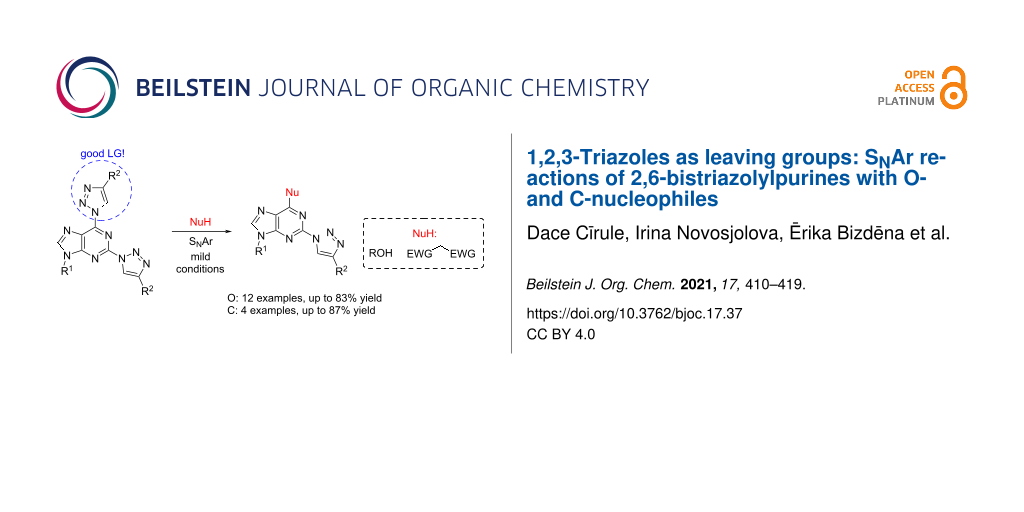
Abstract
A new approach was designed for the synthesis of C6-substituted 2-triazolylpurine derivatives. A series of substituted products was obtained in SNAr reactions between 2,6-bistriazolylpurine derivatives and O- and C-nucleophiles under mild conditions. The products were isolated in yields up to 87%. The developed C–O and C–C bond forming reactions clearly show the ability of the 1,2,3-triazolyl ring at the C6 position of purine to act as leaving group.

Graphical Abstract
Introduction
Modified purine derivatives are an important class of compounds which possess a wide spectrum of biological activities [1-6]. They are often used as antiviral, anticancer and antibacterial agents. Such intensive medicinal chemistry applications demand for constant development of novel synthetic methodologies. Frequently, the purine structure is modified in SNAr reactions with N- [7-11] and S-nucleophiles [12-14] and in metal catalyzed reactions of halopurine derivatives [15-20]. Modifications of purines with O-nucleophiles are based on SNAr reactions between 6-halopurine derivatives and alcohols [21-28] in the presence of a base. Alcohols are used in excess (5–40 equiv) and often play a role of both solvent and reagent. Reactions usually are performed in polar aprotic solvents such as DMF, MeCN or THF using alkoxides NaH, K2CO3 or Na2CO3 as a base, respectively.
Other methods for the introduction of alkyloxy or aryloxy substituents in the purine structure involve substitution reactions of different leaving groups such as: 1) benzotriazolyloxy group (HOBT) [8,29-32]; 2) the alkylimidazolyl group [33,34] and 3) in-situ-generated alkylammonia salts [35-38]. In 1995, the Robins group demonstrated SNAr reactions of 6-(1,2,4-triazol-4-yl)purine with dimethylamine, sodium methoxide and sodium thiomethoxide [39]. Earlier, the use of 6-(1,2,4-triazol-1-yl)purine derivatives in SNAr reactions has been reported [40]. An alternative method for the synthesis of O6-alkylpurines is Pd catalyzed C–O bond formation starting from 6-halopurines [41]. O-Alkylation of guanosine and inosine with Cu(I)-stabilized carbenes derived form α-diazocarbonyl compounds is also known [42]. Alkylation of 6-oxopurine derivatives under Mitsunobu conditions which usually proceeds with O-regioselectivity are mostly described for guanine derivatives [43-50]. In the case of C-nucleophiles there are a few precedents of transition-metal-free substitution of chloro [51-55] or 1,2,4-triazolyl [56] moieties as leaving groups at the C6 position of purine. These transformations usually require prolonged time and elevated temperatures to be completed. Among the widely studied 1,2,3-triazolyl nucleoside conjugates [57,58], the synthesis of 2-triazolylpurine derivatives containing a designed substituent at C6 has been little discussed. 6-N-Substituted purines have been the most studied [11,59-62], but 6-S- [14,63] or 6-O-analogues are less common [61].
Azolylpurine derivatives are important due to their potential as drug candidates. They can be used as agonists and antagonists of adenosine receptors [58,64-66] and against Mycobacterium tuberculosis [60]. They also show useful fluorescent properties [11,67-69] and can be used as metal ion sensors [70]. Therefore, it is important to develop novel methods towards this type of derivatives. To date two approaches have been used to obtain 6-substituted 2-triazolylpurine derivatives (Scheme 1). According to the pathway A, firstly a selected substituent is introduced at the C6 position of the purine ring using SNAr reactions (Ia→II, Scheme 1). If purine contains identical leaving groups at C2 and C6 positions the reactivity order in its SNAr reactions is C6 > C2 [71,72]. Also transition metal catalyzed reactions can be used for C6 functionalization of purine [73-76] or alkylation of inosine or guanosine derivatives (Ib→II, Scheme 1) [30,36]. In the next step, azide can be introduced either by a second SNAr reaction on the C2-halo derivative or by diazotization/azidation at C2. Then, the Cu(I)-catalyzed azide–alkyne cycloaddition (CuAAC) reaction provides the target product IV (Scheme 1, pathway A) [59-61]. Pathway B is designed on the basis of our group investigations on the synthesis of 2,6-bistriazolylpurine derivatives and their application in reactions with N-, S- and P-nucleophiles making use of regioselective SNAr reactions at C(6) (V→VI→IV, Scheme 1) [11,14,62,63,77,78]. The main advantage of pathway B is a straightforward access to 2,6-diazidopurines V and 2,6-bistriazolylpurines VI due to excellent nucleophilic properties of the azide ion and well-established CuAAC reaction. Pathway B also avoids performing of an SNAr process on partially deactivated purines as the introduced nucleophiles are mostly seen as electron-donating substituents (e.g., R2N-, RS-, RO-).
![[1860-5397-17-37-i1]](/bjoc/content/inline/1860-5397-17-37-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthetic pathways for the synthesis of 6-substituted 2-triazolylpurine derivatives IV.
Scheme 1: Synthetic pathways for the synthesis of 6-substituted 2-triazolylpurine derivatives IV.
Herein, we report a synthetic extension of this methodology. We have found that the pronounced leaving group character of 1,2,3-triazoles makes 2,6-bistriazolylpurines excellent substrates for SNAr reactions with O- and C-nucleophiles.
Results and Discussion
Synthesis of 2,6-bistriazolylpurine derivatives and their reactions with O-nucleophiles
The 2,6-diazidopurine derivatives 1a and 1b as strategic starting materials and 2,6-bistriazolylpurine derivatives 2a–c were obtained in the synthetic procedures developed by us before [11,14,67]. The CuAAC reaction was performed between diazide derivatives 1a and 1b and phenylacetylene or methyl propiolate (Scheme 2).
![[1860-5397-17-37-i2]](/bjoc/content/inline/1860-5397-17-37-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of 2,6-bistriazolylpurine derivatives 2a–c.
Scheme 2: Synthesis of 2,6-bistriazolylpurine derivatives 2a–c.
SNAr reactions between bistriazolylpurine derivatives and O-nucleophiles were first performed on N9-alkylated bistriazole 2c. The reactions were carried out with primary and secondary alcohols in the presence of NaH in DMF. The developed transformation required only nearly equimolar loading of an alcohol and a base, and products 3a–f were obtained in yields up to 83% (Scheme 3). In most cases the full conversion of the starting material was reached in 15–30 min at room temperature, which clearly showed the excellent leaving group ability of the triazolyl ring. These SNAr reactions can also be performed in DMSO or DMF in the presence of K2CO3, but the completion of these transformations requires heating the reaction mixtures up to 60 °C for 24 h.
![[1860-5397-17-37-i3]](/bjoc/content/inline/1860-5397-17-37-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of 6-alkyloxy-2-triazolylpurine derivatives 3a–f.
Scheme 3: Synthesis of 6-alkyloxy-2-triazolylpurine derivatives 3a–f.
An SNAr reaction with a non-trivial alcohol was demonstrated on the example of 2',3'-O-isopropylideneuridine and product 3f was isolated after 21 h of heating at 50 °C in 82% yield. It should be noted that tertiary alcohols (e.g., t-BuOH) were inert in SNAr reactions with 2,6-bistriazolylpurines and their attempted reactions resulted in an unidentifiable mixture of byproducts.
