
Abstract
6-Phenylfulvenes bearing (1,3-dioxolan or dioxan)-2-yl substituents at ortho position convert into mixtures of 4- and 9-(hydroxy)alkoxy-substituted benz[f]indenes as result of cascade processes initiated by a thermally activated hydrogen shift. Structurally related fulvenes with non-cyclic acetalic units afforded mixtures of 4- and 9-alkoxybenz[f]indenes under similar thermal conditions. Mechanistic paths promoted by an initial [1,4]-, [1,5]-, [1,7]- or [1,9]-H shift are conceivable for explaining these conversions. Deuterium labelling experiments exclude the [1,4]-hydride shift as the first step. A computational study scrutinized the reaction channels of these tandem conversions starting by [1,5]-, [1,7]- and [1,9]-H shifts, revealing that this first step is the rate-determining one and that the [1,9]-H shift is the one with the lowest energy barrier.
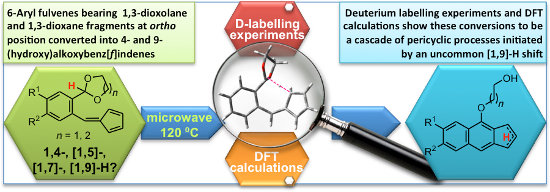
Graphical Abstract
Introduction
Fulvenes (also known as pentafulvenes), [1-4] a unique class of trienes, have intrigued chemists for decades due to their theoretical interest [5,6] and synthetic applications [7-15]. In this latter sense, fulvenes can be involved in multiple modes of cyclization processes such as [4 + 2] [7,8], [6 + 2] [9-11], and [6 + 3] [12-15] cycloaddition reactions resulting in the construction of diverse fused ring systems. Other classical pericyclic processes that may potentially occur in fulvene fragments (electrocyclic and ene reactions, sigmatropic rearrangements and shifts) have received less attention, most probably with the only exception of the Claisen rearrangement [16]. Notably, thermally promoted H-shifts remain, to the best of our knowledge, completely unexplored in fulvene frameworks [17,18].
A part of our recent research focused on showing the special ability of cyclic acetalic functions (1,3-dioxolanes, thiolanes, oxathiolanes, dioxanes, dithianes, oxathianes) for promoting the migration of its acetalic H atom in a hydride-like manner. As result, we have disclosed a variety of tandem processes initiated by [1,5]- and [1,4]-hydride shifts from the acetalic carbon atom toward electrophilic molecular fragments [19-27]. Thus, we have reported that ortho-(1,3-dioxolan-2-yl)benzylidenemalonates 1 undergo tandem hydride shift/cyclization sequences leading to the corresponding indan-1-one-2,2-dicarboxylates 2. Remarkably, the first step of these processes consists of an uncommon [1,4]-hydride shift of the acetalic H atom following the activation of the benzylidenemalonate fragment by scandium(III) triflate as the catalyst (Scheme 1) [27].
![[1860-5397-12-28-i1]](/bjoc/content/inline/1860-5397-12-28-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Lewis acid-catalyzed [1,4]-H transfer/1,5-electrocyclization tandem processes of benzylidenemalonates 1 leading to indan-1-ones 2.
Scheme 1: Lewis acid-catalyzed [1,4]-H transfer/1,5-electrocyclization tandem processes of benzylidenemalonat...
Because fulvenes are widely used as the direct precursors of cyclopentadienyl anions following the addition of nucleophiles, including the hydride anion, to its exocyclic sp2 carbon atom [28,29], we wondered whether acetalic functions could be employed as internal H donors in intramolecular hydride-like shifts, analogous to that highlighted in Scheme 1, toward fulvene frameworks.
With this goal in mind we designed the unknown acetal-fulvenes 3 (Scheme 2) as potential candidates for assaying the [1,4]-hydride shift of its acetalic H atom toward the exocyclic C4 carbon atom of the fulvene fragment (note the numbering in the Scheme 2). At this point it is worth noting that other possible H migrations were not ruled out at the outset of this investigation. For example, C5 and C7 may be the respective termini of sigmatropic [1,5]-H or [1,7]-H shifts, whereas C6 could be also prone to participate in a less common [1,9]-H shift [30-32]. The variety of potential intramolecular H migrations in these reactive species might well justify by itself the research here disclosed.
Results and Discussion
Experimental study
The starting acetal-fulvenes 3 were prepared by the condensation of substituted 2-(1,3-dioxolan-2-yl)benzaldehydes 4 with cyclopentadiene following a well-established synthetic methodology [33]. With the aim of promoting the desired hydride transfer by thermal activation, we first heated the parent acetal-fulvene 3a under a variety of reaction conditions (benzene 110 °C sealed tube; toluene 120 °C sealed tube; DMF 120 °C) but unfortunately without success. Only when a DMSO solution of 3a was heated at 120 °C for 7 h the acetal-fulvene converted into a complex mixture from which we were able to isolate the benz[f]indenes 5a and 6a, in a relative 2:1 ratio and a poor global yield (34%). We next tested the same and similar processes in a microwave apparatus. As presumed, conversions of a series of acetal-fulvenes 3a–f under 120 W microwave irradiation at 120 °C in DMSO required much shorter reaction times (20–40 min) and led in all cases to the isolation of the respective benz[f]indenes 5 and 6, in a 2:1 ratio (Scheme 2). The overall yield of the isomeric mixtures 5 + 6 did not improve significantly with respect to the conventional thermal conditions previously used with 3a, ranging from medium to low as depicted in Table 1. Despite our chromatographic (column, thin-layer) efforts, additional pure products other than 5 and 6 could not be isolated from the complex final reaction mixtures.
![[1860-5397-12-28-i2]](/bjoc/content/inline/1860-5397-12-28-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Preparation of benz[f]indenes 5 and 6. Reagents and conditions: i) cyclopentadiene, pyrrolidine, anhydrous methanol, rt, 10 h; ii) DMSO, microwave, 120 °C, 120 W, 20–40 min.
Scheme 2: Preparation of benz[f]indenes 5 and 6. Reagents and conditions: i) cyclopentadiene, pyrrolidine, an...
The structural determination of the reaction products, the 9- and 4-hydroxyalkoxy regioisomers 5 and 6, was easily accomplished by using the habitual analytical and spectroscopic techniques, whereas the distinction between each two regioisomers is basically supported by 1H NMR NOE difference experiments. For the major isomers 5 enhancements of the signals due to the H-C2 and CH2-OAr protons were observed when the methylenic protons were irradiated. In contrast, similar irradiation experiments with the minor isomers 6 revealed enhancements of H-C2 and H-C9.
