Abstract
Pre-queuosine or queuine (preQ1) is a guanine derivative that is involved in the biosynthetic pathway of the hypermodified tRNA nucleoside queuosine (Que). The core structure of preQ1 is represented by 7-(aminomethyl)-7-deazaguanine (preQ1 base). Here, we report the synthesis of three preQ1 base derivatives with complementary 15N-labeling patterns, utilizing [15N]-KCN, [15N]-phthalimide, and [15N3]-guanidine as cost-affordable 15N sources. Such derivatives are required to explore the binding process of the preQ1 base to RNA targets using advanced NMR spectroscopic methods. PreQ1 base specifically binds to bacterial mRNA domains and thereby regulates genes that are required for queuosine biosynthesis.
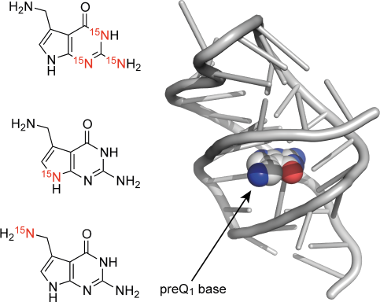
Graphical Abstract
Introduction
The small pyrrolo[2,3-d]pyrimidine 7-(aminomethyl)-7-deazaguanine is a natural product, also termed prequeuosine base (preQ1 base) [1,2]. This guanine derivative is involved in the complex biosynthetic pathway of the hypermodified tRNA nucleoside queuosine [3]. Recently, preQ1 base has attracted considerable attention because this nucleobase specifically binds to bacterial mRNA domains and regulates genes that are required for queuosine biosynthesis, by a so-called riboswitch mechanism [4-8]. To explore the binding process of preQ1 base to the RNA and to shed light on the dynamics underpinning this process advanced NMR spectroscopic methods exist for which 15N-labeled preQ1 base derivatives would be highly beneficial. Here, we report efficient routes for the synthesis of three derivatives with complementary 15N-labeling patterns (Scheme 1).
![[1860-5397-10-199-i1]](/bjoc/content/inline/1860-5397-10-199-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: The hypermodified nucleoside queuosine (Q) and the synthetic targets of preQ1 bases 1 to 3 with complementary 15N labeling patterns for potential NMR spectroscopic applications. Purine and systematic numbering as indicated.
Scheme 1: The hypermodified nucleoside queuosine (Q) and the synthetic targets of preQ1 bases 1 to 3 with com...
The synthesis of preQ1 base has been described first in 1979 by Goto and coworkers from 2-methylthio-6-methoxy-7-methyl-7-deazapurine in 13 steps [9]. Another early, but more efficient procedure was reported by Nishimura in 1988 based on the Mannich reaction using dibenzylamine–formaldehyde and 2-acylaminopyrrolo[2,3-d]pyrimidin-4(3H)-one, which resulted in the selective introduction of the dibenzylaminomethyl group [10]. The following amine exchange reaction of the dibenzylamine function in the Mannich base with ammonia resulted in the preQ1 base. More recently, Carell and coworkers developed a straightforward pathway based on the key reaction of in situ α-brominated 3-phthalimidopropanal with 2,6-diaminopyrimidin-4-one [11], inspired by Grubb’s synthesis of the Q base (queuine) [12]. Alternatively, Klebe and coworkers employed a Michael addition of the same pyrimidinone to the nitroolefin 2-[(2E)-3-nitroprop-2-en-1-yl]-1H-isoindole-1,3(2H)-dione [13], however, this route seemed inconvenient for our purposes because access to the nitroolefin requires several additional steps.
Results and Discussion
Our aim was to develop a robust synthetic pathway to the preQ1 derivatives with the three complementary 15N labeling patterns depicted in Scheme 1. For this undertaking we considered Carell’s synthesis [11] of preQ1 base as a solid foundation that we intended to adapt and modify accordingly, under the premises of efficacy and cost-minimization for 15N incorporation.