The following experiments were performed on 2,6-bistriazolylpurine nucleoside 2b in MeOH, EtOH and PrOH used as solvents and nucleophiles in the presence of NaH (5.0 equiv). The excess of base and alcohol was required due to the cleavage of acetyl protecting groups. Products 3g–i were obtained in yields of up to 79% (Scheme 4). Furthermore, purification of the products 3g–i was complicated due to their poor solubility in organic solvents. The C6 regioselectivity of SNAr reactions was proved by 13C NMR comparison of the products 3a–i with similar compounds from literature [61].
![[1860-5397-17-37-i4]](/bjoc/content/inline/1860-5397-17-37-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of 6-alkyloxy-2-triazolylpurine nucleosides 3g–j.
Scheme 4: Synthesis of 6-alkyloxy-2-triazolylpurine nucleosides 3g–j.
Intriguingly, we were able to conserve the acetate protecting groups in product 3j, when the SNAr reaction was performed in the presence of DBU used as base. The artificial dinucleotide analogue 3j was obtained in 25% isolated yield.
We have explored also reactions of 2,6-bistriazolylpurines 2a and 2c with water in buffered and basic medium, respectively (Scheme 5). The buffered conditions (NaOAc/DMSO/H2O) were sufficiently mild to maintain the acetyl protecting groups in product 4a. Also hydrolysis of 2c into 4b proceeded under mild conditions and only gentle warming to 50 °C was required.
![[1860-5397-17-37-i5]](/bjoc/content/inline/1860-5397-17-37-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: 2,6-Bistriazolylpurine derivatives in SNAr reactions with H2O/HO− as nucleophiles.
Scheme 5: 2,6-Bistriazolylpurine derivatives in SNAr reactions with H2O/HO− as nucleophiles.
2,6-Bistriazolylpurine derivatives in SNAr reactions with C-nucleophiles
Next, SNAr reactions between 2,6-bistriazolylpurine 2c and C-nucleophiles offered an easy way for the C–N bond transformation into a C–C bond. Compounds containing electron-withdrawing groups such as malonitrile, dimedone, ethyl cyanoacetate and diethyl malonate were used as C-nucleophiles. Transformations were performed in DMF in the presence of NaH and the products were obtained in high yields (Scheme 6). The lower yield of compound 5d was obtained due to the ethyl ester hydrolysis and subsequent decarboxylation. Such side reactions were also observed for similar compounds in literature [79,80].
![[1860-5397-17-37-i6]](/bjoc/content/inline/1860-5397-17-37-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Synthesis of C6-substituted 2-triazolylpurine derivatives 5.
Scheme 6: Synthesis of C6-substituted 2-triazolylpurine derivatives 5.
As a limitation of the method we have found that 2,6-bistriazolylpurine 2c was inert to SNAr reactions with deprotonated acetylacetone and diphenylmethane. Even there are reports on SNAr reactions of acetylacetone with purines and pyrimidines [56,80], in our hands only polymerization of acetylacetone was observed. On the other hand, the diphenylmethane anion (pKa 32; DMSO [81]) apparently is too basic and deprotonates purine C(8)–H, thus suspending the SNAr process.
The structures of C6-substituted products 5a–d were elucidated by NMR and IR analysis. These compounds can exist as either C–H acids (A) or N–H acids (B), but dimedone conjugate 5b may possess also an enol form C (Figure 1).
![[1860-5397-17-37-1]](/bjoc/content/figures/1860-5397-17-37-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Possible tautomeric structures of compounds 5a–d.
Figure 1: Possible tautomeric structures of compounds 5a–d.
During the structural studies of cyano group containing products 5a and 5c the cross signals for the C(2’’)–H system were not found using HSQC spectra, excluding the existence of C–H tautomeric forms A. In addition, IR analysis (KBr tablet) indicated absorption bands of cyano groups at 2205 and 2170 cm−1 for product 5a and at 2205 cm−1 for product 5c. These results differ from the absorption in the range of 2260–2240 cm−1, which would be characteristic for a cyano group attached to sp3-hybridized carbon [82]. On the other hand, 13C NMR shifts of the C(2’’) position of purine–malononitrile conjugate 5a and ethyl cyanoacetate–purine conjugate 5c were 40.9 and 61.7 ppm, respectively. This range does not fully correspond to the theoretical values 80–140 ppm, expected for the Csp2 atom of the N–H form B. In compound 5c the N–H form 5cB is possibly the major tautomer in CDCl3 solution as it is stabilized via an intramolecular hydrogen bond. This is supported by a smaller deviation of the C(2’’) chemical shift value (61.7 ppm) in comparison to the theoretical shifts for a Csp2 centre. Similar structural analogues are known in the literature [54,83-85] but their structural analysis was incomplete. As the aforementioned experiments did not determine preference for tautomer A or B of compound 5a, it was analysed in its deprotonated form C (CD3OD/D2O/NaOD). Interestingly, that the 13C NMR spectrum of 5a in basic medium revealed a similar chemical shift for carbon C(2’’) (40.9 ppm) as in neutral CD3OD.
The 13C NMR analysis of purine–dimedone conjugate 5b revealed two downfield shifts of 194.1 and 185.3 ppm. It showed that the structure is not symmetrical and corresponds to either tautomer structure B or C in CDCl3 solution with a theoretical preference for enol form C. Finally, the structure of C–H tautomer 5dA was proved by its HSQC spectrum, in which a cross peak clearly indicated the C(2’’)–H system.
Conclusion
The SNAr reactivity of 2,6-bis(1,2,3-triazol-1-yl)purine derivatives was extended with their substitution with O- and C-nucleophiles. The reactions proceeded under transition metal free conditions and revealed excellent C6 selectivity. The developed synthetic approach provided O-adducts with 25–83% yields and C-adducts with 67–87% yields. The methodology demonstrated the leaving group ability of the 1,2,3-triazolyl substituent at the C6 position of the purine ring.
Experimental
General information
1H and 13C NMR spectra were recorded with a Bruker Avance 300 or a Bruker Avance 500 spectrometer, at 300 and 75.5 MHz or 500 and 125.7 MHz, respectively. The proton signals for residual non-deuterated solvents (δ 7.26 for CDCl3, δ 2.50 for DMSO-d6, δ 3.31 for CD3OD) and the carbon signals (δ 77.1 for CDCl3, δ 39.5 for DMSO-d6, δ 49.0 for CD3OD) were used as an internal reference for 1H and 13C NMR spectra, respectively. Coupling constants are reported in Hz. Chemical shifts of signals are given in ppm and multiplicities are assigned as follows: s – singlet, d – doublet, t – triplet, m – multiplet, brs – broad singlet, tq – triplet of quartets.
Analytical thin-layer chromatography (TLC) was performed on Merck 60 Å silica gel F254 plates. Column chromatography was performed on Merck 40–60 µm 60 Å silica gel. Yields of products refer to chromatographically and spectroscopically homogeneous materials. The solvents used in the reactions were dried with standard drying agents and freshly distilled prior to use. Commercial reagents were used as received.
IR spectra were recorded in KBr tablets with a Perkin–Elmer Spectrum BX FTIR spectrometer (4000–450 cm−1). Wavelengths are given in cm−1.
For HPLC analysis an Agilent Technologies 1200 Series chromatograph equipped with an Agilent XDB-C18 (4.6 × 50 mm, 1.8 µm) column was used. Eluent A: 0.1% TFA solution with 5% v/v MeCN added; eluent B – MeCN. Gradient: 10–95% B 5 min, 95% B 5 min, 95–10% B 2 min. Flow: 1 mL/min. Wavelength of detection was 260 nm.
LC–MS was recorded with a Waters Acquity UPLC system equipped with Acquity UPLC BEH C18 1.7 μm, 2.1 × 50 mm; using 0.1% TFA/H2O and MeCN for mobile phase. HRMS analyses were performed on an Agilent 1290 Infinity series UPLC system equipped with column Extend C18 RRHD 2.1 × 50 mm, 1.8 μm connected to an Agilent 6230 TOF LC/MS mass spectrometer.
General procedures and product characterization
Synthesis of compounds 1a,b and 2a–c and their characterization are described earlier [11,14,67].
