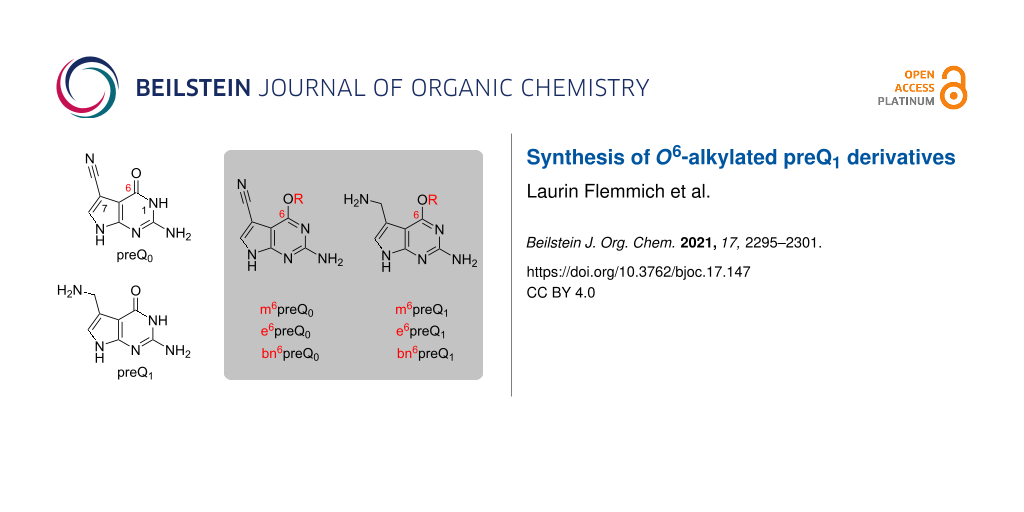
Abstract
A naturally occurring riboswitch can utilize 7-aminomethyl-O6-methyl-7-deazaguanine (m6preQ1) as cofactor for methyl group transfer resulting in cytosine methylation. This recently discovered riboswitch-ribozyme activity opens new avenues for the development of RNA labeling tools based on tailored O6-alkylated preQ1 derivatives. Here, we report a robust synthesis for this class of pyrrolo[2,3-d]pyrimidines starting from readily accessible N2-pivaloyl-protected 6-chloro-7-cyano-7-deazaguanine. Substitution of the 6-chloro atom with the alcoholate of interest proceeds straightforward. The transformation of the 7-cyano substituent into the required aminomethyl group turned out to be challenging and was solved by a hydration reaction sequence on a well-soluble dimethoxytritylated precursor via in situ oxime formation. The synthetic path now provides a solid foundation to access O6-alkylated 7-aminomethyl-7-deazaguanines for the development of RNA labeling tools based on the preQ1 class-I riboswitch scaffold.

Graphical Abstract
Introduction
Methylated preQ1 has attracted much attention recently because this compound has been found to function as cofactor for the conserved fold of a non-coding RNA, namely the preQ1 class-I riboswitch [1]. This riboswitch acts as a ribozyme by using 7-aminomethyl-O6-methyl-7-deazaguanine (m6preQ1) as methyl group donor; it catalyzes self-methylation of a specific cytidine in the aptamer binding pocket, yielding N3-methyl cytidine (m3C) under release of 7-aminomethyl-7-deazaguanine (preQ1) [1]. Thus far, present-day riboswitches have only been known to bind – but not to be able to react – with their ligands [2,3]. This new finding now opens exciting avenues for the development of RNA labeling tools [4], in particular, for RNA methylation, and more generally, for RNA alkylation. To this end, robust synthetic routes towards O6-alkylated 7-aminomethyl-7-deazaguanines are urgently needed and reported here.
Results and Discussion
Biological and synthetic background
Role of preQ1 in queuosine biosynthesis and gene regulation
Queuine (Q base) is a derivative of guanine that is involved in the biosynthetic pathway of the hypermodified tRNA nucleoside queuosine (Q) (Scheme 1) [5]. The core structure of the nucleobase is 7-aminomethyl-7-deazaguanine, a pyrrolo[2,3-d]pyrimidine also termed prequeuosine base (preQ1) [6,7]. In many bacteria, preQ1 binds to specific mRNA domains and thereby regulates genes that are required for queuosine biosynthesis [8-16]. The molecular mechanism behind is called riboswitching. For most riboswitches, ligand binding induces a structural change in the untranslated leader sequence of mRNA by formation (or disruption) of a terminator stem (transcriptional control) or repressor stem (translational control). This conformational event signals on or off to gene expression and represents a feedback-type mechanism that is dependent on cellular ligand concentration [13].
![[1860-5397-17-147-i1]](/bjoc/content/inline/1860-5397-17-147-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Chemical structures of queuine (Q base) and the hypermodified nucleoside queuosine (Q), the natural products dapiramicin A and huimycin, as well as intermediates of queuosine biosynthesis (preQ1 and preQ0), and the major synthetic targets of this study, m6preQ0 (1) and m6preQ1 (2) (grey box).
Scheme 1: Chemical structures of queuine (Q base) and the hypermodified nucleoside queuosine (Q), the natural...
Natural occurrence of alkylated prequeuosines
Evidence for the natural occurrence of methylated prequeuosine bases stems from a recent study that demonstrated that m6preQ0 is produced by Streptomyces [17]. Moreover, the natural products huimycin [18] and dapiramicin contain m6preQ0 as core with their 2-NH2 group linked to a 2'-acetamido-2'-deoxy-ß-ᴅ-glucopyranosyl residue in huimycin and to a 2-[4'-(4''-O-methyl-ß-ᴅ-glucopyranosyl)-6'-deoxy-α-ᴅ-glucopyranosyl] moiety in dapiramicin A [19,20]. In the biosynthetic pathway, the conversion of preQ0 into huimycin requires methylation of preQ0 and attachment of the N-acetylglucosamine moiety as final steps [18]. The methylation reaction is likely to be catalyzed by the product of the gene huiC, which encodes a SAM-dependent methyltransferase [18].
