Abstract
In our natural product screening program from marine fungi, two new aromatic polyketides karimunones A (1) and B (2) and five known compounds (3–7) were isolated from sponge-associated Fusarium sp. KJMT.FP.4.3 which was collected from an Indonesian sponge Xestospongia sp. The structures of these compounds were determined by the analysis of NMR and MS spectroscopic data. The NMR assignment of 1 was assisted by DFT-based theoretical chemical shift calculation. Compound 2 showed antibacterial activity against multidrug resistant Salmonella enterica ser. Typhi with a MIC of 125 µg/mL while 1 was not active.
Graphical Abstract
Introduction
Marine organisms have been known as a potential source of prospective bioactive compounds, and sponges are particularly emphasized as the most promising source among all marine invertebrates [1,2]. However, the collection of sponges in massive amounts leads to environmental disturbance since marine sponges play a key role in building coral reefs [3,4]. As a filter feeder, sponges host an enormous amount of microorganisms including algae, bacteria, actinomycetes, and fungi [5-7]. Many of these microorganisms produce structurally unique secondary metabolites with various biological activities [8,9]. Specifically, sponge-associated fungi are attracting substantial attention because of their high capability of producing a wide range of bioactive compounds [5,10,11]. As a tropical country, Indonesia is known as the second most prospective country for new natural products from marine resources owing to its high biodiversity [12]. Thus far, sponges have also been utilized as the most productive source of novel compounds in Indonesia. According to the latest review by Hanif et al. [13], ca. 500 marine natural products were obtained from Indonesian sponges from January 1970 to December 2017, while less than 50 compounds were reported from marine fungi during the same period. Furthermore, only a few compounds were isolated from fungal symbionts in Indonesian sponges to date [14,15]. This is strongly indicating that sponge-associated fungi from Indonesia are still underevaluated. Therefore, exploration of novel compounds from fungi associating with Indonesian sponges is currently a major subject in our research group [16-18].
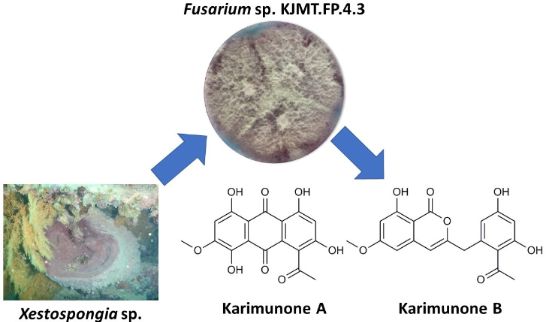
Along this line of study, Fusarium sp. KJMT.FP.4.3 was isolated from a sponge Xestospongia sp. collected in Karimunjawa National Park, Indonesia. This fungus produces a violet pigment in the mycelium and secretes pink to red pigments into the broth medium. Comprehensive chemospectroscopic analysis of the culture extract using HPLC/UV led to the isolation of two new aromatic polyketides, karimunones A (1) and B (2) along with five known compounds, rhodolamprometrin (3) [19], 7-O-methylrhodolamprometrin (4) [20], 6-O-methylSMA93 (5) [21], tricinonoic acid (6) [22], and cyclonerodiol (7) [23] (Figure 1). We herein describe the isolation, structure determination, and biological activity of 1 and 2.
![[1860-5397-15-289-1]](/bjoc/content/figures/1860-5397-15-289-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Results and Discussion
A fungal strain was isolated from a sponge Xestospongia sp. collected in Karimunjawa National Park, Central Java, and identified as a member of Fusarium on the basis of sequence similarity of the internal transcribed spacer (ITS) domain to Fusarium oxysporum strains in the DNA database. HPLC/UV-guided purification of the secondary metabolites from this strain led to the isolation of two new polyketides, karimunones A (1) and B (2), together with five known compounds (3–7, Figure 1).