As the H atom at the acetalic carbon of the initial fulvenes 3 apparently appears at the methylene group of both isomeric reaction products, it seems reasonable to hypothesize that the conversions 3 → 5 + 6 are initiated by an H migration from the acetalic carbon toward the fulvene substructure. If this migration is a [1,4]-hydride shift to the exocyclic C4 carbon atom of the fulvene fragment, as initially postulated, this step (exemplified for the simplest fulvene 3a in Scheme 3) would lead to the dipolar intermediate 7a which reasonably would undergo further cyclization to 8a thus building the benz[f]indene substructure present in the final reaction products. For the conversion of intermediate 8a into the final mixture of 5a and 6a several mechanistic paths are conceivable, each one involving additional H shifts and a fragmentation step, the opening of the dioxolane ring by a formal β-elimination reaction.
![[1860-5397-12-28-i3]](/bjoc/content/inline/1860-5397-12-28-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Postulated reaction path for the conversion 3a → 5a + 6a initiated by a [1,4]-hydride shift.
Scheme 3: Postulated reaction path for the conversion 3a → 5a + 6a initiated by a [1,4]-hydride shift.
A number of additional mechanistic alternatives arise by considering that the experimentally observed transformations of 3 are initiated by other H shifts alternative to the initially expected [1,4] one. Thus, a [1,5]-H shift from the acetalic carbon of 3a to the C5 carbon atom of the cyclopentadiene ring, would lead to the transient ortho-quinodimethane structure 9a, which might transform into two similar intermediates, 10a and 11a, by a sequence of consecutive [1,5]-H shifts around the cyclopentadiene ring. Next, intermediates 10a and 11a would undergo 6π-electrocyclic ring closures (6π-ERC) to the respective dihydrobenzindenes 12a and 13a. Finally, these two species would experiment the acetalic ring opening by a formal β-elimination proccess leading to the respective final products 5a and 6a (Scheme 4).
![[1860-5397-12-28-i4]](/bjoc/content/inline/1860-5397-12-28-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Alternative mechanistic paths for the conversion 3a → 5a + 6a initiated by [1,5]-, [1,7]- or [1,9]-H shifts.
Scheme 4: Alternative mechanistic paths for the conversion 3a → 5a + 6a initiated by [1,5]-, [1,7]- or [1,9]-...
This mechanistic scheme is further complicated when considering that ortho-quinodimethane intermediates 10a and 11a could also result from the respective [1,9]-H and [1,7]-H shifts occurring in the starting acetal-fulvene 3a (Scheme 4). Thus, the different mechanistic paths represented in Scheme 4 for explaining the conversion 3a → 5a + 6a share several common steps, essentially differing in their first hydrogen shift, [1,5]-, [1,7]- or [1,9]-H.
Obviously, any attempt to discern which one is the actual reaction path (if only one!) among the range of potential mechanistic alternatives for these transformations seems a huge task. Seeking for additional experimental data in order to approach such objective, we reasoned that the mechanism initiated by a [1,4]-hydride shift, as summarized in Scheme 3, could be differentiated from those starting by [1,5]-, [1,7]- or [1,9]-H shifts, represented in Scheme 4, by deuterium labelling experiments. Thus, if the conversion of the deuterated acetal-fulvene 14, in which deuterium replaces the proton at the acetalic carbon of 3a, was actually initiated by a [1,4]-deuteride shift, the transformation of the dihydrobenzo[f]indenic species 15, which should form in first instance, would yield the final benz[f]indenes via intermediates 16 and 17. These are labelled with deuterium at C4 of the major product 18 and at C9 of the minor one 19 as well as probably at additional positions of the fused five-membered ring (Scheme 5).
![[1860-5397-12-28-i5]](/bjoc/content/inline/1860-5397-12-28-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Postulated outcome of the conversion 14 → 18 + 19 initiated by a [1,4]-deuteride shift.
Scheme 5: Postulated outcome of the conversion 14 → 18 + 19 initiated by a [1,4]-deuteride shift.
With this in mind we prepared the monodeuterated acetal-fulvene 14 (see Supporting Information File 1) and submitted it to the habitual reaction conditions. As result, a mixture of the monodeuterated benz[f]indenes 18 and 19 was obtained, again in a relative 2:1 ratio (Scheme 6). 1H NMR analyses showed that only protons, not deuteriums, were linked to the C4 atom of 18 and to C9 of 19. Instead, one deuterium atom is found at the methylene group of each regioisomer.
![[1860-5397-12-28-i6]](/bjoc/content/inline/1860-5397-12-28-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Preparation of deuterated benz[f]indenes 18 + 19 and 21 + 22. Reagents and conditions: i) DMSO, microwave, 120 °C, 120 W, 40 min.
Scheme 6: Preparation of deuterated benz[f]indenes 18 + 19 and 21 + 22. Reagents and conditions: i) DMSO, mic...
Moreover, we carried out a second labelling experiment by using now as starting material the monodeuterated acetal-fulvene 20 bearing the deuterium atom at C4. This species converted into a 2:1 mixture of the monodeuterated regioisomers 21 and 22 as the only isolated reaction products (Scheme 6). In both compounds, the deuteration percentage at their respective C4 and C9 positions was determined, by 1H NMR analyses, to be higher than 98%.
This latter result shows that the deuterium atom attached at C4 in the original acetal-fulvene does not migrate in the course of the reaction. In combination, these two labelling experiments are conclusive for discarding an initial [1,4]-deuteride (or hydride) shift as the first step of the previously discussed conversions since, in such a case, we should have found deuterium linked to C4/C9 of the benzindenes 18/19 resulting from the first labelling experiment.
Next we tried to understand why the regioisomeric benz[f]indenes were, in all our reactions, produced in a ratio close to 2:1. It is well known that 1H-indenes are prone to undergo isomerization by H or group migrations at its cyclopentadiene ring [34-47]. Consequently, we postulated that such an isomeric ratio would correspond to the thermodynamic equilibrium between both isomers, 5 and 6, established by two consecutive [1,5]-H shifts around its five-membered ring. To test this hypothesis, we heated a 4:1 mixture of isomeric 5c and 6c in deuterated DMSO solution at 120 °C for 24 h. In this way we could verify by 1H NMR analyses of reaction aliquots that the initial isomeric ratio remained constant over time and heating. Interestingly, the initial 4:1 ratio changed to 2:1 at the end of a related experiment carried out by stirring a DMSO solution of the same isomeric mixture in the presence of a catalytic amount of triethylamine at room temperature for 2 h (Scheme 7).
