Abstract
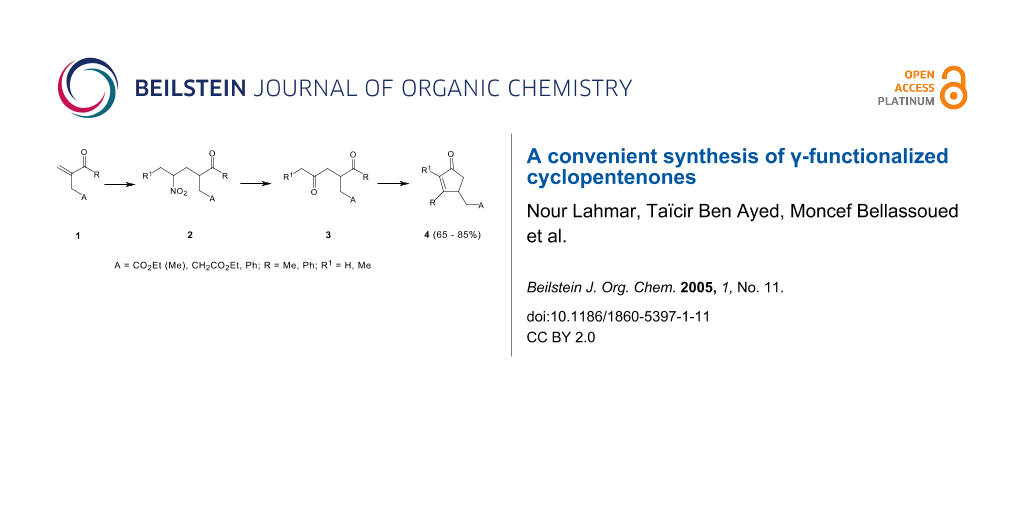
The synthesis of γ-functionalized cyclopentenones was carried out in a few steps, starting firstly with the preparation of nitroketonic intermediates 2, which were readily transformed into 1,4-diketones using the Nef conversion. The intramolecular cyclization of the γ-diketones 3 in a basic medium, led to the functionalized cyclopentenones 4.

Graphical Abstract
Introduction
The cyclopentenones are considered as an important class of compounds because they are present in a large variety of natural products and in many important biologically active compounds [1-6] such as prostanoids, jasmonoids, rethrolones, methylenomycins and allylrethrones. In this paper, we wish to describe a new synthetic procedure for the preparation of γ-functionalized cyclopentenones 4 via a conjugate addition of nitroalkanes to α,β-unsaturated ketones 1 leading to the nitroalkanes derivatives 2 which may be converted into their γ-diketone homologues 3 using Nef reaction. The intramolecular cyclization of 1,4-diketones 3 led to the corresponding cyclopentenones 4.
Results and discussion
As previously reported, the 1,4-addition of primary nitroalkanes [7-12] to functionalized α,β-unsaturated ketones was considered as an appropriate method to prepare multifunctional nitroketonic compounds. These intermediates are important in organic synthesis because the nitro group can be converted into other useful groups such as amines or carbonyls.[13,14] Treatment of α,β-unsaturated ketones 1[15] with primary nitroalkanes was achieved by refluxing the mixture at 50°C in THF using sodium methoxide as base (Scheme 1), resulting in full conversion and yielding the Michael adducts 2 (Table 1).
![[1860-5397-1-11-i1]](/bjoc/content/inline/1860-5397-1-11-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of nitroalkanes derivatives 2 and their corresponding 1,4-diketones 3.
Scheme 1: Synthesis of nitroalkanes derivatives 2 and their corresponding 1,4-diketones 3.
Table 1: Synthesis of nitroalkanes derivatives 2 and their corresponding 1,4-diketones 3
| A | R | R1 | 1,4-Nitroketone | Yield (%) | 1,4-Diketone | Yield (%) |
|---|---|---|---|---|---|---|
| CO2Me | Me | H | 2a | 65 | 3a | 67 |
| CO2Et | Me | H | 2b | 62 | 3a | 64* |
| CH2CO2Et | Me | H | 2c | 61 | 3c | 53* |
| Ph | Me | H | 2d | 76 | 3d | 68 |
| CO2Me | Ph | H | 2e | 68 | 3e | 64 |
| CO2Me | Me | Me | 2f | 61 | 3f | 63 |
| CO2Et | Me | Me | 2g | 62 | 3f | 62* |
| CH2CO2Et | Me | Me | 2h | 61 | 3h | 48* |
| Ph | Me | Me | 2i | 76 | 3i | 65 |
| CO2Me | Ph | Me | 2j | 70 | 3j | 63 |
*This compound undergoes transesterification reaction.