Synthesis of 6-O-substituted 2-triazolylpurines
General procedure A for the SNAr reaction with O-nucleophiles
9-Heptyl-2-(4-phenyl-1H-1,2,3-triazol-1-yl)-6-(prop-1-yl)oxy-9H-purine (3a): To a suspension of 9-heptyl-2,6-bis(4-phenyl-1H-1,2,3-triazol-1-yl)-9H-purine (2c, 188 mg, 0.37 mmol, 1.0 equiv) in anhydrous DMF (2.5 mL) a suspension of n-PrOH (34 μL, 0.45 mmol, 1.2 equiv) and NaH (10 mg, 0.43 mmol, 1.2 equiv) in anhydrous DMF (0.5 mL) was added and the reaction mixture was stirred for 15 min at rt, controlled by HPLC. Then toluene or ethyl acetate (25 mL) was added to the mixture and it was extracted with 5% LiCl solution (3 × 5 mL). The organic phase was dried over anhydrous Na2SO4, filtered and evaporated. Silica gel column chromatography (DCM/MeCN 10:1) gave the product as colourless amorphous solid. Yield 115 mg, 83%. Rf = 0.80 (DCM/MeCN 5:1); HPLC: tR = 7.68 min, purity 98%; IR (KBr) ν (cm−1): 3075, 2965, 2930, 2870, 1745, 1605, 1435, 1415, 1350, 1330, 1245, 1235, 1070; 1H NMR (300 MHz, CDCl3) δ 8.70 (s, 1H, H-C(triazole)), 7.93 (s, 1H, H-C(8)), 7.91 (d, 3J = 7.6 Hz, 2H, Ar), 7.39 (t, 3J = 7.6 Hz, 2H, Ar), 7.30 (t, 3J = 7.6 Hz, 1H, Ar), 4.65 (t, 3J1’’,2’’ = 6.7 Hz, 2H, H2C(1’’)), 4.26 (t, 3J1’,2’ = 7.2 Hz, 2H, H2C(1’)), 1.96 (tq, 3J1’’,2’’= 6.7 Hz, 3J2’’-3’’= 7.4 Hz, 2H, H2C(2’’)), 1.93‒1.82 (m, 2H, H2C(2’)), 1.35‒1.26 (m, 4H, H2C(3’), H2C(4’)), 1.25‒1.17 (m, 4H, H2C(5’), H2C(6’)), 1.08 (t, 3J2’’,3’’= 7.4 Hz, 3H, H3C(3’’)), 0.81 (t, 3J6’-7’ = 6.9 Hz, 3H, H3C(7’)); 13C NMR (75.5 MHz, CDCl3) δ 161.5, 152.8, 148.3, 147.5, 143.0, 130.1, 128.7, 128.3, 125.9, 120.6, 118.6, 69.8, 44.2, 31.5, 29.9, 28.6, 26.5, 22.4, 22.1, 13.9, 10.5; HRESIMS (m/z): [M + H]+ calcd for C23H30N7O, 420.2506; found, 420.2510 (0.95 ppm).
General procedure B for the SNAr reaction with O-nucleophiles
9-(β-ᴅ-Ribofuranosyl)-6-methoxy-2-(4-phenyl-1H-1,2,3-triazol-1-yl)-9H-purine (3g): To a solution of 9-(2’,3’,5’-tri-O-acetyl-β-ᴅ-ribofuranosyl)-2,6-bis(4-phenyl-1H-1,2,3-triazol-1-yl)-9H-purine (2b, 335 mg, 0.50 mmol, 1.0 equiv) in MeOH (6 mL) a suspension of NaH (60 mg, 2.52 mmol, 5.0 equiv) in MeOH (6 mL) was added and the reaction mixture was stirred for 10 min at rt, controlled by HPLC. Then AcOH (0.2 mL) was added and the mixture was partially evaporated. The suspension was centrifuged, the solids were separated and washed with MeOH (4 × 7 mL). Colourless solid. Yield 168 mg, 79%. HPLC: tR = 4.20 min, purity 95%; IR (KBr) ν (cm−1): 3390, 2950, 1605, 1490, 1455, 1400, 1365, 1245, 1035, 1020; 1H NMR (300 MHz, DMSO-d6 + D2O) δ 9.38 (s, 1H, H-C(triazole)), 8.70 (s, 1H, H-C(8)), 8.02 (d, 3J = 7.6 Hz, 2H, Ar), 7.50 (t, 3J = 7.6 Hz, 2H, Ar), 7.39 (t, 3J = 7.6 Hz, 1H, Ar), 6.06 (d, 3J1’,2’ = 5.8 Hz, 1H, H-C(1’)), 4.65 (dd, 3J1’,2’ = 5.8 Hz, 3J2’,3’ = 4.8 Hz, 1H, H-C(2’)), 4.29 (s, 3H, (-OCH3)), 4.22 (dd, 3J2’,3’ = 4.8 Hz, 3J3’,4’= 3.7 Hz, 1H, H-C(3’)), 4.01 (dt, 3J3’,4’ = 3.7 Hz, 3J4’,5a’ = 3J4’,5b’ = 4.0 Hz, 1H, H-C(4’)), 3.71 (dd, 3J4’,5a’ = 4.0 Hz, 2J5a’,5b’ = 12.1 Hz, 1H, Ha-C(5’)), 3.60 (dd, 3J4’,5b’ = 4.0 Hz, 2J5a’,5b’ = 12.1 Hz, 1H, Hb-C(5’)); 13C NMR (75.5 MHz, DMSO-d6 + D2O) δ 161.4, 152.7, 148.0, 147.0, 143.6, 130.0, 129.2, 128.8, 125.8, 120.7, 120.5, 87.8, 86.0, 74.0, 70.4, 61.3, 55.3; HRESIMS (m/z): [M + H]+ calcd for C19H20N7O5, 426.1520; found, 426.1528 (1.88 ppm).
Synthesis of C6-substituted 2-triazolylpurines
General procedure C for the SNAr reaction with C-nucleophiles
2-(9-Heptyl-2-(4-phenyl-1H-1,2,3-triazol-1-yl)-1,9-dihydro-6H-purin-6-ylidene)malononitrile (5a): Under argon atmosphere to a suspension of 9-heptyl-2,6-bis(4-phenyl-1H-1,2,3-triazol-1-yl)-9H-purine (2c, 141 mg, 0.28 mmol, 1 equiv) in anhydrous DMF (2.5 mL) malononitrile (23 mg, 0.35 mmol, 1.3 equiv) and NaH (8 mg, 0.34 mmol, 1.2 equiv) were added and the reaction mixture was stirred for 30 min at rt, controlled by HPLC. Then ethyl acetate (25 mL) was added and the mixture was extracted with 5% LiCl solution (3 × 5 mL). The organic phase was dried over anhydrous Na2SO4, filtered and evaporated. Silica gel column chromatography (toluene/MeCN; gradient 50% → 75%) gave product 5a as a slightly yellow amorphous solid, Rf = 0.17 (toluene/MeCN 1:1). Yield 103 mg, 87%. HPLC: tR = 6.33 min, purity 98%; IR (KBr) ν (cm−1): 3400, 2955, 2925, 2855, 2205, 2170, 1590, 1460, 1430, 1410, 1350, 1235, 1040; 1H NMR (300 MHz, CD3OD + D2O) δ 9.05 (s, 1H, H-C(triazole)), 8.02 (s, 1H, H-C(8)), 7.94 (d, 3J = 7.5 Hz, 2H, Ar), 7.47 (d, 3J = 7.5 Hz, 2H, Ar), 7.37 (t, 3J = 7.5 Hz, 1H, Ar), 4.28 (t, 3J1’,2’ = 7.2 Hz, 2H, H2C(1’)), 1.96‒1.83 (m, 2H, H2C(2’)), 1.40‒1.32 (m, 4H, H2C(3’), H2C(4’)), 1.31‒1.23 (m, 4H, H2C(5’), H2C(6’)), 0.86 (t, 3J6’,7’ = 6.9 Hz, 3H, H3C(7’)); 13C NMR (75.5 MHz, CD3OD) δ (ppm): 161.3, 150.4, 150.2, 148.7, 142.4, 131.4, 130.0, 129.5, 126.9, 125.2, 123.4, 120.7, 44.7, 40.9, 32.9, 31.2, 29.9, 27.6, 23.6, 14.3; HRESIMS (m/z): [M + H]+ calcd for C23H24N9, 426.2149; found, 426.2149 (0 ppm).
Supporting Information
| Supporting Information File 1: Full experimental procedures and copies of 1H, 13C and 1H,13C HSQC NMR spectra. | ||
| Format: PDF | Size: 2.6 MB | Download |
References
-
Dinesh, S.; Shikha, G.; Bhavana, G.; Nidhi, S.; Dileep, S. J. Pharm. Sci. Innovation 2012, 1, 29–34.
