Abstract
Dibromobenzoisofuranone 12, synthesized in six steps, was regiospecifically annulated with 5-substituted cyclohexenones 13/36 in the presence of LiOt-Bu to give brominated anthraquinones 14/38 in good yields. Darzens condensation of 30 was shown to give chain-elongated anthraquinone 32. Alkaline hydrolysis of 38 furnished 39 representing desulfoproisocrinin F.

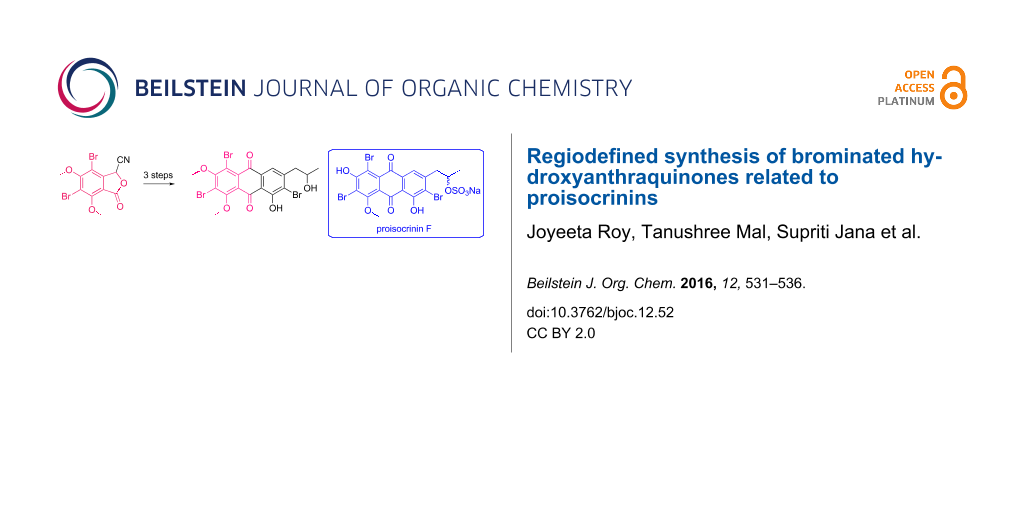
Graphical Abstract
Introduction
Anthraquinones constitute the largest group of naturally occurring quinones [1-5]. Isolated mainly from fungal sources, they display a wide range of biological activities which include anti-inflammatory, antifungal, antiparasidal, and cytotoxic properties [6-11]. Anthraquinones are well-known as colorants in foods, drugs, and textile industries. They are also used as chemical sensors and liquid crystals [1-5]. Halogenated anthraquinones form a minor group of natural pigments [12-15]. 7-Bromoemodic acid (1), isolated from the crinoid Holopus rangii, shows remarkable cytotoxic activities. Topopyrone B (2) stabilizes DNA topoisomerase I and DNA topoisomerase II. Haloemodin (3) acts as an antibacterial agent inhibiting DNA gyrase and bacterial topoisomerase I. 6-O-Methyl-7-chloroaveratin (4) displays potent inhibitory activity against human tumor cell lines SF-268, MCF-7, and NCI-H460, with IC50 values of 7.11, 6.64, and 7.42 μM, respectively [12]. Proisocrinins A–F (6–11), recently isolated from the stalked crinoid Proisocrinus ruberrimus (Figure 1) are the first water soluble natural anthraquinone pigments, and show promising antifeedant properties [16].
![[1860-5397-12-52-1]](/bjoc/content/figures/1860-5397-12-52-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
A brief literature survey revealed that the routes for the synthesis of anthraquinones are primarily based upon five categories, such as Friedel–Crafts reactions, Hauser annulations, Diels–Alder reactions, transition metal-mediated reactions and biomimetic aldol condensations [17-23], and reports on the synthesis of brominated anthraquinones are scare [12-15]. Having inspired by the convergence and the regiochemical integrity of the Hauser annulation [24-30], we explored it for the construction of the bromoanthraquinone scaffolds of proisocrinins 6–11.
Results and Discussion
First synthetic route
Anthraquinone 14 was proposed to be synthesized by the Hauser annulation of cyanophthalide 12 and cyclohexenone 13 (Scheme 1). A functional group manipulation of 14 was expected to give anthraquinone carboxyaldehyde 15. Employment of a Darzens condensation followed by bromination was considered for further elaboration of 15 to 16.
![[1860-5397-12-52-i1]](/bjoc/content/inline/1860-5397-12-52-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Initially proposed synthetic scheme for proisocrinins 6–11.