![[1860-5397-12-28-i7]](/bjoc/content/inline/1860-5397-12-28-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Reagents and conditions: i) triethylamine (10%), DMSO, rt, 2 h.
Scheme 7: Reagents and conditions: i) triethylamine (10%), DMSO, rt, 2 h.
This result seems to indicate that the experimentally recurrent 2:1 proportion between the regioisomeric products 5 and 6, reached by equilibration in the latter experiment, should be due to the presence of adventitious minor amounts of basic species either in the DMSO solutions of the experiments or in the course of the processing of the crude reaction mixtures and the purification steps.
Besides, we also explored the thermally induced transformations of related fulvenes bearing non-cyclic acetalic units (Scheme 8 and Table 2). To this end, benzaldehydes 23 were transformed into fulvenes 24 by the usual procedure, whereas its microwave heating (DMSO, 120 °C, 120 W) yielded a mixture of the benz[f]indenes 25 and 26 in the habitual 2:1 ratio. These conversions most probably occur, in mechanistic terms, similarly to those of the acetal-fulvenes 3 (Scheme 4), although in the present cases with the formal β-elimination of a methanol or ethanol molecule.
![[1860-5397-12-28-i8]](/bjoc/content/inline/1860-5397-12-28-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Preparation of benz[f]indenes 25 and 26. Reagents and conditions: i) cyclopentadiene, pyrrolidine, anhydrous methanol, rt, 10 h; ii) DMSO, microwave, 120 °C, 120 W, 20–40 min.
Scheme 8: Preparation of benz[f]indenes 25 and 26. Reagents and conditions: i) cyclopentadiene, pyrrolidine, ...
These results show that non-cyclic acetalic units are as effective as the cyclic ones on achieving the conversion of acetal-fulvenes into the corresponding benz[f]indenes under microwave irradiation.
Computational study
With the aim of scrutinizing the putative reaction paths leading from the fulvene 3a to the isomeric benz[f]indenes 5a and 6a we have carried out a computational study at the B3LYP/6-31+G** theoretical level. Scheme 9 shows the diversity of the computed reaction paths leading from reactants to products. The geometries of the located transition structures associated to the first mechanistic step of each path, the H shift, are shown in Figure 1 [48].
![[1860-5397-12-28-i9]](/bjoc/content/inline/1860-5397-12-28-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Mechanistic paths for the conversion of fulvene 3a into the benz[f]indenes 5a and 6a showing the energy barriers of each step in kcal·mol−1 as computed at the B3LYP/6-31+G** theoretical level (between parentheses the relative electronic energies of the minima in kcal·mol−1).
Scheme 9: Mechanistic paths for the conversion of fulvene 3a into the benz[f]indenes 5a and 6a showing the en...
![[1860-5397-12-28-1]](/bjoc/content/figures/1860-5397-12-28-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Optimized geometry of transition structures TS1-A, TS1-B, and TS1-C computed at the B3LYP/6-31+G** theoretical level. Distances in Ångstroms, bond angles in degrees.
Figure 1: Optimized geometry of transition structures TS1-A, TS1-B, and TS1-C computed at the B3LYP/6-31+G** ...
We anticipated three general reaction channels, paths A–C (see Scheme 9). In fact, these pathways only differ in the first step. Path A starts by a [1,5]-H shift, path B by a [1,9]-H shift and path C by a [1,7]-H shift. This overall mechanistic scheme is somewhat complicated due to the number of steps of each reaction path and by the fact that some stationary points belong to more than one of these three pathways. Obviously we also envisaged a fourth mechanistic alternative, path D, just that initially conceived starting by the [1,4]-hydride shift of the acetalic H atom to the exocyclic C4 carbon atom of the fulvene unit. All our efforts aimed to locate its corresponding transition structure were unsuccessful. Nevertheless, this latter mechanistic alternative was discarded by the isotopic labelling experiments commented above.
In the following paragraphs we intend to discuss in a simplified way the results of our calculations on the potential surface of the transformations summarized in Scheme 9.
The first step of path A consists of a [1,5]-H shift from the acetalic carbon atom to C5 (see the numbering in Scheme 9). We located the transition structure TS1-A, connecting the fulvene 3a with the ortho-quinodimethane intermediate 9a. As expected TS1-A shows the typical geometry of a suprafacial hydrogen shift (see Figure 1), the computed energy barrier associated to this step being fairly high, 47.5 kcal·mol−1. Intermediate 9a could then experiment two alternative [1,5]-H shifts of its H-C5 proton, migrating either to C6 or to C9, its two vicinal carbon atoms at the cyclopentadiene ring. For the [1,5]-H shift to C9 we located the transition structure TS2, 19.6 kcal·mol−1 above in energy than 9a, connecting it with its isomer 10a. For the alternative [1,5]-H shift to C6 we located the transition structure TS3, 19.0 kcal·mol−1 above in energy than 9a, leading to the isomeric structure 10a’ which is in fact a rotamer of 10a. These two energy barriers are reasonably low and should be easily surmountable under the experimental reaction conditions. Additionally, we were able to locate a transition structure, TS4, connecting 10a and 10a’ by rotation around the C4–C5 single bond. The computed barrier for the conversion of 10 into 10a’ via TS4 is only 7.0 kcal·mol−1, whereas the one for the reverse transformation is 7.9 kcal·mol−1. Accordingly, equilibration between 10a and 10a’ is predicted to occur rapidly by C4–C5 bond rotation rather than by two consecutive [1,5]-H shifts via the isomeric intermediate 9a (see Scheme 9).
Intermediate 10a’ can also convert into a third ortho-quinodimethane isomer 11a via the transition structure TS5 by another [1,5]-H shift from C6 to its vicinal C7 carbon atom at the cyclopentadiene ring. The computed energy barrier for this step is 27.6 kcal·mol−1, significantly higher than those corresponding to the similar [1,5]-H shifts via TS2 and TS3 commented above (19.6 and 19.0 kcal·mol−1). The lower barriers of these two latter transition structures are attributable to its more extended conjugation in comparison with the partially cross-conjugated TS5.
