Abstract
Efficient routes were developed for the diastereoselective synthesis of new O-alkylated and C-branched inositols and their corresponding fluoro analogues. The key steps of the synthesis were the easy accessibility of different types of arms in term of configuration (myo and scyllo), the linking method and length, which could modulate the biological properties. These inositol derivatives, bearing an arm terminated either with a hydroxy group or a fluorine atom, could be interesting candidates for diastereoisomeric intermediates and biological evaluations, especially for PET imaging experiments.

Graphical Abstract
Introduction
Inositol is a trivial name used to describe cyclohexanehexol compounds. Nine stereoisomers exist differing in the relative orientation of the hydroxy groups, the most naturally abounding and biologically important is myo-inositol [1,2]. The chemistry of inositols has been the theme of several investigations in the past few years [3,4]. Indeed inositols and their phosphorylated or glycosylated [5] derivatives are involved in various biological processes [6-19]. In recent years, studies have shown that inositol derivatives interact with the β-amyloid protein (Aβ), characteristic aggregation in Alzheimer disease (AD), and more specifically Aβ42 peptide allowing its conformational change and its stabilization into small, nonfibrillar complexes, less toxic for neuronal cells [20-23]. The most efficient representative, scyllo-inositol, was shown to be able to reduce plaque burden and to improve the performance on memory tasks in animal models of Alzheimer disease [24-29]. Scyllo-inositol was recently used in human clinical trials for the treatment of patients suffering from AD [30,31].
McLaurin has also demonstrated that the corresponding fluoroinositol (1-fluoro-1-deoxy-scyllo-inositol) was also a good candidate to limit this aggregation. However, in vivo studies in animal models of Alzheimer disease with 1-[18F]fluoro-1-deoxy-scyllo-inositol used as radiotracer for positron emission tomography (PET) have shown that this compound crosses the blood brain barrier (BBB) with difficulty [32]. The same PET imaging studies using the diastereoisomeric compound of myo configuration (2-[18F]fluoro-2-deoxy-myo-inositol), did not get better results [33]. On the other hand, these radiotracers have shown interesting features in the diagnosis of breast cancer with this imaging technique [33,34].
Considering the synthetic and biological importance of inositol analogues, the design of new inositol derivatives to study their biological properties and to understand their binding mechanisms with molecular targets, for instance through radiolabeled compounds, seems judicious.
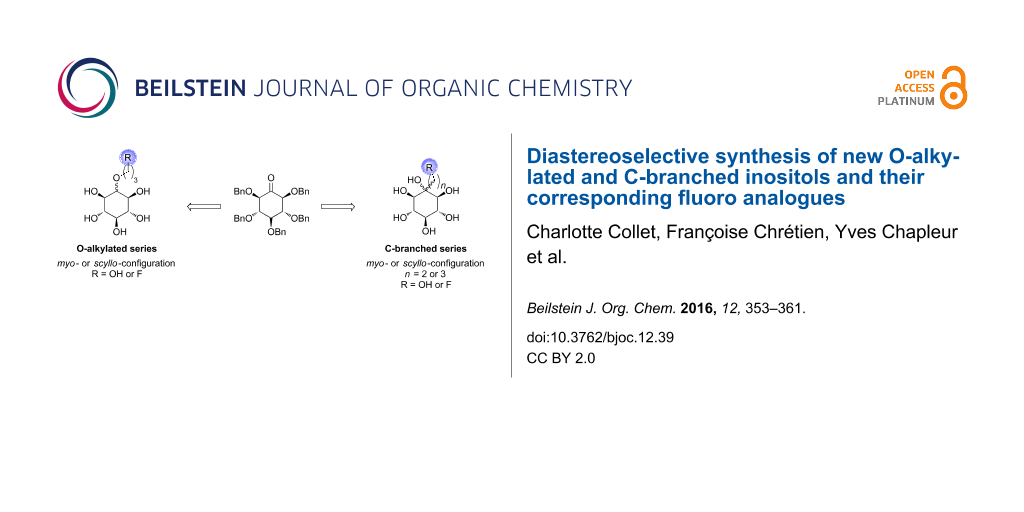
Thus we planned to study new synthetic inositols of myo- and scyllo-configuration equipped with different carbon chains linked either on one hydroxy group, i.e., O-alkylated, or C-branched derivatives retaining the six hydroxy groups of the inositol (Figure 1). These new inositol members could represent attractive tools for the development of further inositol analogues. Indeed, this arm could improve some properties as the lipophilicity for example and should allow the easy anchoring of various groups or molecules, such as carbohydrates for example.
![[1860-5397-12-39-1]](/bjoc/content/figures/1860-5397-12-39-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structures of targeted synthetic inositol derivatives.
Figure 1: Structures of targeted synthetic inositol derivatives.
Two arm lengths of two or three carbons were envisioned, on the C2 for the myo-series and on the C1 for the scyllo-series, ending with a hydroxy group or a fluorine atom. Indeed the addition of fluorine could improve chemical properties including lipophilicity, brain penetration, enhanced binding interaction and metabolic stability [35-40]. Moreover fluorinated inositols could be useful for PET imaging applications.
Results and Discussion
Our strategy to prepare new alkylated inositol compounds and their corresponding fluoro analogues was based on the use of the known benzylated myo-2-inosose 7 [41] that can be easily obtained at large scale from commercially available myo-inositol. The introduction of the arm into the inositol backbone through an O–C linker (O-alkylated) or a C–C linker (C-branched) to afford diastereoselectively myo- and scyllo-derivatives was the key step. In the case of the O-alkylated inositols a three carbon arm was envisioned whereas for the C-branched derivatives two and three carbon atom arms were used. For the fluorinated inositols, we developed a synthetic route with easily removing protecting groups, as acetates, suitable for PET imaging application [42].