Compound 1 was obtained as a red powder. TOF-HRESIMS analysis gave a deprotonated molecule [M − H]− at m/z 343.0452 corresponding to a molecular formula of C17H12O8 (calcd for C17H11O8, 343.0459). Combination of 13C NMR and HSQC analytical data revealed the presence of 17 carbons assignable to three carbonyl carbons (δC 179.8, 186.6, and 201.3), twelve aromatic sp2 carbons (five are oxygenated and two are proton-bearing), and two methyl groups (Table 1). In the 1H NMR spectrum, all resonances were observed as a singlet peak and were assigned to two methyls (δH 2.42 and 3.95), two aromatic methines (δH 6.67 and 6.95), and three exchangeable protons (δH 12.57, 12.65, 12.70). The UV spectrum of 1 with the absorption bands at 234, 266, 313, 499, and 533 nm was closely similar to that for 1-acetyl-2,4,5,7,8-pentahydroxyanthraquinone from a fungus Geosmithia [24], suggesting the presence of a common chromophore in 1.
Table 1: NMR spectroscopic data for karimunone A (1) in DMSO-d6.
| position | δC, typea | δH, multb | HMBCc |
| 1 | 126.3, C | ||
| 2 | 161.5, C | ||
| 3 | 108.5, CH | 6.67, s | 1, 2, 4, 4a |
| 4 | 163.8, C | ||
| 4a | 108.5, C | ||
| 5 | 159.1, C | ||
| 6 | 107.9, CH | 6.95, s | 5, 7, 8, 10a |
| 7 | 157.1, C | ||
| 8 | 149.7, C | ||
| 8a | 111.9, C | ||
| 9 | 179.8, C | ||
| 9a | 130.6, C | ||
| 10 | 186.6, C | ||
| 10a | 104.5, C | ||
| 11 | 201.3, C | ||
| 12 | 30.9, CH3 | 2.42, s | 1, 11 |
| 13 | 56.9, CH3 | 3.95, s | 7 |
| 4-OH | 12.57, s | 3, 4, 4a | |
| OH | 12.65, br.s | ||
| OH | 12.70, br.s | ||
aRecorded at 125 MHz (reference δC 39.5). bRecorded at 500 MHz (reference δH 2.50). cHMBC correlations are from proton(s) stated to the indicated carbon.
Two highly substituted benzene rings were assembled by analysing the HMBC correlation data (Table 1, Figure 2). Strong correlations from H3 to C1 and C4a indicated meta-relationships for C1, C3, and C4a, while weak correlations from H3 to C2 and C4 suggested ortho-positioning of C2 and C4 to C3. An acetyl group at C1 was confirmed by the HMBC correlations from H12 to C11 and C1. HMBC correlations from an exchangeable proton at δH 12.57 (4-OH) to C3, C4, and C4a were also supportive of the substitution pattern in this benzene ring. Strong correlations from H6 to C8 and C10a implied the meta-relationship of C8 and C10a to C6, and similarly the ortho-relationship of C5 and C7 to C6 were deduced from the weak correlations from H6 to C5 and C7. In addition, deshielded resonances for C5, C7, and C8 indicated that these carbons were oxygenated. The methyl protons of the methoxy group (H13) had an HMBC correlation to a carbon at δC 157.1 but it was not possible to determine the site of methoxy substitution only from the available HMBC data. Four-bond correlations detected from H3 and H6 to C10 indicated a carbonyl-bridge between C4a and C10a. Although no further long-range correlations were available, two aromatic carbons (C8a, C9a) and one carbonyl carbon (C9) were placed between the two rings to complete the anthraquinone skeleton in consideration of the UV spectrum and the molecular formula, providing two possible structures a and b for 1 (Figure 2): the methoxy group is positioned at C7 in structure a and at C5 in structure b.
![[1860-5397-15-289-2]](/bjoc/content/figures/1860-5397-15-289-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Key HMBC correlations and two possible structures (a and b) for karimunone A (1).
Figure 2: Key HMBC correlations and two possible structures (a and b) for karimunone A (1).
