Abstract
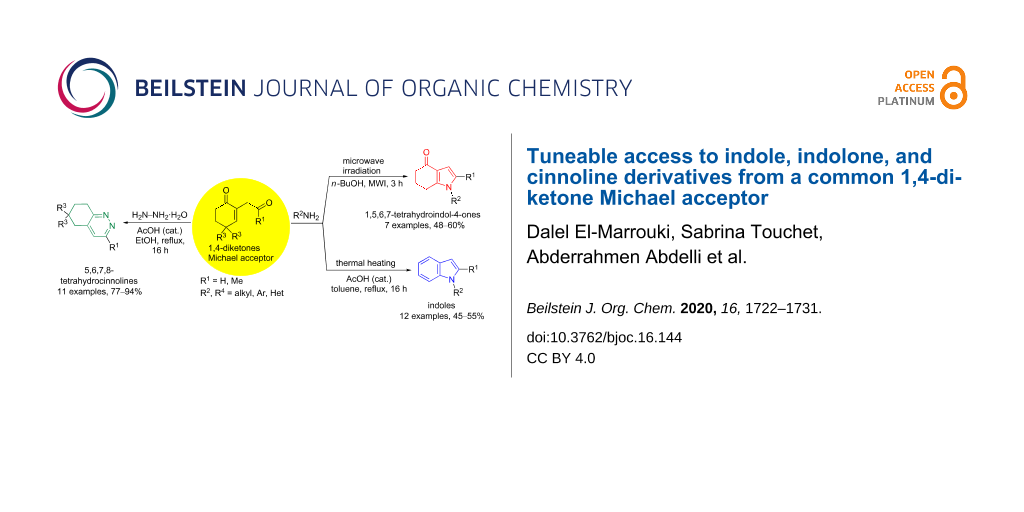
A convergent strategy is reported for the construction of nitrogen-containing heterocycles from common substrates: 1,4-diketones and primary amines. Indeed, by just varying the substrates, the substituents, or the heating mode, it is possible to selectively synthesize indole, indolone (1,5,6,7-tetrahydroindol-4-one), or cinnoline (5,6,7,8-tetrahydrocinnoline) derivatives in moderate to excellent yields.

Graphical Abstract
Introduction
Nitrogen-containing heterocycles are widespread in plenty of molecules of interest, either in materials science, optics, electronics, or biology [1-4]. They are also very useful building blocks to create more sophisticated organic molecules. Therefore, the search for efficient methods for the synthesis of nitrogen-containing heterocycles is crucial to both organic and medicinal chemists. Among these, indole, indolone (1,5,6,7-tetrahydroindol-4-one), and cinnoline (5,6,7,8-tetrahydrocinnoline) derivatives are important classes of functionalized compounds, having biological and medicinal activities of interest (Figure 1) [5-8].
![[1860-5397-16-144-1]](/bjoc/content/figures/1860-5397-16-144-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Examples of bioactive nitrogen-containing heterocycles (indole [9], indolone [10], and cinnoline [11] derivatives).
Figure 1: Examples of bioactive nitrogen-containing heterocycles (indole [9], indolone [10], and cinnoline [11] derivati...
Indeed, indole ring-containing compounds have various biological and pharmacological activities and are part of many marketed drugs used as anticancer, antiemetic, antihypertensive, antidepressant, anti-inflammatory, or anti-HIV agents, among others [9]. In contrast, concerning indolone and cinnoline derivatives, there are very few marketed drugs, but many molecules are under investigations for their activities as antibacterial, antifungal, anticancer, or anti-inflammatory agents or even on the central nervous system [7,12,13].
Several routes have been reported to access these key compounds, the most developed being for the indole [14] derivatives using the Fischer indole synthesis involving sigmatropic rearrangements [15-18], nucleophilic and electrophilic cyclizations [19-25], reductive and oxidative cyclizations [26,27], and transition-metal catalysis [28-37]. There are fewer ways to access indolone derivatives, mainly based on the use of di- [12,38,39] or triketones [10,13] and enaminones [40-43] as starting materials. For the synthesis of cinnoline derivatives, aryldiazenes and aryltriazenes are substrates of choice for transition-metal-catalyzed (Rh, Pd, Cu) cross-coupling reactions, followed by intramolecular cyclizations [44-47]. Moreover, arylhydrazones and arylhydrazines/hydrazines can be used as well, respectively, as partners in [4 + 2] cyclization reactions [48-51] or by reacting mostly with carbonyl derivatives [52-55].
From the state-of-the-art, a strategy that promotes the synthesis of indole, indolone, or cinnoline derivatives from the same starting material is not yet available. To reach this goal, the Michael reaction between 1,4-diketones and primary amines seems particularly attractive because of its straightforward and metal-free properties, and because they can be performed under air. Herein, we report our investigations on this reaction, and we have shown that it can be selectively directed towards the synthesis of indole, indolone, or cinnoline derivatives by just changing the substrates, substituents, or heating mode (Scheme 1).
![[1860-5397-16-144-i1]](/bjoc/content/inline/1860-5397-16-144-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: General strategy to access indole, indolone, and cinnoline derivatives from 1,4-diketones.
Scheme 1: General strategy to access indole, indolone, and cinnoline derivatives from 1,4-diketones.
Results and Discussion
The synthesis of the target compounds required the prior preparation of a panel of variously substituted 1,4-diketones 5. The 1,4-diketones 5 have been prepared either by a Nef reaction [56] from the corresponding nitroenone 3 or a Wittig reaction [57] from 1,2-cyclohexanedione and the corresponding ylide 4 (Scheme 2).