Various synthetic methods were developed to obtain 1,4-diketones [16-20] since these compounds were valuable intermediates in the synthesis of cyclopentenone ring system. Obviously the most straightforward route was the Nef reaction. The transformation of the nitro derivatives to carbonyls by the formation of the nitronate anion in the presence of sodium methoxide followed by its addition to concentrated sulfuric acid at -50°C, allowed the formation of the 1,4-diketones 3 in fair to good yields (Table 1).
During recent years, some cyclopentenones derivatives were known to exhibit biological and pharmaceutical properties and have been widely studied [21-23]. In order to contribute to the synthesis of this family of products, we present in this work a strategy of preparation of a new γ-functionalized cyclopentenones 4. Thus, we showed that intramolecular cyclization of the functionalized 1,4-diketones 3 could be conducted in the presence of anhydrous potassium carbonate (K2CO3) in the refluxing methanol to afford the pure cyclopentenones 4 with satisfactory yields (Table 2, Scheme 2).
![[1860-5397-1-11-i2]](/bjoc/content/inline/1860-5397-1-11-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: γ-Functionalized cyclopentenones 4.
Scheme 2: γ-Functionalized cyclopentenones 4.
Conclusion
A convenient procedure has been developed for the synthesis of γ-functionalized cyclopentenones 4 using the reaction between nitroalkanes and α,β-unsaturated ketones 1 to afford γ-nitroketonic intermediates 2 which may be converted into their γ-diketones homologues 3 using Nef reaction. The intramolecular cyclization of diketones 3 leads to the corresponding cyclopentenones 4. The easy workup procedure and short reaction steps, impart greater merit of this methodology over those existing.
Experimental
All reactions are carried out under a nitrogen atmosphere. Unless otherwise noted, all products are purified before use. 1H-NMR spectra (300 MHz) and 13C NMR spectra (75 MHz) were recorded on Bruker AC 300 MHz spectrophotometers using CDCl3 as solvent. Chemical shifts (δ) are given from TMS (0 ppm) as internal standard for 1H-NMR and CDCl3 (77.0 ppm) for 13C NMR. Flash chromatography was done on Merck grade 60 silica gel (230–400 mesh) using mixtures of hexane and ethyl acetate as eluent.
Synthesis of nitroketones (2): General procedure
To a mixture of nitroalkanes (32 mmol) and sodium methoxide (32 mmol) in anhydrous THF (20 mL) was added vinylketone 1 (a-j) (8 mmol), the solution was stirred for 13 h at 50°C, the mixture was treated with H2O and extracted with Et2O (3 × 20 mL). The organic layer was washed with brine and dried (MgSO4). The solvent was removed to leave an oil which was purified by column chromatography on silica gel.
3-Acetyl-5-nitrohexanoic acid methyl ester (2a)
Purified by column chromatography (hexane/AcOEt, 7/3). 1H NMR (300 MHz, CDCl3) δ 1.55 (d, 3H, J = 6.7 Hz), 2.29 (s, 3H), 2.37–2.79 (2 m, 4 H), 2.92 (m, 1H), 3.67 (s, 3H), 4.55 (m, 1H); 13C NMR (75 MHz, CDCl3) δ 19.8, 29.8, 34.4, 36.2, 44.7, 52.0, 81.6, 171.4, 208.4. HRMS Calcd C9H15NO5: 217.0950. Found: 217.0932.
Preparation of functionalized 1,4-diketones (3): General procedure
At the solution of sodium (7.5 mmol) in 15 mL of methanol, was added at room temperature the nitro intermediate 2 (5 mmol). After one hour, a solution of H2SO4 (3 mL) in 15 mL of methanol at -50°C was added. The mixture was treated with H2O, the methanol was evaporated, and the residue was extracted with dichloromethane. The organic layer was washed by a solution of NaOH 1% and a solution of NaCl and dried (MgSO4). The solvent was removed to leave oil, which was purified by column chromatography on silica gel.
3-Acetyl-5-oxohexanoic acid methyl ester (3a)
Purified by column chromatography (hexane/AcOEt, 7/3). 1H NMR (300 MHz, CDCl3) δ 2.15 (s, 3H), 2.28 (s, 3H), 2.43; 2.65 (ABd, 2H, JAB = 16.5 Hz, JA-H = 7 Hz, JB-H = 6.6 Hz), 2.60; 2.93 (ABd, 2H, JAB = 18.1 Hz, JA-H = 8 Hz, JB-H = 5.5 Hz), 3.36 (m, 1H), 3.67 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 29.2, 29.8, 35.0, 42.9, 44.4, 51.9, 172.0, 206.2, 209.5. HRMS Calcd C9H14O4: 186.0892. Found: 186.0877.