Return to citation in text: [1] -
Parker, W. B. Chem. Rev. 2009, 109, 2880–2893. doi:10.1021/cr900028p
Return to citation in text: [1] -
Shelton, J.; Lu, X.; Hollenbaugh, J. A.; Cho, J. H.; Amblard, F.; Schinazi, R. F. Chem. Rev. 2016, 116, 14379–14455. doi:10.1021/acs.chemrev.6b00209
Return to citation in text: [1] -
Seley-Radtke, K. L.; Yates, M. K. Antiviral Res. 2018, 154, 66–86. doi:10.1016/j.antiviral.2018.04.004
Return to citation in text: [1] -
Yates, M. K.; Seley-Radtke, K. L. Antiviral Res. 2019, 162, 5–21. doi:10.1016/j.antiviral.2018.11.016
Return to citation in text: [1] -
Liang, Y.; Wen, Z.; Cabrera, M.; Howlader, A. H.; Wnuk, S. F. Purines (Update 2020). In SOS Knowledge Updates 2020/1; Christmann, M.; Huang, Z.; Jiang, X.; Li, J. J.; Oestreich, M.; Petersson, E. J.; Schaumann, E.; Wang, M., Eds.; Georg Thieme Verlag: Stuttgart, Germany, 2020. doi:10.1055/sos-sd-116-01081
Return to citation in text: [1] -
Véliz, E. A.; Beal, P. A. J. Org. Chem. 2001, 66, 8592–8598. doi:10.1021/jo016078v
Return to citation in text: [1] -
Lakshman, M. K.; Frank, J. Org. Biomol. Chem. 2009, 7, 2933–2940. doi:10.1039/b905298d
Return to citation in text: [1] [2] -
Manvar, A.; Shah, A. Tetrahedron 2013, 69, 8105–8127. doi:10.1016/j.tet.2013.06.034
Return to citation in text: [1] -
Manvar, A.; Shah, A. Tetrahedron 2013, 69, 680–691. doi:10.1016/j.tet.2012.10.079
Return to citation in text: [1] -
Kovaļovs, A.; Novosjolova, I.; Bizdēna, Ē.; Bižāne, I.; Skardziute, L.; Kazlauskas, K.; Jursenas, S.; Turks, M. Tetrahedron Lett. 2013, 54, 850–853. doi:10.1016/j.tetlet.2012.11.095
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Aleksandrova, E. V. Pharm. Chem. J. 2003, 37, 645–652. doi:10.1023/b:phac.0000022083.93211.2c
Return to citation in text: [1] -
Aleksandrova, E. V.; Kochergin, P. M. Pharm. Chem. J. 2013, 46, 612–621. doi:10.1007/s11094-013-0856-y
Return to citation in text: [1] -
Novosjolova, I.; Bizdēna, Ē.; Turks, M. Tetrahedron Lett. 2013, 54, 6557–6561. doi:10.1016/j.tetlet.2013.09.095
Return to citation in text: [1] [2] [3] [4] [5] -
Gurram, V.; Pottabathini, N.; Garlapati, R.; Chaudhary, A. B.; Patro, B.; Lakshman, M. K. Chem. – Asian J. 2012, 7, 1853–1861. doi:10.1002/asia.201200093
Return to citation in text: [1] -
Lakshman, M. K.; Gunda, P.; Pradhan, P. J. Org. Chem. 2005, 70, 10329–10335. doi:10.1021/jo0513764
Return to citation in text: [1] -
Havelková, M.; Hocek, M.; Česnek, M.; Dvořák, D. Synlett 1999, 1145–1147. doi:10.1055/s-1999-2753
Return to citation in text: [1] -
Havelková, M.; Dvořak, D.; Hocek, M. Synthesis 2001, 1704–1710. doi:10.1055/s-2001-16765
Return to citation in text: [1] -
Hocek, M.; Masojídková, M.; Holý, A. Collect. Czech. Chem. Commun. 1997, 62, 136–146. doi:10.1135/cccc19970136
Return to citation in text: [1] -
Hasník, Z.; Pohl, R.; Hocek, M. Synthesis 2009, 1309–1317. doi:10.1055/s-0028-1088038
Return to citation in text: [1] -
Guo, H.-M.; Xin, P.-Y.; Niu, H.-Y.; Wang, D.-C.; Jiang, Y.; Qu, G.-R. Green Chem. 2010, 12, 2131–2134. doi:10.1039/c0gc00517g
Return to citation in text: [1] -
Pathak, A. K.; Pathak, V.; Seitz, L. E.; Suling, W. J.; Reynolds, R. C. Bioorg. Med. Chem. 2013, 21, 1685–1695. doi:10.1016/j.bmc.2013.01.054
Return to citation in text: [1] -
McGuigan, C.; Madela, K.; Aljarah, M.; Gilles, A.; Battina, S. K.; Ramamurty, C. V. S.; Srinivas Rao, C.; Vernachio, J.; Hutchins, J.; Hall, A.; Kolykhalov, A.; Henson, G.; Chamberlain, S. Bioorg. Med. Chem. Lett. 2011, 21, 6007–6012. doi:10.1016/j.bmcl.2011.06.013
Return to citation in text: [1] -
Vijay Kumar, D.; Hoarau, C.; Bursavich, M.; Slattum, P.; Gerrish, D.; Yager, K.; Saunders, M.; Shenderovich, M.; Roth, B. L.; McKinnon, R.; Chan, A.; Cimbora, D. M.; Bradford, C.; Reeves, L.; Patton, S.; Papac, D. I.; Williams, B. L.; Carlson, R. O. Bioorg. Med. Chem. Lett. 2012, 22, 4377–4385. doi:10.1016/j.bmcl.2012.04.131
Return to citation in text: [1] -
Hulpia, F.; Balzarini, J.; Schols, D.; Andrei, G.; Snoeck, R.; Van Calenbergh, S. Bioorg. Med. Chem. Lett. 2016, 26, 1970–1972. doi:10.1016/j.bmcl.2016.03.005
Return to citation in text: [1] -
Bonnac, L. F.; Dreis, C. D.; Geraghty, R. J. Bioorg. Med. Chem. Lett. 2020, 30, 126819. doi:10.1016/j.bmcl.2019.126819
Return to citation in text: [1] -
Besada, P.; Costas, T.; Teijeira, M.; Terán, C. Eur. J. Med. Chem. 2010, 45, 6114–6119. doi:10.1016/j.ejmech.2010.09.046
Return to citation in text: [1] -
Daumar, P.; Zeglis, B. M.; Ramos, N.; Divilov, V.; Sevak, K. K.; Pillarsetty, N.; Lewis, J. S. Eur. J. Med. Chem. 2014, 86, 769–781. doi:10.1016/j.ejmech.2014.09.019
Return to citation in text: [1] -
Bae, S.; Lakshman, M. K. J. Am. Chem. Soc. 2007, 129, 782–789. doi:10.1021/ja064682n
Return to citation in text: [1] -
Kokatla, H. P.; Lakshman, M. K. Org. Lett. 2010, 12, 4478–4481. doi:10.1021/ol101655h
Return to citation in text: [1] [2] -
Basava, V.; Yang, L.; Pradhan, P.; Lakshman, M. K. Org. Biomol. Chem. 2016, 14, 7069–7083. doi:10.1039/c6ob01170e
Return to citation in text: [1] -
Bae, S.; Lakshman, M. K. J. Org. Chem. 2008, 73, 1311–1319. doi:10.1021/jo7021795
Return to citation in text: [1] -
Zhong, M.; Nowak, I.; Robins, M. J. Org. Lett. 2005, 7, 4601–4603. doi:10.1021/ol051573p
Return to citation in text: [1] -
Zhong, M.; Nowak, I.; Robins, M. J. J. Org. Chem. 2006, 71, 7773–7779. doi:10.1021/jo061282+
Return to citation in text: [1] -
Linn, J. A.; McLean, E. W.; Kelley, J. L. J. Chem. Soc., Chem. Commun. 1994, 913–914. doi:10.1039/c39940000913
Return to citation in text: [1] -
Lakshman, M. K.; Ngassa, F. N.; Keeler, J. C.; Dinh, Y. Q. V.; Hilmer, J. H.; Russon, L. M. Org. Lett. 2000, 2, 927–930. doi:10.1021/ol005564m
Return to citation in text: [1] [2] -
Schirrmacher, R.; Wängler, B.; Schirrmacher, E.; August, T.; Rösch, F. Synthesis 2002, 538–542. doi:10.1055/s-2002-20970
Return to citation in text: [1] -
Schirrmacher, R.; Mühlhausen, U.; Wängler, B.; Schirrmacher, E.; Reinhard, J.; Nagel, G.; Kaina, B.; Piel, M.; Wießler, M.; Rösch, F. Tetrahedron Lett. 2002, 43, 6301–6304. doi:10.1016/s0040-4039(02)01394-1