To the best of our knowledge, in contrast to m6preQ0 [17] the reduced counterpart 7-aminomethyl-O6-methyl-7-deazaguanine m6preQ1 has not yet been reported to be isolated from natural sources.
Earlier syntheses of preQ1, preQ0 and m6preQ0
The synthesis of preQ1 has been first described by Goto starting from 2-methylthio-6-methoxy-7-methyl-7-deazapurine and requiring more than ten steps [21]. More efficient was a procedure reported by Nishimura applying a Mannich reaction with dibenzylamine–formaldehyde and 2-acylaminopyrrolo[2,3-d]pyrimidin-4(3H)-one as key step, thereby selectively installing a dibenzylaminomethyl moiety [22]. Exchange of the dibenzylamine group in the Mannich base with NH3 provided preQ1 [22]. Alternatively, Klebe demonstrated a Michael addition of 2,6-diaminopyrimidin-4-one to the nitroolefin 2-[(2E)-3-nitroprop-2-en-1-yl]-1H-isoindole-1,3(2H)-dione [23]. Finally, Carell reported a cycloaddition route relying on α-brominated 3-phthalimidopropanal and diaminopyrimidin-4-one [24,25]. We further optimized this path for the synthesis of 15N-labeled prequeuosine nucleobase derivatives [26] required for advanced NMR spectroscopic applications [27], and for the syntheses of azido- or amino-functionalized preQ1 derivatives needed for cellular applications with engineered riboswitches [28]. Finally, we point out that only a single synthetic route has been published to a potential O6-methylated precursor of m6preQ1, namely N9-trimethysilylethyl protected m6preQ0 [20]. This synthesis, however, is based on methylation using diazomethane resulting in a mixture of N1 and O6 methylated products, and we therefore did not further consider this path. Finally, one route was described for m6preQ0 1 [29] which is similar to the first step we developed for the synthesis of m6preQ1 2 as outlined below.
Synthesis of m6preQ1, e6preQ1, and bn6preQ1
Our initial attempts to site-specifically methylate trimethylsilylated preQ1 (which was generated in situ with N,O-bis[trimethylsilyl]acetamide) by trimethyloxonium tetrafluoroborate in apolar solvents resulted in the recovery of starting material only. Next, we tested a cyclocondensation reaction between 2-chloro-3-cyanopropan-1-al and 2,6-diamino-4-methoxypyrimidine [30], however, target compound 1 (m6preQ0) was obtained in yields of 21% (Scheme 2) which is significantly lower compared to the cyclocondensations with 2,6-diamino-4-pyrimidin-4-one mentioned above [23-27].
![[1860-5397-17-147-i2]](/bjoc/content/inline/1860-5397-17-147-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of compound 1 (m6preQ0) by cyclocondensation using a 4-methoxypyrimidine derivative resulted in unsatisfying yields.
Scheme 2: Synthesis of compound 1 (m6preQ0) by cyclocondensation using a 4-methoxypyrimidine derivative resul...
We therefore envisaged a path involving 6-chloro-7-deazapurine derivative 3 (Scheme 3) as this compound is readily available from cheap starting materials following published procedures. Chloroacetonitrile and methyl formate gave 2-chloro-2-cyanoacetaldehyde which was then reacted with 2,6-diaminopyrimidin-4-one to provide preQ0 in good yields [31]. Protection of the exocyclic amino group using pivaloyl chloride was optimized from a published procedure [32] and gave nearly quantitative yields of N2-pivaloyl preQ0 in our hands. Finally, transformation of the 6-carbonyl group by using phosphorus oxychloride gave 6-chloro-7-deazapurine derivative 3 [33]. Notably, attempts to directly transform preQ0 (without N2 protection) into 6-chloro-7-cyano-7-deazaguanine failed in our hands.
![[1860-5397-17-147-i3]](/bjoc/content/inline/1860-5397-17-147-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of compound 3 following known procedures [31-33].
Scheme 3: Synthesis of compound 3 following known procedures [31-33].
The 6-chloro atom of compound 3 was substituted using sodium methoxide under concomitant cleavage of the pivaloyl group to yield the desired O6-methylated compound 1, m6preQ0 (Scheme 4). After dissolving this compound under strong silylating conditions in the presence of N,O-bis(trimethylsilyl)acetamide [34], simultaneous tritylation of N9 and the N2 atoms was achieved using 4,4'-dimethoxytrityl chloride in pyridine. The obtained derivative 4 was amenable to nitrile reduction using diisobutylaluminium hydride (DIBAL-H) in dichloromethane at −78 °C, followed by workup with potassium sodium tartrate solution (Rochelle salt) to furnish the aldehyde 5. Then, transformation of the 7-formyl into the 7-aminomethyl group proceeded via oxime formation, applying hydroxylamine hydrochloride in methanolic ammonia, followed by reduction with Raney nickel to yield the tritylated precursor 6. Finally, the auxiliary functions were cleaved using trifluoroacetic acid (TFA) in dichloromethane and the target compound 2, m6preQ1, was isolated as TFA salt.
![[1860-5397-17-147-i4]](/bjoc/content/inline/1860-5397-17-147-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: The five-step synthesis of m6preQ1 2 from compound 3 required derivatization to make the intermediates soluble in organic solvents for a controllable reaction sequence to reduce the cyano group; overall yield: 30%.