A series of reaction steps starting from intermediates 10a and 11a can lead respectively to the final benzindenes 5a and 6a. Thus, intermediate 11a undergoes a disrotatory 6π-electrocyclic ring closure [49] via the transition structure TS6 to give the tricyclic species 13a. By an analogous electrocyclic ring closure through TS7, compound 10a is converted into the isomeric spirotricycle 12a. The computed energy barriers for these processes are relatively small, 13.9 and 17.0 kcal·mol−1 respectively. Again the differences in the extent of the electronic conjugation in these electrocyclization transition states can give account of the relative stabilities of TS6 and TS7.
Two transition structures, TS8 and TS9 were located for the respective transformations of 13a and 12a into the final benzindenes 5a and 6a, involving each one the opening of the acetalic ring with simultaneous transfer of an hydrogen to one of the oxygen atoms (in other words, a concerted β-elimination along a C–C single bond), with the concomitant aromatization of the central ring. The computed energy barriers for these concerted β-eliminations are high, 41.7 and 40.8 kcal·mol−1 respectively [50].
Concerning the alternative reaction paths B and C, we have located essentially the same stationary points that in path A with the sole difference of the respective first mechanistic steps. Path B starts with a [1,9]-H sigmatropic rearrangement through TS1-B leading to intermediate 10a’, which then transforms via the mechanistic paths commented above. The geometry of TS1-B (see Figure 1) is in accordance with a suprafacial transfer of the H atom between the acetalic carbon and C6. The calculated energy barrier associated to this step is 41.4 kcal·mol−1, 6.1 kcal·mol−1 lower in energy than the initial [1,5]-H shift of path A. This difference could be rationalized attending to the geometries of both transition structures TS1-A and TS1-B, more specifically to the distance between the two carbon atom termini of the H migration, shorter in TS1-B (2.63 Å) than in TS1-A (2.67 Å). Therefore, TS1-B is earlier than TS1-A. The geometry of TS1-B also accounts for its greater conjugation as the spatial positioning of the cyclopentadiene ring allows its orbital overlapping with the rest of the π system. Moreover, TS1-B is less sterically congested and also less distorted than TS1-A (see the bond distances and bond angles displayed in Figure 1).
For the first step of path C we have located a transition structure, TS1-C, connecting fulvene 3a with the intermediate 11a by a [1,7]-H sigmatropic shift (see Scheme 9). The computed energy barrier is very high, 64.3 kcal·mol−1. This large value is probably due to the heptatrienic fragment not being able of adopting the helical all s-cis conformation, optimal for an antarafacial [1,7]-H shift, as result of the conformational restrictions imposed by the cyclopentadiene ring (Figure 1). As a consequence, the distance between the two carbon atom termini of the H migration is considerably long (2.88 Å), thus accounting for the high computed energy barrier.
To summarize so far, by comparing the energy barriers associated to the three alternative H shifts, this study predicts that path B is the one involving the lowest energy barrier and, in accordance, the calculations predict that the transformation of fulvene 3a into the benzindenes 5a and 6a should take place via an initial [1,9]-H shift.
Moreover, we also considered that 5a and 6a could equilibrate by two consecutive [1,5]-H shifts occurring at the five-membered ring. By exploring the potential energy surface associated to these transformations we were able to locate transition structures TS10 and TS11 connecting 5a and 6a through the intermediate 27a (see Scheme 9). The computed energy barriers for the conversions 5a → 27a, and 6a → 27a are fairly high, 43.2 and 42.6 kcal·mol−1, respectively, as expected on going from a fully aromatic central ring to an ortho-quinoid structure, whereas those calculated for the reverse conversions are considerably lower (13.3 kcal·mol−1 in both cases).
By analysing the overall picture showing the different mechanistic paths connecting 3a with the final benzindenes 5a and 6a we can extract the following conclusions:
1) On going from 3a to the two tricyclic intermediates 12a and 13a, the rate determining reaction step is predicted to be the first one, i.e. the initial hydrogen migration, and this study predicts that a [1,9]-H shift is less costly in terms of energy than a [1,5]-H or [1,7]-H one. The more extended conjugation, the lower steric hindrance and the shorter C–C distance between the two carbon atoms termini of the H migration in TS1-B, the transition structure of the [1,9]-H shift, can account for its lower energy when compared with those of the alternative two other H shifts. Nevertheless, the higher electronic conjugation in TS1-B, in comparison with those of TS1-A and TS1-C, could be also decisive in accounting for the differences in the respective energy barriers.
2) Concerning the two key polyenes 10a and 11a, precursors of the tricycles 12a and 13a, respectively, the computed energy barriers of this study show that i) 10a most probably forms by an easy rotational isomerization of 10a’, instead of the alternative path involving the [1,5]-H shift from 9a; and ii) 11a will form mainly from 10a’ rather than directly from 3a. That is, 10a and 11a should form via intermediate 10a’ resulting from the [1,9]-H shift, which then transforms into 11a by a [1,5]-H shift (barrier of 27.6 kcal·mol−1) or equilibrates to its rotamer 10a (barrier of 7.0 kcal·mol−1).
3) The two 6π-electrocyclic ring closures converting respectively 10a and 11a into 12a and 13a involve low energy barriers, that corresponding to the conversion of 11a into 13a being lower than that of 10a into 12a (13.9 and 17.0 kcal·mol−1, respectively).
4) The overall processes 3a → 5a and 3a → 6a are exothermic by 31.3 and 30.7 kcal·mol−1, respectively. The interconversion between 5a and 6a is predicted to take place via two consecutive [1,5]-H shifts at the pentagonal ring through transient intermediate 27a.
As a final point, we have also explored the potential energy surface associated to these conversions by considering the effect of the solvent used in the experimental study, DMSO. The computed energy barriers in the gas phase and in DMSO are depicted in Table 3.
Table 3: Electronic energy barriers in kcal·mol–1 for the conversions of acetal fulvene 3a into indenes 5a and 6a calculated at the B3LYP/6-31+G** theoretical level.a
| 3a → 5a + 6a | Gas | DMSO |
|---|---|---|
| ΔE1A | 47.5 | 47.3 |
| ΔE2 | 19.6 | 19.0 |
| ΔE3 | 19.0 | 17.6 |
| ΔE1B | 41.4 | 39.9 |
| ΔE4 | 7.0 | 6.9 |
| ΔE5 | 27.6 | 26.6 |
| ΔE1C | 64.3 | 63.6 |
| ΔE6 | 13.9 | 14.4 |
| ΔE7 | 17.0 | 17.5 |
| ΔE8 | 41.7 | 36.8 |
| ΔE9 | 40.8 | 37.3 |
| ΔE10 | 43.2 | 42.3 |
| ΔE11 | 42.6 | 41.4 |
aSee Scheme 9 for the notation of the energy barriers.