Scheme 1: Initially proposed synthetic scheme for proisocrinins 6–11.
For the synthesis of key synthon 12 (Scheme 2), we started from cyclohexenone 19, which was prepared by base-catalyzed condensation of methyl acetoacetate with methyl crotonate [24-30]. It was then treated with bromine in AcOH to afford 3,5-dibromoorsellinate 20 in 81% yield [31-33]. Subsequent O-methylation of 20 (using CH3I, K2CO3), and benzylic bromination of 21 with NBS followed by lactonization of 22 in a refluxing mixture of dioxane and water afforded phthalide 23 in 61% yield. NBS bromination of 23 afforded the 3-bromophthalide [33], which on treatment with dioxane/water furnished phthalaldehydic acid 24 in 69% yield over two steps [34]. Treatment of 24 with KCN furnished 3-cyanophathalide 12 in 75% yield analogously as described in references [35-37]. The structure of phthalide 12 was confirmed by the appearance of a singlet at δ 5.84 (s, 1H) in the 1H NMR spectrum and the appearance of a characteristic band for the C≡N stretching frequency at 2260 cm−1 in the IR spectrum. The characteristic carbon for the cyano functionality appeared at δ 111.7 ppm in the 13C NMR spectrum.
![[1860-5397-12-52-i2]](/bjoc/content/inline/1860-5397-12-52-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of cyanophthalide 12.
Scheme 2: Synthesis of cyanophthalide 12.
The Michael acceptor 13 was prepared according to the literature procedure starting from cyclohex-3-enecarbaldehyde (25) [38]. The diol 26 was oxidized with activated MnO2, leading to selective oxidation of the secondary alcohol forming 27 in 83% yield. The cyclohexenone 27 was acetylated with acetyl chloride and pyridine to furnish 13 as an oil in 62% yield (Scheme 3).
![[1860-5397-12-52-i3]](/bjoc/content/inline/1860-5397-12-52-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
In the next stage, Hauser annulation of cyanophthalide 12 with cyclohexenone 13 was carried out in the presence of LiOt-Bu (LTB) in THF at −60 °C to furnish quinol A [39-42]. Due to its sensitivity to aerial oxidation; it was directly aromatized by bubbling O2 through its DMF solution to give anthraquinone 28 in the manner described in [43]. The acetate group in 28 was cleaved with an aqueous alkaline solution to furnish 29 in 80% yield. The alcohol 29 was oxidized to the corresponding aldehyde 30 using PCC in dichloroethane. It was derivatized to its MOM derivative 31 using MOMCl and DIEPA in DCM. Darzens glycidic ester condensation of 31 with methyl 2-chloroacetate and sodium methoxide in methanol (Scheme 4) afforded the desired epoxide 32 [44]. The epoxide 32 was characterized by the signals corresponding to two protons of the epoxide at δ 4.18 and 3.54 [44]. Since the yield of 32 was low, we considered a Horner–Wadsworth–Emmons reaction of aldehyde 31 with triethyl phosphonoacetate as an alternative. Unfortunately, it was not successful, probably due to the interference of the anthraquinone moiety in 31.
![[1860-5397-12-52-i4]](/bjoc/content/inline/1860-5397-12-52-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Darzens condensation route to proisocrinins.
Scheme 4: Darzens condensation route to proisocrinins.
Second synthetic route
Keeping in view the problems of functionalization of the aldehyde group in 31, we contemplated the use of already homologated cyclohexenone 36 as the acceptor. Bicyclic lactone 33 [45] was treated with DIBAL-H to afford lactol 34 in 85% yield [46]. Treatment of lactol 34 with methylmagnesium bromide afforded diol 35 in 72% yield. Selective oxidation of the allylic alcohol group in 35 with MnO2, followed by acetylation of the secondary hydroxy group with acetyl chloride, triethylamine and DMAP furnished cyclohexenone 36 (Scheme 5).
![[1860-5397-12-52-i5]](/bjoc/content/inline/1860-5397-12-52-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
The Hauser annulation of cyanophthalide 12 with acceptor 36 formed hydroquinone 37, which was directly treated with bromine in DCM to give tribrominated quinone 38 in 58% yield (over two steps) (Scheme 6). The structure of bromo compound 38 was proposed on the basis of the high chemical shift (δ = 7.63 ppm) of the proton attached to the C-4 carbon of the anthraquinone, and its comparison with that in similar structural analogs [47,48]. All attempts to demethylate 38 with BBr3 or HBr failed to give the monomethyl analog of 38 [49-52]. The acetate 38 was treated with sodium hydroxide in THF/water (1:1) to give tribromoanthraquinone 39.