For [15N1,15N3,H215N(C2)]-7-(aminomethyl)-7-deazaguanine (1), we started with the reaction of methyl cyanoacetate (4) and [15N3]-guanidine hydrochloride (5) under basic conditions to give the corresponding [15N1,15N3,H215N(C2)]-2,6-diaminopyrimidin-4-one (6) in high purity after work-up and reversed-phase column chromatography (C18) (Scheme 2) [14]. Then, the α-bromo aldehyde 7 was obtained in two steps from commercially available 3-phthalimidopropan-1-ol that was oxidized using Dess–Martin periodinane. Subsequent in situ bromination of the 3-phthalimidopropan-1-al with CH3SiBr, described previously by others [11,12], did not work reliable in our hands.
![[1860-5397-10-199-i2]](/bjoc/content/inline/1860-5397-10-199-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of [15N1,15N3,H215N(C2)]-preQ1 base (1). a) CH3ONa (10 equiv) CH3OH, reflux, 10 h, RP C18 chromatography, 35%. b) NaOAc·3H2O (2 equiv), acetonitrile/water, 40 °C, 4 h, 63%. c) H2N-NH2·H2O (10 equiv), ethanol, reflux, 14 h, RP C18 chromatography, 75%. Compound 1 was isolated as salt of trifluoroacetic acid (TFA).
Scheme 2: Synthesis of [15N1,15N3,H215N(C2)]-preQ1 base (1). a) CH3ONa (10 equiv) CH3OH, reflux, 10 h, RP C18...
Therefore, in anlogy to Grubb [15] and a more detailed protocol by Yamaguchi [16], we applied 5,5-dibromobarbituric acid [17] to obtain the α-bromo aldehyde 7 which was well stable during purification by column chromatography on SiO2 and isolated in good yields. The pyrrolo[2,3-d]pyrimidine ring system of preQ1 base was built in good yields via the cyclocondensation reaction between [15N1,15N3,H215N(C2)]-2,6-diaminopyrimidin-4-one (6) and the 2-bromo-3-phthalimidopropan-1-al (7). Finally, deprotection was performed with hydrazine hydrate. The previously published route [11] recommended N-Boc functionalization of the preQ1 base in the crude reaction mixture to enable flash chromatography on SiO2 followed by cleavage of the auxiliary function, however, although robust in handling, the yields were rather modest. We therefore decided to directly purify the crude product by reversed-phase column chromatography (HPLC) and obtained compound 1 in excellent yield and purity. We mention that compound 1 was isolated as salt of trifluoroacetic acid (TFA) using 1% TFA in the eluent and it was assumed to exist in 1:1 stoichiometry (preQ1:TFA) based on 1H NMR spectra and consideration of pKa values. However, it is noteworthy that a crystal structure of the preQ1·TFA salt that was crystallized from saturated aqueous solution showed the co-existence of mono- (N(C'7)) and dications (N(C'7), N3) in the crystal [11].
The synthetic track for the [15N3]-preQ1 base (1) was designed with the concept in mind to access the complementary 15N patterns of [15N9]-preQ1 base (2) and [H215N(C7')]-preQ1 base (3) by employing the same key steps. In this sense, the key intermediate [H215N(C6)]-2,6-diaminopyrimidin-4-one (13) for target 2 was accessible by first synthesizing ethyl [15N]-2-cyanoacetate (11) from 2-bromoacetic acid (9) and potassium cyanide [15N]-KCN, followed by esterification (Scheme 3).
![[1860-5397-10-199-i3]](/bjoc/content/inline/1860-5397-10-199-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of [15N9]-preQ1 base (2). a) [15N]-KCN (1 equiv), Na2CO3, H2O, pH 9, 80 °C, 3 h, then room temperature, 20 h, 90%. b) Ethanol (5 equiv), H2SO4 cat., reflux, 18 h, 92%. c) CH3ONa (10 equiv) CH3OH, reflux, 10 h, RP C18 chromatography, 40%. d) NaOAc·3H2O (2 equiv), acetonitrile/water, 40 °C, 4 h, 52%. e) H2N-NH2·H2O (10 equiv), ethanol, reflux, 14 h, RP C18 chromatography, 92%. Compound 2 was isolated as salt of trifluoroacetic acid (TFA).