Synthesis of O-alkylated myo- and scyllo-inositol derivatives bearing a hydroxylated arm
The synthesis of the targeted hydroxylated O-alkylated inositols myo-1 and scyllo-1 (Scheme 1, see Supporting Information File 1 for full experimental data) started with a sodium borohydride reduction of the benzylated myo-2-inosose 7 in isopropyl alcohol at room temperature to give a mixture of the myo- and scyllo-inositol diastereoisomers myo-8 and scyllo-8. The mixture was separated by column chromatography to afford the pure myo- and scyllo-inositol derivatives in 58% and 30% yields, respectively. The selectivity of this reduction was lower compared to previously published results [43]. A diastereoisomeric ratio of 4:1 myo-8/scyllo-8 was estimated by 1H NMR when the same reduction was done in a dichloromethane/methanol mixture. This selectivity can be explained by a chelation of the sodium ion with the two OBn adjoining the ketone, which hindered the upper face. For our strategy having a moderate selectivity is of great advantage to continue the synthesis with both isolated diastereoisomers myo-8 and scyllo-8. Next, the ether link was created by the reaction of alcohols myo-8 and scyllo-8 with allyl bromide under classical conditions in the presence of NaH at reflux [44] to afford myo-9 and scyllo-9 in good yield of 80% and 82%, respectively. The hydroxylated arm was obtained by hydroboration using BH3 on the unsaturated compounds myo-9 and scyllo-9 followed by an oxidation with H2O2/NaOH allowing the access to myo-10 and scyllo-10 in 78% and 75% yield, respectively. The O-alkylated inositol derivatives myo-1 and scyllo-1 were obtained quantitatively by removing of the benzyl protecting groups of compounds myo-10 and scyllo-10 by hydrogenation under pressure [45] using palladium hydroxide as catalyst.
![[1860-5397-12-39-i1]](/bjoc/content/inline/1860-5397-12-39-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of O-alkylated inositol derivatives 1. Reagents and conditions: a) NaBH4, iPrOH, rt, 2 h, 58 and 30%; b) allyl bromide, NaH, 70 °C, 2 h, 80–82%; c) BH3, THF, rt, 6 h then H2O2, NaOH, rt, 12 h, 75–78%; d) Pd(OH)2, MeOH/CH2Cl2/H2O 10:10:1 (v/v/v), 45 psi, 18 h, quantitative.
Scheme 1: Synthesis of O-alkylated inositol derivatives 1. Reagents and conditions: a) NaBH4, iPrOH, rt, 2 h,...
Synthesis of O-alkylated myo- and scyllo-inositol derivatives bearing a fluorinated arm
The fluoro analogues of myo-1 and scyllo-1 were then prepared through a synthetic strategy that involves easily removable acetates on the inositol ring (Scheme 2, see Supporting Information File 1 for full experimental data).
![[1860-5397-12-39-i2]](/bjoc/content/inline/1860-5397-12-39-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of O-alkylated fluorinated inositol derivatives 2.
Scheme 2: Synthesis of O-alkylated fluorinated inositol derivatives 2.
The compounds myo-11 and scyllo-11 were obtained in 80% and 76% yield, respectively in a one pot reaction by the treatment of myo-1 and scyllo-1 with triphenylmethyl chloride in pyridine and a catalytic amount of DMAP at 40 °C for three days followed by the addition of acetic anhydride [46]. The trityl group in myo-11 and scyllo-11 was cleaved without acetyl migration using FeCl3 [47] to afford myo-12 and scyllo-12 in good yields. The primary hydroxy groups can be easily converted into fluorine by action of DAST in dichloromethane in less than 5 minutes to give myo-13 and scyllo-13 in 95 and 92% yield, respectively [48]. All protecting groups of myo-13 and scyllo-13 were removed by the treatment with Na0 in MeOH [49], which gave quantitatively the fully deprotected fluorinated O-alkylated inositols myo-2 and scyllo-2.
Synthesis of C-branched myo- and scyllo-inositol derivatives bearing a hydroxylated arm
The synthesis of C-branched inositols was investigated using two different arm lengths, with either two or three carbon atoms. The introduction of these substituents was performed by a Grignard reaction in THF with vinyl- or allylmagnesium bromide on benzylated myo-2-inosose 7 (Scheme 3, see Supporting Information File 1 for full experimental data). The reaction gave in both cases diastereoisomeric myo- and scyllo-inositols, in a 60:40 proportion, respectively. The myo-inositol diastereoisomers were obtained in a larger proportion, due to the favored equatorial attack as compared to the axial one. Fortunately, this poor selectivity was associated with an easy separation by column chromatography giving myo-14 and scyllo-14 bearing a two carbon chain in 56% and 37% yield, respectively. The corresponding compounds myo-15 and scyllo-15 having an allyl substituent were obtained in 53% and 38% yields, respectively.
![[1860-5397-12-39-i3]](/bjoc/content/inline/1860-5397-12-39-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of C-alkenylated inositol intermediates.
Scheme 3: Synthesis of C-alkenylated inositol intermediates.
The configuration of the diastereoisomers was determined with 1H and nOe NMR experiments (Figure 2). For compounds myo-14 and myo-15 correlations between H1/H3 and the alkenyl proton of the arm were observed confirming the equatorial orientation of the substituent and therefore the myo-inositol configuration. The same experiments were performed with compounds scyllo-14 and scyllo-15 and showed correlations between H3/H5 and the hydrogen on the vinylic or allylic arm respectively fully in agreement with a scyllo-inositol configuration.
![[1860-5397-12-39-2]](/bjoc/content/figures/1860-5397-12-39-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: nOe correlations for C-alkenylated inositol intermediates.
Figure 2: nOe correlations for C-alkenylated inositol intermediates.
Having the C-alkenylated inositols in hand, the synthesis of C-branched inositol compounds with hydroxylated two or three carbon atom arms could be achieved (Scheme 4, see Supporting Information File 1 for full experimental data).
![[1860-5397-12-39-i4]](/bjoc/content/inline/1860-5397-12-39-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of C-branched inositol derivatives 3 and 4.
Scheme 4: Synthesis of C-branched inositol derivatives 3 and 4.
Unsaturated C-branched inositols 14 and 15 lead to the corresponding hydroxylated compounds 16 and 17, after hydroboration using BH3 in THF at 0 °C followed by conventional oxidation using H2O2/NaOH, in 70–80% yield. Subsequent catalytic hydrogenation under pressure with Pd(OH)2 as the catalyst [45] allowed to obtaining quantitatively the fully hydroxylated inositols myo-3, scyllo-3, myo-4 and scyllo-4 as a new hydroxylated inositol family.