![[1860-5397-12-52-i6]](/bjoc/content/inline/1860-5397-12-52-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Synthesis of the proisocrinin core structure.
Scheme 6: Synthesis of the proisocrinin core structure.
Conclusion
The Hauser annulation of a dibromophthalide with 5-(2-acetoxypropyl)cyclohexenone has been shown to provide a regiospecific route to the scaffold of proisocrinin F. Further studies on the completion of the synthesis of proisocrinins 6–11 are underway.
Supporting Information
| Supporting Information File 1: Detailed experimental procedures, characterization data and copies of 1H and 13C NMR for all new compounds. | ||
| Format: PDF | Size: 1.8 MB | Download |
References
-
Dollendorf, C.; Kreth, S. K.; Choi, S. W.; Ritter, H. Beilstein J. Org. Chem. 2013, 9, 453–459. doi:10.3762/bjoc.9.48
Return to citation in text: [1] [2] -
Park, S.; Park, J.; Lee, S.; Park, J. J. Nanosci. Nanotechnol. 2014, 14, 6435–6437. doi:10.1166/jnn.2014.8809
Return to citation in text: [1] [2] -
Mapari, S. A. S.; Nielsen, K. F.; Larsen, T. O.; Frisvad, J. C.; Meyer, A. S.; Thrane, U. Curr. Opin. Biotechnol. 2005, 16, 231–238. doi:10.1016/j.copbio.2005.03.004
Return to citation in text: [1] [2] -
Shin, M.-G.; Kim, S. O.; Park, H. T.; Park, S. J.; Yu, H. S.; Kim, Y.-H.; Kwon, S.-K. Dyes Pigm. 2012, 92, 1075–1082. doi:10.1016/j.dyepig.2011.03.002
Return to citation in text: [1] [2] -
Tietze, L. F.; Gericke, K. M.; Schuberth, I. Eur. J. Org. Chem. 2007, 4563–4577. doi:10.1002/ejoc.200700418
Return to citation in text: [1] [2] -
Hu, Y.; Martinez, E. D.; MacMillan, J. B. J. Nat. Prod. 2012, 75, 1759–1764. doi:10.1021/np3004326
Return to citation in text: [1] -
Sturdy, M.; Krunic, A.; Cho, S.; Franzblau, S.; Orjala, J. J. Nat. Prod. 2010, 73, 1441–1443. doi:10.1021/np100299v
Return to citation in text: [1] -
Batista, R. M. F.; Oliveira, E.; Costa, S. P. G.; Lodeiro, C.; Raposo, M. M. M. Org. Lett. 2007, 9, 3201–3204. doi:10.1021/ol071029b
Return to citation in text: [1] -
Kalogerakis, A.; Groth, U. Org. Lett. 2003, 5, 843–844. doi:10.1021/ol0274920
Return to citation in text: [1] -
Abou-Elkhair, R. A. I.; Dixon, D. W.; Netzel, T. L. J. Org. Chem. 2009, 74, 4712–4719. doi:10.1021/jo900306g
Return to citation in text: [1] -
Akar, K. B.; Cakmak, O.; Büyükgüngör, O.; Sahin, E. Beilstein J. Org. Chem. 2011, 7, 1036–1045. doi:10.3762/bjoc.7.118
Return to citation in text: [1] -
Huang, H.; Wang, F.; Luo, M.; Chen, Y.; Song, Y.; Zhang, W.; Zhang, S.; Ju, J. J. Nat. Prod. 2012, 75, 1346–1352. doi:10.1021/np3002699
Return to citation in text: [1] [2] [3] -
Zaleski, P. A.; Maini, R.; Leiris, S. J.; Elban, M. A.; Hecht, S. M. J. Nat. Prod. 2012, 75, 577–585. doi:10.1021/np200777z
Return to citation in text: [1] [2] -
Wangun, H. V. K.; Wood, A.; Fiorilla, C.; Reed, J. K.; McCarthy, P. J.; Wright, A. E. J. Nat. Prod. 2010, 73, 712–715. doi:10.1021/np900526y
Return to citation in text: [1] [2] -
Duan, F.; Li, X.; Cai, S.; Xin, G.; Wang, Y.; Du, D.; He, S.; Huang, B.; Guo, X.; Zhao, H.; Zhang, R.; Ma, L.; Liu, Y.; Du, Q.; Wei, Z.; Xing, Z.; Liang, Y.; Wu, X.; Fan, C.; Ji, C.; Zeng, D.; Chen, Q.; He, Y.; Liu, X.; Huang, W. J. Med. Chem. 2014, 57, 3707–3714. doi:10.1021/jm401685f
Return to citation in text: [1] [2] -
Wolkenstein, K.; Schoefberger, W.; Müller, N.; Oji, T. J. Nat. Prod. 2009, 72, 2036–2039. doi:10.1021/np900171h
Return to citation in text: [1] -
Krohn, K. Angew. Chem., Int. Ed. Engl. 1986, 25, 790–807. doi:10.1002/anie.198607901
Return to citation in text: [1] -
Krohn, K. Prog. Chem. Org. Nat. Prod. 1989, 55, 37–88.