![[1860-5397-16-144-i2]](/bjoc/content/inline/1860-5397-16-144-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of the 1,4-diketones 5a–k via the Nef reaction or the Wittig reaction. i) HCHO (aq), DMAP, THF, rt, 24 h; ii) Ac2O, Et3N, DMAP, 0 °C, then 2 h, rt; iii) R1–CH2–NO2, Et3N, EtOH, reflux, 24 h; iv) EtONa, EtOH, rt, 3 h, then H2SO4, −50 °C, 1 h; v) toluene or DCM, reflux, 48 h, then rt, 2 d. aVia the Nef reaction. bVia the Wittig reaction.
Scheme 2: Synthesis of the 1,4-diketones 5a–k via the Nef reaction or the Wittig reaction. i) HCHO (aq), DMAP...
The nitrenones 3a–d were obtained in three steps from the appropriate commercially available cyclohexenones (Scheme 2). First, a Baylis–Hillman reaction between cyclohexanone and formaldehyde led to the formation of the corresponding Baylis–Hillman alcohols 1a/b in good yield [58], followed by a DMAP-catalyzed acetylation of these alcohols, which gave the corresponding acetates 2a–d [59]. The nitrenones 3a–d were finally obtained in an acceptable yield by reacting the acetate derivatives with the appropriate nitroalkanes [60]. The next step was the transformation of the nitro group of 3a–d via the Nef reaction, using sodium ethoxide in ethanol, followed by the hydrolysis with concentrated sulfuric acid at a low temperature [56], leading to the corresponding new γ-diketones 5a–d in 61–87% yield (Scheme 2).
Despite the efficiency of the Nef reaction, the diversity at the R1 position via this synthetic route remains limited in terms of chemical diversity as it depends on the availability of the corresponding nitro derivative. It was thus decided to move on to the Wittig reaction [57], offering a much more straightforward and efficient route to a panel of new diketones, 5e–k, from 1,2-cyclohexanedione and the corresponding Wittig ylides 4a–g (readily accessible from the corresponding halogenated derivatives, Scheme 2). Indeed, the Wittig reaction leads exclusively to the isomerized products 5.
With the 1,4-diketones 5 in hand, we first investigated the synthesis of indole and indolone derivatives. The reaction mechanism shown in Scheme 3 involves the formation of an imine upon the reaction of the primary amine with the most reactive carbonyl moiety (nonconjugated and exocyclic carbonyl function). Then, depending on the reaction conditions, the imine can react following a 1,2- or 1,4-addition process, leading respectively to an indole 6 (after dehydration and aromatization) or an indolone 7. The reaction was first investigated by mixing the diketone 5b as the Michael acceptor and benzylamine under various conditions (Table 1).
![[1860-5397-16-144-i3]](/bjoc/content/inline/1860-5397-16-144-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Mechanism of the formation of indole and indolone derivatives.
Scheme 3: Mechanism of the formation of indole and indolone derivatives.
Table 1: Effects of solvent and heating mode on the 6b:7b ratio.
![[Graphic 1]](/bjoc/content/inline/1860-5397-16-144-i10.svg?max-width=637&scale=1.0)
|
||||||
| entrya | catalyst | solvent | t (h) | 6b (%)b | 7b (%)b | 6b:7b |
| 1 | TfOH (3%) | toluene | 16 h | – | – | – |
| 2 | AgOTf (3%) | toluene | 16 h | – | – | – |
| 3 | TFA (3%) | toluene | 16 h | – | – | – |
| 4 | pTsOH (3%) | toluene | 16 h | – | – | – |
| 5 | AcOH (3%) | toluene | 16 h | 47 | 10 | 83:17 |
| 6 | – | toluene | 16 h | 33 | 8 | 80:20 |
| 7 | – | CH2Cl2 | 16 h | – | – | – |
| 8 | – | THF | 16 h | – | – | – |
| 9 | – | ethanol | 16 h | 13 | 16 | 45:55 |
| 10 | – | propanol | 16 h | 6 | 31 | 16:84 |
| 11 | – | butanol | 16 h | 10 | 43 | 19:81 |
| 12 | – | pentanol | 16 h | 7 | 28 | 20:80 |
| 13c | – | butanol | MWI 3 h | – | 60 | 0:100 |
aReaction conditions: 5b (0.54 mmol), primary amine (0.81 mmol), 4 mL solvent, and catalyst (0.02 mmol) unless otherwise specified (in column 2, the catalyst percentage corresponds to 0.02 mmol). bIsolated yield. c13 mL of butanol, MWI 100 °C.
We first investigated the reactivity in the presence of a set of catalysts with different acidities (Table 1, entries 1–5). Among them, only acetic acid afforded reaction products, while the others only produced complex mixtures of degradation products. Under the conditions of entry 5, the indole 6b was isolated in 47%, and the indolone 7b was also formed concomitantly in 10% yield. The experiments in Table 1, entries 6–11 were next performed with the aim to favor the 1,4-addition process, and thus to form the indolone 7b. Removing the acid catalyst from the reaction mixture (Table 1, entry 6) did not affect the 6b:7b ratio obtained in Table 1, entry 5. While in aprotic solvents, other than toluene (Table 1, entries 7 and 8), the reaction produced complex mixtures of degradation products, and using alcohols had a notable impact on the reaction contents. Indeed, going from ethanol to propanol, and thus increasing the refluxing temperature, led to the indolone 7b as the main product, with the best yield being obtained in butanol (43%, Table 1, entry 11), however, with the formation of 6b occurring only in 10% yield. Switching to microwave irradiation formed exclusively 7b in 60% yield after 3 h (Table 1, entry 13). Note, that to check the effect of a shorter reaction time on the reaction outcome, we reduced the time to 3 h also under the classical refluxing conditions from Table 1, entry 11 and obtained a partial conversion of the starting diketone 5b. Despite the side formation of the indolone 7b in Table 1, entry 5, these conditions were applied to several amines, producing the corresponding substituted indoles 6a and 6c–f in 41–54% yield (Scheme 4). The yield of the indolones 7a and 7c–f was found almost constant (10–14%) with all amines involved. It is worthy of note that the two compounds were easily separated using usual chromatographic techniques.