In general, the values of the energy barriers do not vary noticeably in DMSO when compared with those in gas phase. Only ΔE8 and ΔE9 are appreciably lower in DMSO with respect to those in gas phase by 3.9 and 3.5 kcal·mol−1, respectively (Table 3). Consequently, according to these calculations, the rate determining step in the transformations 3a → 5a and 3a → 6a in DMSO should be the first one, i.e. the [1,9]-H shift, with an energy barrier slightly lower than that calculated in the gas phase.
In summary, this computational study shows that the conversion of fulvene 3a into the benzindenes 5a and 6a could take place by a variety of alternative reaction paths according to a complicated mechanistic scheme. By analysing in detail the energy barriers computed for each mechanistic step, the energetically preferred path starts with a [1,9]-H sigmatropic rearrangement of the acetalic hydrogen atom leading to an ortho-quinodimethane intermediate, further transforming into the isomeric final products by two alternative reaction channels. These two latter pathways may involve up to three consecutive steps such as [1,5]-H shifts, 6π-electrocyclic ring closures, C–C rotations and formal β-eliminations. The interconversion between the isomeric benzindenes 5a and 6a could also occur by means of two consecutive [1,5]-H shifts through an unstable benzisoindene intermediate.
Conclusion
The ability of benzofulvenes bearing 1,3-dioxolane or -dioxane units in ortho position for undergoing cascade processes initiated by an H shift step has been tested. Such acetal-fulvenes, under thermal activation, transformed into mixtures of the corresponding 4- and 9-(hydroxy)alkoxy-substituted benz[f]indenes in a 1:2 ratio. Analogous fulvenes bearing non-cyclic dialkoxymethyl units when submitted to similar thermal conditions also afforded 1:2 mixtures of the respective 4 and 9-alkoxybenz[f]indenes. Such 1:2 ratio has been interpreted as the one corresponding to the thermodynamic equilibrium established between both isomers. Mechanistic paths initiated by an initial [1,4]-, [1,5]-, [1,7]- or [1,9]-H shift are conceivable for explaining these cascade transformations leading to benz[f]indenes. The results of deuterium labelling experiments excluded a [1,4]-hydride shift as the initial step. The reaction of the unsubstituted 1,3-dioxolane-fulvene has been computationally studied by DFT methods. The results of this study revealed that the first mechanistic step, the H shift, is the rate-determining one and that, among the alternative [1,5]-, [1,7]- or [1,9]-H migrations, the energy barrier of the [1,9]-H shift is the lowest one, a fact that is rationalised attending to some key structural and electronic characteristics of the respective transition states. The calculations have also shown that the tandem conversions of the starting fulvenes into benz[f]indenes are exergonics, the 9-substituted regioisomer being the thermodynamically-controlled major product, in accordance with the experimental results.
References
-
Neuenschwander, M. Helv. Chim. Acta 2015, 98, 731–762. doi:10.1002/hlca.201400210
Return to citation in text: [1] [2] -
Neuenschwander, M. In Double-Bonded Functional Groups; Patai, S., Ed.; John Wiley & Sons: Chichester, 1989; Vol. 2, pp 1131–1286. doi:10.1002/9780470772256.ch4
Return to citation in text: [1] -
Bergmann, E. D. Chem. Rev. 1968, 68, 41–84. doi:10.1021/cr60251a002
Return to citation in text: [1] -
Day, J. H. Chem. Rev. 1953, 53, 167–189. doi:10.1021/cr60165a002
Return to citation in text: [1] [2] -
Dahlstrand, C.; Rosenberg, M.; Kilsa, K.; Ottosson, H. J. Phys. Chem. A 2012, 116, 5008–5017. doi:10.1021/jp3032397
Return to citation in text: [1] -
Oziminsky, W. P.; Krygowski, T. M.; Fowler, P. W.; Soncini, A. Org. Lett. 2010, 12, 4880–4883. doi:10.1021/ol102037e
Return to citation in text: [1] -
Hong, B.-C.; Chen, F.-L.; Chen, S.-H.; Liao, J.-H.; Lee, G.-H. Org. Lett. 2005, 7, 557–560. doi:10.1021/ol047730m
Return to citation in text: [1] [2] -
Gleiter, R.; Borzyk, O. Angew. Chem., Int. Ed. Engl. 1995, 34, 1001–1003. doi:10.1002/anie.199510011
And references therein.
Return to citation in text: [1] [2] -
Hong, B.-C.; Shr, Y.-J.; Wu, J.-L.; Gupta, A. K.; Lin, K.-J. Org. Lett. 2002, 4, 2249–2252. doi:10.1021/ol026103z
Return to citation in text: [1] [2] -
Wu, T. C.; Houk, K. N. J. Am. Chem. Soc. 1985, 107, 5308–5309. doi:10.1021/ja00304a065
Return to citation in text: [1] [2] -
Hafner, K.; Suda, M. Angew. Chem., Int. Ed. Engl. 1976, 15, 314–315. doi:10.1002/anie.197603141
Return to citation in text: [1] [2] -
He, Z.-L.; Teng, H.-L.; Wang, C.-J. Angew. Chem., Int. Ed. 2013, 52, 2934–2938. doi:10.1002/anie.201208799
Return to citation in text: [1] [2] -
Potowski, M.; Antonchick, A. P.; Waldmann, H. Chem. Commun. 2013, 49, 7800–7802. doi:10.1039/c3cc43824d
Return to citation in text: [1] [2] -
Hong, B.-C.; Gupta, A. K.; Wu, M.-F.; Liao, J.-H.; Lee, G.-H. Org. Lett. 2003, 5, 1689–1692. doi:10.1021/ol034329b
Return to citation in text: [1] [2] -
Barluenga, J.; Martinez, S.; Suárez-Sobrino, A. L.; Tomás, M. J. Am. Chem. Soc. 2001, 123, 11113–11114. doi:10.1021/ja011600g
Return to citation in text: [1] [2] -
Olsson, T.; Wennerstroem, O. Acta Chem. Scand., Ser. B 1979, B33, 256–260.