Return to citation in text: [1] -
Thomas, G. J. In Recent progress in the chemical synthesis of antibiotics and related microbial products; Lukacs, G., Ed.; Springer: Berlin, Heidelberg, 1993; Vol. 2, pp 677–749.
Return to citation in text: [1] -
Kelly, T. R. Annu. Rep. Med. Chem. 1979, 14, 288–298. doi:10.1016/S0065-7743(08)61373-1
Return to citation in text: [1] -
Tapia, R. A.; Venegas, J.; Cantuarias, L. B. Synth. Commun. 2009, 40, 151–156. doi:10.1080/00397910902963421
Return to citation in text: [1] -
Devi, A. R.; Rajaram, S. Synth. Commun. 1999, 29, 591–597. doi:10.1080/00397919908085807
Return to citation in text: [1] -
Kotha, S.; Gunta, R. Beilstein J. Org. Chem. 2015, 11, 1727–1731. doi:10.3762/bjoc.11.188
Return to citation in text: [1] -
Mal, D.; Pahari, P.; De, S. R. Tetrahedron 2007, 63, 11781–11792. doi:10.1016/j.tet.2007.08.048
Return to citation in text: [1] [2] -
Mal, D.; Pahari, P. Chem. Rev. 2007, 107, 1892–1918. doi:10.1021/cr068398q
Return to citation in text: [1] [2] -
Mal, D.; Ray, S.; Sharma, I. J. Org. Chem. 2007, 72, 4981–4984. doi:10.1021/jo062271j
Return to citation in text: [1] [2] -
Naysmith, B. J.; Brimble, M. A. Org. Lett. 2013, 15, 2006–2009. doi:10.1021/ol400686f
Return to citation in text: [1] [2] -
Brimble, M. A.; Hassan, N. P. S.; Naysmith, B. J.; Sperry, J. J. Org. Chem. 2014, 79, 7169–7178. doi:10.1021/jo501344c
Return to citation in text: [1] [2] -
Nicolaou, K. C.; Becker, J.; Lim, Y. H.; Lemire, A.; Neubauer, T.; Montero, A. J. Am. Chem. Soc. 2009, 131, 14812–14826. doi:10.1021/ja9073694
Return to citation in text: [1] [2] -
Mal, D.; Ghosh, K.; Chakraborty, S. Synthesis 2015, 2473–2484. doi:10.1055/s-0034-1380656
Return to citation in text: [1] [2] -
Gramatica, P.; Gianotti, M. P.; Speranza, G.; Manitto, P. Heterocycles 1986, 24, 743–750. doi:10.3987/R-1986-03-0743
Return to citation in text: [1] -
Nouguier, R.; Bertrand, M. P.; Picon, P.; Perfetti, P. Tetrahedron Lett. 1994, 35, 8171–8172. doi:10.1016/0040-4039(94)88274-6
Return to citation in text: [1] -
Allison, W. R.; Newbold, G. T. J. Chem. Soc. 1959, 3335–3340. doi:10.1039/jr9590003335
Return to citation in text: [1] [2] -
Roy, J.; Mal, D. Eur. J. Org. Chem. 2014, 1873–1881. doi:10.1002/ejoc.201301652
Return to citation in text: [1] -
Freskos, J. N.; Morrow, G. W.; Swenton, J. S. J. Org. Chem. 1985, 50, 805–810. doi:10.1021/jo00206a016
Return to citation in text: [1] -
Zhuang, Z.; Hu, Z.-P.; Liao, W.-W. Org. Lett. 2014, 16, 3380–3383. doi:10.1021/ol501427h
Return to citation in text: [1] -
Karmakar, R.; Mal, D. J. Org. Chem. 2012, 77, 10235–10248. doi:10.1021/jo301712b
Return to citation in text: [1] -
Hauser, F. M.; Mal, D. J. Am. Chem. Soc. 1984, 106, 1862–1863. doi:10.1021/ja00318a065
Return to citation in text: [1] -