![[1860-5397-16-144-i4]](/bjoc/content/inline/1860-5397-16-144-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of the indoles 6a–f and the corresponding side product indolones 7a–f.
Scheme 4: Synthesis of the indoles 6a–f and the corresponding side product indolones 7a–f.
The reaction was also applied to a diamine (Scheme 5). When 1,3-diaminopropane was used, the bisindole 6g was isolated in 46% yield. Interestingly, the mixed indolone/indole compound 9 was also obtained as a side product. However, no traces of the bisindolone derivative were detected.
![[1860-5397-16-144-i5]](/bjoc/content/inline/1860-5397-16-144-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
We then succeeded in directing the reaction exclusively towards indole formation by reacting the diketone with appropriate substrates, combining a primary amine with a tertiary amine or a pyridine separated by several spacer arms (Scheme 6).
![[1860-5397-16-144-i6]](/bjoc/content/inline/1860-5397-16-144-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Under these conditions, the functional indoles 6h–l were obtained exclusively in 45–55% yield. We assume that the tertiary amine would interact with the protonated intermediate, and thus promoting the 1,2-addition (Scheme 6). However, for the pyridine substituent (compounds 6k and 6l), another intermediate may be involved as the shape of this diamine does not allow enough flexibility to achieve the conformation of the proposed intermediate.
We next examined the preparation of a set of indolones under the microwave conditions determined in Table 1, entry 13. These conditions were applied to several amines, producing exclusively the corresponding substituted indolones 7d and 7g–k in 48–56% yield (Scheme 7).
![[1860-5397-16-144-i7]](/bjoc/content/inline/1860-5397-16-144-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Synthesis of the indolone derivatives 7b, 7d, and 7g–k.
Scheme 7: Synthesis of the indolone derivatives 7b, 7d, and 7g–k.
Here again, the amount of indolone was found to be almost constant, whatever the amine involved was, suggesting that the reaction is not dependent on the nature of the amine. It is worth to notice that, for a substrate combining a primary amine with a pyridine separated by a spacer arm, only the indolone derivative is obtained in those conditions as well (compound 7i, Scheme 7). Based on these results, we found it important to check whether the indole 6b resulted from a 1,2-addition and not from a degradation of the indolone 7b. For this purpose, the indolone 7b was refluxed overnight with acetic acid in toluene, under these conditions producing mainly the indole (Table 1, entry 5). The indolone 7b was found unchanged, with no trace of the indole 6b being detected (see Supporting Information File 1, chapter I), indicating that the indole was formed intramolecularly by a 1,2-addition of the intermediately formed imine to the Michael acceptor (Scheme 3).
We then investigated the synthesis of cinnoline derivatives by mixing the diketone 5a and hydrazine monohydrate under various conditions (Table 2). We first investigated the reactivity in ethanol, as a protic solvent, at room temperature (Table 2, entry 1). Under these conditions, the expected cinnoline 8a was obtained in a low yield of 20% that could be increased up to 40% upon refluxing the mixture (Table 2, entry 2). Switching to toluene (Table 2, entry 3) did not improve the reaction outcome. However, the addition of a catalytic amount of acetic acid in refluxing ethanol while shortening the reaction time dramatically increased the yield of 8a up to 82% (Table 2, entry 4).
Table 2: Optimization of the reaction conditions for the synthesis of the cinnolines 8.
![[Graphic 2]](/bjoc/content/inline/1860-5397-16-144-i11.svg?max-width=637&scale=1.0)
|
|||||
| entrya | catalyst | solvent | T (°C) | t (h) | 8a (%)b |
| 1 | – | EtOH | rt | 48 | 20 |
| 2 | – | EtOH | reflux | 48 | 40 |
| 3 | – | toluene | reflux | 48 | 35 |
| 4 | AcOH (3 mol %) | EtOH | reflux | 16 | 82 |
aReaction conditions: 5a (1 mmol), hydrazine monohydrate (1.5 mmol), solvent (6 mL), catalyst (0.03 mmol), unless otherwise specified. The reported catalyst percentage exactly corresponds to 0.03 mmol. bIsolated yield.
These optimized conditions were then applied to the previously synthesized 1,4-diketones 5a–k (Scheme 8). As a general observation, the reaction was found to be efficient for producing the expected cinnoline derivatives 8a–k in good to excellent yield (77–94%) and tolerated alkyl, aromatic, and heteroaromatic groups as R1.
![[1860-5397-16-144-i8]](/bjoc/content/inline/1860-5397-16-144-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Synthesis of the cinnoline derivatives 8a–k.
Scheme 8: Synthesis of the cinnoline derivatives 8a–k.
The success of our convergent strategy here can be explained through the mechanism suggested in Scheme 9. The synthetic pathway leading to the formation of the indolone 7 starts with an imine formation between the secondary amine and the nonconjugated carbonyl from the 1,4-diketone. After an imine–enamine equilibrium, an intramolecular 1,4-addition to the Michael acceptor part of the molecule occurs, followed by a prototropy, leading to an intermediate enol that, after a keto–enol equilibrium and aromatization, gives the indolone 7. For the indole 6 and the cinnoline 8, the synthesis starts with the protonation of the oxygen atom of the conjugated carbonyl group of the 1,4-diketone, followed by an imine formation between the secondary amine and the nonconjugated carbonyl unit. Next, an intramolecular 1,2-addition to the Michael acceptor part of the molecule, previously activated by acid catalysis, takes place (after an imine–enamine equilibrium for the indole pathway), followed by a prototropy, the release of a water molecule, the recovery of the proton catalyst, and atmospheric oxygen aromatization, leading to the indole 6 or the cinnoline 8.
![[1860-5397-16-144-i9]](/bjoc/content/inline/1860-5397-16-144-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Proposed mechanism for the preparation of the compounds 6, 7, and 8.