Scheme 3: Synthesis of [15N9]-preQ1 base (2). a) [15N]-KCN (1 equiv), Na2CO3, H2O, pH 9, 80 °C, 3 h, then roo...
All further steps were conducted in direct analogy as described for target 1, namely reaction with guanidine hydrochloride to furnish compound 13, followed by cyclocondensation with 2-bromo-3-phthalimidopropan-1-al (7) to give the protected [15N9]-preQ1 base 14 for subsequent deprotection yielding the desired [15N9]-preQ1 base (2) (Scheme 3).
Also for the third target, [H215N(C7')]-preQ1 base (3), our strategy turn out to be highly convenient. First, we prepared the 15N-labeled aldehyde 18 as the key intermediate (Scheme 4). This was achieved by reaction of 3-chloropropanol (15) with [15N]-phthalimide 16 to give [15N]-3-phthalimidopropan-1-ol (17). All further steps were in direct analogy as described for targets 1 and 2, namely reaction with 5,5-dibromobarbituric acid [17] to obtain [15N]-2-bromo-3-phthalimidopropan-1-al (19), followed by cyclocondensation with commercially available 2,6-diaminopyrimidin-4-one (20) to give the protected [15N(C7')]-preQ1 base 21 for subsequent deprotection yielding the desired [15N(C7')]-preQ1 base (3) (Scheme 4).
![[1860-5397-10-199-i4]](/bjoc/content/inline/1860-5397-10-199-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of [H215N(C7')] preQ1 base (3). a) K2CO3 (1.5 equiv), DMF, 70 °C, 14 h, 47%. b) Dess–Martin periodinane (1.5 equiv), CH2Cl2, 3 h, room temperature. c) 5,5-dibromobarbituric acid (0.6 equiv), acetonitrile, reflux, 2 h, 45%. d) NaOAc·3H2O (2 equiv), acetonitrile/water, 40 °C, 4 h, 58%. e) H2N-NH2·H2O (10 equiv), ethanol, reflux, 14 h, RP C18 chromatography, 82%. Compound 3 was isolated as salt of trifluoroacetic acid (TFA).
Scheme 4: Synthesis of [H215N(C7')] preQ1 base (3). a) K2CO3 (1.5 equiv), DMF, 70 °C, 14 h, 47%. b) Dess–Mart...
Finally, a direct comparison of 1H NMR spectra of the three 15N labeled preQ1 bases synthesized here is provided in Figure 1.
![[1860-5397-10-199-1]](/bjoc/content/figures/1860-5397-10-199-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Comparison of 1H NMR spectra of the preQ1 bases 1, 2 and 3 with complementary 15N labeling patterns. Conditions: cpreQ1 base = 1 mM; DMSO-d6, 298 K.
Figure 1: Comparison of 1H NMR spectra of the preQ1 bases 1, 2 and 3 with complementary 15N labeling patterns...
Conclusion
In this short note, an efficient and cost-minimizing route for 15N labeled preQ1 base derivatives has been described. The synthesis of the pyrrolo[2,3-d]pyrimidine ring system is based on the cyclocondensation reaction between α-bromoaldehydes and 2,6-diaminopyrimidin-4-ones and utilizes [15N]-KCN, [15N]-phthalimide, and [15N3]-guanidine for 15N sources to achieve three complementary labeling patterns that cover all five nitrogen atoms of preQ1 base. The new derivatives carry the potential for modern NMR spectroscopic applications to study the recognition process of these small molecules with RNA aptamer domains from the three preQ1 riboswitch classes known to this date [4,5,18].