Scheme 4: The five-step synthesis of m6preQ1 2 from compound 3 required derivatization to make the intermedia...
The here established path to synthesize m6preQ1 offers high flexibility with respect to the O6 substituent. We demonstrate this by complementing the set of preQ1-derived alkylating cofactors with e6preQ1 (2a) and bn6preQ1 (2b) that were synthesized following the same path with overall yields of 23% and 34%, respectively (Scheme 5 and Supporting Information File 1).
![[1860-5397-17-147-i5]](/bjoc/content/inline/1860-5397-17-147-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: The synthesis of e6preQ1 (2a) and bn6preQ1 (2b) was performed in analogy to the route outlined in Scheme 4 with an overall yield of 23% and 34%, respectively. For details see the Supporting Information File 1.
Scheme 5: The synthesis of e6preQ1 (2a) and bn6preQ1 (2b) was performed in analogy to the route outlined in Scheme 4 ...
Conclusion
We developed a robust synthesis for O6-alkylated 7-aminomethyl-7-deazaguanines that starts from a readily accessible 6-chloro-7-cyano-7-deazaguanine derivative. The chloro atom is smoothly substituted by the alcoholate of interest, followed by N9 and N2 dimethoxytritylation to obtain a well-soluble intermediate. The reduction of the 7-cyano substituent into the target aminomethyl group is accomplished by a hydration reaction sequence via in situ oxime formation. The route now provides a solid basis to generate O6-alkylated preQ1 derivatives for the development of RNA labeling tools utilizing the RNA scaffold of the preQ1 class-I riboswitch.
Experimental
General. Chemical reagents and solvents were purchased from commercial suppliers (Sigma-Aldrich) and used without further purification. Organic solvents for reactions were dried overnight over freshly activated molecular sieves (4 Å). The reactions were carried out under argon atmosphere. Analytical thin-layer chromatography (TLC) was performed on Marchery-Nagel Polygram SIL G/UV254 plates. Column chromatography was done on silica gel 60 (70–230 mesh). 1H, and 13C NMR spectra were recorded on Bruker DRX 300 MHz, Bruker Avance 4 Neo 400 MHz, and Bruker Avance 4 Neo 700 MHz instruments. Chemical shifts (δ) are reported relative to tetramethylsilane (TMS) and referenced to the residual proton or carbon signal of the deuterated solvent: CDCl3 (7.26 ppm) or DMSO-d6 (2.49 ppm) for 1H NMR; CDCl3 (77.0 ppm) or DMSO-d6 (39.5 ppm) for 13C NMR spectra. 1H and 13C assignments are based on COSY, HSQC, and HMBC experiments. MS experiments were performed on a Thermo Fisher QExactive Classic. Samples were analyzed in the positive-ion mode.
O6-Methyl preQ0 (1) (m6preQ0). Procedure A: 2-Chloro-3-oxoproanenitrile [29] (6.80 g, 65.6 mmol) was added to a solution of sodium acetate (10.77 g, 131.3 mmol) and 6-methoxypyrimidine-2,4-diamine [33] (9.20 g, 65.6 mmol) in water (270 mL) at 50 °C. After 16 hours, the solution was refluxed for an additional hour and allowed to cool to room temperature. Filtration gave 2.61 g of compound 1 (21%) as grey solid. Procedure B: Sodium (42 mg, 1.8 mmol) was dissolved in 0.8 mL methanol at 0 °C. Compound 3 [29-31] (200 mg, 0.720 mmol) was added and the mixture was heated to 110 °C in a pressure tube for 24 h. After neutralization with glacial acetic acid the volatiles were removed in vacuo. The crude product was dry-loaded onto silica gel and purified via flash column chromatography (5–20% methanol in dichloromethane) to give 104 mg of compound 1 (76%) as a beige solid. TLC: 10% methanol in dichloromethane, Rf 0.37; 1H NMR (300 MHz, DMSO-d6) δ 12.08 (bs, 1H, HN(9)), 7.81 (s, 1H, HC(8)), 6.45 (s, 2H, H2N(2)), 3.96 (s, 3H, H3CO(6)) ppm; 13C NMR (75 MHz, DMSO-d6) δ 162.7 (C(6)), 160.7 (C(2)), 154.9 (C(4)), 130.3 (C(8)), 116.1 & 95.4 & 82.4 (C(5) & C(7) & CN), 53.26 (H3CO(6)); ESIMS (m/z): [M + H]+ calcd, 190.0723; found, 190.0721.