Return to citation in text: [1] -
Prinzbach, V. H.; Rosswog, W. Tetrahedron Lett. 1963, 4, 1217–1221. doi:10.1016/S0040-4039(01)90806-8
See for a base-catalyzed [1,5]-H shift in cycloheptatrienyl-substituted fulvenes.
Return to citation in text: [1] -
Van de Ven, L. J. M.; Keulemans-Lebbink, J. L. M.; de Haan, J. W.; Kloosterziel, H. J. Chem. Soc. D 1970, 1509a. doi:10.1039/c2970001509a
See for the preparation of a fulvene via a photochemical [1,7]-H shift.
Return to citation in text: [1] -
Alajarin, M.; Bonillo, B.; Ortin, M.-M.; Sanchez-Andrada, P.; Vidal, A. Org. Lett. 2006, 8, 5645–5648. doi:10.1021/ol062373w
Return to citation in text: [1] -
Alajarin, M.; Bonillo, B.; Ortin, M.-M.; Sanchez-Andrada, P.; Vidal, A. Eur. J. Org. Chem. 2011, 1896–1913. doi:10.1002/ejoc.201001372
Return to citation in text: [1] -
Alajarin, M.; Bonillo, B.; Sanchez-Andrada, P.; Vidal, A.; Bautista, D. Org. Lett. 2009, 11, 1365–1368. doi:10.1021/ol9001416
Return to citation in text: [1] -
Alajarin, M.; Bonillo, B.; Sanchez-Andrada, P.; Vidal, A. J. Org. Chem. 2010, 75, 3737–3750. doi:10.1021/jo100502p
Return to citation in text: [1] -
Alajarin, M.; Bonillo, B.; Marin-Luna, M.; Sanchez-Andrada, P.; Vidal, A.; Orenes, R.-A. Tetrahedron 2012, 68, 4672–4681. doi:10.1016/j.tet.2012.04.021
Return to citation in text: [1] -
Alajarin, M.; Bonillo, B.; Orenes, R.-A.; Ortin, M.-M.; Vidal, A. Org. Biomol. Chem. 2012, 10, 9523–9537. doi:10.1039/c2ob27010b
Return to citation in text: [1] -
Vidal, A.; Marin-Luna, M.; Alajarin, M. Eur. J. Org. Chem. 2014, 878–886. doi:10.1002/ejoc.201301501
Return to citation in text: [1] -
Alajarin, M.; Marin-Luna, M.; Ortin, M.-M.; Sanchez-Andrada, P.; Vidal, A. Tetrahedron 2011, 67, 5590–5595. doi:10.1016/j.tet.2011.05.119
Return to citation in text: [1] -
Alajarin, M.; Marin-Luna, M.; Vidal, A. Adv. Synth. Catal. 2011, 353, 553–557. doi:10.1002/adsc.201000812
Return to citation in text: [1] [2] -
Strohfeldt, K.; Tacke, M. Chem. Soc. Rev. 2008, 37, 1174–1187. doi:10.1039/b707310k
Return to citation in text: [1] -
Suzuka, T.; Ogasawara, M.; Hayashi, T. J. Org. Chem. 2002, 67, 3355–3359. doi:10.1021/jo0111199
Return to citation in text: [1] -
Ho, T.-I.; Ho, J.-H.; Wu, J.-Y. J. Am. Chem. Soc. 2000, 122, 8575–8576. doi:10.1021/ja0011562
Return to citation in text: [1] -
Ho, T.-I.; Wu, J.-Y.; Wang, S.-L. Angew. Chem., Int. Ed. 1999, 38, 2558–2560. doi:10.1002/(SICI)1521-3773(19990903)38:17<2558::AID-ANIE2558>3.0.CO;2-E
Return to citation in text: [1] -
Heller, H. G.; Jenkins, G. A. J. Chem. Soc., Perkin Trans. 1 1984, 2871–2875. doi:10.1039/p19840002871
Return to citation in text: [1] -
Stone, K. J.; Little, R. D. J. Org. Chem. 1984, 49, 1849–1853. doi:10.1021/jo00185a001
Return to citation in text: [1] -
Roth, W. R. Tetrahedron Lett. 1964, 1009–1013. doi:10.1016/S0040-4039(00)90422-2
Return to citation in text: [1] -
Koelsch, C. F.; Johnson, P. R. J. Am. Chem. Soc. 1943, 65, 567–573. doi:10.1021/ja01244a021
Return to citation in text: [1] -
Miller, L. L.; Greisinger, R.; Boyer, R. F. J. Am. Chem. Soc. 1969, 91, 1578–1580. doi:10.1021/ja01034a076
Return to citation in text: [1] -
Rakita, P. E.; Taylor, G. A. Inorg. Chem. 1972, 11, 2136–2141. doi:10.1021/ic50115a029
Return to citation in text: [1] -
Almy, J.; Cram, D. J. J. Am. Chem. Soc. 1970, 92, 4316–4320. doi:10.1021/ja00717a031
Return to citation in text: [1] -
Rakita, P. E.; Taylor, G. A. J. Organomet. Chem. 1973, 61, 71–81. doi:10.1016/S0022-328X(00)86536-5
Return to citation in text: [1] -
Jones, D. W.; Marmon, R. J. J. Chem. Soc., Perkin Trans. 1 1993, 681–690. doi:10.1039/p19930000681
Return to citation in text: [1] -
Rupert, K. C.; Liu, C. C.; Nguyen, T. T.; Whitener, M. A.; Sowa, J. R. Organometallics 2002, 21, 144–149. doi:10.1021/om010731n
Return to citation in text: [1] -
Ohlsson, L.; Wallmark, I.; Bergson, G. Acta Chem. Scand. 1966, 20, 750–753. doi:10.3891/acta.chem.scand.20-0750
Return to citation in text: [1] -
Weidler, A. M.; Bergson, G. Acta Chem. Scand. 1964, 18, 1487–1497. doi:10.3891/acta.chem.scand.18-1487
Return to citation in text: [1] -
Friedrich, E. C.; Taggart, D. B. J. Org. Chem. 1975, 40, 720–723. doi:10.1021/jo00894a012
Return to citation in text: [1] -
Bergson, G.; Weidler, A. M. Acta Chem. Scand. 1963, 17, 862–864. doi:10.3891/acta.chem.scand.17-0862
Return to citation in text: [1] -
Bergson, G.; Weidler, A. M. Acta Chem. Scand. 1963, 17, 1798–1799. doi:10.3891/acta.chem.scand.17-1798
Return to citation in text: [1] -
Bergson, G. Acta Chem. Scand. 1963, 17, 2691–2700. doi:10.3891/acta.chem.scand.17-2691
Return to citation in text: [1] -
The relative electronic and Gibbs energies and the energy barriers computed in gas phase and in DMSO are gathered in Table S2 of Supporting Information File 2.