Scheme 9: Proposed mechanism for the preparation of the compounds 6, 7, and 8.
Conclusion
In summary, we have successfully developed a straightforward and metal-free strategy for the synthesis of nitrogen-containing heterocyclic moieties of biological interest; indoles 6, indolones 7, and cinnolines 8, starting from common substrates 1,4-diketones 5 and primary amines. The protocols developed here used mild conditions, were functional-group tolerant, transition-metal-free, proceeded in moderate to good yield, and could therefore easily be used in medicinal chemistry projects for the rapid access to a wide range of variously substituted compounds for structure–activity relationship studies. The biological activity of the molecules is currently being studied.
Supporting Information
| Supporting Information File 1: Experimental procedures, characterization data, and copies of the spectra of all compounds. | ||
| Format: PDF | Size: 7.9 MB | Download |
References
-
Kumar, D.; Kumar Jain, S. Curr. Med. Chem. 2016, 23, 4338–4394. doi:10.2174/0929867323666160809093930
Return to citation in text: [1] -
Liu, J.-C.; Narva, S.; Zhou, K.; Zhang, W. Mini-Rev. Med. Chem. 2019, 19, 1517–1530. doi:10.2174/1389557519666190312152358
Return to citation in text: [1] -
Wang, S.-Y.; Shi, X.-C.; Laborda, P. Eur. J. Med. Chem. 2020, 185, 111847. doi:10.1016/j.ejmech.2019.111847
Return to citation in text: [1] -
Rubina, B.; Dharam P, P.; Garima, K.; Ravi, K.; Manni, D. Indian J. Pharm. Educ. Res. 2019, 53, s1–s16. doi:10.5530/ijper.53.2s.44
Return to citation in text: [1] -
Singh, T. P.; Singh, O. M. Mini-Rev. Med. Chem. 2018, 18, 9–25. doi:10.2174/1389557517666170807123201
Return to citation in text: [1] -
Thanikachalam, P. V.; Maurya, R. K.; Garg, V.; Monga, V. Eur. J. Med. Chem. 2019, 180, 562–612. doi:10.1016/j.ejmech.2019.07.019
Return to citation in text: [1] -
Lewgowd, W.; Stanczak, A. Arch. Pharm. (Weinheim, Ger.) 2007, 340, 65–80. doi:10.1002/ardp.200500194
Return to citation in text: [1] [2] -
Awad, E. D.; El-Abadelah, M. M.; Matar, S.; Zihlif, M. A.; Naffa, R. G.; Al-Momani, E. Q.; Mubarak, M. S. Molecules 2012, 17, 227–239. doi:10.3390/molecules17010227
Return to citation in text: [1] -
Kaushik, N. K.; Kaushik, N.; Attri, P.; Kumar, N.; Kim, C. H.; Verma, A. K.; Choi, E. H. Molecules 2013, 18, 6620–6662. doi:10.3390/molecules18066620
Return to citation in text: [1] [2] -
Martínez, R.; Clara-Sosa, A.; Ramírez Apan, M. T. Bioorg. Med. Chem. 2007, 15, 3912–3918. doi:10.1016/j.bmc.2006.12.018
Return to citation in text: [1] [2] -
Yuvaraj, T. V.; Unnissa, S. H.; Surendiran, N. S.; Ur rahman, M. A.; Binumon, V. Asian J. Res. Chem. 2010, 3, 853–858.
Return to citation in text: [1] -
Batchu, H.; Batra, S. Eur. J. Org. Chem. 2012, 2935–2944. doi:10.1002/ejoc.201200107
Return to citation in text: [1] [2] -
Khalafy, J.; Badparvar, F.; Mariani, A. P. J. Chil. Chem. Soc. 2016, 61, 3112–3115. doi:10.4067/s0717-97072016000300021
Return to citation in text: [1] [2] -
Gribble, G. W. J. Chem. Soc., Perkin Trans. 1 2000, 1045–1075. doi:10.1039/a909834h
Return to citation in text: [1] -
Vuong, H.; Duarte, S.; Klumpp, D. A. Top. Catal. 2018, 61, 685–688. doi:10.1007/s11244-018-0908-3
Return to citation in text: [1] -
Ghiyasabadi, Z.; Bahadorikhalili, S.; Saeedi, M.; Karimi‐Niyazagheh, M.; Mirfazli, S. S. J. Heterocycl. Chem. 2020, 57, 606–610. doi:10.1002/jhet.3790
Return to citation in text: [1] -
Irgashev, R. A.; Steparuk, A. S.; Rusinov, G. L. Tetrahedron Lett. 2019, 60, 151185. doi:10.1016/j.tetlet.2019.151185
Return to citation in text: [1] -
Yu, J.; Xu, J.; Yu, Z.; Jin, Y.; Li, J.; Lv, Y. J. Flow Chem. 2017, 7, 33–36. doi:10.1556/1846.2017.00004
Return to citation in text: [1] -
Chesnokov, G. A.; Ageshina, A. A.; Topchiy, M. A.; Nechaev, M. S.; Asachenko, A. F. Eur. J. Org. Chem. 2019, 4844–4854. doi:10.1002/ejoc.201900772