Pahari, P.; Senapati, B.; Mal, D. Tetrahedron Lett. 2007, 48, 2635–2638. doi:10.1016/j.tetlet.2007.01.159
Return to citation in text: [1] -
Nicolaou, K. C.; Lim, Y. H.; Piper, J. L.; Papageorgiou, C. D. J. Am. Chem. Soc. 2007, 129, 4001–4013. doi:10.1021/ja0685708
Return to citation in text: [1] -
Snider, B. B.; Gao, X. J. Org. Chem. 2005, 70, 6863–6869. doi:10.1021/jo0508898
Return to citation in text: [1] -
Huang, J.-K.; Lauderdale, T.-L. Y.; Shia, K.-S. Org. Lett. 2015, 17, 4248–4251. doi:10.1021/acs.orglett.5b02039
Return to citation in text: [1] -
Senapati, B.; Mal, D. Int. J. Org. Chem. 2015, 5, 63–74. doi:10.4236/ijoc.2015.52008
Return to citation in text: [1] -
Cannon, J. G.; True, C. D.; Long, J. P.; Bhatnagar, R. K.; Leonard, P.; Flynn, J. R. J. Med. Chem. 1989, 32, 2210–2214. doi:10.1021/jm00129a029
Return to citation in text: [1] [2] -
Carroll, F. I.; Abraham, P.; Pitner, J. B.; Jablonski, S. D.; Singh, P.; Kwon, Y. W.; Triggle, D. J. J. Chem. Soc., Chem. Commun. 1992, 795–796. doi:10.1039/c39920000795
Return to citation in text: [1] -
Ibrahim, A. A.; Golonka, A. N.; Lopez, A. M.; Stockdill, J. L. Org. Lett. 2014, 16, 1072–1075. doi:10.1021/ol4034868
Return to citation in text: [1] -
Tietze, L. F.; Gericke, K. M.; Singidi, R. R.; Schuberth, I. Org. Biomol. Chem. 2007, 5, 1191–1200. doi:10.1039/b700838d
Return to citation in text: [1] -
Alexander, J.; Bhatia, A. V.; Mitscher, L. A.; Omoto, S.; Suzuki, T. J. Org. Chem. 1980, 45, 20–24. doi:10.1021/jo01289a004
Return to citation in text: [1] -
Carvalho, C. F.; Sargent, M. V. J. Chem. Soc., Chem. Commun. 1984, 227–229. doi:10.1039/c39840000227
Return to citation in text: [1] -
Wang, Y.-H.; Bailey, J. F.; Petersen, J. L.; Wan, K. K. Beilstein J. Org. Chem. 2011, 7, 496–502. doi:10.3762/bjoc.7.58
Return to citation in text: [1] -
Jones, K.; Roset, X.; Rossiter, S.; Whitfield, P. Org. Biomol. Chem. 2003, 1, 4380–4383. doi:10.1039/b311281k
Return to citation in text: [1] -
Leyva-Pérez, A.; Cómbita-Merchán, D.; Cabrero-Antonino, J. R.; Al-Resayes, S. I.; Corma, A. ACS Catal. 2013, 3, 250–258. doi:10.1021/cs300644s
Return to citation in text: [1]
| 44. | Cannon, J. G.; True, C. D.; Long, J. P.; Bhatnagar, R. K.; Leonard, P.; Flynn, J. R. J. Med. Chem. 1989, 32, 2210–2214. doi:10.1021/jm00129a029 |
| 43. | Senapati, B.; Mal, D. Int. J. Org. Chem. 2015, 5, 63–74. doi:10.4236/ijoc.2015.52008 |
| 44. | Cannon, J. G.; True, C. D.; Long, J. P.; Bhatnagar, R. K.; Leonard, P.; Flynn, J. R. J. Med. Chem. 1989, 32, 2210–2214. doi:10.1021/jm00129a029 |
| 1. | Dollendorf, C.; Kreth, S. K.; Choi, S. W.; Ritter, H. Beilstein J. Org. Chem. 2013, 9, 453–459. doi:10.3762/bjoc.9.48 |
| 2. | Park, S.; Park, J.; Lee, S.; Park, J. J. Nanosci. Nanotechnol. 2014, 14, 6435–6437. doi:10.1166/jnn.2014.8809 |