Experimental
General.Chemical reagents and solvents were purchased from commercial suppliers (Sigma-Aldrich) and used without further purification. Organic solvents for reactions were dried overnight over freshly activated molecular sieves (4 Å). The reactions were carried out under argon atmosphere. Analytical thin-layer chromatography (TLC) was carried out on Marchery-Nagel Polygram SIL G/UV254 plates. Column chromatography was carried out on silica gel 60 (70–230 mesh). Reversed-phase column chromatography was performed on a GE Healthcare Äktaprime system using a commercial Götec-Labortechnik GmbH 310-25 LiChroprep RP-18 (40–63 µm) column (Merck Lobar compatible). The LC separation was monitored by ultraviolet (UV) detection at 280 nm. Solvent systems were as described below for the individual compounds. 1H and 13C NMR spectra were recorded on Bruker DRX 300 MHz and Bruker Avance II+ 600 MHz instruments. The chemical shifts (δ) are reported relative to tetramethylsilane (TMS) and referenced to the residual proton or carbon signal of the deuterated solvent: CDCl3 (7.26 ppm), DMSO-d6 (2.49 ppm), for 1H NMR spectra; CDCl3 (77.0 ppm) or DMSO-d6 (39.5 ppm) for 13C NMR spectra. 1H and 13C assignments are based on COSY and HSQC experiments. MS experiments were performed on a Bruker 7T FT-ICR instrument with an electrospray ion source. Samples were analyzed in the positive-ion mode.
[15N1,15N3,H215N(C2)]-2,6-Diaminopyrimidin-4(3H)-one (6). To a solution of methyl cyanoacetate (4, 90 µL, 1.02 mmol) and [15N3]-guanidine hydrochloride (5, 100 mg, 1.02 mmol) in methanol (6.6 mL) was added dropwise NaOCH3 (0.53 g, 9.85 mmol) in methanol (4.1 mL). After the addition was complete, the mixture was refluxed for 10 hours and allowed to cool to room temperature. The mixture was filtrated, and the filtrate evaporated to dryness. The residue was redissolved in water (1 mL) at 90 °C. The yellow solution was then acidified to pH 6 by acetic acid. The crude product was purified by reversed-phase (C18) column chromatography (eluent A: water, eluent B: acetonitrile; 0–15% B in 40 min, 5 mL/min) as eluent to yield 46 mg of compound 6 (35%) as a yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 4.41 (s, 1H, CH), 5.82 (s, 2H, NH2), 6.70 (d, J = 89.03 Hz, 2H, 15NH2), 7.91 (d, J = 89.51 Hz, 1H, 15NH) ppm.
[15N1,15N3,H215N(C2)]-2-[(2-Amino-4,7-dihydro-4-oxo-1H-pyrrolo[2,3-d]pyrimidin-5-yl)methyl]-1,3-dihydro-2H-isoindole-1,3-dione (8). Compound 6 (46 mg, 0.37 mmol) and α-bromoaldehyde 7 [15] (100 mg, 0.37 mmol) were suspended in a mixture of acetonitrile and water (1.6 mL; 1:1). Sodium acetate trihydrate (97 mg, 0.72 mmol) was added, and the suspension stirred at 40 °C. After 10 minutes, all solids were disolved and a yellow solution was obtained which rapidly turned into a suspension again, indicating that product 8 started to precipitate. The mixture was stirred for 4 hours, then cooled to room temperature and filtered. The residue was dried under reduced pressure to yield 70 mg of compound 8 (63%) as yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 4.84 (s, 2H, CH2-N), 6.16 (d, J = 88.96 Hz, 2H, 15NH2-C2), 6.36 (s, 1H, H-C8), 7.85–7.87 (m, 4H, arom H), 10.42 (d, J = 89.21 Hz, 1H, 15N1-H), 10.81 (s, 1H, NH) ppm.
Trifluoroacetate salt of [15N1,15N3,H215N(C2)]-7-(aminomethyl)-7-deazaguanine (1). Compound 8 (70 mg, 0.22 mmol) was added to a solution containing hydrazine hydrate (111 µL, 2.2 mmol) and ethanol (3 mL). The mixture was refluxed overnight and evaporated to dryness. The residue was purified by reversed-phase (C18) column chromatograpy (eluent A: 1% trifluoroacetic acid in H2O; eluent B: acetonitrile; 0–20% B in 50 min, 4 mL/min) to yield 49 mg of compound 1 (75%; calculated as mono TFA salt) as a light yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 4.01 (s, 2H,CH2), 6.30 (d, J = 89.1 Hz, 2H, 15NH2-C2), 6.78 (s, 1H, H-C8), 8.18 (s, 3H, NH3+), 10.82 (d, J = 89.6 Hz, 1H, 15N1-H), 11.20 (s, 1H, NH) ppm; 13C NMR (75 MHz, D2O) δ 34.57 (CH2NH3+), 98.32 (C7), 110.29 (C5), 113.88 (C2), 118.55 (C8), 141.05 (C4), 150.60 (C6=O), 159.09 (COO−), 162.18 (CF3) ppm; HRMS–ESI m/z: [M + H]+ calcd for C7H9N215N3O, 183.07909; found, 183.07865.