Synthesis of C-branched myo- and scyllo-inositol derivatives bearing a fluorinated two carbons arm
For the synthesis of fluorinated C-branched inositols with a two carbon atoms arm, a similar strategy as developed for the O-alkylated inositols was chosen to access the fluorinated compounds bearing easily removable acetyl protecting groups (Scheme 5, see Supporting Information File 1 for full experimental data). Starting from the fully hydroxylated inositols myo-3 and scyllo-3, the first step was the protection of the primary hydroxy group with trityl chloride, followed by peracetylation with acetic anhydride in a one pot reaction [46]. Myo-18 and scyllo-18 were obtained in 74 and 70% yield, respectively. It is worthy to note that the tertiary hydroxy group in myo-18 and scyllo-18 remained unprotected under these conditions. The literature data are rather contradictory concerning the acetylation of this tertiary alcohol using acetic anhydride [50,51]. With regard to a PET application, the protection of this hydroxy group is of paramount importance since the free hydroxy group could impair the incorporation of the radioactive fluorine. Therefor several methods of acetylation were carried out on myo-14, which was used as model. The best results for the acetylation of the tertiary alcohol in good yield was obtained with the transesterification method using isopropenyl acetate as acylating agent and p-toluenesulfonic acid as catalyst at 80 °C for 2 h [52]. Unfortunately, when these reaction conditions were applied on the tritylated compounds myo-18 and scyllo-18 we were unable to obtain the fully protected products myo-19 and scyllo-19; we only got several compounds without trityl group due to the acidic reaction conditions. So we decided to cleanly remove this troublesome group using FeCl3 in dichloromethane for 18 h at room temperature [47]. Under these conditions the compound scyllo-20 was obtained in a good yield (72%) while myo-20 was obtained only in a poor yield (15%) due to the formation of byproducts proceeding from cyclization and/or acetate migration. By reducing the reaction time to 15 minutes the byproducts formation could be lowered to 10% and myo-2 was obtained in 45% isolated yield. It is worthy to note that 41% of the starting material was recovered.
![[1860-5397-12-39-i5]](/bjoc/content/inline/1860-5397-12-39-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Synthesis of C-branched fluorinated inositol derivatives 5. Reagents and conditions: a) TrCl, DMAP (cat), pyridine, 3 days, 70 °C, then Ac2O, rt, 2 h, 70–74%; b) isopropenyl acetate, p-TSA, 80 °C, 2 h; c) FeCl3, CH2Cl2, rt, 18 h or 15 min, 45–72%; d) DAST, CH2Cl2, rt; e) TsCl, Et3N, CH2Cl2, rt, 3 h, 70–71%; f) isopropenyl acetate, p-TSA, 80 °C, 2 h, 78–99%; g) KF, 18-crown-6, CH3CN or TBAF, CH3CN or KHF2, 18-crown-6, CH3CN; h) KF; 18-crown-6; CH3CN, rt, 2 h, 70%; i) Na0 (cat), MeOH, rt, 2 h, quantitative.
Scheme 5: Synthesis of C-branched fluorinated inositol derivatives 5. Reagents and conditions: a) TrCl, DMAP ...
At this stage, the fluorination was tested using DAST [48] on myo-20 and scyllo-20. Unfortunately, the desired fluorinated products myo-21 and scyllo-21 were not obtained; instead elimination reaction and the formation of unidentified products were observed. Hence, tosylation was next performed, in order to allow the nucleophilic substitution with fluoride at a later stage. For this, compounds myo-20 and scyllo-20 were reacted with tosyl chloride in the presence of a catalytic amount of triethylamine at room temperature for 3 h to give tosylated myo-22 and scyllo-22 in 70 and 71% yield, respectively. Next, acetylation of the tertiary alcohol was performed before fluorination. The utilization of isopropenyl acetate as solvent in the presence of p-TSA at 80 °C for 2 h [52] gave the corresponding acetylated compounds myo-23 and scyllo-23 in good to excellent yields. The access to fluorinated compounds by nucleophilic substitution was investigated starting from these fully acetylated inositols. The fluorination reactions using potassium fluoride, 18-crown-6 in acetonitrile at 70 °C [53], TBAF in acetonitrile at 60 °C for 2 h [54] or KHF2 and 18-crown-6 in acetonitrile for 2 h at 60 °C [55] were performed. However, 19F NMR analyses of the reaction mixtures did not show any fluorine incorporation. Various types of compounds resulting from the migration of the tertiary acetate to the primary position, elimination reaction or cyclization were identified depending on the experimental procedure.
From these results, the fluorination was finally investigated on myo-22 and scyllo-22 derivatives, which were not acetylated on the tertiary position; in order to avoid the acyl migration. The treatment of myo-22 with KF, 18-crown-6 in acetonitrile [53] at room temperature (optimal conditions) furnished the fluorinated compound myo-25 in 70% yield. The 19F NMR spectrum showed only one signal at −217 ppm (triplet of triplet, J = 48 Hz, J = 28 Hz) establishing the formation of the expected fluorinated product. Unfortunately, in the case of the isomeric scyllo-22 whatever the conditions used, the desired fluoro compound scyllo-25 could not be obtained; three different cyclized byproducts were formed. Finally, Zemplen [49] deprotection was performed on compound myo-25 to afford myo-5 quantitatively.
Synthesis of C-branched myo- and scyllo-inositol derivatives bearing a fluorinated three carbon atoms arm
Having in hand myo-4 and scyllo-4, the development of C-branched inositols fluorinated on the three carbon atoms arm was carried out by the same methodology as used for the C-branched two carbon atoms chain (Scheme 6, see Supporting Information File 1 for full experimental data).
![[1860-5397-12-39-i6]](/bjoc/content/inline/1860-5397-12-39-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Synthesis of C-branched fluorinated inositol derivatives 6. Reagents and conditions: a) TrCl, DMAP (cat), pyridine, 3 d, rt, then Ac2O, rt, 2 h, 67–70%; b) isopropenyl acetate, p-TSA, 80 °C, 2 h, 52–56%; c) FeCl3, CH2Cl2, rt, 18 h, 63–70%; d) DAST, CH2Cl2, rt, 5 min, 91–95%; e) Na0 (cat), MeOH, rt, 2 h, quantitative.
Scheme 6: Synthesis of C-branched fluorinated inositol derivatives 6. Reagents and conditions: a) TrCl, DMAP ...