N2,9-Bis(4,4'-dimethoxytrityl)-O6-methyl preQ0 (4). Compound 1 (300 mg, 1.59 mmol) was suspended in N,N-dimethylformamide (12 mL). N,O-Bis(trimethylsilyl)acetamide (820 µL, 3.33 mmol) was added dropwise and the reaction mixture was stirred for three hours at room temperature upon which a solution was obtained. Afterwards, the volatile components were removed under reduced pressure and the residue was coevaporated three times with toluene and twice with pyridine. The residue was dissolved in pyridine (3.8 mL) and 4,4'-dimethoxytrityl chloride (1.18 g, 3.50 mmol) was added in portions. The solution was stirred for 18 h at 40 °C, subsequently poured into 5% aqueous sodium bicarbonate solution and the suspension was extracted three times with dichloromethane. The combined organic layers were washed with brine and dried over magnesium sulfate. The solvents were removed and the remaining crude product was purified by flash column chromatography on silica gel (10–30% ethyl acetate in cyclohexane) to give 1.00 g of compound 4 (79%) as a white foam. TLC: 40% ethyl acetate in cyclohexane, Rf = 0.68; 1H NMR (300 MHz, CDCl3) δ 7.32–6.94 (m, 19H, HC(aromatic, DMTr) & HC(8)), 6.83–6.65 (m, 8H, HC(arom, DMTr)), 5.54 (s, 1H, HN(2)), 3.80 & 3.77 (s, 12H, H3CO(DMTr), 3.37 (s, 1H, H3CO(O6) ppm; 13C NMR (101 MHz, CDCl3) δ 162.4 (C(6)), 158.7 & 158.4 & 158.0 (C(aromatic, DMTr)), 154.8 (C(4)), 146.3 (C(2)), 142.15 & 138.4 & 134.3 (C(aromatic, DMTr)), 133.1 (C(8)), 131.4 & 130.4 & 130.3 & 130.2 & 129.80 & 129.1 & 127.8 & 127.5 & 127.4 & 126.4 (C(aromatic, DMTr)), 115.8 (C(5)/C(7)), 113.3 & 113.1 & 112.7 (C(aromatic, DMTr)), 99.2 (C(5)/C(7)), 83.1 (CN(nitrile)), 76.1 & 70.5 (CAr3(DMTr)), 55.4 & 55.3 (H3CO(DMTr)), 53.88 (H3CO(6)) ppm; ESIMS (m/z): [M + H]+ calcd, 794.3337; found, 794.3321.
7-Formyl-N2,9-bis(4,4'-dimethoxytrityl)-O6-methyl-7-deazaguanine (5). To a cooled solution (–78 °C) of compound 4 (1.00 g, 1.26 mmol) in dichloromethane (8 mL) diisobutylaluminium hydride (1 M in dichloromethane, 1.6 mL, 2.57 mmol) was added dropwise. The reaction was continued for three hours, quenched by the addition of ethyl acetate (4 mL) and allowed to come to room temperature. Half-saturated potassium sodium tartrate solution was added (4 mL) and the biphasic mixture was stirred vigorously for one and a half hour until satisfactory phase separation was achieved. The aqueous layer was separated and subsequently extracted three times with ethyl acetate. The combined organic layers were washed with brine, dried over magnesium sulfate and evaporated. The residue was purified by flash column chromatography on silica gel (10–25 % ethyl acetate in cyclohexane) to give 823 mg of compound 5 (82%) as a white foam. TLC: 30% ethyl acetate in cyclohexane Rf 0.51; 1H NMR (CDCl3, 400 MHz) δ 9.91 (s, 1H, CHO), 7.49 (s, 1H, HC(8)), 7.29–7.23 (m, 2H, HC(aromatic, DMTr), 7.20–7.11 (m, 6H, HC(aromatic, DMTr), 7.10–7.06 (m, 2H, HC(aromatic, DMTr)), 7.05–6.92 (m, 8H, HC(aromatic, DMTr)), 6.81–6.76 (m, 4H, HC(aromatic, DMTr)), 6.72–6.68 (m, 4H, HC(aromatic, DMTr)), 5.51 (s, 1H, HN(2)), 3.80 (s, 6H, H3CO(DMTr)), 3.77 (s, 6H, H3CO(DMTr)), 3.38 (s, 3H, H3CO(6)) ppm; 13C NMR (CDCl3, 101 MHz) δ 185.7 (CHO), 163.2 (C(6)), 158.7 & 158.0 & 156.5 (C(aromatic, DMTr)), 156.5 (C(4)), 146.4 (C(2)), 142.3 & 138.6 & 134.5 (C(aromatic, DMTr)), 131.9 (C(8)), 131.5 & 130.3 & 129.9 & 129.1 & 127.7 & 127.4 & 127.3 & 126.4 (C(aromatic, DMTr)), 115.7 (C(5)/C(7)), 113.1 & 112.7 (C(aromatic, DMTr)), 98.0 (C(5))/C(7)), 76.0 (CAr3 (DMTr)), 70.5 (DMTr), 55.4 & 33.3 (H3CO(DMTr)), 54.0 (H3CO(6)) ppm; ESIMS (m/z): [M + H]+ calcd, 797.3334; found, 797.3315.
7-Aminomethyl-N2,9-bis(4,4'-dimethoxytrityl)-O6-methyl-7-deazaguanine (6). To a suspension of compound 5 (200 mg, 0.251 mmol) in 7 M methanolic ammonia (6 mL) hydroxylamine hydrochloride (21 mg, 0.300 mmol) was added. A clear solution was obtained shortly thereafter. The reaction was stirred for two hours at room temperature. Tetrahydrofuran (4 mL) and damp Raney-Nickel (approximately 200 mg) were introduced and the reaction was continued for one hour. The reaction mixture was filtered over a Celite pad and the filtrate was evaporated. The residue was taken up in 3% methanol in dichloromethane and passed over a short, deactivated silica pad and evaporated once more to give 163 mg of compound 6 (81%) as a white foam. TLC: 6% MeOH, 1% NEt3 in dichloromethane, Rf 0.26; 1H NMR (CDCl3, 400 MHz) δ 7.24–7.20 (m, 3H, HC(aromatic, DMTr)), 7.18–7.08 (m, 7H, HC(aromatic, DMTr)), 7.06–7.00 (m, 8H, HC(aromatic, DMTr)), 6.77–6.67 (m, 7H, HC(aromatic, DMTr)), 6.44 (s, 1H, HC(8)), 5.46 (s, 1H, HN(2)), 3.79 (s, 6H, H3CO(DMTr)), 3.77 (s, 6H, H3CO(DMTr)), 3.67 (s, 2H, H2CC(7)), 3.33 (s, 3H, H3CO(6)), 1.81 (bs, 2H, H2N) ppm; 13C NMR (CDCl3, 101 MHz) δ 162.3 (C(6)), 158.3 & 157.8 & 157.2 & (C(arom, DMTr)), 155.9 (C(4)), 146.8 (C(2)), 143.6 & 139.0 & 135.9 & 131.4 130.4 & 130.0 & 129.2 & 127.4 & 127.3 & 126.8 & 126.2 (C(arom, DMTr)), 121.4 (C(8)), 115.9 (C(5)) or (C(7)), 112.71 & 112.6 (C(arom, DMTr)), 74.5 (CAr3(DMTr)), 70.3 (CAr3(DMTr)), 55.3 (H3CO(DMTr)), 53.4 (H3CO(6)), 38.9 (H2CC(7)) ppm; ESIMS (m/z): [M + H]+ calcd, 798.3650; found, 798.3629.