| 3. | Mapari, S. A. S.; Nielsen, K. F.; Larsen, T. O.; Frisvad, J. C.; Meyer, A. S.; Thrane, U. Curr. Opin. Biotechnol. 2005, 16, 231–238. doi:10.1016/j.copbio.2005.03.004 |
| 4. | Shin, M.-G.; Kim, S. O.; Park, H. T.; Park, S. J.; Yu, H. S.; Kim, Y.-H.; Kwon, S.-K. Dyes Pigm. 2012, 92, 1075–1082. doi:10.1016/j.dyepig.2011.03.002 |
| 5. | Tietze, L. F.; Gericke, K. M.; Schuberth, I. Eur. J. Org. Chem. 2007, 4563–4577. doi:10.1002/ejoc.200700418 |
| 12. | Huang, H.; Wang, F.; Luo, M.; Chen, Y.; Song, Y.; Zhang, W.; Zhang, S.; Ju, J. J. Nat. Prod. 2012, 75, 1346–1352. doi:10.1021/np3002699 |
| 38. | Hauser, F. M.; Mal, D. J. Am. Chem. Soc. 1984, 106, 1862–1863. doi:10.1021/ja00318a065 |
| 12. | Huang, H.; Wang, F.; Luo, M.; Chen, Y.; Song, Y.; Zhang, W.; Zhang, S.; Ju, J. J. Nat. Prod. 2012, 75, 1346–1352. doi:10.1021/np3002699 |
| 13. | Zaleski, P. A.; Maini, R.; Leiris, S. J.; Elban, M. A.; Hecht, S. M. J. Nat. Prod. 2012, 75, 577–585. doi:10.1021/np200777z |
| 14. | Wangun, H. V. K.; Wood, A.; Fiorilla, C.; Reed, J. K.; McCarthy, P. J.; Wright, A. E. J. Nat. Prod. 2010, 73, 712–715. doi:10.1021/np900526y |
| 15. | Duan, F.; Li, X.; Cai, S.; Xin, G.; Wang, Y.; Du, D.; He, S.; Huang, B.; Guo, X.; Zhao, H.; Zhang, R.; Ma, L.; Liu, Y.; Du, Q.; Wei, Z.; Xing, Z.; Liang, Y.; Wu, X.; Fan, C.; Ji, C.; Zeng, D.; Chen, Q.; He, Y.; Liu, X.; Huang, W. J. Med. Chem. 2014, 57, 3707–3714. doi:10.1021/jm401685f |
| 39. | Pahari, P.; Senapati, B.; Mal, D. Tetrahedron Lett. 2007, 48, 2635–2638. doi:10.1016/j.tetlet.2007.01.159 |
| 40. | Nicolaou, K. C.; Lim, Y. H.; Piper, J. L.; Papageorgiou, C. D. J. Am. Chem. Soc. 2007, 129, 4001–4013. doi:10.1021/ja0685708 |
| 41. | Snider, B. B.; Gao, X. J. Org. Chem. 2005, 70, 6863–6869. doi:10.1021/jo0508898 |
| 42. | Huang, J.-K.; Lauderdale, T.-L. Y.; Shia, K.-S. Org. Lett. 2015, 17, 4248–4251. doi:10.1021/acs.orglett.5b02039 |
| 1. | Dollendorf, C.; Kreth, S. K.; Choi, S. W.; Ritter, H. Beilstein J. Org. Chem. 2013, 9, 453–459. doi:10.3762/bjoc.9.48 |
| 2. | Park, S.; Park, J.; Lee, S.; Park, J. J. Nanosci. Nanotechnol. 2014, 14, 6435–6437. doi:10.1166/jnn.2014.8809 |
| 3. | Mapari, S. A. S.; Nielsen, K. F.; Larsen, T. O.; Frisvad, J. C.; Meyer, A. S.; Thrane, U. Curr. Opin. Biotechnol. 2005, 16, 231–238. doi:10.1016/j.copbio.2005.03.004 |
| 4. | Shin, M.-G.; Kim, S. O.; Park, H. T.; Park, S. J.; Yu, H. S.; Kim, Y.-H.; Kwon, S.-K. Dyes Pigm. 2012, 92, 1075–1082. doi:10.1016/j.dyepig.2011.03.002 |
| 5. | Tietze, L. F.; Gericke, K. M.; Schuberth, I. Eur. J. Org. Chem. 2007, 4563–4577. doi:10.1002/ejoc.200700418 |
| 34. | Roy, J.; Mal, D. Eur. J. Org. Chem. 2014, 1873–1881. doi:10.1002/ejoc.201301652 |
| 6. | Hu, Y.; Martinez, E. D.; MacMillan, J. B. J. Nat. Prod. 2012, 75, 1759–1764. doi:10.1021/np3004326 |
| 7. | Sturdy, M.; Krunic, A.; Cho, S.; Franzblau, S.; Orjala, J. J. Nat. Prod. 2010, 73, 1441–1443. doi:10.1021/np100299v |