The selective protection of the primary hydroxy group with trityl chloride, followed by peracetylation with acetic anhydride in one pot was carried out [46]. Compounds myo-26 and scyllo-26 were obtained in 67% and 70% yields, respectively. However, as already observed for the two carbon-atoms-arm-branched compounds, the tertiary hydroxy group remained unprotected. As explained above, the protection of this hydroxy group is mandatory for labeling with [18F]-fluoride with regard to a PET application. Therefore the acetylation of myo-26 and scyllo-26 using the transacetylization protocol [52] gave the fully acetylated products myo-27 and scyllo-27 in 56% and 52% isolated yields, respectively. The moderate yields could be explained by the presence of detritylated fully acetylated byproducts due to the acidic reaction conditions. Next, deprotection of the trityl group from myo-27 and scyllo-27 was performed using FeCl3 in dry dichloromethane at room temperature overnight [47]. The hydroxylated products myo-28 and scyllo-28 were obtained in 63% and 70% yields, respectively. The fluorination of compounds 28 was carried out with DAST as fluorinating agent in dichloromethane at room temperature for 5 min [48] and the fluoro-compounds myo-29 and scyllo-29 were obtained in excellent yields of 95% and 91%, respectively. Finally, Zemplen deprotection [49] of the acetyl groups was carried out to furnish quantitatively the fully deprotected fluoro-inositols myo-6 and scyllo-6. The obtained diastereoisomeric compounds myo-6 and scyllo-6 having a C–C linked three carbon-atoms arm represent a new class of inositols.
The above results demonstrate that the new O-alkylated and C-branched inositols with myo- and scyllo-configuration were available in a highly diastereoselective way. Different types of arm were easily introduced on the inositol ring. For the O-alkylated series, the hydroxylated (myo-1 and scyllo-1) as well as fluorinated derivatives (myo-2 and scyllo-2) were synthesized in a simple and efficient manner. Concerning the C-branched fluorinated inositols, the synthesis of compounds having a three carbon atoms arm (myo-6 and scyllo-6) was easier than the corresponding compounds substituted with the two carbon atoms arm where only compound myo-5 could be obtained. Indeed with the chain being longer, the formation of byproducts originating from cyclization and migration, favored by the proximity of the inositol ring and preferential transition states, was avoided. The synthetic routes are convenient and could provide sufficient material for biological study.
Conclusion
In conclusion, novel families of O-alkylated and C-branched inositols with a two or three carbon atoms arm and their corresponding fluoro analogues were synthesized in an efficient and diastereoselective manner. Noteworthy aspects of this methodology are the easy accessibility of different types of arms in term of configuration (myo and scyllo), linking method and length, which could modulate the biological properties. The series bearing an arm terminated by a hydroxy group could provide interesting candidates for biological evaluations but also as useful diastereoisomeric intermediates to anchor various groups such as carbohydrates, for example, opening up new avenues for the total synthesis of new inositol derivatives. The synthesized inositol derivatives with fluorinated arms could be more specifically interesting candidates for PET imaging experiments. Their potential use as radiotracers is under current investigation.
Supporting Information
| Supporting Information File 1: Experimental procedures, characterization data and copies of 1H, 13C, and 19F NMR spectra of all new compounds. | ||
| Format: PDF | Size: 1.7 MB | Download |
References
-
Posternak, T. The cyclitols; Holden-Day: San Francisco, 1962.
Return to citation in text: [1] -
Anderson, L. The Cyclitols. In The Carbohydrates, 2nd ed.; Pigman, W.; Horton, D., Eds.; Academic Press: San Diego, 1972; Vol. 1A, pp 519–579.
Return to citation in text: [1] -
Kılbaş, B.; Balci, M. Tetrahedron 2011, 67, 2355–2389. doi:10.1016/j.tet.2011.01.012
And references cited therein.
Return to citation in text: [1] -
Potter, B. V. L.; Lampe, D. Angew. Chem., Int. Ed. Engl. 1995, 34, 1933–1972. doi:10.1002/anie.199519331
And references cited therein.
Return to citation in text: [1] -
McConville, M. J.; Ferguson, M. A. Biochem. J. 1993, 294, 305–324. doi:10.1042/bj2940305
See for a comprehensive review on structure and biosynthesis of GPI anchors.
Return to citation in text: [1] -
Bleasdale, J. E.; Eichberg, J.; Hauser, G., Eds. Inositol and Phophoinositides; Humana: Clifton, NJ, 1985.
Return to citation in text: [1] -
Billington, D. C. The inositol Phosphates. Chemical Synthesis and Biological Significance; Wiley VCH: New York, 1993.
Return to citation in text: [1] -
Dalko, O. P.; Sinay, P. In Organic Synthesis Highlights V; Schmalz, H. G.; Wirth, T., Eds.; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2003; pp 1–14.
Return to citation in text: [1] -
Michell, R. H.; Drumond, A. H.; Downes, C. P., Eds. Inositol Lipids in Cell Signalling; Academic Press: San Diego, 1989.
Return to citation in text: [1] -
Schedler, D. J. A.; Baker, D. C. Carbohydr. Res. 2004, 339, 1585–1595. doi:10.1016/j.carres.2004.03.030
Return to citation in text: [1] -
Roussel, F.; Moitessier, N.; Hilly, M.; Chrétien, F.; Mauger, J.-P.; Chapleur, Y. Bioorg. Med. Chem. 2002, 10, 759–768. doi:10.1016/S0968-0896(01)00329-7
Return to citation in text: [1] -
Bennett, M.; Onnebo, S. M. N.; Azevedo, C.; Saiardi, A. Cell. Mol. Life Sci. 2006, 63, 552–564. doi:10.1007/s00018-005-5446-z
Return to citation in text: [1] -
Barker, C. J.; Illies, C.; Gaboardi, G. C.; Berggren, P.-O. Cell. Mol. Life Sci. 2009, 66, 3851–3871. doi:10.1007/s00018-009-0115-2
Return to citation in text: [1] -
Roussel, F.; Hilly, M.; Chrétien, F.; Mauger, J.-P.; Chapleur, Y. J. Carbohydr. Chem. 1999, 18, 697–707. doi:10.1080/07328309908544031
Return to citation in text: [1] -
Hilton, J. M.; Plomann, M.; Ritter, B.; Modregger, J.; Freeman, H. N.; Falck, J. R.; Krishna, U. M.; Tobin, A. B. J. Biol. Chem. 2001, 276, 16341–16347. doi:10.1074/jbc.M011122200
Return to citation in text: [1] -
Tsui, M. M.; York, J. D. Adv. Enzyme Regul. 2010, 50, 324–337. doi:10.1016/j.advenzreg.2009.12.002
Return to citation in text: [1] -
Vajanaphanich, M.; Schultz, C.; Rudolf, M. T.; Wasserman, M.; Enyedi, P.; Craxton, A.; Shears, S. B.; Tsien, R. Y.; Barrett, K. E.; Traynor-Kaplan, A. Nature 1994, 371, 711–714. doi:10.1038/371711a0
Return to citation in text: [1] -
Shi, Y.; Azab, A. N.; Thompson, M. N.; Greenberg, M. L. Biology of Inositol phosphates and phosphoinositides. In Subcellular Biochemistry; Majumder, A. L.; Biswas, B. B., Eds.; Springer Verlag: USA, 2006; Vol. 39, pp 265–292.