Return to citation in text: [1] -
Miles, R. W.; Samano, V.; Robins, M. J. J. Am. Chem. Soc. 1995, 117, 5951–5957. doi:10.1021/ja00127a007
Return to citation in text: [1] -
Clivio, P.; Fourrey, J.-L.; Favre, A. J. Chem. Soc., Perkin Trans. 1 1993, 2585–2590. doi:10.1039/p19930002585
Return to citation in text: [1] -
Caner, J.; Vilarrasa, J. J. Org. Chem. 2010, 75, 4880–4883. doi:10.1021/jo100808w
Return to citation in text: [1] -
Geigle, S. N.; Wyss, L. A.; Sturla, S. J.; Gillingham, D. G. Chem. Sci. 2017, 8, 499–506. doi:10.1039/c6sc03502g
Return to citation in text: [1] -
Vincent, S. P.; Mioskowski, C.; Lebean, L. Nucleosides Nucleotides 1999, 18, 2127–2139. doi:10.1080/07328319908044869
Return to citation in text: [1] -
Woo, J.; Sigurdsson, S. T.; Hopkins, P. B. J. Am. Chem. Soc. 1993, 115, 3407–3415. doi:10.1021/ja00062a002
Return to citation in text: [1] -
Wilds, C. J.; Booth, J. D.; Noronha, A. M. Tetrahedron Lett. 2006, 47, 9125–9128. doi:10.1016/j.tetlet.2006.10.074
Return to citation in text: [1] -
Wang, L.; Spratt, T. E.; Liu, X.-K.; Hecht, S. S.; Pegg, A. E.; Peterson, L. A. Chem. Res. Toxicol. 1997, 10, 562–567. doi:10.1021/tx9602067
Return to citation in text: [1] -
Giordano, C.; Pedone, F.; Fattibene, P.; Cellai, L. Nucleosides, Nucleotides Nucleic Acids 2000, 19, 1301–1310. doi:10.1080/15257770008033053
Return to citation in text: [1] -
Cooper, M. D.; Hodge, R. P.; Tamura, P. J.; Wilkinson, A. S.; Harris, C. M.; Harris, T. M. Tetrahedron Lett. 2000, 41, 3555–3558. doi:10.1016/s0040-4039(00)00461-5
Return to citation in text: [1] -
Harwood, E. A.; Hopkins, P. B.; Sigurdsson, S. T. J. Org. Chem. 2000, 65, 2959–2964. doi:10.1021/jo991501+
Return to citation in text: [1] -
De Napoli, L.; Di Fabio, G.; Messere, A.; Montesarchio, D.; Piccialli, G.; Varra, M. J. Chem. Soc., Perkin Trans. 1 1999, 3489–3493. doi:10.1039/a906195i
Return to citation in text: [1] -
Zhang, Y.; Wang, L.; Zhang, Q.; Zhu, G.; Zhang, Z.; Zhou, X.; Chen, Y.; Lu, T.; Tang, W. J. Chem. Inf. Model. 2017, 57, 1439–1452. doi:10.1021/acs.jcim.6b00795
Return to citation in text: [1] -
Hamamichi, N.; Miyasaka, T. J. Org. Chem. 1994, 59, 1525–1531. doi:10.1021/jo00085a046
Return to citation in text: [1] -
Hamamichi, N.; Miyasaka, T. Tetrahedron Lett. 1985, 26, 4743–4746. doi:10.1016/s0040-4039(00)94939-6
Return to citation in text: [1] -
Hamamichi, N.; Miyasaka, T. J. Heterocycl. Chem. 1990, 27, 835–838. doi:10.1002/jhet.5570270403
Return to citation in text: [1] [2] -
Hamamichi, N. Tetrahedron Lett. 1991, 32, 7415–7418. doi:10.1016/0040-4039(91)80121-l
Return to citation in text: [1] -
Timoshchuk, V. Nucleosides, Nucleotides Nucleic Acids 2005, 24, 1043–1046. doi:10.1081/ncn-200059763
Return to citation in text: [1] [2] -
Amblard, F.; Cho, J. H.; Schinazi, R. F. Chem. Rev. 2009, 109, 4207–4220. doi:10.1021/cr9001462
Return to citation in text: [1] -
Novosjolova, I.; Bizdēna, Ē.; Turks, M. Eur. J. Org. Chem. 2015, 3629–3649. doi:10.1002/ejoc.201403527
Return to citation in text: [1] [2] -
Cosyn, L.; Palaniappan, K. K.; Kim, S.-K.; Duong, H. T.; Gao, Z.-G.; Jacobson, K. A.; Van Calenbergh, S. J. Med. Chem. 2006, 49, 7373–7383. doi:10.1021/jm0608208
Return to citation in text: [1] [2] -
Gupte, A.; Boshoff, H. I.; Wilson, D. J.; Neres, J.; Labello, N. P.; Somu, R. V.; Xing, C.; Barry, C. E.; Aldrich, C. C. J. Med. Chem. 2008, 51, 7495–7507. doi:10.1021/jm8008037
Return to citation in text: [1] [2] [3] -
Lakshman, M. K.; Kumar, A.; Balachandran, R.; Day, B. W.; Andrei, G.; Snoeck, R.; Balzarini, J. J. Org. Chem. 2012, 77, 5870–5883. doi:10.1021/jo300628y
Return to citation in text: [1] [2] [3] [4] -
Cīrule, D.; Ozols, K.; Platnieks, O.; Bizdēna, Ē.; Māliņa, I.; Turks, M. Tetrahedron 2016, 72, 4177–4185. doi:10.1016/j.tet.2016.05.043
Return to citation in text: [1] [2] -
Novosjolova, I.; Bizdēna, Ē.; Turks, M. Phosphorus, Sulfur Silicon Relat. Elem. 2015, 190, 1236–1241. doi:10.1080/10426507.2014.989435
Return to citation in text: [1] [2] -
Palle, V. P.; Elzein, E. O.; Gothe, S. A.; Li, Z.; Gao, Z.; Meyer, S.; Blackburn, B.; Zablocki, J. A. Bioorg. Med. Chem. Lett. 2002, 12, 2935–2939. doi:10.1016/s0960-894x(02)00609-1
Return to citation in text: [1] -
Gao, Z.; Li, Z.; Baker, S. P.; Lasley, R. D.; Meyer, S.; Elzein, E.; Palle, V.; Zablocki, J.; Blackburn, B.; Belardinelli, L. J. Pharmacol. Exp. Ther. 2001, 298, 209–218.
Return to citation in text: [1] -
Elzein, E.; Kalla, R.; Li, X.; Perry, T.; Marquart, T.; Micklatcher, M.; Li, Y.; Wu, Y.; Zeng, D.; Zablocki, J. Bioorg. Med. Chem. Lett. 2007, 17, 161–166. doi:10.1016/j.bmcl.2006.09.065
Return to citation in text: [1] -
Šišuļins, A.; Bucevičius, J.; Tseng, Y.-T.; Novosjolova, I.; Traskovskis, K.; Bizdēna, Ē.; Chang, H.-T.; Tumkevičius, S.; Turks, M. Beilstein J. Org. Chem. 2019, 15, 474–489. doi:10.3762/bjoc.15.41
Return to citation in text: [1] [2] [3] -
Zayas, J.; Annoual, M.; Das, J. K.; Felty, Q.; Gonzalez, W. G.; Miksovska, J.; Sharifai, N.; Chiba, A.; Wnuk, S. F. Bioconjugate Chem. 2015, 26, 1519–1532. doi:10.1021/acs.bioconjchem.5b00300
Return to citation in text: [1] -
Dyrager, C.; Börjesson, K.; Dinér, P.; Elf, A.; Albinsson, B.; Wilhelmsson, L. M.; Grøtli, M. Eur. J. Org. Chem. 2009, 1515–1521. doi:10.1002/ejoc.200900018
Return to citation in text: [1] -
Jovaisaite, J.; Cīrule, D.; Jeminejs, A.; Novosjolova, I.; Turks, M.; Baronas, P.; Komskis, R.; Tumkevicius, S.; Jonusauskas, G.; Jursenas, S. Phys. Chem. Chem. Phys. 2020, 22, 26502–26508. doi:10.1039/d0cp04091f
Return to citation in text: [1] -
Joule, J. A.; Mills, K. Purines. Heterocyclic Chemistry at a Glance; John Wiley & Sons: Chichester, UK, 2012; pp 122–131. doi:10.1002/9781118380208.ch13
Return to citation in text: [1] -
Eicher, T.; Hauptmann, S.; Speicher, A. The Chemistry of Heterocycles: Structure, Reactions, Synthesis, and Applications, 3rd ed.; Wiley-VCH: Weinheim, Germany, 2012. doi:10.1002/352760183x
Return to citation in text: [1] -
Gunda, P.; Russon, L. M.; Lakshman, M. K. Angew. Chem., Int. Ed. 2004, 43, 6372–6377. doi:10.1002/anie.200460782