In order to eliminate one of the two possible structures for 1, the experimental chemical shifts of 1 were compared with the NMR chemical shifts calculated for structures a and b at mPW1PW91/6-31G+(d,p)-PCM(DMSO) level of theory. Since only five resonances including one exchangeable phenolic proton were available for 1H chemical shifts, 13C chemical shifts were used for comparison. The theoretical 13C NMR data of structure a showed better agreement with the experimental 13C NMR data of 1. Specifically, the chemical shift difference between the experimental and theoretical values for the carbons spatially close to the methoxy group (C5, C6, C7, C8, C8a, C9, C10, and C10a) were significantly smaller for structure a than structure b (Table 2). Then, DP4+ was used to quantify this calculated result [25]. DP4+ probability analysis provides reliable guidance on the correct structure among several possible isomers. The developers recommend to use the combination of 1H and 13C data for enhancement of DP4+ performance. In this study, the DP4+ probability using only 1H data or only 13C data gave an inconsistent result, but the DP4+ probability using both 1H and 13C data supported the structure a, in which the methoxy group was present at C7, as a correct structure for 1 (100% for structure a and 0.0% for structure b, Table 2).
Table 2: DFT-calculated NMR chemical shifts of two possible structures a and b for karimunone A (1) at the mPW1PW91/6-31G+(d,p)-PCM(DMSO) level.
| karimunone A (1) | structure a | structure b | structure a | structure b | ||||
| position | δC(exp) | δH(exp) | δC(calc) | δH(calc) | δC(calc) | δH(calc) | |δC(exp) – δc(calc)| | |δC(exp) – δc(calc)| |
| 1 | 126.3 | 122.8 | 121.7 | 3.5 | 4.6 | |||
| 2 | 161.5 | 156.7 | 155.6 | 4.8 | 5.9 | |||
| 3 | 108.5 | 6.67 | 107.5 | 6.89 | 108.1 | 6.90 | 1.0 | 0.4 |
| 4 | 163.8 | 162.3 | 162.4 | 1.5 | 1.4 | |||
| 4a | 108.5 | 110.2 | 110.9 | 1.7 | 2.4 | |||
| 5 | 159.1 | 157.1 | 155.2 | 2.0 | 3.9 | |||
| 6 | 107.9 | 6.95 | 106.6 | 6.93 | 104.1 | 7.27 | 1.3 | 3.8 |
| 7 | 157.1 | 155.4 | 151.3 | 1.7 | 5.8 | |||
| 8 | 149.7 | 148.3 | 142.0 | 1.4 | 7.7 | |||
| 8a | 111.9 | 111.9 | 114.7 | 0.0 | 2.8 | |||
| 9 | 179.8 | 183.4 | 185.7 | 3.6 | 5.9 | |||
| 9a | 130.6 | 133.8 | 132.1 | 3.2 | 1.5 | |||
| 10 | 186.6 | 182.8 | 181.1 | 3.8 | 5.5 | |||
| 10a | 104.5 | 104.3 | 109.2 | 0.2 | 4.7 | |||
| 11 | 201.3 | 204.8 | 204.7 | 3.5 | 3.4 | |||
| 12 | 30.9 | 2.42 | 33.7 | 2.58 | 33.8 | 2.58 | 2.8 | 2.9 |
| 13 | 56.9 | 3.95 | 56.6 | 4.08 | 55.9 | 4.05 | 0.3 | 1.0 |
| 4-OH | 12.57 | 12.53* | 13.51* | |||||
| structure a | structure b | |||||||
| DP4+ (1H data) | 1.5% | 98.5% | ||||||
| DP4+ (13C data) | 100.0% | 0.0% | ||||||
| DP4+ (1H and 13C data) | 100.0% | 0.0% | ||||||
*Exchangeable signals are not included for evaluation.