Return to citation in text: [1] -
Chen, W.; Deng, Z.; Chen, K.; Dou, D.; Song, F.; Li, L.; Xi, Z. Eur. J. Med. Chem. 2015, 93, 172–181. doi:10.1016/j.ejmech.2015.01.064
Return to citation in text: [1] -
McLaurin, J.; Golomb, R.; Jurewicz, A.; Antel, J. P.; Fraser, P. E. J. Biol. Chem. 2000, 275, 18495–18502. doi:10.1074/jbc.M906994199
Return to citation in text: [1] -
Nitz, M.; Fenili, D.; Darabie, A. A.; Wu, L.; Cousins, J. E.; McLaurin, J. FEBS J. 2008, 275, 1663–1674. doi:10.1111/j.1742-4658.2008.06321.x
Return to citation in text: [1] -
Hawkes, C. A.; Ng, V.; McLaurin, J. Drug Dev. Res. 2009, 70, 111–124. doi:10.1002/ddr.20290
Return to citation in text: [1] -
Sun, Y.; Zhang, G.; Hawkes, C. A.; Shaw, J. E.; McLaurin, J.; Nitz, M. Bioorg. Med. Chem. 2008, 16, 7177–7184. doi:10.1016/j.bmc.2008.06.045
Return to citation in text: [1] -
McLaurin, J.; Kierstead, M. E.; Brown, M. E.; Hawkes, C. A.; Lambermon, M. H.; Phinney, A. L.; Darabie, A. A.; Cousins, J. E.; French, J. E.; Lan, M. F.; Chen, F.; Wong, S. S. N.; Mount, H. T. J.; Fraser, P. E.; Westaway, D.; St George-Hyslop, P. Nat. Med. 2006, 12, 801–808. doi:10.1038/nm1423
Return to citation in text: [1] -
Townsend, M.; Cleary, J. P.; Mehta, T.; Hofmeister, J.; Lesne, S.; O'Hare, E.; Walsh, D. M.; Selkoe, D. J. Ann. Neurol. 2006, 60, 668–676. doi:10.1002/ana.21051
Return to citation in text: [1] -
Choi, J.-K.; Carreras, I.; Dedeoglu, A.; Jenkins, B. G. Neuropharmacology 2010, 59, 353–357. doi:10.1016/j.neuropharm.2010.03.011
Return to citation in text: [1] -
Aytan, N.; Choi, J.-K.; Carreras, I.; Kowall, N. W.; Jenkins, B. G.; Dedeoglu, A. Exp. Neurol. 2013, 250, 228–238. doi:10.1016/j.expneurol.2013.10.001
Return to citation in text: [1] -
Fenili, D.; Brown, M.; Rappaport, R.; McLaurin, J. J. Mol. Med. (Heidelberg, Ger.) 2007, 85, 603–611. doi:10.1007/s00109-007-0156-7
Return to citation in text: [1] -
Fenili, D.; Keran, M. A.; Mclaurin, J. scyllo-Inositol: A Potential Therapeutic for Alzheimer’s Disease. In Emerging Drugs and Targets for Alzheimer’s Disease, Vol. 2: Neuronal Plasticity, Neuronal Protection and Other Miscellaneous Strategies; Martinez, A., Ed.; RSC Drug Discovery Series; Royal Society of Chemistry: UK, 2010; pp 94–116.
Return to citation in text: [1] -
Salloway, S.; Sperling, R.; Keren, R.; Porsteinsson, A. P.; van Dyck, C. H.; Tariot, P. N.; Gilman, S.; Arnold, D.; Abushakra, S.; Hernandez, C.; Crans, G.; Liang, E.; Quinn, G.; Bairu, M.; Pastrak, A.; Cedarbaum, J. M. Neurology 2011, 77, 1253–1262. doi:10.1212/WNL.0b013e3182309fa5
Return to citation in text: [1] -
Ma, K.; Thomason, L. A. M.; McLaurin, J. Adv. Pharmacol. (San Diego, CA, U. S.) 2012, 64, 177–212. doi:10.1016/B978-0-12-394816-8.00006-4
Return to citation in text: [1] -
Vasdev, N.; Chio, J.; van Oosten, E. M.; Nitz, M.; McLaurin, J.; Vines, D. C.; Houle, S.; Reilly, R. M.; Wilson, A. A. Chem. Commun. 2009, 5527–5529. doi:10.1039/b913317h
Return to citation in text: [1] -
Carroll, L.; Perumal, M.; Vasdev, N.; Robins, E.; Aboagye, E. O. Bioorg. Med. Chem. Lett. 2012, 22, 6148–6150. doi:10.1016/j.bmcl.2012.08.022
Return to citation in text: [1] [2] -
McLarty, K.; Moran, M. D.; Scollard, D. A.; Chan, C.; Sabha, N.; Mukherjee, J.; Guha, A.; McLaurin, J.; Nitz, M.; Houle, S.; Wilson, A. A.; Reilly, R. M.; Vasdev, N. Nucl. Med. Biol. 2011, 38, 953–959. doi:10.1016/j.nucmedbio.2011.02.017
Return to citation in text: [1] -
Hiyama, T. In Organofluorine Compounds – Chemistry and Applications; Yamamoto, H., Ed.; Springer Verlag: Berlin, Heidelberg, 2000.
Return to citation in text: [1] -
Yokohama, M. Carbohydr. Res. 2000, 327, 5–14. doi:10.1016/S0008-6215(99)00324-9
And references cited therein.
Return to citation in text: [1] -
Toshima, K. Carbohydr. Res. 2000, 327, 15–26. doi:10.1016/S0008-6215(99)00325-0
And references cited therein.
Return to citation in text: [1] -
Dax, K.; Albert, M.; Ortner, J.; Paul, B. J. Carbohydr. Res. 2000, 327, 47–86. doi:10.1016/S0008-6215(00)00022-7
And references cited therein.
Return to citation in text: [1] -
Plantier-Royon, R.; Portelle, C. Carbohydr. Res. 2000, 327, 119–146. doi:10.1016/S0008-6215(00)00034-3
And references cited therein.
Return to citation in text: [1] -
Hagmann, W. K. J. Med. Chem. 2008, 51, 4359–4369. doi:10.1021/jm800219f
And references cited therein.