Return to citation in text: [1] -
Lagisetty, P.; Russon, L. M.; Lakshman, M. K. Angew. Chem., Int. Ed. 2006, 45, 3660–3663. doi:10.1002/anie.200504565
Return to citation in text: [1] -
Thomson, P. F.; Lagisetty, P.; Balzarini, J.; De Clercq, E.; Lakshman, M. K. Adv. Synth. Catal. 2010, 352, 1728–1735. doi:10.1002/adsc.200900728
Return to citation in text: [1] -
Liang, Y.; Wnuk, S. F. C-H Bond Functionalization Strategies for Modification of Nucleosides. Palladium-Catalyzed Modification of Nucleosides, Nucleotides and Oligonucleotides; Elsevier, 2018; pp 197–246. doi:10.1016/b978-0-12-811292-2.00007-6
Return to citation in text: [1] -
Kriķis, K.-Ē.; Novosjolova, I.; Mishnev, A.; Turks, M. Beilstein J. Org. Chem. 2021, 17, 193–202. doi:10.3762/bjoc.17.19
Return to citation in text: [1] -
Kapilinskis, Z.; Novosjolova, I.; Bizdēna, Ē.; Turks, M. Chem. Heterocycl. Compd. 2021, 57, 55–62. doi:10.1007/s10593-021-02867-w
Return to citation in text: [1] -
Qu, G.-R.; Mao, Z.-J.; Niu, H.-Y.; Wang, D.-C.; Xia, C.; Guo, H.-M. Org. Lett. 2009, 11, 1745–1748. doi:10.1021/ol9002256
Return to citation in text: [1] -
Guo, H.-M.; Zhang, Y.; Niu, H.-Y.; Wang, D.-C.; Chu, Z.-L.; Qu, G.-R. Org. Biomol. Chem. 2011, 9, 2065–2068. doi:10.1039/c0ob01213k
Return to citation in text: [1] [2] -
Bordwell, F. G. Acc. Chem. Res. 1988, 21, 456–463. doi:10.1021/ar00156a004
Return to citation in text: [1] -
Pretsch, E.; Bühlmann, P.; Badertscher, M. Structure Determination of Organic Compounds; Springer: Berlin, Heidelberg, 2009. doi:10.1007/978-3-540-93810-1
Return to citation in text: [1] -
Hamamichi, N.; Miyasaka, T. J. Heterocycl. Chem. 1991, 28, 397–400. doi:10.1002/jhet.5570280236
Return to citation in text: [1] -
Odijk, W. M.; Koomen, G. J. Tetrahedron 1985, 41, 1893–1904. doi:10.1016/s0040-4020(01)96552-4
Return to citation in text: [1] -
Zaki, M. E. A.; Proença, M. F.; Booth, B. L. J. Org. Chem. 2003, 68, 276–282. doi:10.1021/jo020347f
Return to citation in text: [1]
| 11. | Kovaļovs, A.; Novosjolova, I.; Bizdēna, Ē.; Bižāne, I.; Skardziute, L.; Kazlauskas, K.; Jursenas, S.; Turks, M. Tetrahedron Lett. 2013, 54, 850–853. doi:10.1016/j.tetlet.2012.11.095 |
| 14. | Novosjolova, I.; Bizdēna, Ē.; Turks, M. Tetrahedron Lett. 2013, 54, 6557–6561. doi:10.1016/j.tetlet.2013.09.095 |
| 62. | Cīrule, D.; Ozols, K.; Platnieks, O.; Bizdēna, Ē.; Māliņa, I.; Turks, M. Tetrahedron 2016, 72, 4177–4185. doi:10.1016/j.tet.2016.05.043 |
| 63. | Novosjolova, I.; Bizdēna, Ē.; Turks, M. Phosphorus, Sulfur Silicon Relat. Elem. 2015, 190, 1236–1241. doi:10.1080/10426507.2014.989435 |
| 77. | Kriķis, K.-Ē.; Novosjolova, I.; Mishnev, A.; Turks, M. Beilstein J. Org. Chem. 2021, 17, 193–202. doi:10.3762/bjoc.17.19 |
| 78. | Kapilinskis, Z.; Novosjolova, I.; Bizdēna, Ē.; Turks, M. Chem. Heterocycl. Compd. 2021, 57, 55–62. doi:10.1007/s10593-021-02867-w |
| 11. | Kovaļovs, A.; Novosjolova, I.; Bizdēna, Ē.; Bižāne, I.; Skardziute, L.; Kazlauskas, K.; Jursenas, S.; Turks, M. Tetrahedron Lett. 2013, 54, 850–853. doi:10.1016/j.tetlet.2012.11.095 |
| 14. | Novosjolova, I.; Bizdēna, Ē.; Turks, M. Tetrahedron Lett. 2013, 54, 6557–6561. doi:10.1016/j.tetlet.2013.09.095 |
| 67. | Šišuļins, A.; Bucevičius, J.; Tseng, Y.-T.; Novosjolova, I.; Traskovskis, K.; Bizdēna, Ē.; Chang, H.-T.; Tumkevičius, S.; Turks, M. Beilstein J. Org. Chem. 2019, 15, 474–489. doi:10.3762/bjoc.15.41 |
| 61. | Lakshman, M. K.; Kumar, A.; Balachandran, R.; Day, B. W.; Andrei, G.; Snoeck, R.; Balzarini, J. J. Org. Chem. 2012, 77, 5870–5883. doi:10.1021/jo300628y |
| 1. | Dinesh, S.; Shikha, G.; Bhavana, G.; Nidhi, S.; Dileep, S. J. Pharm. Sci. Innovation 2012, 1, 29–34. |
| 2. | Parker, W. B. Chem. Rev. 2009, 109, 2880–2893. doi:10.1021/cr900028p |
| 3. | Shelton, J.; Lu, X.; Hollenbaugh, J. A.; Cho, J. H.; Amblard, F.; Schinazi, R. F. Chem. Rev. 2016, 116, 14379–14455. doi:10.1021/acs.chemrev.6b00209 |
| 4. | Seley-Radtke, K. L.; Yates, M. K. Antiviral Res. 2018, 154, 66–86. doi:10.1016/j.antiviral.2018.04.004 |
| 5. | Yates, M. K.; Seley-Radtke, K. L. Antiviral Res. 2019, 162, 5–21. doi:10.1016/j.antiviral.2018.11.016 |
| 6. | Liang, Y.; Wen, Z.; Cabrera, M.; Howlader, A. H.; Wnuk, S. F. Purines (Update 2020). In SOS Knowledge Updates 2020/1; Christmann, M.; Huang, Z.; Jiang, X.; Li, J. J.; Oestreich, M.; Petersson, E. J.; Schaumann, E.; Wang, M., Eds.; Georg Thieme Verlag: Stuttgart, Germany, 2020. doi:10.1055/sos-sd-116-01081 |
| 21. | Guo, H.-M.; Xin, P.-Y.; Niu, H.-Y.; Wang, D.-C.; Jiang, Y.; Qu, G.-R. Green Chem. 2010, 12, 2131–2134. doi:10.1039/c0gc00517g |
| 22. | Pathak, A. K.; Pathak, V.; Seitz, L. E.; Suling, W. J.; Reynolds, R. C. Bioorg. Med. Chem. 2013, 21, 1685–1695. doi:10.1016/j.bmc.2013.01.054 |
| 23. | McGuigan, C.; Madela, K.; Aljarah, M.; Gilles, A.; Battina, S. K.; Ramamurty, C. V. S.; Srinivas Rao, C.; Vernachio, J.; Hutchins, J.; Hall, A.; Kolykhalov, A.; Henson, G.; Chamberlain, S. Bioorg. Med. Chem. Lett. 2011, 21, 6007–6012. doi:10.1016/j.bmcl.2011.06.013 |
| 24. | Vijay Kumar, D.; Hoarau, C.; Bursavich, M.; Slattum, P.; Gerrish, D.; Yager, K.; Saunders, M.; Shenderovich, M.; Roth, B. L.; McKinnon, R.; Chan, A.; Cimbora, D. M.; Bradford, C.; Reeves, L.; Patton, S.; Papac, D. I.; Williams, B. L.; Carlson, R. O. Bioorg. Med. Chem. Lett. 2012, 22, 4377–4385. doi:10.1016/j.bmcl.2012.04.131 |
| 25. | Hulpia, F.; Balzarini, J.; Schols, D.; Andrei, G.; Snoeck, R.; Van Calenbergh, S. Bioorg. Med. Chem. Lett. 2016, 26, 1970–1972. doi:10.1016/j.bmcl.2016.03.005 |
| 26. | Bonnac, L. F.; Dreis, C. D.; Geraghty, R. J. Bioorg. Med. Chem. Lett. 2020, 30, 126819. doi:10.1016/j.bmcl.2019.126819 |
| 27. | Besada, P.; Costas, T.; Teijeira, M.; Terán, C. Eur. J. Med. Chem. 2010, 45, 6114–6119. doi:10.1016/j.ejmech.2010.09.046 |
| 28. | Daumar, P.; Zeglis, B. M.; Ramos, N.; Divilov, V.; Sevak, K. K.; Pillarsetty, N.; Lewis, J. S. Eur. J. Med. Chem. 2014, 86, 769–781. doi:10.1016/j.ejmech.2014.09.019 |
| 56. | Timoshchuk, V. Nucleosides, Nucleotides Nucleic Acids 2005, 24, 1043–1046. doi:10.1081/ncn-200059763 |