Compound 2 was also isolated as a reddish powder. TOF-HRESIMS analysis gave an [M − H]− ion peak at m/z 355.0823 corresponding to a molecular formula of C19H16O7 (calcd for C19H15O7, 355.0818). The IR spectrum showed a strong absorption band at 1683 cm−1, indicating the presence of carbonyl functionality. The 13C NMR exhibited 19 carbon signals that could be assigned to eleven nonprotonated sp2 carbons (seven are oxygenated), five sp2 methine carbons, one methylene carbon, and two methyl carbons from HSQC spectral data (Table 3). Mutual HMBC correlations between two aromatic methines H5 and H7 and their correlations to C8a established their meta-relationship in a benzene ring system. A methoxy group was placed at C6 on the basis of HMBC correlations from H5, H7, and H10 to C6. A sharp singlet resonance at δH 10.94 was deduced to be a hydrogen-bonded phenolic proton that showed HMBC correlations to C7, C8, and C8a, completing the tetrasubstituted benzene ring with a hydroxy and a methoxy substituent. To this ring were connected a carbonyl carbon C1 at C8a and a three-carbon fragment C4–C3–C9 at C4a by the HMBC correlations from 8-OH to C1 and H4 to C3, C4a, C5, C8a, and C9, respectively. Another tetrasubstituted benzene ring was deduced also from the HMBC analysis. Mutual HMBC correlations of two aromatic protons H4’ and H6’ and their correlations to C2’ revealed the meta-relationship among C2’, C4’ and C6’. Shielded chemical shifts of C2’, C4’, and C6’ and deshielded chemical shifts of C3’ and C5’ suggested the presence of hydroxy groups at C3’ and C5’. HMBC correlations from H12 to C2’ and C11 as well as a highly deshielded carbon chemical shift for C11 (δC 203.5) indicated the acetyl substituent at C2’. This benzene ring was then connected to the methylene carbon C9 by the correlations from H9 to C1’, C2’, and C6’. Finally, consideration of the deshielded chemical shift of C3 and the molecular formula allowed the linkage between C1 and C3 via an oxygen atom to complete the structure of 2 (Figure 3).
Table 3: NMR spectroscopic data for karimunone B (2) in CDCl3.
| position | δC, typea | δH, mult.b | HMBCc |
| 1 | 166.0, C | ||
| 3 | 155.9, C | ||
| 4 | 105.9, CH | 5.98, s | 3, 4a, 5, 8a, 9 |
| 4a | 138.8, C | ||
| 5 | 102.0, CH | 6.27, s | 4, 6, 7, 8a |
| 6 | 167.3, C | ||
| 7 | 101.2, CH | 6.48, s | 5, 6, 8, 8a |
| 8 | 163.9, C | ||
| 8a | 99.9, C | ||
| 9 | 39.8, CH2 | 4.07, s | 3, 4, 1', 2', 6' |
| 10 | 56.0, CH3 | 3.85, s | 6 |
| 11 | 203.5, C | ||
| 12 | 32.3, CH3 | 2.61, s | 11, 2' |
| 1' | 138.6, C | ||
| 2' | 115.7, C | ||
| 3' | 166.8, C | ||
| 4' | 103.7, CH | 6.41, s | 2', 3', 5', 6' |
| 5' | 161.5, C | ||
| 6' | 112.9, CH | 6.34, s | 2', 4', 5', 9 |
| 8-OH | 10.94, s | 1, 6, 7, 8, 8a | |
aRecorded at 125 MHz (reference δC 77.0). bRecorded at 500 MHz (reference δH 7.27). cHMBC correlations are from proton(s) stated to the indicated carbon.
![[1860-5397-15-289-3]](/bjoc/content/figures/1860-5397-15-289-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Key HMBC correlations for karimunone B (2).
Figure 3: Key HMBC correlations for karimunone B (2).
Antibacterial activities of compounds 1–7 were tested against clinical pathogen Salmonella enterica ser. Typhi strain MDR from General Hospital Dr. Kariadi, Semarang, Indonesia. Compounds 2–7 were weakly active with MIC values of 125 µg/mL, while 1 was not active in the same concentration range.