Return to citation in text: [1] -
Yao, T.; Yue, D.; Larock, R. C. J. Org. Chem. 2005, 70, 9985–9989. doi:10.1021/jo0517038
Return to citation in text: [1] -
Zhao, H.; Yuan, H.; Zhang, Y.; Li, R.; Li, W. Org. Lett. 2019, 21, 6557–6561. doi:10.1021/acs.orglett.9b02507
Return to citation in text: [1] -
Yue, D.; Larock, R. C. Org. Lett. 2004, 6, 1037–1040. doi:10.1021/ol0498996
Return to citation in text: [1] -
Chen, Y.; Cho, C.-H.; Shi, F.; Larock, R. C. J. Org. Chem. 2009, 74, 6802–6811. doi:10.1021/jo9014003
Return to citation in text: [1] -
Yue, D.; Yao, T.; Larock, R. C. J. Org. Chem. 2006, 71, 62–69. doi:10.1021/jo051549p
Return to citation in text: [1] -
Zhao, G.; Roudaut, C.; Gandon, V.; Alami, M.; Provot, O. Green Chem. 2019, 21, 4204–4210. doi:10.1039/c9gc01880h
Return to citation in text: [1] -
Sathishkumar, P. N.; Prabha, P. S.; Bhuvanesh, N. S. P.; Karvembu, R. J. Organomet. Chem. 2020, 908, 121087. doi:10.1016/j.jorganchem.2019.121087
Return to citation in text: [1] -
Rode, N. D.; Abdalghani, I.; Arcadi, A.; Aschi, M.; Chiarini, M.; Marinelli, F. J. Org. Chem. 2018, 83, 6354–6362. doi:10.1021/acs.joc.8b00508
Return to citation in text: [1] -
He, Y.-P.; Wu, H.; Wang, Q.; Zhu, J. Angew. Chem., Int. Ed. 2020, 59, 2105–2109. doi:10.1002/anie.201914049
Return to citation in text: [1] -
Arcadi, A.; Cacchi, S.; Fabrizi, G.; Goggiamani, A.; Iazzetti, A.; Marinelli, F. Org. Biomol. Chem. 2013, 11, 545–548. doi:10.1039/c2ob27125g
Return to citation in text: [1] -
Yao, B.; Wang, Q.; Zhu, J. Angew. Chem., Int. Ed. 2012, 51, 5170–5174. doi:10.1002/anie.201201640
Return to citation in text: [1] -
Yao, B.; Wang, Q.; Zhu, J. Angew. Chem., Int. Ed. 2013, 52, 12992–12996. doi:10.1002/anie.201307738
Return to citation in text: [1] -
Zhao, G.; Bignon, J.; Levaique, H.; Dubois, J.; Alami, M.; Provot, O. J. Org. Chem. 2018, 83, 15323–15332. doi:10.1021/acs.joc.8b02563
Return to citation in text: [1] -
Krüger (née Alex), K.; Tillack, A.; Beller, M. Adv. Synth. Catal. 2008, 350, 2153–2167. doi:10.1002/adsc.200800409
Return to citation in text: [1] -
Ho, H. E.; Oniwa, K.; Yamamoto, Y.; Jin, T. Org. Lett. 2016, 18, 2487–2490. doi:10.1021/acs.orglett.6b01067
Return to citation in text: [1] -
Sperança, A.; Godoi, B.; Menezes, P.; Zeni, G. Synlett 2013, 24, 1125–1132. doi:10.1055/s-0033-1338427
Return to citation in text: [1] -
Zeng, X.; Kinjo, R.; Donnadieu, B.; Bertrand, G. Angew. Chem., Int. Ed. 2010, 49, 942–945. doi:10.1002/anie.200905341
Return to citation in text: [1] -
Filatova, E. A.; Pozharskii, A. F.; Gulevskaya, A. V.; Ozeryanskii, V. A. J. Org. Chem. 2015, 80, 872–881. doi:10.1021/jo502363t
Return to citation in text: [1] -
Heravi, M. M.; Baghernejad, B.; Oskooie, H. A.; Hekmatshoar, R. Tetrahedron Lett. 2008, 49, 6101–6103. doi:10.1016/j.tetlet.2008.08.012
Return to citation in text: [1] -
Wu, S.; Fluxe, A.; Janusz, J. M.; Sheffer, J. B.; Browning, G.; Blass, B.; Cobum, K.; Hedges, R.; Murawsky, M.; Fang, B.; Fadayel, G. M.; Hare, M.; Djandjighian, L. Bioorg. Med. Chem. Lett. 2006, 16, 5859–5863. doi:10.1016/j.bmcl.2006.08.057
Return to citation in text: [1] -
To, Q. H.; Lee, Y. R.; Kim, S. H. Tetrahedron 2014, 70, 8108–8113. doi:10.1016/j.tet.2014.08.011
Return to citation in text: [1] -
Lan, X.-C.; Chen, T.-T.; Zhao, Y.; Wu, Y.; Wang, J.; Tu, S.-J.; Jiang, B.; Hao, W.-J. Tetrahedron Lett. 2017, 58, 1519–1522. doi:10.1016/j.tetlet.2017.03.009
Return to citation in text: [1] -
Jiang, B.; Yi, M.-S.; Shi, F.; Tu, S.-J.; Pindi, S.; McDowell, P.; Li, G. Chem. Commun. 2012, 48, 808–810. doi:10.1039/c1cc15913e
Return to citation in text: [1] -
Jiang, B.; Li, Q.-Y.; Zhang, H.; Tu, S.-J.; Pindi, S.; Li, G. Org. Lett. 2012, 14, 700–703. doi:10.1021/ol203166c
Return to citation in text: [1] -