| 15. | Gurram, V.; Pottabathini, N.; Garlapati, R.; Chaudhary, A. B.; Patro, B.; Lakshman, M. K. Chem. – Asian J. 2012, 7, 1853–1861. doi:10.1002/asia.201200093 |
| 16. | Lakshman, M. K.; Gunda, P.; Pradhan, P. J. Org. Chem. 2005, 70, 10329–10335. doi:10.1021/jo0513764 |
| 17. | Havelková, M.; Hocek, M.; Česnek, M.; Dvořák, D. Synlett 1999, 1145–1147. doi:10.1055/s-1999-2753 |
| 18. | Havelková, M.; Dvořak, D.; Hocek, M. Synthesis 2001, 1704–1710. doi:10.1055/s-2001-16765 |
| 19. | Hocek, M.; Masojídková, M.; Holý, A. Collect. Czech. Chem. Commun. 1997, 62, 136–146. doi:10.1135/cccc19970136 |
| 20. | Hasník, Z.; Pohl, R.; Hocek, M. Synthesis 2009, 1309–1317. doi:10.1055/s-0028-1088038 |
| 57. | Amblard, F.; Cho, J. H.; Schinazi, R. F. Chem. Rev. 2009, 109, 4207–4220. doi:10.1021/cr9001462 |
| 58. | Novosjolova, I.; Bizdēna, Ē.; Turks, M. Eur. J. Org. Chem. 2015, 3629–3649. doi:10.1002/ejoc.201403527 |
| 12. | Aleksandrova, E. V. Pharm. Chem. J. 2003, 37, 645–652. doi:10.1023/b:phac.0000022083.93211.2c |
| 13. | Aleksandrova, E. V.; Kochergin, P. M. Pharm. Chem. J. 2013, 46, 612–621. doi:10.1007/s11094-013-0856-y |
| 14. | Novosjolova, I.; Bizdēna, Ē.; Turks, M. Tetrahedron Lett. 2013, 54, 6557–6561. doi:10.1016/j.tetlet.2013.09.095 |
| 43. | Vincent, S. P.; Mioskowski, C.; Lebean, L. Nucleosides Nucleotides 1999, 18, 2127–2139. doi:10.1080/07328319908044869 |
| 44. | Woo, J.; Sigurdsson, S. T.; Hopkins, P. B. J. Am. Chem. Soc. 1993, 115, 3407–3415. doi:10.1021/ja00062a002 |
| 45. | Wilds, C. J.; Booth, J. D.; Noronha, A. M. Tetrahedron Lett. 2006, 47, 9125–9128. doi:10.1016/j.tetlet.2006.10.074 |
| 46. | Wang, L.; Spratt, T. E.; Liu, X.-K.; Hecht, S. S.; Pegg, A. E.; Peterson, L. A. Chem. Res. Toxicol. 1997, 10, 562–567. doi:10.1021/tx9602067 |
| 47. | Giordano, C.; Pedone, F.; Fattibene, P.; Cellai, L. Nucleosides, Nucleotides Nucleic Acids 2000, 19, 1301–1310. doi:10.1080/15257770008033053 |
| 48. | Cooper, M. D.; Hodge, R. P.; Tamura, P. J.; Wilkinson, A. S.; Harris, C. M.; Harris, T. M. Tetrahedron Lett. 2000, 41, 3555–3558. doi:10.1016/s0040-4039(00)00461-5 |
| 49. | Harwood, E. A.; Hopkins, P. B.; Sigurdsson, S. T. J. Org. Chem. 2000, 65, 2959–2964. doi:10.1021/jo991501+ |
| 50. | De Napoli, L.; Di Fabio, G.; Messere, A.; Montesarchio, D.; Piccialli, G.; Varra, M. J. Chem. Soc., Perkin Trans. 1 1999, 3489–3493. doi:10.1039/a906195i |
| 54. | Hamamichi, N.; Miyasaka, T. J. Heterocycl. Chem. 1990, 27, 835–838. doi:10.1002/jhet.5570270403 |
| 83. | Hamamichi, N.; Miyasaka, T. J. Heterocycl. Chem. 1991, 28, 397–400. doi:10.1002/jhet.5570280236 |
| 84. | Odijk, W. M.; Koomen, G. J. Tetrahedron 1985, 41, 1893–1904. doi:10.1016/s0040-4020(01)96552-4 |
| 85. | Zaki, M. E. A.; Proença, M. F.; Booth, B. L. J. Org. Chem. 2003, 68, 276–282. doi:10.1021/jo020347f |
| 7. | Véliz, E. A.; Beal, P. A. J. Org. Chem. 2001, 66, 8592–8598. doi:10.1021/jo016078v |
| 8. | Lakshman, M. K.; Frank, J. Org. Biomol. Chem. 2009, 7, 2933–2940. doi:10.1039/b905298d |
| 9. | Manvar, A.; Shah, A. Tetrahedron 2013, 69, 8105–8127. doi:10.1016/j.tet.2013.06.034 |
| 10. | Manvar, A.; Shah, A. Tetrahedron 2013, 69, 680–691. doi:10.1016/j.tet.2012.10.079 |
| 11. | Kovaļovs, A.; Novosjolova, I.; Bizdēna, Ē.; Bižāne, I.; Skardziute, L.; Kazlauskas, K.; Jursenas, S.; Turks, M. Tetrahedron Lett. 2013, 54, 850–853. doi:10.1016/j.tetlet.2012.11.095 |
| 51. | Zhang, Y.; Wang, L.; Zhang, Q.; Zhu, G.; Zhang, Z.; Zhou, X.; Chen, Y.; Lu, T.; Tang, W. J. Chem. Inf. Model. 2017, 57, 1439–1452. doi:10.1021/acs.jcim.6b00795 |
| 52. | Hamamichi, N.; Miyasaka, T. J. Org. Chem. 1994, 59, 1525–1531. doi:10.1021/jo00085a046 |
| 53. | Hamamichi, N.; Miyasaka, T. Tetrahedron Lett. 1985, 26, 4743–4746. doi:10.1016/s0040-4039(00)94939-6 |
| 54. | Hamamichi, N.; Miyasaka, T. J. Heterocycl. Chem. 1990, 27, 835–838. doi:10.1002/jhet.5570270403 |
| 55. | Hamamichi, N. Tetrahedron Lett. 1991, 32, 7415–7418. doi:10.1016/0040-4039(91)80121-l |
| 11. | Kovaļovs, A.; Novosjolova, I.; Bizdēna, Ē.; Bižāne, I.; Skardziute, L.; Kazlauskas, K.; Jursenas, S.; Turks, M. Tetrahedron Lett. 2013, 54, 850–853. doi:10.1016/j.tetlet.2012.11.095 |
| 14. | Novosjolova, I.; Bizdēna, Ē.; Turks, M. Tetrahedron Lett. 2013, 54, 6557–6561. doi:10.1016/j.tetlet.2013.09.095 |
| 67. | Šišuļins, A.; Bucevičius, J.; Tseng, Y.-T.; Novosjolova, I.; Traskovskis, K.; Bizdēna, Ē.; Chang, H.-T.; Tumkevičius, S.; Turks, M. Beilstein J. Org. Chem. 2019, 15, 474–489. doi:10.3762/bjoc.15.41 |
| 39. | Miles, R. W.; Samano, V.; Robins, M. J. J. Am. Chem. Soc. 1995, 117, 5951–5957. doi:10.1021/ja00127a007 |
| 41. | Caner, J.; Vilarrasa, J. J. Org. Chem. 2010, 75, 4880–4883. doi:10.1021/jo100808w |
| 35. | Linn, J. A.; McLean, E. W.; Kelley, J. L. J. Chem. Soc., Chem. Commun. 1994, 913–914. doi:10.1039/c39940000913 |
| 36. | Lakshman, M. K.; Ngassa, F. N.; Keeler, J. C.; Dinh, Y. Q. V.; Hilmer, J. H.; Russon, L. M. Org. Lett. 2000, 2, 927–930. doi:10.1021/ol005564m |
| 37. | Schirrmacher, R.; Wängler, B.; Schirrmacher, E.; August, T.; Rösch, F. Synthesis 2002, 538–542. doi:10.1055/s-2002-20970 |
| 38. | Schirrmacher, R.; Mühlhausen, U.; Wängler, B.; Schirrmacher, E.; Reinhard, J.; Nagel, G.; Kaina, B.; Piel, M.; Wießler, M.; Rösch, F. Tetrahedron Lett. 2002, 43, 6301–6304. doi:10.1016/s0040-4039(02)01394-1 |
| 42. | Geigle, S. N.; Wyss, L. A.; Sturla, S. J.; Gillingham, D. G. Chem. Sci. 2017, 8, 499–506. doi:10.1039/c6sc03502g |
| 82. | Pretsch, E.; Bühlmann, P.; Badertscher, M. Structure Determination of Organic Compounds; Springer: Berlin, Heidelberg, 2009. doi:10.1007/978-3-540-93810-1 |
| 33. | Zhong, M.; Nowak, I.; Robins, M. J. Org. Lett. 2005, 7, 4601–4603. doi:10.1021/ol051573p |
| 34. | Zhong, M.; Nowak, I.; Robins, M. J. J. Org. Chem. 2006, 71, 7773–7779. doi:10.1021/jo061282+ |
| 79. | Qu, G.-R.; Mao, Z.-J.; Niu, H.-Y.; Wang, D.-C.; Xia, C.; Guo, H.-M. Org. Lett. 2009, 11, 1745–1748. doi:10.1021/ol9002256 |
| 80. | Guo, H.-M.; Zhang, Y.; Niu, H.-Y.; Wang, D.-C.; Chu, Z.-L.; Qu, G.-R. Org. Biomol. Chem. 2011, 9, 2065–2068. doi:10.1039/c0ob01213k |