Return to citation in text: [1] -
Lowe, G.; McPhee, F. J. Chem. Soc., Perkin Trans. 1 1991, 1249–1253. doi:10.1039/P19910001249
Return to citation in text: [1] -
Chapleur, Y.; Lamande-Langle, S.; Collet, C.; Chretien, F. Radiotracer, in particular for Alzheimer's disease. WO Patent WO2013/150117A1, Oct 10, 2013.
Return to citation in text: [1] -
Jagdhane, R. C.; Patil, M. T.; Krishnaswamy, S.; Shashidhar, M. S. Tetrahedron 2013, 69, 5144–5151. doi:10.1016/j.tet.2013.04.081
Return to citation in text: [1] -
Koto, S.; Hirooka, M.; Yoshida, T.; Takenaka, K.; Asai, C.; Nagamitsu, T.; Sakuma, H.; Sakurai, M.; Masuzawa, S.; Komiya, M.; Sato, T.; Zen, S.; Yago, K.; Tomonaga, F. Bull. Chem. Soc. Jpn. 2000, 73, 2521–2529. doi:10.1246/bcsj.73.2521
Return to citation in text: [1] -
Vishwakarma, R. A.; Vehring, S.; Mehta, A.; Sinha, A.; Pomorski, T.; Herrmann, A.; Menon, A. K. Org. Biomol. Chem. 2005, 3, 1275–1283. doi:10.1039/b500300h
Return to citation in text: [1] [2] -
Eguchi, T.; Sasaki, S.; Huang, Z.; Kakinuma, K. J. Org. Chem. 2002, 67, 3979–3984. doi:10.1021/jo011107n
Return to citation in text: [1] [2] [3] -
Zhang, F.; Zhang, W.; Zhang, Y.; Curran, D. P.; Liu, G. J. Org. Chem. 2009, 74, 2594–2597. doi:10.1021/jo9000993
Return to citation in text: [1] [2] [3] -
Kováč, P. Carbohydr. Res. 1986, 153, 168–170. doi:10.1016/S0008-6215(00)90209-X
Return to citation in text: [1] [2] [3] -
Zemplén, G.; Pacsu, E. Chem. Ber. 1929, 62B, 1613–1614. doi:10.1002/cber.19290620640
Return to citation in text: [1] [2] [3] -
Kiely, D. E.; Cantrell, C. E. Carbohydr. Res. 1972, 23, 155–157. doi:10.1016/S0008-6215(00)81589-X
Return to citation in text: [1] -
Sarmah, M. P.; Shashidhar, M. S.; Sureshan, K. M.; Gonnade, R. G.; Bhadbhade, M. M. Tetrahedron 2005, 61, 4437–4446. doi:10.1016/j.tet.2005.02.073
Return to citation in text: [1] -
Gula, M. J.; Vitale, D. E.; Dostal, J. M.; Trometer, J. D.; Spencer, T. A. J. Am. Chem. Soc. 1988, 110, 4400–4405. doi:10.1021/ja00221a047
Return to citation in text: [1] [2] [3] -
Vogel, P.; Figueira, S.; Muthukrishnan, S.; Mack, J. Tetrahedron Lett. 2009, 50, 55–56. doi:10.1016/j.tetlet.2008.10.079
Return to citation in text: [1] [2] -
Kim, K.-Y.; Kim, B. C.; Lee, H. B.; Shin, H. J. Org. Chem. 2008, 73, 8106–8108. doi:10.1021/jo8015659
Return to citation in text: [1] -
Yang, S. S.; Min, J. M.; Beattie, T. R. Synth. Commun. 1988, 18, 899–905. doi:10.1080/00397918808060873
Return to citation in text: [1]
| 53. | Vogel, P.; Figueira, S.; Muthukrishnan, S.; Mack, J. Tetrahedron Lett. 2009, 50, 55–56. doi:10.1016/j.tetlet.2008.10.079 |
| 54. | Kim, K.-Y.; Kim, B. C.; Lee, H. B.; Shin, H. J. Org. Chem. 2008, 73, 8106–8108. doi:10.1021/jo8015659 |
| 55. | Yang, S. S.; Min, J. M.; Beattie, T. R. Synth. Commun. 1988, 18, 899–905. doi:10.1080/00397918808060873 |
| 1. | Posternak, T. The cyclitols; Holden-Day: San Francisco, 1962. |
| 2. | Anderson, L. The Cyclitols. In The Carbohydrates, 2nd ed.; Pigman, W.; Horton, D., Eds.; Academic Press: San Diego, 1972; Vol. 1A, pp 519–579. |
| 20. | McLaurin, J.; Golomb, R.; Jurewicz, A.; Antel, J. P.; Fraser, P. E. J. Biol. Chem. 2000, 275, 18495–18502. doi:10.1074/jbc.M906994199 |
| 21. | Nitz, M.; Fenili, D.; Darabie, A. A.; Wu, L.; Cousins, J. E.; McLaurin, J. FEBS J. 2008, 275, 1663–1674. doi:10.1111/j.1742-4658.2008.06321.x |
| 22. | Hawkes, C. A.; Ng, V.; McLaurin, J. Drug Dev. Res. 2009, 70, 111–124. doi:10.1002/ddr.20290 |
| 23. | Sun, Y.; Zhang, G.; Hawkes, C. A.; Shaw, J. E.; McLaurin, J.; Nitz, M. Bioorg. Med. Chem. 2008, 16, 7177–7184. doi:10.1016/j.bmc.2008.06.045 |
| 44. | Koto, S.; Hirooka, M.; Yoshida, T.; Takenaka, K.; Asai, C.; Nagamitsu, T.; Sakuma, H.; Sakurai, M.; Masuzawa, S.; Komiya, M.; Sato, T.; Zen, S.; Yago, K.; Tomonaga, F. Bull. Chem. Soc. Jpn. 2000, 73, 2521–2529. doi:10.1246/bcsj.73.2521 |
| 49. | Zemplén, G.; Pacsu, E. Chem. Ber. 1929, 62B, 1613–1614. doi:10.1002/cber.19290620640 |
| 6. | Bleasdale, J. E.; Eichberg, J.; Hauser, G., Eds. Inositol and Phophoinositides; Humana: Clifton, NJ, 1985. |
| 7. | Billington, D. C. The inositol Phosphates. Chemical Synthesis and Biological Significance; Wiley VCH: New York, 1993. |