Conclusion
In summary, our chemical analysis of a sponge-derived fungus of the genus Fusarium led to the identification of two new and three known aromatic polyketides (1 and 2, and 3–5, respectively) and two sesquiterpenes (6 and 7). Although secondary metabolites of Fusarium were extensively studied in the past [26-28], it is still possible to obtain additional new compounds, implying the existence of unstudied Fusarium species capable of producing unknown metabolites in marine symbiotic environments. Interestingly, 4 and its 5-O-methylated congener were previously reported from the Australian marine crinoid Comatula rotalaria [20]. Compound 4, an 8-deoxy congener of 1, is presumably a biosynthetic precursor for 1. Our finding of 1, 3, and 4 from a sponge-associated fungus is suggestive of the production of these anthraquinones by symbiotic fungi in crinoids. The 7-hydroxy congener of 1 is known as a metabolite of the fungus Geosmithia [24]. Plant metabolites, feralolide and its glycoside, possessing the same skeleton as 2 were reported from the medicinal herb Aloe vera [29], but this carbon skeleton is novel as a fungal secondary metabolite.
Experimental
General experimental procedures
NMR spectra were recorded on a Bruker AVANCE II 500 spectrometer (Bruker Biospin K. K., Yokohama, Japan) and mass spectra were measured on a Bruker micrOTOF (Bruker Daltonics K. K., Yokohama, Japan). IR spectra were recorded on a Shimadzu FT-IR-300 spectrophotometer (Shimadzu Corp., Kyoto, Japan) and UV spectra on a Shimadzu UV-1800 (Shimadzu Corp., Kyoto, Japan).
Microorganism
Fusarium sp. KJMT.FP.4.3 was isolated from a sponge Xestospongia sp. collected from Karimunjawa National Park, Indonesia with the permission number 1096/T.34/TU/SIMAKSI/7/2017. This fungus was isolated using a surface sterilization method [14,30]. Strain KJMT.FP.4.3 was identified as Fusarium on the basis of the gene sequence analytical data of the ITS domain of partial 18S rRNA gene and 28S rRNA gene. Strain KJMT.FP.4.3 showed 99.7% similarity of ITS domain of partial 18S rRNA gene and 28S rRNA gene (555 nucleotides, GeneBank accession number MK393925.1) to Fusarium oxysporum strain NZZCDHL (549 nucleotides, GeneBank accession number KU939031.1) and 99.5% similarity of ITS domain of partial 18S rRNA gene and 28S rRNA gene to Fusarium oxysporum isolate DG-2 (548 nucleotides, GeneBank accession number MK429839.1).
Fermentation
Strain KJMT.FP.4.3 cultured on PDA agar was inoculated into 500 mL K-1 flasks each containing V22 seed medium consisting of soluble starch 1%, glucose 0.5%, NZ-case (Sigma-Aldrich, Co., LLC.) 0.3%, yeast extract (Kyokuto Pharmaceutical Industrial, Co., Ltd.) 0.2%, Tryptone (Difco Laboratories) 0.5%, K2HPO4 0.1%, MgSO4·7H2O 0.05%, and CaCO3 0.3% (pH 7.0). The flasks were shaken at 30 °C for 4 days on a rotary shaker (200 rpm). The seed culture (3 mL) was transferred into 500 mL K-1 flasks each containing 100 mL of A3M production medium (pH 7.0) consisting of 2.0% soluble starch, 0.5% glucose, 2.0% glycerol, 0.3% yeast extract, 1.5% Pharmamedia (Traders Protein), and 1% Diaion HP-20 (Mitsubishi Chemical Co.). The inoculated flasks were placed on a rotary shaker (200 rpm) at 30 °C for 7 days.