O6-Methyl preQ1 (trifluoroacetate salt) (2). Compound 6 (120 mg, 150 µmol) was dissolved in 500 µL dichloromethane. Trifluoroacetic acid (60 µL, 0.75 mmol) and 10 µL water were added. After 45 minutes the reaction was quenched by the addition of 100 µL methanol. Afterwards the solvents were removed in vacuo. The residue was triturated five times with dichloromethane and dried on high vacuum to give 35 mg of compound 2 (76%) as a white solid. TLC: 15% MeOH, 1% NEt3 in dichloromethane, Rf 0.56; 1H NMR (CDCl3, 400 MHz) δ 11.64 (bs, 1H, HN(9), 8.07 (bs, 3H, H3N+), 6.99 (d, 1H, JHH = 2.0 Hz, HC(8)), 4.06 (q, 2H, H2CC(7)), 3.98 (s, 3H, H3CO) ppm; 13C NMR (CDCl3, 101 MHz) δ 164.1 (C(6)), 158.4 (q, JCF = 34.0 Hz, CF3COO−), 158.1 (C(2)), 150.7 (C(4)), 120.0 (C(8)), 116.3 (q, JCF = 295.0 Hz, CF3COO−), 107.5 & 96.2 ((C(5) & C(7)), 53.7 (H3CO), 34.8 (CH2CC(7)) ppm; ESIMS (m/z): [M + H – NH3]+ calcd, 177.0771; found, 177.0767; [M + H]+ calcd, 194.1036; found, 194.1032.
Supporting Information
| Supporting Information File 1: Synthetic procedures for compounds 1a–6a, and 1b–6b, and 1H and 13C NMR spectra of all compounds. 1H,13C HSQC and 1H,13C HMBC spectra of all final products 1, 1a, 1b, 2, 2a, and 2b. | ||
| Format: PDF | Size: 4.0 MB | Download |
References
-
Flemmich, L.; Heel, S.; Moreno, S.; Breuker, K.; Micura, R. Nat. Commun. 2021, 12, 3877. doi:10.1038/s41467-021-24193-7
Return to citation in text: [1] [2] -
Breaker, R. R. ACS Chem. Biol. 2020, 15, 2020–2030. doi:10.1021/acschembio.0c00214
Return to citation in text: [1] -
Batey, R. T. RNA 2015, 21, 560–563. doi:10.1261/rna.050765.115
Return to citation in text: [1] -
Micura, R.; Höbartner, C. Chem. Soc. Rev. 2020, 49, 7331–7353. doi:10.1039/d0cs00617c
Return to citation in text: [1] -
McCarty, R. M.; Bandarian, V. Bioorg. Chem. 2012, 43, 15–25. doi:10.1016/j.bioorg.2012.01.001
Return to citation in text: [1] -
Iwata-Reuyl, D. Bioorg. Chem. 2003, 31, 24–43. doi:10.1016/s0045-2068(02)00513-8
Return to citation in text: [1] -
Vinayak, M.; Pathak, C. Biosci. Rep. 2010, 30, 135–148. doi:10.1042/bsr20090057
Return to citation in text: [1] -
Roth, A.; Winkler, W. C.; Regulski, E. E.; Lee, B. W. K.; Lim, J.; Jona, I.; Barrick, J. E.; Ritwik, A.; Kim, J. N.; Welz, R.; Iwata-Reuyl, D.; Breaker, R. R. Nat. Struct. Mol. Biol. 2007, 14, 308–317. doi:10.1038/nsmb1224
Return to citation in text: [1] -
Meyer, M. M.; Roth, A.; Chervin, S. M.; Garcia, G. A.; Breaker, R. R. RNA 2008, 14, 685–695. doi:10.1261/rna.937308
Return to citation in text: [1] -
McCown, P. J.; Liang, J. J.; Weinberg, Z.; Breaker, R. R. Chem. Biol. 2014, 21, 880–889. doi:10.1016/j.chembiol.2014.05.015
Return to citation in text: [1] -
Jenkins, J. L.; Krucinska, J.; McCarty, R. M.; Bandarian, V.; Wedekind, J. E. J. Biol. Chem. 2011, 286, 24626–24637. doi:10.1074/jbc.m111.230375
Return to citation in text: [1] -
Chatterjee, S.; Chauvier, A.; Dandpat, S. S.; Artsimovitch, I.; Walter, N. G. Proc. Natl. Acad. Sci. U. S. A. 2021, 118, e2023426118. doi:10.1073/pnas.2023426118
Return to citation in text: [1] -
Serganov, A.; Nudler, E. Cell 2013, 152, 17–24. doi:10.1016/j.cell.2012.12.024
Return to citation in text: [1] [2] -
Zallot, R.; Yuan, Y.; de Crécy-Lagard, V. Biomolecules 2017, 7, 12. doi:10.3390/biom7010012
Return to citation in text: [1] -
Iwata-Reuyl, D. Curr. Opin. Chem. Biol. 2008, 12, 126–133. doi:10.1016/j.cbpa.2008.01.041
Return to citation in text: [1] -
Gaur, R.; Varshney, U. J. Bacteriol. 2005, 187, 6893–6901. doi:10.1128/jb.187.20.6893-6901.2005