UV spectroscopic analysis of unlabeled 7-(aminomethyl)-7-deazaguanine (preQ1 base): UV (H2O) λmax (ε) = 218 (13630), 258 (7940) nm. For comparison, see UV spectroscopic data and extinction coefficients of 7-deazaguanine, N9-methyl-7-deazaguanine and preQ1 nucleoside in references [19-21].
Supporting Information
1H and 13C NMR spectra are provided in Supporting Information File 1. Synthetic procedures for the syntheses of compounds 2, 3, 10, 11, 13, 14, 17–19, and 21.
| Supporting Information File 1: Synthetic procedures and NMR spectra of the most typical compounds. | ||
| Format: PDF | Size: 938.8 KB | Download |
Acknowledgements
The authors thank Marina Frener (Micura group) for synthetic contributions to 2-bromo-3-phthalimidopropan-1-al (7), Kathrin Breuker for FT ICR mass spectrometric analysis, Christoph Kreutz and Tobias Santner for discussions. Funding by the Austrian Science Fund FWF (P21641, I1040) is gratefully acknowledged.
References
-
Iwata-Reuyl, D. Bioorg. Chem. 2003, 31, 24–43. doi:10.1016/S0045-2068(02)00513-8
Return to citation in text: [1] -
Vinayak, M.; Pathak, C. Biosci. Rep. 2009, 30, 135–148. doi:10.1042/BSR20090057
Return to citation in text: [1] -
McCarty, R. M.; Bandarian, V. Bioorg. Chem. 2012, 43, 15–25. doi:10.1016/j.bioorg.2012.01.001
Return to citation in text: [1] -
Roth, A.; Winkler, W. C.; Regulski, E. E.; Lee, B. W. K.; Lim, J.; Jona, I.; Barrick, J. E.; Ritwik, A.; Kim, J. N.; Welz, R.; Iwata-Reuyl, D.; Breaker, R. R. Nat. Struct. Mol. Biol. 2007, 14, 308–317. doi:10.1038/nsmb1224
Return to citation in text: [1] [2] -
Meyer, M. M.; Roth, A.; Chervin, S. M.; Garcia, G. A.; Breaker, R. R. RNA 2008, 14, 685–695. doi:10.1261/rna.937308
Return to citation in text: [1] [2] -
Santner, T.; Rieder, U.; Kreutz, C.; Micura, R. J. Am. Chem. Soc. 2012, 134, 11928–11931. doi:10.1021/ja3049964
Return to citation in text: [1] -
Soulière, M. F.; Altman, R. B.; Schwarz, V.; Haller, A.; Blanchard, S. C.; Micura, R. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, E3256–E3264. doi:10.1073/pnas.1304585110
Return to citation in text: [1] -
Kang, M.; Eichhorn, C. D.; Feigon, J. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, E663–E671. doi:10.1073/pnas.1400126111
Return to citation in text: [1] -
Ohgi, T.; Kondo, T.; Goto, T. Chem. Lett. 1979, 8, 1283–1286. doi:10.1246/cl.1979.1283
Return to citation in text: [1] -
Akimoto, H.; Imamiya, E.; Hitaka, T.; Nomura, H.; Nishimura, S. J. Chem. Soc., Perkin Trans. 1 1988, 1637–1644. doi:10.1039/p19880001637
Return to citation in text: [1] -
Klepper, F.; Polborn, K.; Carell, T. Helv. Chim. Acta 2005, 88, 2610–2616. doi:10.1002/hlca.200590201
Return to citation in text: [1] [2] [3] [4] [5] -
Barnett, C. J.; Grubb, L. M. Tetrahedron 2000, 56, 9221–9225. doi:10.1016/S0040-4020(00)00895-4
Return to citation in text: [1] [2] -
Gerber, H.-D.; Klebe, G. Org. Biomol. Chem. 2012, 10, 8660–8668. doi:10.1039/c2ob26387d
Return to citation in text: [1] -
Lolli, M.; Medana, C.; Romagnano, S.; Castoldi, F.; Pozzoli, S.; Vago, F.; Fanelli, R.; Airoldi, L. J. Labelled Compd. Radiopharm. 1998, 41, 243–252. doi:10.1002/(SICI)1099-1344(199803)41:3<243::AID-JLCR73>3.0.CO;2-H
Return to citation in text: [1] -
Barnett, C. J.; Grubb, L. M. Tetrahedron Lett. 2000, 41, 9741–9745. doi:10.1016/S0040-4039(00)01768-8
Return to citation in text: [1] [2] -
Yamaguchi, H. Thiazolimine compound and oxazolimine compound. Eur. Pat. Appl. EP1640369 A1, March 29, 2006; pp 26–27.