Return to citation in text: [1] -
We have also found one additional transition structure for each one of the electrocyclizations 10a→12a and 11a→13a, those corresponding to conrotatory processes. As presumed, they are higher in energy than its disrotatory counterparts. With the aim of simplifying, they have been omitted from this discussion.
Return to citation in text: [1] -
The assistance of one or more water molecules in a concerted process has been also envisaged but not computationally scrutinized.
Return to citation in text: [1]
| 49. | We have also found one additional transition structure for each one of the electrocyclizations 10a→12a and 11a→13a, those corresponding to conrotatory processes. As presumed, they are higher in energy than its disrotatory counterparts. With the aim of simplifying, they have been omitted from this discussion. |
| 50. | The assistance of one or more water molecules in a concerted process has been also envisaged but not computationally scrutinized. |
| 1. | Neuenschwander, M. Helv. Chim. Acta 2015, 98, 731–762. doi:10.1002/hlca.201400210 |
| 2. | Neuenschwander, M. In Double-Bonded Functional Groups; Patai, S., Ed.; John Wiley & Sons: Chichester, 1989; Vol. 2, pp 1131–1286. doi:10.1002/9780470772256.ch4 |
| 3. | Bergmann, E. D. Chem. Rev. 1968, 68, 41–84. doi:10.1021/cr60251a002 |
| 4. | Day, J. H. Chem. Rev. 1953, 53, 167–189. doi:10.1021/cr60165a002 |
| 9. | Hong, B.-C.; Shr, Y.-J.; Wu, J.-L.; Gupta, A. K.; Lin, K.-J. Org. Lett. 2002, 4, 2249–2252. doi:10.1021/ol026103z |
| 10. | Wu, T. C.; Houk, K. N. J. Am. Chem. Soc. 1985, 107, 5308–5309. doi:10.1021/ja00304a065 |
| 11. | Hafner, K.; Suda, M. Angew. Chem., Int. Ed. Engl. 1976, 15, 314–315. doi:10.1002/anie.197603141 |
| 34. | Roth, W. R. Tetrahedron Lett. 1964, 1009–1013. doi:10.1016/S0040-4039(00)90422-2 |
| 35. | Koelsch, C. F.; Johnson, P. R. J. Am. Chem. Soc. 1943, 65, 567–573. doi:10.1021/ja01244a021 |
| 36. | Miller, L. L.; Greisinger, R.; Boyer, R. F. J. Am. Chem. Soc. 1969, 91, 1578–1580. doi:10.1021/ja01034a076 |
| 37. | Rakita, P. E.; Taylor, G. A. Inorg. Chem. 1972, 11, 2136–2141. doi:10.1021/ic50115a029 |
| 38. | Almy, J.; Cram, D. J. J. Am. Chem. Soc. 1970, 92, 4316–4320. doi:10.1021/ja00717a031 |
| 39. | Rakita, P. E.; Taylor, G. A. J. Organomet. Chem. 1973, 61, 71–81. doi:10.1016/S0022-328X(00)86536-5 |
| 40. | Jones, D. W.; Marmon, R. J. J. Chem. Soc., Perkin Trans. 1 1993, 681–690. doi:10.1039/p19930000681 |
| 41. | Rupert, K. C.; Liu, C. C.; Nguyen, T. T.; Whitener, M. A.; Sowa, J. R. Organometallics 2002, 21, 144–149. doi:10.1021/om010731n |
| 42. | Ohlsson, L.; Wallmark, I.; Bergson, G. Acta Chem. Scand. 1966, 20, 750–753. doi:10.3891/acta.chem.scand.20-0750 |
| 43. | Weidler, A. M.; Bergson, G. Acta Chem. Scand. 1964, 18, 1487–1497. doi:10.3891/acta.chem.scand.18-1487 |
| 44. | Friedrich, E. C.; Taggart, D. B. J. Org. Chem. 1975, 40, 720–723. doi:10.1021/jo00894a012 |
| 45. | Bergson, G.; Weidler, A. M. Acta Chem. Scand. 1963, 17, 862–864. doi:10.3891/acta.chem.scand.17-0862 |
| 46. | Bergson, G.; Weidler, A. M. Acta Chem. Scand. 1963, 17, 1798–1799. doi:10.3891/acta.chem.scand.17-1798 |
| 47. | Bergson, G. Acta Chem. Scand. 1963, 17, 2691–2700. doi:10.3891/acta.chem.scand.17-2691 |
| 7. | Hong, B.-C.; Chen, F.-L.; Chen, S.-H.; Liao, J.-H.; Lee, G.-H. Org. Lett. 2005, 7, 557–560. doi:10.1021/ol047730m |
| 8. |
Gleiter, R.; Borzyk, O. Angew. Chem., Int. Ed. Engl. 1995, 34, 1001–1003. doi:10.1002/anie.199510011
And references therein. |
| 48. | The relative electronic and Gibbs energies and the energy barriers computed in gas phase and in DMSO are gathered in Table S2 of Supporting Information File 2. |
| 7. | Hong, B.-C.; Chen, F.-L.; Chen, S.-H.; Liao, J.-H.; Lee, G.-H. Org. Lett. 2005, 7, 557–560. doi:10.1021/ol047730m |
| 8. |
Gleiter, R.; Borzyk, O. Angew. Chem., Int. Ed. Engl. 1995, 34, 1001–1003. doi:10.1002/anie.199510011
And references therein. |
| 9. | Hong, B.-C.; Shr, Y.-J.; Wu, J.-L.; Gupta, A. K.; Lin, K.-J. Org. Lett. 2002, 4, 2249–2252. doi:10.1021/ol026103z |