| 8. | Lakshman, M. K.; Frank, J. Org. Biomol. Chem. 2009, 7, 2933–2940. doi:10.1039/b905298d |
| 29. | Bae, S.; Lakshman, M. K. J. Am. Chem. Soc. 2007, 129, 782–789. doi:10.1021/ja064682n |
| 30. | Kokatla, H. P.; Lakshman, M. K. Org. Lett. 2010, 12, 4478–4481. doi:10.1021/ol101655h |
| 31. | Basava, V.; Yang, L.; Pradhan, P.; Lakshman, M. K. Org. Biomol. Chem. 2016, 14, 7069–7083. doi:10.1039/c6ob01170e |
| 32. | Bae, S.; Lakshman, M. K. J. Org. Chem. 2008, 73, 1311–1319. doi:10.1021/jo7021795 |
| 40. | Clivio, P.; Fourrey, J.-L.; Favre, A. J. Chem. Soc., Perkin Trans. 1 1993, 2585–2590. doi:10.1039/p19930002585 |
| 56. | Timoshchuk, V. Nucleosides, Nucleotides Nucleic Acids 2005, 24, 1043–1046. doi:10.1081/ncn-200059763 |
| 80. | Guo, H.-M.; Zhang, Y.; Niu, H.-Y.; Wang, D.-C.; Chu, Z.-L.; Qu, G.-R. Org. Biomol. Chem. 2011, 9, 2065–2068. doi:10.1039/c0ob01213k |
| 61. | Lakshman, M. K.; Kumar, A.; Balachandran, R.; Day, B. W.; Andrei, G.; Snoeck, R.; Balzarini, J. J. Org. Chem. 2012, 77, 5870–5883. doi:10.1021/jo300628y |
| 11. | Kovaļovs, A.; Novosjolova, I.; Bizdēna, Ē.; Bižāne, I.; Skardziute, L.; Kazlauskas, K.; Jursenas, S.; Turks, M. Tetrahedron Lett. 2013, 54, 850–853. doi:10.1016/j.tetlet.2012.11.095 |
| 59. | Cosyn, L.; Palaniappan, K. K.; Kim, S.-K.; Duong, H. T.; Gao, Z.-G.; Jacobson, K. A.; Van Calenbergh, S. J. Med. Chem. 2006, 49, 7373–7383. doi:10.1021/jm0608208 |
| 60. | Gupte, A.; Boshoff, H. I.; Wilson, D. J.; Neres, J.; Labello, N. P.; Somu, R. V.; Xing, C.; Barry, C. E.; Aldrich, C. C. J. Med. Chem. 2008, 51, 7495–7507. doi:10.1021/jm8008037 |
| 61. | Lakshman, M. K.; Kumar, A.; Balachandran, R.; Day, B. W.; Andrei, G.; Snoeck, R.; Balzarini, J. J. Org. Chem. 2012, 77, 5870–5883. doi:10.1021/jo300628y |
| 62. | Cīrule, D.; Ozols, K.; Platnieks, O.; Bizdēna, Ē.; Māliņa, I.; Turks, M. Tetrahedron 2016, 72, 4177–4185. doi:10.1016/j.tet.2016.05.043 |
| 14. | Novosjolova, I.; Bizdēna, Ē.; Turks, M. Tetrahedron Lett. 2013, 54, 6557–6561. doi:10.1016/j.tetlet.2013.09.095 |
| 63. | Novosjolova, I.; Bizdēna, Ē.; Turks, M. Phosphorus, Sulfur Silicon Relat. Elem. 2015, 190, 1236–1241. doi:10.1080/10426507.2014.989435 |
| 30. | Kokatla, H. P.; Lakshman, M. K. Org. Lett. 2010, 12, 4478–4481. doi:10.1021/ol101655h |
| 36. | Lakshman, M. K.; Ngassa, F. N.; Keeler, J. C.; Dinh, Y. Q. V.; Hilmer, J. H.; Russon, L. M. Org. Lett. 2000, 2, 927–930. doi:10.1021/ol005564m |
| 59. | Cosyn, L.; Palaniappan, K. K.; Kim, S.-K.; Duong, H. T.; Gao, Z.-G.; Jacobson, K. A.; Van Calenbergh, S. J. Med. Chem. 2006, 49, 7373–7383. doi:10.1021/jm0608208 |
| 60. | Gupte, A.; Boshoff, H. I.; Wilson, D. J.; Neres, J.; Labello, N. P.; Somu, R. V.; Xing, C.; Barry, C. E.; Aldrich, C. C. J. Med. Chem. 2008, 51, 7495–7507. doi:10.1021/jm8008037 |
| 61. | Lakshman, M. K.; Kumar, A.; Balachandran, R.; Day, B. W.; Andrei, G.; Snoeck, R.; Balzarini, J. J. Org. Chem. 2012, 77, 5870–5883. doi:10.1021/jo300628y |
| 71. | Joule, J. A.; Mills, K. Purines. Heterocyclic Chemistry at a Glance; John Wiley & Sons: Chichester, UK, 2012; pp 122–131. doi:10.1002/9781118380208.ch13 |
| 72. | Eicher, T.; Hauptmann, S.; Speicher, A. The Chemistry of Heterocycles: Structure, Reactions, Synthesis, and Applications, 3rd ed.; Wiley-VCH: Weinheim, Germany, 2012. doi:10.1002/352760183x |
| 73. | Gunda, P.; Russon, L. M.; Lakshman, M. K. Angew. Chem., Int. Ed. 2004, 43, 6372–6377. doi:10.1002/anie.200460782 |
| 74. | Lagisetty, P.; Russon, L. M.; Lakshman, M. K. Angew. Chem., Int. Ed. 2006, 45, 3660–3663. doi:10.1002/anie.200504565 |
| 75. | Thomson, P. F.; Lagisetty, P.; Balzarini, J.; De Clercq, E.; Lakshman, M. K. Adv. Synth. Catal. 2010, 352, 1728–1735. doi:10.1002/adsc.200900728 |
| 76. | Liang, Y.; Wnuk, S. F. C-H Bond Functionalization Strategies for Modification of Nucleosides. Palladium-Catalyzed Modification of Nucleosides, Nucleotides and Oligonucleotides; Elsevier, 2018; pp 197–246. doi:10.1016/b978-0-12-811292-2.00007-6 |
| 11. | Kovaļovs, A.; Novosjolova, I.; Bizdēna, Ē.; Bižāne, I.; Skardziute, L.; Kazlauskas, K.; Jursenas, S.; Turks, M. Tetrahedron Lett. 2013, 54, 850–853. doi:10.1016/j.tetlet.2012.11.095 |
| 67. | Šišuļins, A.; Bucevičius, J.; Tseng, Y.-T.; Novosjolova, I.; Traskovskis, K.; Bizdēna, Ē.; Chang, H.-T.; Tumkevičius, S.; Turks, M. Beilstein J. Org. Chem. 2019, 15, 474–489. doi:10.3762/bjoc.15.41 |
| 68. | Zayas, J.; Annoual, M.; Das, J. K.; Felty, Q.; Gonzalez, W. G.; Miksovska, J.; Sharifai, N.; Chiba, A.; Wnuk, S. F. Bioconjugate Chem. 2015, 26, 1519–1532. doi:10.1021/acs.bioconjchem.5b00300 |
| 69. | Dyrager, C.; Börjesson, K.; Dinér, P.; Elf, A.; Albinsson, B.; Wilhelmsson, L. M.; Grøtli, M. Eur. J. Org. Chem. 2009, 1515–1521. doi:10.1002/ejoc.200900018 |
| 70. | Jovaisaite, J.; Cīrule, D.; Jeminejs, A.; Novosjolova, I.; Turks, M.; Baronas, P.; Komskis, R.; Tumkevicius, S.; Jonusauskas, G.; Jursenas, S. Phys. Chem. Chem. Phys. 2020, 22, 26502–26508. doi:10.1039/d0cp04091f |
| 58. | Novosjolova, I.; Bizdēna, Ē.; Turks, M. Eur. J. Org. Chem. 2015, 3629–3649. doi:10.1002/ejoc.201403527 |
| 64. | Palle, V. P.; Elzein, E. O.; Gothe, S. A.; Li, Z.; Gao, Z.; Meyer, S.; Blackburn, B.; Zablocki, J. A. Bioorg. Med. Chem. Lett. 2002, 12, 2935–2939. doi:10.1016/s0960-894x(02)00609-1 |
| 65. | Gao, Z.; Li, Z.; Baker, S. P.; Lasley, R. D.; Meyer, S.; Elzein, E.; Palle, V.; Zablocki, J.; Blackburn, B.; Belardinelli, L. J. Pharmacol. Exp. Ther. 2001, 298, 209–218. |
| 66. | Elzein, E.; Kalla, R.; Li, X.; Perry, T.; Marquart, T.; Micklatcher, M.; Li, Y.; Wu, Y.; Zeng, D.; Zablocki, J. Bioorg. Med. Chem. Lett. 2007, 17, 161–166. doi:10.1016/j.bmcl.2006.09.065 |
| 60. | Gupte, A.; Boshoff, H. I.; Wilson, D. J.; Neres, J.; Labello, N. P.; Somu, R. V.; Xing, C.; Barry, C. E.; Aldrich, C. C. J. Med. Chem. 2008, 51, 7495–7507. doi:10.1021/jm8008037 |
© 2021 Cīrule et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the author(s) and source are credited and that individual graphics may be subject to special legal provisions.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc/terms)