Return to citation in text: [1] -
Grundke, G.; Keese, W.; Rimpler, M. Chem. Ber. 1985, 118, 4288–4291. doi:10.1002/cber.19851181037
Return to citation in text: [1] [2] -
McCown, P. J.; Liang, J. J.; Weinberg, Z.; Breaker, R. R. Chem. Biol. 2014, 21, 880–889. doi:10.1016/j.chembiol.2014.05.015
Return to citation in text: [1] -
Seela, F.; Lüpke, U. Chem. Ber. 1977, 110, 1462–1469. doi:10.1002/cber.19771100428
Return to citation in text: [1] -
Seela, F.; Kehne, A.; Winkeler, H.-D. Liebigs Ann. Chem. 1983, 137–146. doi:10.1002/jlac.198319830113
Return to citation in text: [1] -
Kondo, T.; Okamoto, K.; Ohgi, T.; Goto, T. Tetrahedron 1986, 42, 207–213. doi:10.1016/S0040-4020(01)87419-6
Return to citation in text: [1]
| 15. | Barnett, C. J.; Grubb, L. M. Tetrahedron Lett. 2000, 41, 9741–9745. doi:10.1016/S0040-4039(00)01768-8 |
| 17. | Grundke, G.; Keese, W.; Rimpler, M. Chem. Ber. 1985, 118, 4288–4291. doi:10.1002/cber.19851181037 |
| 4. | Roth, A.; Winkler, W. C.; Regulski, E. E.; Lee, B. W. K.; Lim, J.; Jona, I.; Barrick, J. E.; Ritwik, A.; Kim, J. N.; Welz, R.; Iwata-Reuyl, D.; Breaker, R. R. Nat. Struct. Mol. Biol. 2007, 14, 308–317. doi:10.1038/nsmb1224 |
| 5. | Meyer, M. M.; Roth, A.; Chervin, S. M.; Garcia, G. A.; Breaker, R. R. RNA 2008, 14, 685–695. doi:10.1261/rna.937308 |
| 18. | McCown, P. J.; Liang, J. J.; Weinberg, Z.; Breaker, R. R. Chem. Biol. 2014, 21, 880–889. doi:10.1016/j.chembiol.2014.05.015 |
| 1. | Iwata-Reuyl, D. Bioorg. Chem. 2003, 31, 24–43. doi:10.1016/S0045-2068(02)00513-8 |
| 2. | Vinayak, M.; Pathak, C. Biosci. Rep. 2009, 30, 135–148. doi:10.1042/BSR20090057 |
| 10. | Akimoto, H.; Imamiya, E.; Hitaka, T.; Nomura, H.; Nishimura, S. J. Chem. Soc., Perkin Trans. 1 1988, 1637–1644. doi:10.1039/p19880001637 |
| 11. | Klepper, F.; Polborn, K.; Carell, T. Helv. Chim. Acta 2005, 88, 2610–2616. doi:10.1002/hlca.200590201 |
| 9. | Ohgi, T.; Kondo, T.; Goto, T. Chem. Lett. 1979, 8, 1283–1286. doi:10.1246/cl.1979.1283 |
| 11. | Klepper, F.; Polborn, K.; Carell, T. Helv. Chim. Acta 2005, 88, 2610–2616. doi:10.1002/hlca.200590201 |
| 4. | Roth, A.; Winkler, W. C.; Regulski, E. E.; Lee, B. W. K.; Lim, J.; Jona, I.; Barrick, J. E.; Ritwik, A.; Kim, J. N.; Welz, R.; Iwata-Reuyl, D.; Breaker, R. R. Nat. Struct. Mol. Biol. 2007, 14, 308–317. doi:10.1038/nsmb1224 |