| 8. | Batista, R. M. F.; Oliveira, E.; Costa, S. P. G.; Lodeiro, C.; Raposo, M. M. M. Org. Lett. 2007, 9, 3201–3204. doi:10.1021/ol071029b |
| 9. | Kalogerakis, A.; Groth, U. Org. Lett. 2003, 5, 843–844. doi:10.1021/ol0274920 |
| 10. | Abou-Elkhair, R. A. I.; Dixon, D. W.; Netzel, T. L. J. Org. Chem. 2009, 74, 4712–4719. doi:10.1021/jo900306g |
| 11. | Akar, K. B.; Cakmak, O.; Büyükgüngör, O.; Sahin, E. Beilstein J. Org. Chem. 2011, 7, 1036–1045. doi:10.3762/bjoc.7.118 |
| 35. | Freskos, J. N.; Morrow, G. W.; Swenton, J. S. J. Org. Chem. 1985, 50, 805–810. doi:10.1021/jo00206a016 |
| 36. | Zhuang, Z.; Hu, Z.-P.; Liao, W.-W. Org. Lett. 2014, 16, 3380–3383. doi:10.1021/ol501427h |
| 37. | Karmakar, R.; Mal, D. J. Org. Chem. 2012, 77, 10235–10248. doi:10.1021/jo301712b |
| 24. | Mal, D.; Pahari, P.; De, S. R. Tetrahedron 2007, 63, 11781–11792. doi:10.1016/j.tet.2007.08.048 |
| 25. | Mal, D.; Pahari, P. Chem. Rev. 2007, 107, 1892–1918. doi:10.1021/cr068398q |
| 26. | Mal, D.; Ray, S.; Sharma, I. J. Org. Chem. 2007, 72, 4981–4984. doi:10.1021/jo062271j |
| 27. | Naysmith, B. J.; Brimble, M. A. Org. Lett. 2013, 15, 2006–2009. doi:10.1021/ol400686f |
| 28. | Brimble, M. A.; Hassan, N. P. S.; Naysmith, B. J.; Sperry, J. J. Org. Chem. 2014, 79, 7169–7178. doi:10.1021/jo501344c |
| 29. | Nicolaou, K. C.; Becker, J.; Lim, Y. H.; Lemire, A.; Neubauer, T.; Montero, A. J. Am. Chem. Soc. 2009, 131, 14812–14826. doi:10.1021/ja9073694 |
| 30. | Mal, D.; Ghosh, K.; Chakraborty, S. Synthesis 2015, 2473–2484. doi:10.1055/s-0034-1380656 |
| 31. | Gramatica, P.; Gianotti, M. P.; Speranza, G.; Manitto, P. Heterocycles 1986, 24, 743–750. doi:10.3987/R-1986-03-0743 |
| 32. | Nouguier, R.; Bertrand, M. P.; Picon, P.; Perfetti, P. Tetrahedron Lett. 1994, 35, 8171–8172. doi:10.1016/0040-4039(94)88274-6 |
| 33. | Allison, W. R.; Newbold, G. T. J. Chem. Soc. 1959, 3335–3340. doi:10.1039/jr9590003335 |
| 47. | Tietze, L. F.; Gericke, K. M.; Singidi, R. R.; Schuberth, I. Org. Biomol. Chem. 2007, 5, 1191–1200. doi:10.1039/b700838d |
| 48. | Alexander, J.; Bhatia, A. V.; Mitscher, L. A.; Omoto, S.; Suzuki, T. J. Org. Chem. 1980, 45, 20–24. doi:10.1021/jo01289a004 |
| 12. | Huang, H.; Wang, F.; Luo, M.; Chen, Y.; Song, Y.; Zhang, W.; Zhang, S.; Ju, J. J. Nat. Prod. 2012, 75, 1346–1352. doi:10.1021/np3002699 |
| 13. | Zaleski, P. A.; Maini, R.; Leiris, S. J.; Elban, M. A.; Hecht, S. M. J. Nat. Prod. 2012, 75, 577–585. doi:10.1021/np200777z |
| 14. | Wangun, H. V. K.; Wood, A.; Fiorilla, C.; Reed, J. K.; McCarthy, P. J.; Wright, A. E. J. Nat. Prod. 2010, 73, 712–715. doi:10.1021/np900526y |