Preparation of γ-functionalized cyclopentenones (4): General procedure
To a solution of 1,4-diketone 3 (5 mmol) in MeOH (10 mL) was added 1 equivalent of K2CO3, the mixture was bring to reflux during one hour. After workup, the product 4 was purified by column chromatography on silica gel (EtOAc/hexane).
4-Benzyl-3-methylcyclopent-2-enone (4d)
Purified by column chromatography (hexane/AcOEt, 8/2). 1H NMR (300 MHz, CDCl3) δ 2.09 (s, 3H), 2.26–2.59 (m, 2H), 2.71 (m, 1H), 3.21 (m, 2H), 5.92 (s, 1H), 7.17–7.28 (m, 5H); 13C NMR (75 MHz, CDCl3) δ 19.4, 37.0, 39.0, 48.0, 126.3, 128.4, 128.8, 129.8, 139.6, 177.6, 211.0. HRMS Calcd C13H14O: 186.1045. Found: 186.1029
References
-
Mikolajczyk, M.; Grzejszczak, S.; Midura, W.; Zatorski, A. Phosphorus Sulfur Relat. Elem. 1983, 18, 175–178.
Return to citation in text: [1] -
Romanet, R. F.; Schlessinger, R. H. J. Am. Chem. Soc. 1974, 96, 3701–3702. doi:10.1021/ja00818a082
Return to citation in text: [1] -
Ballini, R. Synthesis 1993, 687–688. doi:10.1055/s-1993-25922
Return to citation in text: [1] -
Mikolajczyk, M.; Mikina, M.; Zurawinski, R. Pure Appl. Chem. 1999, 71, 473–480.
Return to citation in text: [1] -
Mikolajczyk, M.; Grzejszczak, S.; Lyzwa, P. Tetrahedron Lett. 1982, 23, 2237–2240. doi:10.1016/S0040-4039(00)87310-4
Return to citation in text: [1] -
Rezgui, F.; Amri, H.; El Gaïed, M. M. Tetrahedron 2003, 59, 1369–1380. doi:10.1016/S0040-4020(03)00035-8
Return to citation in text: [1] -
Ballini, R.; Bosica, G.; Petrelli, L.; Petrini, M. Synthesis 1999, 1236–1240. doi:10.1055/s-1999-3533
Return to citation in text: [1] -
Ballini, R.; Bosica, G.; Fiorini, D.; Righi, P. Synthesis 2002, 681–685. doi:10.1055/s-2002-23548
Return to citation in text: [1] -
Nef, J. U. Liebigs Ann. Chem. 1894, 280, 263–291.
Return to citation in text: [1] -
Ballini, R.; Bosica, G.; Livi, D. Synthesis 2001, 1519–1522. doi:10.1055/s-2001-16098
Return to citation in text: [1] -
Chamakh, A.; M'hirsi, M.; Villiéras, J.; Lebreton, J.; Amri, H. Synthesis 2000, 2, 295–299. doi:10.1055/s-2000-6257
Return to citation in text: [1] -
Hbaïeb, S.; Amri, H. J. Soc. Chim. Tunis. 2000, 671–681.
Return to citation in text: [1] -
Seebach, D.; Colvin, E. W.; Lehr, F.; Weller, T. Chimia 1979, 33, 1–18.
Return to citation in text: [1] -
Kende, A. S.; Mendoza, J. S. Tetrahedron Lett. 1991, 32, 1699–1702. doi:10.1016/S0040-4039(00)74307-3
Return to citation in text: [1] -
Ben Ayed, T.; Amri, H. Synth. Commun. 1995, 25, 3813–3819.