Zhu, C.; Yamane, M. Tetrahedron 2011, 67, 4933–4938. doi:10.1016/j.tet.2011.04.079
Return to citation in text: [1] -
Zhao, D.; Wu, Q.; Huang, X.; Song, F.; Lv, T.; You, J. Chem. – Eur. J. 2013, 19, 6239–6244. doi:10.1002/chem.201300155
Return to citation in text: [1] -
Muralirajan, K.; Cheng, C.-H. Chem. – Eur. J. 2013, 19, 6198–6202. doi:10.1002/chem.201300922
Return to citation in text: [1] -
Danilkina, N. A.; Bukhtiiarova, N. S.; Govdi, A. I.; Vasileva, A. A.; Rumyantsev, A. M.; Volkov, A. A.; Sharaev, N. I.; Povolotskiy, A. V.; Boyarskaya, I. A.; Kornyakov, I. V.; Tokareva, P. V.; Balova, I. A. Molecules 2019, 24, 2386. doi:10.3390/molecules24132386
Return to citation in text: [1] -
Attanasi, O. A.; Favi, G.; Filippone, P.; Perrulli, F. R.; Santeusanio, S. Org. Lett. 2009, 11, 309–312. doi:10.1021/ol802432z
Return to citation in text: [1] -
South, M. S.; Jakuboski, T. L.; Westmeyer, M. D.; Dukesherer, D. R. Tetrahedron Lett. 1996, 37, 1351–1354. doi:10.1016/0040-4039(96)00020-2
Return to citation in text: [1] -
South, M. S.; Jakuboski, T. L.; Westmeyer, M. D.; Dukesherer, D. R. J. Org. Chem. 1996, 61, 8921–8934. doi:10.1021/jo960029e
Return to citation in text: [1] -
South, M. S.; Jakuboski, T. L. Tetrahedron Lett. 1995, 36, 5703–5706. doi:10.1016/0040-4039(95)01128-5
Return to citation in text: [1] -
Chang, M.-Y.; Lu, Y.-J.; Cheng, Y.-C. Tetrahedron 2015, 71, 6840–6845. doi:10.1016/j.tet.2015.07.025
Return to citation in text: [1] -
Fukumoto, S.; Imamiya, E.; Kusumoto, K.; Fujiwara, S.; Watanabe, T.; Shiraishi, M. J. Med. Chem. 2002, 45, 3009–3021. doi:10.1021/jm0104567
Return to citation in text: [1] -
Khalafy, J.; Rimaz, M.; Ezzati, M.; Poursattar Marjani, A. Curr. Chem. Lett. 2013, 2, 43–48. doi:10.5267/j.ccl.2012.11.002
Return to citation in text: [1] -
Altomare, C.; Cellamare, S.; Summo, L.; Catto, M.; Carotti, A.; Thull, U.; Carrupt, P.-A.; Testa, B.; Stoeckli-Evans, H. J. Med. Chem. 1998, 41, 3812–3820. doi:10.1021/jm981005y
Return to citation in text: [1] -
Chamakh, A.; M'hirsi, M.; Villiéras, J.; Lebreton, J.; Amri, H. Synthesis 2000, 295–299. doi:10.1055/s-2000-6257
Return to citation in text: [1] [2] -
Mawaziny, S.; Lawny, A. M. Phosphorus, Sulfur Silicon Relat. Elem. 2000, 163, 99–120. doi:10.1080/10426500008046614
Return to citation in text: [1] [2] -
Rezgui, F.; El Gaied, M. M. Tetrahedron Lett. 1998, 39, 5965–5966. doi:10.1016/s0040-4039(98)01206-4
Return to citation in text: [1] -
Rezgui, F.; El Gaïed, M. M. Tetrahedron 1997, 53, 15711–15716. doi:10.1016/s0040-4020(97)10023-0
Return to citation in text: [1] -
Mhasni, O.; Rezgui, F. Tetrahedron Lett. 2010, 51, 586–587. doi:10.1016/j.tetlet.2009.11.053
Return to citation in text: [1]
| 1. | Kumar, D.; Kumar Jain, S. Curr. Med. Chem. 2016, 23, 4338–4394. doi:10.2174/0929867323666160809093930 |
| 2. | Liu, J.-C.; Narva, S.; Zhou, K.; Zhang, W. Mini-Rev. Med. Chem. 2019, 19, 1517–1530. doi:10.2174/1389557519666190312152358 |
| 3. | Wang, S.-Y.; Shi, X.-C.; Laborda, P. Eur. J. Med. Chem. 2020, 185, 111847. doi:10.1016/j.ejmech.2019.111847 |
| 4. | Rubina, B.; Dharam P, P.; Garima, K.; Ravi, K.; Manni, D. Indian J. Pharm. Educ. Res. 2019, 53, s1–s16. doi:10.5530/ijper.53.2s.44 |
| 11. | Yuvaraj, T. V.; Unnissa, S. H.; Surendiran, N. S.; Ur rahman, M. A.; Binumon, V. Asian J. Res. Chem. 2010, 3, 853–858. |
| 40. | To, Q. H.; Lee, Y. R.; Kim, S. H. Tetrahedron 2014, 70, 8108–8113. doi:10.1016/j.tet.2014.08.011 |
| 41. | Lan, X.-C.; Chen, T.-T.; Zhao, Y.; Wu, Y.; Wang, J.; Tu, S.-J.; Jiang, B.; Hao, W.-J. Tetrahedron Lett. 2017, 58, 1519–1522. doi:10.1016/j.tetlet.2017.03.009 |
| 42. | Jiang, B.; Yi, M.-S.; Shi, F.; Tu, S.-J.; Pindi, S.; McDowell, P.; Li, G. Chem. Commun. 2012, 48, 808–810. doi:10.1039/c1cc15913e |
| 43. | Jiang, B.; Li, Q.-Y.; Zhang, H.; Tu, S.-J.; Pindi, S.; Li, G. Org. Lett. 2012, 14, 700–703. doi:10.1021/ol203166c |
