888. doi:10.1016/j.jpowsour.2007.06.103 |
| 13. | Watanabe, Y.; Kinoshita, S.-i.; Wada, S.; Hoshino, K.; Morimoto, H.; Tobishima, S.-i. J. Power Sources 2008, 179, 770–779. doi:10.1016/j.jpowsour.2008.01.006 |
© 2013 Khasanova et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Nanotechnology terms and conditions: (http://www.beilstein-journals.org/bjnano)