| 10. | Wu, T. C.; Houk, K. N. J. Am. Chem. Soc. 1985, 107, 5308–5309. doi:10.1021/ja00304a065 |
| 11. | Hafner, K.; Suda, M. Angew. Chem., Int. Ed. Engl. 1976, 15, 314–315. doi:10.1002/anie.197603141 |
| 12. | He, Z.-L.; Teng, H.-L.; Wang, C.-J. Angew. Chem., Int. Ed. 2013, 52, 2934–2938. doi:10.1002/anie.201208799 |
| 13. | Potowski, M.; Antonchick, A. P.; Waldmann, H. Chem. Commun. 2013, 49, 7800–7802. doi:10.1039/c3cc43824d |
| 14. | Hong, B.-C.; Gupta, A. K.; Wu, M.-F.; Liao, J.-H.; Lee, G.-H. Org. Lett. 2003, 5, 1689–1692. doi:10.1021/ol034329b |
| 15. | Barluenga, J.; Martinez, S.; Suárez-Sobrino, A. L.; Tomás, M. J. Am. Chem. Soc. 2001, 123, 11113–11114. doi:10.1021/ja011600g |
| 33. | Stone, K. J.; Little, R. D. J. Org. Chem. 1984, 49, 1849–1853. doi:10.1021/jo00185a001 |
| 5. | Dahlstrand, C.; Rosenberg, M.; Kilsa, K.; Ottosson, H. J. Phys. Chem. A 2012, 116, 5008–5017. doi:10.1021/jp3032397 |
| 6. | Oziminsky, W. P.; Krygowski, T. M.; Fowler, P. W.; Soncini, A. Org. Lett. 2010, 12, 4880–4883. doi:10.1021/ol102037e |
| 1. | Neuenschwander, M. Helv. Chim. Acta 2015, 98, 731–762. doi:10.1002/hlca.201400210 |
| 4. | Day, J. H. Chem. Rev. 1953, 53, 167–189. doi:10.1021/cr60165a002 |
| 19. | Alajarin, M.; Bonillo, B.; Ortin, M.-M.; Sanchez-Andrada, P.; Vidal, A. Org. Lett. 2006, 8, 5645–5648. doi:10.1021/ol062373w |
| 20. | Alajarin, M.; Bonillo, B.; Ortin, M.-M.; Sanchez-Andrada, P.; Vidal, A. Eur. J. Org. Chem. 2011, 1896–1913. doi:10.1002/ejoc.201001372 |
| 21. | Alajarin, M.; Bonillo, B.; Sanchez-Andrada, P.; Vidal, A.; Bautista, D. Org. Lett. 2009, 11, 1365–1368. doi:10.1021/ol9001416 |
| 22. | Alajarin, M.; Bonillo, B.; Sanchez-Andrada, P.; Vidal, A. J. Org. Chem. 2010, 75, 3737–3750. doi:10.1021/jo100502p |
| 23. | Alajarin, M.; Bonillo, B.; Marin-Luna, M.; Sanchez-Andrada, P.; Vidal, A.; Orenes, R.-A. Tetrahedron 2012, 68, 4672–4681. doi:10.1016/j.tet.2012.04.021 |
| 24. | Alajarin, M.; Bonillo, B.; Orenes, R.-A.; Ortin, M.-M.; Vidal, A. Org. Biomol. Chem. 2012, 10, 9523–9537. doi:10.1039/c2ob27010b |
| 25. | Vidal, A.; Marin-Luna, M.; Alajarin, M. Eur. J. Org. Chem. 2014, 878–886. doi:10.1002/ejoc.201301501 |
| 26. | Alajarin, M.; Marin-Luna, M.; Ortin, M.-M.; Sanchez-Andrada, P.; Vidal, A. Tetrahedron 2011, 67, 5590–5595. doi:10.1016/j.tet.2011.05.119 |
| 27. | Alajarin, M.; Marin-Luna, M.; Vidal, A. Adv. Synth. Catal. 2011, 353, 553–557. doi:10.1002/adsc.201000812 |
| 28. | Strohfeldt, K.; Tacke, M. Chem. Soc. Rev. 2008, 37, 1174–1187. doi:10.1039/b707310k |
| 29. | Suzuka, T.; Ogasawara, M.; Hayashi, T. J. Org. Chem. 2002, 67, 3355–3359. doi:10.1021/jo0111199 |
| 17. |
Prinzbach, V. H.; Rosswog, W. Tetrahedron Lett. 1963, 4, 1217–1221. doi:10.1016/S0040-4039(01)90806-8
See for a base-catalyzed [1,5]-H shift in cycloheptatrienyl-substituted fulvenes. |
| 18. |
Van de Ven, L. J. M.; Keulemans-Lebbink, J. L. M.; de Haan, J. W.; Kloosterziel, H. J. Chem. Soc. D 1970, 1509a. doi:10.1039/c2970001509a
See for the preparation of a fulvene via a photochemical [1,7]-H shift. |
| 30. | Ho, T.-I.; Ho, J.-H.; Wu, J.-Y. J. Am. Chem. Soc. 2000, 122, 8575–8576. doi:10.1021/ja0011562 |
| 31. | Ho, T.-I.; Wu, J.-Y.; Wang, S.-L. Angew. Chem., Int. Ed. 1999, 38, 2558–2560. doi:10.1002/(SICI)1521-3773(19990903)38:17<2558::AID-ANIE2558>3.0.CO;2-E |
| 32. | Heller, H. G.; Jenkins, G. A. J. Chem. Soc., Perkin Trans. 1 1984, 2871–2875. doi:10.1039/p19840002871 |
| 12. | He, Z.-L.; Teng, H.-L.; Wang, C.-J. Angew. Chem., Int. Ed. 2013, 52, 2934–2938. doi:10.1002/anie.201208799 |
| 13. | Potowski, M.; Antonchick, A. P.; Waldmann, H. Chem. Commun. 2013, 49, 7800–7802. doi:10.1039/c3cc43824d |
| 14. | Hong, B.-C.; Gupta, A. K.; Wu, M.-F.; Liao, J.-H.; Lee, G.-H. Org. Lett. 2003, 5, 1689–1692. doi:10.1021/ol034329b |
| 15. | Barluenga, J.; Martinez, S.; Suárez-Sobrino, A. L.; Tomás, M. J. Am. Chem. Soc. 2001, 123, 11113–11114. doi:10.1021/ja011600g |
| 27. | Alajarin, M.; Marin-Luna, M.; Vidal, A. Adv. Synth. Catal. 2011, 353, 553–557. doi:10.1002/adsc.201000812 |
© 2016 Alajarin et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)