| 10. | Martínez, R.; Clara-Sosa, A.; Ramírez Apan, M. T. Bioorg. Med. Chem. 2007, 15, 3912–3918. doi:10.1016/j.bmc.2006.12.018 |
| 44. | Zhu, C.; Yamane, M. Tetrahedron 2011, 67, 4933–4938. doi:10.1016/j.tet.2011.04.079 |
| 45. | Zhao, D.; Wu, Q.; Huang, X.; Song, F.; Lv, T.; You, J. Chem. – Eur. J. 2013, 19, 6239–6244. doi:10.1002/chem.201300155 |
| 46. | Muralirajan, K.; Cheng, C.-H. Chem. – Eur. J. 2013, 19, 6198–6202. doi:10.1002/chem.201300922 |
| 47. | Danilkina, N. A.; Bukhtiiarova, N. S.; Govdi, A. I.; Vasileva, A. A.; Rumyantsev, A. M.; Volkov, A. A.; Sharaev, N. I.; Povolotskiy, A. V.; Boyarskaya, I. A.; Kornyakov, I. V.; Tokareva, P. V.; Balova, I. A. Molecules 2019, 24, 2386. doi:10.3390/molecules24132386 |
| 9. | Kaushik, N. K.; Kaushik, N.; Attri, P.; Kumar, N.; Kim, C. H.; Verma, A. K.; Choi, E. H. Molecules 2013, 18, 6620–6662. doi:10.3390/molecules18066620 |
| 12. | Batchu, H.; Batra, S. Eur. J. Org. Chem. 2012, 2935–2944. doi:10.1002/ejoc.201200107 |
| 38. | Heravi, M. M.; Baghernejad, B.; Oskooie, H. A.; Hekmatshoar, R. Tetrahedron Lett. 2008, 49, 6101–6103. doi:10.1016/j.tetlet.2008.08.012 |
| 39. | Wu, S.; Fluxe, A.; Janusz, J. M.; Sheffer, J. B.; Browning, G.; Blass, B.; Cobum, K.; Hedges, R.; Murawsky, M.; Fang, B.; Fadayel, G. M.; Hare, M.; Djandjighian, L. Bioorg. Med. Chem. Lett. 2006, 16, 5859–5863. doi:10.1016/j.bmcl.2006.08.057 |
| 5. | Singh, T. P.; Singh, O. M. Mini-Rev. Med. Chem. 2018, 18, 9–25. doi:10.2174/1389557517666170807123201 |
| 6. | Thanikachalam, P. V.; Maurya, R. K.; Garg, V.; Monga, V. Eur. J. Med. Chem. 2019, 180, 562–612. doi:10.1016/j.ejmech.2019.07.019 |
| 7. | Lewgowd, W.; Stanczak, A. Arch. Pharm. (Weinheim, Ger.) 2007, 340, 65–80. doi:10.1002/ardp.200500194 |
| 8. | Awad, E. D.; El-Abadelah, M. M.; Matar, S.; Zihlif, M. A.; Naffa, R. G.; Al-Momani, E. Q.; Mubarak, M. S. Molecules 2012, 17, 227–239. doi:10.3390/molecules17010227 |
| 10. | Martínez, R.; Clara-Sosa, A.; Ramírez Apan, M. T. Bioorg. Med. Chem. 2007, 15, 3912–3918. doi:10.1016/j.bmc.2006.12.018 |
| 13. | Khalafy, J.; Badparvar, F.; Mariani, A. P. J. Chil. Chem. Soc. 2016, 61, 3112–3115. doi:10.4067/s0717-97072016000300021 |
| 15. | Vuong, H.; Duarte, S.; Klumpp, D. A. Top. Catal. 2018, 61, 685–688. doi:10.1007/s11244-018-0908-3 |
| 16. | Ghiyasabadi, Z.; Bahadorikhalili, S.; Saeedi, M.; Karimi‐Niyazagheh, M.; Mirfazli, S. S. J. Heterocycl. Chem. 2020, 57, 606–610. doi:10.1002/jhet.3790 |
| 17. | Irgashev, R. A.; Steparuk, A. S.; Rusinov, G. L. Tetrahedron Lett. 2019, 60, 151185. doi:10.1016/j.tetlet.2019.151185 |
| 18. | Yu, J.; Xu, J.; Yu, Z.; Jin, Y.; Li, J.; Lv, Y. J. Flow Chem. 2017, 7, 33–36. doi:10.1556/1846.2017.00004 |
| 26. | Sathishkumar, P. N.; Prabha, P. S.; Bhuvanesh, N. S. P.; Karvembu, R. J. Organomet. Chem. 2020, 908, 121087. doi:10.1016/j.jorganchem.2019.121087 |
| 27. | Rode, N. D.; Abdalghani, I.; Arcadi, A.; Aschi, M.; Chiarini, M.; Marinelli, F. J. Org. Chem. 2018, 83, 6354–6362. doi:10.1021/acs.joc.8b00508 |
| 14. | Gribble, G. W. J. Chem. Soc., Perkin Trans. 1 2000, 1045–1075. doi:10.1039/a909834h |
| 28. | He, Y.-P.; Wu, H.; Wang, Q.; Zhu, J. Angew. Chem., Int. Ed. 2020, 59, 2105–2109. doi:10.1002/anie.201914049 |
| 29. | Arcadi, A.; Cacchi, S.; Fabrizi, G.; Goggiamani, A.; Iazzetti, A.; Marinelli, F. Org. Biomol. Chem. 2013, 11, 545–548. doi:10.1039/c2ob27125g |
| 30. | Yao, B.; Wang, Q.; Zhu, J. Angew. Chem., Int. Ed. 2012, 51, 5170–5174. doi:10.1002/anie.201201640 |
| 31. | Yao, B.; Wang, Q.; Zhu, J. Angew. Chem., Int. Ed. 2013, 52, 12992–12996. doi:10.1002/anie.201307738 |
| 32. | Zhao, G.; Bignon, J.; Levaique, H.; Dubois, J.; Alami, M.; Provot, O. J. Org. Chem. 2018, 83, 15323–15332. doi:10.1021/acs.joc.8b02563 |
| 33. | Krüger (née Alex), K.; Tillack, A.; Beller, M. Adv. Synth. Catal. 2008, 350, 2153–2167. doi:10.1002/adsc.200800409 |