Return to citation in text: [1] -
Mc murry, J. E.; Melton, J. J. Am. Chem. Soc. 1971, 93, 5309–5311. doi:10.1021/ja00749a086
Return to citation in text: [1] -
Mc Murry, J. E.; Melton, J. J. Org. Chem. 1973, 38, 4367–4373. doi:10.1021/jo00965a004
Return to citation in text: [1] -
Bartlett, P. A.; Green, F. R., III; Webb, T. R. Tetrahedron Lett. 1977, 331–334. doi:10.1016/S0040-4039(01)92629-2
Return to citation in text: [1] -
Bellassoued, M.; Dardoise, F.; Gaudemar, M. J. Organomet. Chem. 1979, 177, 35–38. doi:10.1016/S0022-328X(00)92329-5
Return to citation in text: [1] -
Ballini, R.; Bartoli, G. Synthesis 1993, 965–967. doi:10.1055/s-1993-25981
Return to citation in text: [1] -
Pohmakotr, M.; Chancharunee, S. Tetrahedron Lett. 1984, 25, 4141–4144. doi:10.1016/S0040-4039(01)90204-7
Return to citation in text: [1] -
Welch, S. C.; Assercq, J. M.; Loh, J. P.; Glase, S. A. J. Org. Chem. 1987, 52, 1440–1450. doi:10.1021/jo00384a012
Return to citation in text: [1] -
Ballini, R.; Petrini, M.; Marcantoni, E.; Rosini, G. Synthesis 1988, 231–233. doi:10.1055/s-1988-27524
Return to citation in text: [1]
| 1. | Mikolajczyk, M.; Grzejszczak, S.; Midura, W.; Zatorski, A. Phosphorus Sulfur Relat. Elem. 1983, 18, 175–178. |
| 2. | Romanet, R. F.; Schlessinger, R. H. J. Am. Chem. Soc. 1974, 96, 3701–3702. doi:10.1021/ja00818a082 |
| 3. | Ballini, R. Synthesis 1993, 687–688. doi:10.1055/s-1993-25922 |
| 4. | Mikolajczyk, M.; Mikina, M.; Zurawinski, R. Pure Appl. Chem. 1999, 71, 473–480. |
| 5. | Mikolajczyk, M.; Grzejszczak, S.; Lyzwa, P. Tetrahedron Lett. 1982, 23, 2237–2240. doi:10.1016/S0040-4039(00)87310-4 |
| 6. | Rezgui, F.; Amri, H.; El Gaïed, M. M. Tetrahedron 2003, 59, 1369–1380. doi:10.1016/S0040-4020(03)00035-8 |
| 16. | Mc murry, J. E.; Melton, J. J. Am. Chem. Soc. 1971, 93, 5309–5311. doi:10.1021/ja00749a086 |
| 17. | Mc Murry, J. E.; Melton, J. J. Org. Chem. 1973, 38, 4367–4373. doi:10.1021/jo00965a004 |
| 18. | Bartlett, P. A.; Green, F. R., III; Webb, T. R. Tetrahedron Lett. 1977, 331–334. doi:10.1016/S0040-4039(01)92629-2 |
| 19. | Bellassoued, M.; Dardoise, F.; Gaudemar, M. J. Organomet. Chem. 1979, 177, 35–38. doi:10.1016/S0022-328X(00)92329-5 |
| 20. | Ballini, R.; Bartoli, G. Synthesis 1993, 965–967. doi:10.1055/s-1993-25981 |
| 13. | Seebach, D.; Colvin, E. W.; Lehr, F.; Weller, T. Chimia 1979, 33, 1–18. |
| 14. | Kende, A. S.; Mendoza, J. S. Tetrahedron Lett. 1991, 32, 1699–1702. doi:10.1016/S0040-4039(00)74307-3 |
| 7. | Ballini, R.; Bosica, G.; Petrelli, L.; Petrini, M. Synthesis 1999, 1236–1240. doi:10.1055/s-1999-3533 |
| 8. | Ballini, R.; Bosica, G.; Fiorini, D.; Righi, P. Synthesis 2002, 681–685. doi:10.1055/s-2002-23548 |
| 9. | Nef, J. U. Liebigs Ann. Chem. 1894, 280, 263–291. |
| 10. | Ballini, R.; Bosica, G.; Livi, D. Synthesis 2001, 1519–1522. doi:10.1055/s-2001-16098 |
| 11. | Chamakh, A.; M'hirsi, M.; Villiéras, J.; Lebreton, J.; Amri, H. Synthesis 2000, 2, 295–299. doi:10.1055/s-2000-6257 |
| 12. | Hbaïeb, S.; Amri, H. J. Soc. Chim. Tunis. 2000, 671–681. |
| 21. | Pohmakotr, M.; Chancharunee, S. Tetrahedron Lett. 1984, 25, 4141–4144. doi:10.1016/S0040-4039(01)90204-7 |
| 22. | Welch, S. C.; Assercq, J. M.; Loh, J. P.; Glase, S. A. J. Org. Chem. 1987, 52, 1440–1450. doi:10.1021/jo00384a012 |
| 23. | Ballini, R.; Petrini, M.; Marcantoni, E.; Rosini, G. Synthesis 1988, 231–233. doi:10.1055/s-1988-27524 |
© 2005 Lahmar et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)