| 8. | Dalko, O. P.; Sinay, P. In Organic Synthesis Highlights V; Schmalz, H. G.; Wirth, T., Eds.; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2003; pp 1–14. |
| 9. | Michell, R. H.; Drumond, A. H.; Downes, C. P., Eds. Inositol Lipids in Cell Signalling; Academic Press: San Diego, 1989. |
| 10. | Schedler, D. J. A.; Baker, D. C. Carbohydr. Res. 2004, 339, 1585–1595. doi:10.1016/j.carres.2004.03.030 |
| 11. | Roussel, F.; Moitessier, N.; Hilly, M.; Chrétien, F.; Mauger, J.-P.; Chapleur, Y. Bioorg. Med. Chem. 2002, 10, 759–768. doi:10.1016/S0968-0896(01)00329-7 |
| 12. | Bennett, M.; Onnebo, S. M. N.; Azevedo, C.; Saiardi, A. Cell. Mol. Life Sci. 2006, 63, 552–564. doi:10.1007/s00018-005-5446-z |
| 13. | Barker, C. J.; Illies, C.; Gaboardi, G. C.; Berggren, P.-O. Cell. Mol. Life Sci. 2009, 66, 3851–3871. doi:10.1007/s00018-009-0115-2 |
| 14. | Roussel, F.; Hilly, M.; Chrétien, F.; Mauger, J.-P.; Chapleur, Y. J. Carbohydr. Chem. 1999, 18, 697–707. doi:10.1080/07328309908544031 |
| 15. | Hilton, J. M.; Plomann, M.; Ritter, B.; Modregger, J.; Freeman, H. N.; Falck, J. R.; Krishna, U. M.; Tobin, A. B. J. Biol. Chem. 2001, 276, 16341–16347. doi:10.1074/jbc.M011122200 |
| 16. | Tsui, M. M.; York, J. D. Adv. Enzyme Regul. 2010, 50, 324–337. doi:10.1016/j.advenzreg.2009.12.002 |
| 17. | Vajanaphanich, M.; Schultz, C.; Rudolf, M. T.; Wasserman, M.; Enyedi, P.; Craxton, A.; Shears, S. B.; Tsien, R. Y.; Barrett, K. E.; Traynor-Kaplan, A. Nature 1994, 371, 711–714. doi:10.1038/371711a0 |
| 18. | Shi, Y.; Azab, A. N.; Thompson, M. N.; Greenberg, M. L. Biology of Inositol phosphates and phosphoinositides. In Subcellular Biochemistry; Majumder, A. L.; Biswas, B. B., Eds.; Springer Verlag: USA, 2006; Vol. 39, pp 265–292. |
| 19. | Chen, W.; Deng, Z.; Chen, K.; Dou, D.; Song, F.; Li, L.; Xi, Z. Eur. J. Med. Chem. 2015, 93, 172–181. doi:10.1016/j.ejmech.2015.01.064 |
| 45. | Vishwakarma, R. A.; Vehring, S.; Mehta, A.; Sinha, A.; Pomorski, T.; Herrmann, A.; Menon, A. K. Org. Biomol. Chem. 2005, 3, 1275–1283. doi:10.1039/b500300h |
| 5. |
McConville, M. J.; Ferguson, M. A. Biochem. J. 1993, 294, 305–324. doi:10.1042/bj2940305
See for a comprehensive review on structure and biosynthesis of GPI anchors. |
| 42. | Chapleur, Y.; Lamande-Langle, S.; Collet, C.; Chretien, F. Radiotracer, in particular for Alzheimer's disease. WO Patent WO2013/150117A1, Oct 10, 2013. |
| 47. | Zhang, F.; Zhang, W.; Zhang, Y.; Curran, D. P.; Liu, G. J. Org. Chem. 2009, 74, 2594–2597. doi:10.1021/jo9000993 |
| 3. |
Kılbaş, B.; Balci, M. Tetrahedron 2011, 67, 2355–2389. doi:10.1016/j.tet.2011.01.012
And references cited therein. |
| 4. |
Potter, B. V. L.; Lampe, D. Angew. Chem., Int. Ed. Engl. 1995, 34, 1933–1972. doi:10.1002/anie.199519331
And references cited therein. |
| 43. | Jagdhane, R. C.; Patil, M. T.; Krishnaswamy, S.; Shashidhar, M. S. Tetrahedron 2013, 69, 5144–5151. doi:10.1016/j.tet.2013.04.081 |
| 48. | Kováč, P. Carbohydr. Res. 1986, 153, 168–170. doi:10.1016/S0008-6215(00)90209-X |
| 33. | Carroll, L.; Perumal, M.; Vasdev, N.; Robins, E.; Aboagye, E. O. Bioorg. Med. Chem. Lett. 2012, 22, 6148–6150. doi:10.1016/j.bmcl.2012.08.022 |
| 35. | Hiyama, T. In Organofluorine Compounds – Chemistry and Applications; Yamamoto, H., Ed.; Springer Verlag: Berlin, Heidelberg, 2000. |
| 36. |
Yokohama, M. Carbohydr. Res. 2000, 327, 5–14. doi:10.1016/S0008-6215(99)00324-9
And references cited therein. |
| 37. |
Toshima, K. Carbohydr. Res. 2000, 327, 15–26. doi:10.1016/S0008-6215(99)00325-0
And references cited therein. |
| 38. |
Dax, K.; Albert, M.; Ortner, J.; Paul, B. J. Carbohydr. Res. 2000, 327, 47–86. doi:10.1016/S0008-6215(00)00022-7
And references cited therein. |
| 39. |
Plantier-Royon, R.; Portelle, C. Carbohydr. Res. 2000, 327, 119–146. doi:10.1016/S0008-6215(00)00034-3
And references cited therein. |
| 40. |
Hagmann, W. K. J. Med. Chem. 2008, 51, 4359–4369. doi:10.1021/jm800219f
And references cited therein. |
| 46. | Eguchi, T.; Sasaki, S.; Huang, Z.; Kakinuma, K. J. Org. Chem. 2002, 67, 3979–3984. doi:10.1021/jo011107n |
| 32. | Vasdev, N.; Chio, J.; van Oosten, E. M.; Nitz, M.; McLaurin, J.; Vines, D. C.; Houle, S.; Reilly, R. M.; Wilson, A. A. Chem. Commun. 2009, 5527–5529. doi:10.1039/b913317h |