Return to citation in text: [1] -
Iijima, M.; Kubota, Y.; Sawa, R.; Kubota, Y.; Hatano, M.; Igarashi, M.; Kawada, M.; Momose, I.; Takekawa, M.; Shibasaki, M. J. Antibiot. 2018, 71, 135–138. doi:10.1038/ja.2017.100
Return to citation in text: [1] [2] -
Shuai, H.; Myronovskyi, M.; Nadmid, S.; Luzhetskyy, A. Biomolecules 2020, 10, 1074. doi:10.3390/biom10071074
Return to citation in text: [1] [2] [3] -
Nishizawa, N.; Kondo, Y.; Koyama, M.; Omoto, S.; Iwata, M.; Tsuruoka, T.; Inouye, S. J. Antibiot. 1984, 37, 1–5. doi:10.7164/antibiotics.37.1
Return to citation in text: [1] -
Ohno, H.; Terui, T.; Kitawaki, T.; Chida, N. Tetrahedron Lett. 2006, 47, 5747–5750. doi:10.1016/j.tetlet.2006.06.001
Return to citation in text: [1] [2] -
Ohgi, T.; Kondo, T.; Goto, T. Chem. Lett. 1979, 8, 1283–1286. doi:10.1246/cl.1979.1283
Return to citation in text: [1] -
Akimoto, H.; Imamiya, E.; Hitaka, T.; Nomura, H.; Nishimura, S. J. Chem. Soc., Perkin Trans. 1 1988, 1637–1644. doi:10.1039/p19880001637
Return to citation in text: [1] [2] -
Gerber, H.-D.; Klebe, G. Org. Biomol. Chem. 2012, 10, 8660–8668. doi:10.1039/c2ob26387d
Return to citation in text: [1] [2] -
Klepper, F.; Polborn, K.; Carell, T. Helv. Chim. Acta 2005, 88, 2610–2616. doi:10.1002/hlca.200590201
Return to citation in text: [1] [2] -
Barnett, C. J.; Grubb, L. M. Tetrahedron 2000, 56, 9221–9225. doi:10.1016/s0040-4020(00)00895-4
Return to citation in text: [1] [2] -
Levic, J.; Micura, R. Beilstein J. Org. Chem. 2014, 10, 1914–1918. doi:10.3762/bjoc.10.199
Return to citation in text: [1] [2] -
Moschen, T.; Wunderlich, C. H.; Spitzer, R.; Levic, J.; Micura, R.; Tollinger, M.; Kreutz, C. Angew. Chem., Int. Ed. 2015, 54, 560–563. doi:10.1002/anie.201409779
Return to citation in text: [1] [2] -
Neuner, E.; Frener, M.; Lusser, A.; Micura, R. RNA Biol. 2018, 15, 1376–1383. doi:10.1080/15476286.2018.1534526
Return to citation in text: [1] -
Khalaf, A. I.; Huggan, J. K.; Suckling, C. J.; Gibson, C. L.; Stewart, K.; Giordani, F.; Barrett, M. P.; Wong, P. E.; Barrack, K. L.; Hunter, W. N. J. Med. Chem. 2014, 57, 6479–6494. doi:10.1021/jm500483b
Return to citation in text: [1] [2] [3] -
Chang, L.; Lee, S.-Y.; Leonczak, P.; Rozenski, J.; De Jonghe, S.; Hanck, T.; Müller, C. E.; Herdewijn, P. J. Med. Chem. 2014, 57, 10080–10100. doi:10.1021/jm501434y
Return to citation in text: [1] [2] -
Zhu, G.; Liu, Z.; Xu, Y.; Mao, Z. Heterocycles 2008, 75, 1631–1638. doi:10.3987/com-08-11323
Return to citation in text: [1] [2] [3] -
Wilding, B.; Winkler, M.; Petschacher, B.; Kratzer, R.; Egger, S.; Steinkellner, G.; Lyskowski, A.; Nidetzky, B.; Gruber, K.; Klempier, N. Chem. – Eur. J. 2013, 19, 7007–7012. doi:10.1002/chem.201300163
Return to citation in text: [1] [2] -
Brückl, T.; Klepper, F.; Gutsmiedl, K.; Carell, T. Org. Biomol. Chem. 2007, 5, 3821–3825. doi:10.1039/b713309j
Return to citation in text: [1] [2] [3] -
Brooks, A. F.; Garcia, G. A.; Showalter, H. D. H. Tetrahedron Lett. 2010, 51, 4163–4165. doi:10.1016/j.tetlet.2010.06.008
Return to citation in text: [1]
| 32. | Wilding, B.; Winkler, M.; Petschacher, B.; Kratzer, R.; Egger, S.; Steinkellner, G.; Lyskowski, A.; Nidetzky, B.; Gruber, K.; Klempier, N. Chem. – Eur. J. 2013, 19, 7007–7012. doi:10.1002/chem.201300163 |
| 33. | Brückl, T.; Klepper, F.; Gutsmiedl, K.; Carell, T. Org. Biomol. Chem. 2007, 5, 3821–3825. doi:10.1039/b713309j |
| 31. | Zhu, G.; Liu, Z.; Xu, Y.; Mao, Z. Heterocycles 2008, 75, 1631–1638. doi:10.3987/com-08-11323 |