| 5. | Meyer, M. M.; Roth, A.; Chervin, S. M.; Garcia, G. A.; Breaker, R. R. RNA 2008, 14, 685–695. doi:10.1261/rna.937308 |
| 6. | Santner, T.; Rieder, U.; Kreutz, C.; Micura, R. J. Am. Chem. Soc. 2012, 134, 11928–11931. doi:10.1021/ja3049964 |
| 7. | Soulière, M. F.; Altman, R. B.; Schwarz, V.; Haller, A.; Blanchard, S. C.; Micura, R. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, E3256–E3264. doi:10.1073/pnas.1304585110 |
| 8. | Kang, M.; Eichhorn, C. D.; Feigon, J. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, E663–E671. doi:10.1073/pnas.1400126111 |
| 16. | Yamaguchi, H. Thiazolimine compound and oxazolimine compound. Eur. Pat. Appl. EP1640369 A1, March 29, 2006; pp 26–27. |
| 3. | McCarty, R. M.; Bandarian, V. Bioorg. Chem. 2012, 43, 15–25. doi:10.1016/j.bioorg.2012.01.001 |
| 17. | Grundke, G.; Keese, W.; Rimpler, M. Chem. Ber. 1985, 118, 4288–4291. doi:10.1002/cber.19851181037 |
| 11. | Klepper, F.; Polborn, K.; Carell, T. Helv. Chim. Acta 2005, 88, 2610–2616. doi:10.1002/hlca.200590201 |
| 11. | Klepper, F.; Polborn, K.; Carell, T. Helv. Chim. Acta 2005, 88, 2610–2616. doi:10.1002/hlca.200590201 |
| 12. | Barnett, C. J.; Grubb, L. M. Tetrahedron 2000, 56, 9221–9225. doi:10.1016/S0040-4020(00)00895-4 |
| 13. | Gerber, H.-D.; Klebe, G. Org. Biomol. Chem. 2012, 10, 8660–8668. doi:10.1039/c2ob26387d |
| 15. | Barnett, C. J.; Grubb, L. M. Tetrahedron Lett. 2000, 41, 9741–9745. doi:10.1016/S0040-4039(00)01768-8 |
| 12. | Barnett, C. J.; Grubb, L. M. Tetrahedron 2000, 56, 9221–9225. doi:10.1016/S0040-4020(00)00895-4 |
| 19. | Seela, F.; Lüpke, U. Chem. Ber. 1977, 110, 1462–1469. doi:10.1002/cber.19771100428 |
| 20. | Seela, F.; Kehne, A.; Winkeler, H.-D. Liebigs Ann. Chem. 1983, 137–146. doi:10.1002/jlac.198319830113 |
| 21. | Kondo, T.; Okamoto, K.; Ohgi, T.; Goto, T. Tetrahedron 1986, 42, 207–213. doi:10.1016/S0040-4020(01)87419-6 |
| 11. | Klepper, F.; Polborn, K.; Carell, T. Helv. Chim. Acta 2005, 88, 2610–2616. doi:10.1002/hlca.200590201 |
| 14. | Lolli, M.; Medana, C.; Romagnano, S.; Castoldi, F.; Pozzoli, S.; Vago, F.; Fanelli, R.; Airoldi, L. J. Labelled Compd. Radiopharm. 1998, 41, 243–252. doi:10.1002/(SICI)1099-1344(199803)41:3<243::AID-JLCR73>3.0.CO;2-H |
© 2014 Levic and Micura; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)