| 41. | Lowe, G.; McPhee, F. J. Chem. Soc., Perkin Trans. 1 1991, 1249–1253. doi:10.1039/P19910001249 |
| 52. | Gula, M. J.; Vitale, D. E.; Dostal, J. M.; Trometer, J. D.; Spencer, T. A. J. Am. Chem. Soc. 1988, 110, 4400–4405. doi:10.1021/ja00221a047 |
| 30. | Salloway, S.; Sperling, R.; Keren, R.; Porsteinsson, A. P.; van Dyck, C. H.; Tariot, P. N.; Gilman, S.; Arnold, D.; Abushakra, S.; Hernandez, C.; Crans, G.; Liang, E.; Quinn, G.; Bairu, M.; Pastrak, A.; Cedarbaum, J. M. Neurology 2011, 77, 1253–1262. doi:10.1212/WNL.0b013e3182309fa5 |
| 31. | Ma, K.; Thomason, L. A. M.; McLaurin, J. Adv. Pharmacol. (San Diego, CA, U. S.) 2012, 64, 177–212. doi:10.1016/B978-0-12-394816-8.00006-4 |
| 53. | Vogel, P.; Figueira, S.; Muthukrishnan, S.; Mack, J. Tetrahedron Lett. 2009, 50, 55–56. doi:10.1016/j.tetlet.2008.10.079 |
| 24. | McLaurin, J.; Kierstead, M. E.; Brown, M. E.; Hawkes, C. A.; Lambermon, M. H.; Phinney, A. L.; Darabie, A. A.; Cousins, J. E.; French, J. E.; Lan, M. F.; Chen, F.; Wong, S. S. N.; Mount, H. T. J.; Fraser, P. E.; Westaway, D.; St George-Hyslop, P. Nat. Med. 2006, 12, 801–808. doi:10.1038/nm1423 |
| 25. | Townsend, M.; Cleary, J. P.; Mehta, T.; Hofmeister, J.; Lesne, S.; O'Hare, E.; Walsh, D. M.; Selkoe, D. J. Ann. Neurol. 2006, 60, 668–676. doi:10.1002/ana.21051 |
| 26. | Choi, J.-K.; Carreras, I.; Dedeoglu, A.; Jenkins, B. G. Neuropharmacology 2010, 59, 353–357. doi:10.1016/j.neuropharm.2010.03.011 |
| 27. | Aytan, N.; Choi, J.-K.; Carreras, I.; Kowall, N. W.; Jenkins, B. G.; Dedeoglu, A. Exp. Neurol. 2013, 250, 228–238. doi:10.1016/j.expneurol.2013.10.001 |
| 28. | Fenili, D.; Brown, M.; Rappaport, R.; McLaurin, J. J. Mol. Med. (Heidelberg, Ger.) 2007, 85, 603–611. doi:10.1007/s00109-007-0156-7 |
| 29. | Fenili, D.; Keran, M. A.; Mclaurin, J. scyllo-Inositol: A Potential Therapeutic for Alzheimer’s Disease. In Emerging Drugs and Targets for Alzheimer’s Disease, Vol. 2: Neuronal Plasticity, Neuronal Protection and Other Miscellaneous Strategies; Martinez, A., Ed.; RSC Drug Discovery Series; Royal Society of Chemistry: UK, 2010; pp 94–116. |
| 33. | Carroll, L.; Perumal, M.; Vasdev, N.; Robins, E.; Aboagye, E. O. Bioorg. Med. Chem. Lett. 2012, 22, 6148–6150. doi:10.1016/j.bmcl.2012.08.022 |
| 34. | McLarty, K.; Moran, M. D.; Scollard, D. A.; Chan, C.; Sabha, N.; Mukherjee, J.; Guha, A.; McLaurin, J.; Nitz, M.; Houle, S.; Wilson, A. A.; Reilly, R. M.; Vasdev, N. Nucl. Med. Biol. 2011, 38, 953–959. doi:10.1016/j.nucmedbio.2011.02.017 |
| 49. | Zemplén, G.; Pacsu, E. Chem. Ber. 1929, 62B, 1613–1614. doi:10.1002/cber.19290620640 |
| 48. | Kováč, P. Carbohydr. Res. 1986, 153, 168–170. doi:10.1016/S0008-6215(00)90209-X |
| 46. | Eguchi, T.; Sasaki, S.; Huang, Z.; Kakinuma, K. J. Org. Chem. 2002, 67, 3979–3984. doi:10.1021/jo011107n |
| 47. | Zhang, F.; Zhang, W.; Zhang, Y.; Curran, D. P.; Liu, G. J. Org. Chem. 2009, 74, 2594–2597. doi:10.1021/jo9000993 |
| 48. | Kováč, P. Carbohydr. Res. 1986, 153, 168–170. doi:10.1016/S0008-6215(00)90209-X |
| 52. | Gula, M. J.; Vitale, D. E.; Dostal, J. M.; Trometer, J. D.; Spencer, T. A. J. Am. Chem. Soc. 1988, 110, 4400–4405. doi:10.1021/ja00221a047 |
| 52. | Gula, M. J.; Vitale, D. E.; Dostal, J. M.; Trometer, J. D.; Spencer, T. A. J. Am. Chem. Soc. 1988, 110, 4400–4405. doi:10.1021/ja00221a047 |
| 47. | Zhang, F.; Zhang, W.; Zhang, Y.; Curran, D. P.; Liu, G. J. Org. Chem. 2009, 74, 2594–2597. doi:10.1021/jo9000993 |
| 46. | Eguchi, T.; Sasaki, S.; Huang, Z.; Kakinuma, K. J. Org. Chem. 2002, 67, 3979–3984. doi:10.1021/jo011107n |
| 50. | Kiely, D. E.; Cantrell, C. E. Carbohydr. Res. 1972, 23, 155–157. doi:10.1016/S0008-6215(00)81589-X |
| 51. | Sarmah, M. P.; Shashidhar, M. S.; Sureshan, K. M.; Gonnade, R. G.; Bhadbhade, M. M. Tetrahedron 2005, 61, 4437–4446. doi:10.1016/j.tet.2005.02.073 |
| 49. | Zemplén, G.; Pacsu, E. Chem. Ber. 1929, 62B, 1613–1614. doi:10.1002/cber.19290620640 |
| 45. | Vishwakarma, R. A.; Vehring, S.; Mehta, A.; Sinha, A.; Pomorski, T.; Herrmann, A.; Menon, A. K. Org. Biomol. Chem. 2005, 3, 1275–1283. doi:10.1039/b500300h |
© 2016 Collet et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)