Extraction and isolation
Extraction and isolation of secondary metabolites from strain KJMT.FP.4.3 were carried out in a similar manner as described previously [31]. At the end of the fermentation period, 50 mL of 1-butanol was added to each flask, and the flasks were shaken for 1 h. The mixture was centrifuged at 6000 rpm for 10 min and the organic layer was separated from the aqueous layer containing the mycelium. Evaporation of the solvent gave 3.37 g of crude extract from 1.5 L of culture. The crude extract (3.37 g) was subjected to silica gel column chromatography with a step gradient of CHCl3/MeOH 1:0, 20:1, 10:1, 4:1, 2:1, 1:1, and 0:1 (v/v). Fraction 3 (10:1) was concentrated to provide 0.97 g of brown oil, which was further purified by reversed-phase ODS column chromatography with a gradient of MeCN–H2O 2:8, 3:7, 4:6, 5:5, 6:4, 7:3, and 8:2 (v/v). Fraction 5 (6:4) was evaporated and the remaining aqueous solution was extracted with EtOAc. After drying with anhydrous Na2SO4, the organic layer was concentrated to dryness. The residual solid (53 mg) was applied to the preparative HPLC (Cosmosil Cholester Packed Column, 10 × 250 mm, Nacalai Tesque) using a linear gradient of 40 to 70% MeCN in 0.1% HCO2H over 25 min at a flow rate of 4 mL/min, yielding karimunone A (1, 7.3 mg, tR 15.6 min) and B (2, 4.2 mg, tR 12.0 min), together with 7-O-methylrhodolamprometrin (4, 7.2 mg, tR 17.4 min), O-methylSMA93 (5, 7.4 mg, tR 13.2 min), tricinonoic acid (6, 3.7 mg, tR 13.1 min), and cyclonerodiol (7, 5.4 mg, tR 11.3 min) after evaporation and extraction with EtOAc. Rhodolamprometrin (3, 4.0 mg, tR 15.2 min) was isolated from fraction 4 of ODS chromatography (5:5), which was derived from fraction 3 of silica gel fractionation (10:1), using a linear gradient of 40 to 70% MeCN in 0.1% HCO2H over 25 min at a flow rate of 4 mL/min. Physicochemical data including 1H and 13C NMR spectroscopic data of compounds 3–7 were in good accordance with those described in the literatures [19-23].
Karimunone A (1): red powder; UV (MeOH) λmax (log ε) 234 (3.44), 266 (3.28), 313 (3.04), 499 (2.81), 533 (2.68) nm; IR νmax 3111, 2926, 1712, 1679, 1601 cm−1; see Table 1 for 1H NMR and 13C NMR data; TOF-HRESIMS [M − H]− m/z 343.0452 (calcd for C17H11O8, 343.0459).
Karimunone B (2): red powder; UV (MeOH) λmax (log ε) 244 (3.49), 277 (2.97), 324 (2.85) nm; IR νmax 3100, 1683, 1615 cm−1; see Table 3 for 1H NMR and 13C NMR data; TOF-HRESIMS [M − H]− m/z 355.0823 (calcd for C19H15O7, 355.0818).
Computational procedure
General information
Conformational search was performed with MacroModel version 12.1 in the Maestro 11.7 software package [32,33]. All DFT-based calculations were performed with the Gaussian 16 Rev B.01 program [34]. A part of these computations were conducted using a Fujitsu PRIMERGY CX400 multi-node server (Information Technology Center of Nagoya University). Molecular structures were visualized using Maestro 11.7 software package. DP4+ analysis was performed with the Excel spreadsheet [25] made by Sarotti et al. Cartesian coordinates of the structures described in this paper are included in Supporting Information File 2.
Computational search and NMR calculations of structures a and b
The conformational search on structure a began by applying 100,000 steps of the Monte-Carlo Multiple Minimum (MCMM) method with PRCG energy minimization using the OPLS3e force field (gas phase) to obtain 42 conformational isomers within 35 kcal/mol from the minimum energy conformer. The next optimizations were performed at the M06-2X/6-31G(d) level of theory and solvation effects were included using the PCM solvation model (DMSO). Frequency calculations were carried out at the same level of theory to confirm the absence of imaginary frequencies and to obtain thermal corrections to the Gibbs free energies at 1 atm, 298.15 K. The duplicate structures with RMSDs of 0.01 Å were removed. Single-point energies were calculated at the M06-2X/6-311+G(d,p) level of theory and solvation effects were included using the PCM solvation model (DMSO). The NMR chemical shifts were simulated by GIAO method at the mPW1PW91/6-31G+(d,p)-PCM(DMSO) level of theory. The chemical shifts (δcalc) were calculated using tetramethylsilane (TMS) as a reference standard according to δcalc = σ0 − σx, where σx is the Boltzmann-averaged shielding tensor of the most stable 4 conformers within 3.5 kcal/mol and σ0 is the shielding tensor of TMS calculated at the same level of theory with σx. The NMR shifts of structure b was similarly calculated using 50 structures as the OPLS3e-minimized structures and 4 structures as the DFT-optimized structures for the NMR calculation.