| 15. | Duan, F.; Li, X.; Cai, S.; Xin, G.; Wang, Y.; Du, D.; He, S.; Huang, B.; Guo, X.; Zhao, H.; Zhang, R.; Ma, L.; Liu, Y.; Du, Q.; Wei, Z.; Xing, Z.; Liang, Y.; Wu, X.; Fan, C.; Ji, C.; Zeng, D.; Chen, Q.; He, Y.; Liu, X.; Huang, W. J. Med. Chem. 2014, 57, 3707–3714. doi:10.1021/jm401685f |
| 33. | Allison, W. R.; Newbold, G. T. J. Chem. Soc. 1959, 3335–3340. doi:10.1039/jr9590003335 |
| 49. | Carvalho, C. F.; Sargent, M. V. J. Chem. Soc., Chem. Commun. 1984, 227–229. doi:10.1039/c39840000227 |
| 50. | Wang, Y.-H.; Bailey, J. F.; Petersen, J. L.; Wan, K. K. Beilstein J. Org. Chem. 2011, 7, 496–502. doi:10.3762/bjoc.7.58 |
| 51. | Jones, K.; Roset, X.; Rossiter, S.; Whitfield, P. Org. Biomol. Chem. 2003, 1, 4380–4383. doi:10.1039/b311281k |
| 52. | Leyva-Pérez, A.; Cómbita-Merchán, D.; Cabrero-Antonino, J. R.; Al-Resayes, S. I.; Corma, A. ACS Catal. 2013, 3, 250–258. doi:10.1021/cs300644s |
| 17. | Krohn, K. Angew. Chem., Int. Ed. Engl. 1986, 25, 790–807. doi:10.1002/anie.198607901 |
| 18. | Krohn, K. Prog. Chem. Org. Nat. Prod. 1989, 55, 37–88. |
| 19. | Thomas, G. J. In Recent progress in the chemical synthesis of antibiotics and related microbial products; Lukacs, G., Ed.; Springer: Berlin, Heidelberg, 1993; Vol. 2, pp 677–749. |
| 20. | Kelly, T. R. Annu. Rep. Med. Chem. 1979, 14, 288–298. doi:10.1016/S0065-7743(08)61373-1 |
| 21. | Tapia, R. A.; Venegas, J.; Cantuarias, L. B. Synth. Commun. 2009, 40, 151–156. doi:10.1080/00397910902963421 |
| 22. | Devi, A. R.; Rajaram, S. Synth. Commun. 1999, 29, 591–597. doi:10.1080/00397919908085807 |
| 23. | Kotha, S.; Gunta, R. Beilstein J. Org. Chem. 2015, 11, 1727–1731. doi:10.3762/bjoc.11.188 |
| 45. | Carroll, F. I.; Abraham, P.; Pitner, J. B.; Jablonski, S. D.; Singh, P.; Kwon, Y. W.; Triggle, D. J. J. Chem. Soc., Chem. Commun. 1992, 795–796. doi:10.1039/c39920000795 |
| 16. | Wolkenstein, K.; Schoefberger, W.; Müller, N.; Oji, T. J. Nat. Prod. 2009, 72, 2036–2039. doi:10.1021/np900171h |
| 24. | Mal, D.; Pahari, P.; De, S. R. Tetrahedron 2007, 63, 11781–11792. doi:10.1016/j.tet.2007.08.048 |
| 25. | Mal, D.; Pahari, P. Chem. Rev. 2007, 107, 1892–1918. doi:10.1021/cr068398q |
| 26. | Mal, D.; Ray, S.; Sharma, I. J. Org. Chem. 2007, 72, 4981–4984. doi:10.1021/jo062271j |
| 27. | Naysmith, B. J.; Brimble, M. A. Org. Lett. 2013, 15, 2006–2009. doi:10.1021/ol400686f |
| 28. | Brimble, M. A.; Hassan, N. P. S.; Naysmith, B. J.; Sperry, J. J. Org. Chem. 2014, 79, 7169–7178. doi:10.1021/jo501344c |
| 29. | Nicolaou, K. C.; Becker, J.; Lim, Y. H.; Lemire, A.; Neubauer, T.; Montero, A. J. Am. Chem. Soc. 2009, 131, 14812–14826. doi:10.1021/ja9073694 |
| 30. | Mal, D.; Ghosh, K.; Chakraborty, S. Synthesis 2015, 2473–2484. doi:10.1055/s-0034-1380656 |
| 46. | Ibrahim, A. A.; Golonka, A. N.; Lopez, A. M.; Stockdill, J. L. Org. Lett. 2014, 16, 1072–1075. doi:10.1021/ol4034868 |
© 2016 Roy et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)