| 32. | Wilding, B.; Winkler, M.; Petschacher, B.; Kratzer, R.; Egger, S.; Steinkellner, G.; Lyskowski, A.; Nidetzky, B.; Gruber, K.; Klempier, N. Chem. – Eur. J. 2013, 19, 7007–7012. doi:10.1002/chem.201300163 |
| 33. | Brückl, T.; Klepper, F.; Gutsmiedl, K.; Carell, T. Org. Biomol. Chem. 2007, 5, 3821–3825. doi:10.1039/b713309j |
| 1. | Flemmich, L.; Heel, S.; Moreno, S.; Breuker, K.; Micura, R. Nat. Commun. 2021, 12, 3877. doi:10.1038/s41467-021-24193-7 |
| 5. | McCarty, R. M.; Bandarian, V. Bioorg. Chem. 2012, 43, 15–25. doi:10.1016/j.bioorg.2012.01.001 |
| 21. | Ohgi, T.; Kondo, T.; Goto, T. Chem. Lett. 1979, 8, 1283–1286. doi:10.1246/cl.1979.1283 |
| 4. | Micura, R.; Höbartner, C. Chem. Soc. Rev. 2020, 49, 7331–7353. doi:10.1039/d0cs00617c |
| 22. | Akimoto, H.; Imamiya, E.; Hitaka, T.; Nomura, H.; Nishimura, S. J. Chem. Soc., Perkin Trans. 1 1988, 1637–1644. doi:10.1039/p19880001637 |
| 2. | Breaker, R. R. ACS Chem. Biol. 2020, 15, 2020–2030. doi:10.1021/acschembio.0c00214 |
| 3. | Batey, R. T. RNA 2015, 21, 560–563. doi:10.1261/rna.050765.115 |
| 18. | Shuai, H.; Myronovskyi, M.; Nadmid, S.; Luzhetskyy, A. Biomolecules 2020, 10, 1074. doi:10.3390/biom10071074 |
| 1. | Flemmich, L.; Heel, S.; Moreno, S.; Breuker, K.; Micura, R. Nat. Commun. 2021, 12, 3877. doi:10.1038/s41467-021-24193-7 |
| 17. | Iijima, M.; Kubota, Y.; Sawa, R.; Kubota, Y.; Hatano, M.; Igarashi, M.; Kawada, M.; Momose, I.; Takekawa, M.; Shibasaki, M. J. Antibiot. 2018, 71, 135–138. doi:10.1038/ja.2017.100 |
| 17. | Iijima, M.; Kubota, Y.; Sawa, R.; Kubota, Y.; Hatano, M.; Igarashi, M.; Kawada, M.; Momose, I.; Takekawa, M.; Shibasaki, M. J. Antibiot. 2018, 71, 135–138. doi:10.1038/ja.2017.100 |
| 19. | Nishizawa, N.; Kondo, Y.; Koyama, M.; Omoto, S.; Iwata, M.; Tsuruoka, T.; Inouye, S. J. Antibiot. 1984, 37, 1–5. doi:10.7164/antibiotics.37.1 |
| 20. | Ohno, H.; Terui, T.; Kitawaki, T.; Chida, N. Tetrahedron Lett. 2006, 47, 5747–5750. doi:10.1016/j.tetlet.2006.06.001 |
| 33. | Brückl, T.; Klepper, F.; Gutsmiedl, K.; Carell, T. Org. Biomol. Chem. 2007, 5, 3821–3825. doi:10.1039/b713309j |
| 13. | Serganov, A.; Nudler, E. Cell 2013, 152, 17–24. doi:10.1016/j.cell.2012.12.024 |
| 18. | Shuai, H.; Myronovskyi, M.; Nadmid, S.; Luzhetskyy, A. Biomolecules 2020, 10, 1074. doi:10.3390/biom10071074 |
| 29. | Khalaf, A. I.; Huggan, J. K.; Suckling, C. J.; Gibson, C. L.; Stewart, K.; Giordani, F.; Barrett, M. P.; Wong, P. E.; Barrack, K. L.; Hunter, W. N. J. Med. Chem. 2014, 57, 6479–6494. doi:10.1021/jm500483b |
| 30. | Chang, L.; Lee, S.-Y.; Leonczak, P.; Rozenski, J.; De Jonghe, S.; Hanck, T.; Müller, C. E.; Herdewijn, P. J. Med. Chem. 2014, 57, 10080–10100. doi:10.1021/jm501434y |
| 31. | Zhu, G.; Liu, Z.; Xu, Y.; Mao, Z. Heterocycles 2008, 75, 1631–1638. doi:10.3987/com-08-11323 |
| 8. | Roth, A.; Winkler, W. C.; Regulski, E. E.; Lee, B. W. K.; Lim, J.; Jona, I.; Barrick, J. E.; Ritwik, A.; Kim, J. N.; Welz, R.; Iwata-Reuyl, D.; Breaker, R. R. Nat. Struct. Mol. Biol. 2007, 14, 308–317. doi:10.1038/nsmb1224 |
| 9. | Meyer, M. M.; Roth, A.; Chervin, S. M.; Garcia, G. A.; Breaker, R. R. RNA 2008, 14, 685–695. doi:10.1261/rna.937308 |
| 10. | McCown, P. J.; Liang, J. J.; Weinberg, Z.; Breaker, R. R. Chem. Biol. 2014, 21, 880–889. doi:10.1016/j.chembiol.2014.05.015 |
| 11. | Jenkins, J. L.; Krucinska, J.; McCarty, R. M.; Bandarian, V.; Wedekind, J. E. J. Biol. Chem. 2011, 286, 24626–24637. doi:10.1074/jbc.m111.230375 |