| 34. | Ho, H. E.; Oniwa, K.; Yamamoto, Y.; Jin, T. Org. Lett. 2016, 18, 2487–2490. doi:10.1021/acs.orglett.6b01067 |
| 35. | Sperança, A.; Godoi, B.; Menezes, P.; Zeni, G. Synlett 2013, 24, 1125–1132. doi:10.1055/s-0033-1338427 |
| 36. | Zeng, X.; Kinjo, R.; Donnadieu, B.; Bertrand, G. Angew. Chem., Int. Ed. 2010, 49, 942–945. doi:10.1002/anie.200905341 |
| 37. | Filatova, E. A.; Pozharskii, A. F.; Gulevskaya, A. V.; Ozeryanskii, V. A. J. Org. Chem. 2015, 80, 872–881. doi:10.1021/jo502363t |
| 7. | Lewgowd, W.; Stanczak, A. Arch. Pharm. (Weinheim, Ger.) 2007, 340, 65–80. doi:10.1002/ardp.200500194 |
| 12. | Batchu, H.; Batra, S. Eur. J. Org. Chem. 2012, 2935–2944. doi:10.1002/ejoc.201200107 |
| 13. | Khalafy, J.; Badparvar, F.; Mariani, A. P. J. Chil. Chem. Soc. 2016, 61, 3112–3115. doi:10.4067/s0717-97072016000300021 |
| 9. | Kaushik, N. K.; Kaushik, N.; Attri, P.; Kumar, N.; Kim, C. H.; Verma, A. K.; Choi, E. H. Molecules 2013, 18, 6620–6662. doi:10.3390/molecules18066620 |
| 19. | Chesnokov, G. A.; Ageshina, A. A.; Topchiy, M. A.; Nechaev, M. S.; Asachenko, A. F. Eur. J. Org. Chem. 2019, 4844–4854. doi:10.1002/ejoc.201900772 |
| 20. | Yao, T.; Yue, D.; Larock, R. C. J. Org. Chem. 2005, 70, 9985–9989. doi:10.1021/jo0517038 |
| 21. | Zhao, H.; Yuan, H.; Zhang, Y.; Li, R.; Li, W. Org. Lett. 2019, 21, 6557–6561. doi:10.1021/acs.orglett.9b02507 |
| 22. | Yue, D.; Larock, R. C. Org. Lett. 2004, 6, 1037–1040. doi:10.1021/ol0498996 |
| 23. | Chen, Y.; Cho, C.-H.; Shi, F.; Larock, R. C. J. Org. Chem. 2009, 74, 6802–6811. doi:10.1021/jo9014003 |
| 24. | Yue, D.; Yao, T.; Larock, R. C. J. Org. Chem. 2006, 71, 62–69. doi:10.1021/jo051549p |
| 25. | Zhao, G.; Roudaut, C.; Gandon, V.; Alami, M.; Provot, O. Green Chem. 2019, 21, 4204–4210. doi:10.1039/c9gc01880h |
| 56. | Chamakh, A.; M'hirsi, M.; Villiéras, J.; Lebreton, J.; Amri, H. Synthesis 2000, 295–299. doi:10.1055/s-2000-6257 |
| 48. | Attanasi, O. A.; Favi, G.; Filippone, P.; Perrulli, F. R.; Santeusanio, S. Org. Lett. 2009, 11, 309–312. doi:10.1021/ol802432z |
| 49. | South, M. S.; Jakuboski, T. L.; Westmeyer, M. D.; Dukesherer, D. R. Tetrahedron Lett. 1996, 37, 1351–1354. doi:10.1016/0040-4039(96)00020-2 |
| 50. | South, M. S.; Jakuboski, T. L.; Westmeyer, M. D.; Dukesherer, D. R. J. Org. Chem. 1996, 61, 8921–8934. doi:10.1021/jo960029e |
| 51. | South, M. S.; Jakuboski, T. L. Tetrahedron Lett. 1995, 36, 5703–5706. doi:10.1016/0040-4039(95)01128-5 |
| 52. | Chang, M.-Y.; Lu, Y.-J.; Cheng, Y.-C. Tetrahedron 2015, 71, 6840–6845. doi:10.1016/j.tet.2015.07.025 |
| 53. | Fukumoto, S.; Imamiya, E.; Kusumoto, K.; Fujiwara, S.; Watanabe, T.; Shiraishi, M. J. Med. Chem. 2002, 45, 3009–3021. doi:10.1021/jm0104567 |
| 54. | Khalafy, J.; Rimaz, M.; Ezzati, M.; Poursattar Marjani, A. Curr. Chem. Lett. 2013, 2, 43–48. doi:10.5267/j.ccl.2012.11.002 |
| 55. | Altomare, C.; Cellamare, S.; Summo, L.; Catto, M.; Carotti, A.; Thull, U.; Carrupt, P.-A.; Testa, B.; Stoeckli-Evans, H. J. Med. Chem. 1998, 41, 3812–3820. doi:10.1021/jm981005y |
| 56. | Chamakh, A.; M'hirsi, M.; Villiéras, J.; Lebreton, J.; Amri, H. Synthesis 2000, 295–299. doi:10.1055/s-2000-6257 |
| 57. | Mawaziny, S.; Lawny, A. M. Phosphorus, Sulfur Silicon Relat. Elem. 2000, 163, 99–120. doi:10.1080/10426500008046614 |
| 59. | Rezgui, F.; El Gaïed, M. M. Tetrahedron 1997, 53, 15711–15716. doi:10.1016/s0040-4020(97)10023-0 |
| 60. | Mhasni, O.; Rezgui, F. Tetrahedron Lett. 2010, 51, 586–587. doi:10.1016/j.tetlet.2009.11.053 |
| 57. | Mawaziny, S.; Lawny, A. M. Phosphorus, Sulfur Silicon Relat. Elem. 2000, 163, 99–120. doi:10.1080/10426500008046614 |
| 58. | Rezgui, F.; El Gaied, M. M. Tetrahedron Lett. 1998, 39, 5965–5966. doi:10.1016/s0040-4039(98)01206-4 |
© 2020 El-Marrouki et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)