| 12. | Chatterjee, S.; Chauvier, A.; Dandpat, S. S.; Artsimovitch, I.; Walter, N. G. Proc. Natl. Acad. Sci. U. S. A. 2021, 118, e2023426118. doi:10.1073/pnas.2023426118 |
| 13. | Serganov, A.; Nudler, E. Cell 2013, 152, 17–24. doi:10.1016/j.cell.2012.12.024 |
| 14. | Zallot, R.; Yuan, Y.; de Crécy-Lagard, V. Biomolecules 2017, 7, 12. doi:10.3390/biom7010012 |
| 15. | Iwata-Reuyl, D. Curr. Opin. Chem. Biol. 2008, 12, 126–133. doi:10.1016/j.cbpa.2008.01.041 |
| 16. | Gaur, R.; Varshney, U. J. Bacteriol. 2005, 187, 6893–6901. doi:10.1128/jb.187.20.6893-6901.2005 |
| 34. | Brooks, A. F.; Garcia, G. A.; Showalter, H. D. H. Tetrahedron Lett. 2010, 51, 4163–4165. doi:10.1016/j.tetlet.2010.06.008 |
| 6. | Iwata-Reuyl, D. Bioorg. Chem. 2003, 31, 24–43. doi:10.1016/s0045-2068(02)00513-8 |
| 7. | Vinayak, M.; Pathak, C. Biosci. Rep. 2010, 30, 135–148. doi:10.1042/bsr20090057 |
| 18. | Shuai, H.; Myronovskyi, M.; Nadmid, S.; Luzhetskyy, A. Biomolecules 2020, 10, 1074. doi:10.3390/biom10071074 |
| 29. | Khalaf, A. I.; Huggan, J. K.; Suckling, C. J.; Gibson, C. L.; Stewart, K.; Giordani, F.; Barrett, M. P.; Wong, P. E.; Barrack, K. L.; Hunter, W. N. J. Med. Chem. 2014, 57, 6479–6494. doi:10.1021/jm500483b |
| 24. | Klepper, F.; Polborn, K.; Carell, T. Helv. Chim. Acta 2005, 88, 2610–2616. doi:10.1002/hlca.200590201 |
| 25. | Barnett, C. J.; Grubb, L. M. Tetrahedron 2000, 56, 9221–9225. doi:10.1016/s0040-4020(00)00895-4 |
| 22. | Akimoto, H.; Imamiya, E.; Hitaka, T.; Nomura, H.; Nishimura, S. J. Chem. Soc., Perkin Trans. 1 1988, 1637–1644. doi:10.1039/p19880001637 |
| 23. | Gerber, H.-D.; Klebe, G. Org. Biomol. Chem. 2012, 10, 8660–8668. doi:10.1039/c2ob26387d |
| 23. | Gerber, H.-D.; Klebe, G. Org. Biomol. Chem. 2012, 10, 8660–8668. doi:10.1039/c2ob26387d |
| 24. | Klepper, F.; Polborn, K.; Carell, T. Helv. Chim. Acta 2005, 88, 2610–2616. doi:10.1002/hlca.200590201 |
| 25. | Barnett, C. J.; Grubb, L. M. Tetrahedron 2000, 56, 9221–9225. doi:10.1016/s0040-4020(00)00895-4 |
| 26. | Levic, J.; Micura, R. Beilstein J. Org. Chem. 2014, 10, 1914–1918. doi:10.3762/bjoc.10.199 |
| 27. | Moschen, T.; Wunderlich, C. H.; Spitzer, R.; Levic, J.; Micura, R.; Tollinger, M.; Kreutz, C. Angew. Chem., Int. Ed. 2015, 54, 560–563. doi:10.1002/anie.201409779 |
| 31. | Zhu, G.; Liu, Z.; Xu, Y.; Mao, Z. Heterocycles 2008, 75, 1631–1638. doi:10.3987/com-08-11323 |
| 29. | Khalaf, A. I.; Huggan, J. K.; Suckling, C. J.; Gibson, C. L.; Stewart, K.; Giordani, F.; Barrett, M. P.; Wong, P. E.; Barrack, K. L.; Hunter, W. N. J. Med. Chem. 2014, 57, 6479–6494. doi:10.1021/jm500483b |
| 30. | Chang, L.; Lee, S.-Y.; Leonczak, P.; Rozenski, J.; De Jonghe, S.; Hanck, T.; Müller, C. E.; Herdewijn, P. J. Med. Chem. 2014, 57, 10080–10100. doi:10.1021/jm501434y |
| 28. | Neuner, E.; Frener, M.; Lusser, A.; Micura, R. RNA Biol. 2018, 15, 1376–1383. doi:10.1080/15476286.2018.1534526 |
| 20. | Ohno, H.; Terui, T.; Kitawaki, T.; Chida, N. Tetrahedron Lett. 2006, 47, 5747–5750. doi:10.1016/j.tetlet.2006.06.001 |
| 26. | Levic, J.; Micura, R. Beilstein J. Org. Chem. 2014, 10, 1914–1918. doi:10.3762/bjoc.10.199 |
| 27. | Moschen, T.; Wunderlich, C. H.; Spitzer, R.; Levic, J.; Micura, R.; Tollinger, M.; Kreutz, C. Angew. Chem., Int. Ed. 2015, 54, 560–563. doi:10.1002/anie.201409779 |
© 2021 Flemmich et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the author(s) and source are credited and that individual graphics may be subject to special legal provisions.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc/terms)