Acknowledgements
The authors would like to acknowledge Balai Taman Nasional Karimunjawa for the research permit with the number 1096/T.34/TU/SIMAKSI/7/2017. This research was partly funded by The Ministry of Research, Technology and Higher Education, Republic Indonesia through Program Pendidikan Magister Menuju Doktor Untuk Sarjana Unggul (PMDSU) scheme with contract number 315-06/UN7.5.1/PP/2017 and the travel grant through Peningkatan Kualitas Publikasi Internasional (PKPI) scheme.
References
-
Blunt, J. W.; Carroll, A. R.; Copp, B. R.; Davis, R. A.; Keyzers, R. A.; Prinsep, M. R. Nat. Prod. Rep. 2018, 35, 8–53. doi:10.1039/c7np00052a
Return to citation in text: [1] -
Carroll, A. R.; Copp, B. R.; Davis, R. A.; Keyzers, R. A.; Prinsep, M. R. Nat. Prod. Rep. 2019, 36, 122–173. doi:10.1039/c8np00092a
Return to citation in text: [1] -
Hunt, B.; Vincent, A. C. J. Ambio 2006, 35, 57–64. doi:10.1579/0044-7447(2006)35[57:sasomb]2.0.co;2
Return to citation in text: [1] -
Bell, J. J. Estuarine, Coastal Shelf Sci. 2008, 79, 341–353. doi:10.1016/j.ecss.2008.05.002
Return to citation in text: [1] -
Indraningrat, A. A. G.; Smidt, H.; Sipkema, D. Mar. Drugs 2016, 14, No. 87. doi:10.3390/md14050087
Return to citation in text: [1] [2] -
Tian, R.-M.; Sun, J.; Cai, L.; Zhang, W.-P.; Zhou, G.-W.; Qiu, J.-W.; Qian, P.-Y. Environ. Microbiol. 2016, 18, 2481–2494. doi:10.1111/1462-2920.13161
Return to citation in text: [1] -
Kiran, G. S.; Sekar, S.; Ramasamy, P.; Thinesh, T.; Hassan, S.; Lipton, A. N.; Ninawe, A. S.; Selvin, J. Mar. Environ. Res. 2018, 140, 169–179. doi:10.1016/j.marenvres.2018.04.017
Return to citation in text: [1] -
Blockley, A.; Elliott, D. R.; Roberts, A. P.; Sweet, M. Diversity 2017, 9, 49. doi:10.3390/d9040049
Return to citation in text: [1] -
Romano, G.; Costantini, M.; Sansone, C.; Lauritano, C.; Ruocco, N.; Ianora, A. Mar. Environ. Res. 2017, 128, 58–69. doi:10.1016/j.marenvres.2016.05.002
Return to citation in text: [1] -
Zhang, J.; Yuan, B.; Liu, D.; Gao, S.; Proksch, P.; Lin, W. Front. Chem. (Lausanne, Switz.) 2018, 6, No. 314. doi:10.3389/fchem.2018.00314
Return to citation in text: [1] -
Kumla, D.; Dethoup, T.; Gales, L.; Pereira, J. A.; Freitas-Silva, J.; Costa, P. M.; Silva, A. M. S.; Pinto, M. M. M.; Kijjoa, A. Molecules 2019, 24, No. 208. doi:10.3390/molecules24010208
Return to citation in text: [1] -
Mehbub, M. F.; Lei, J.; Franco, C.; Zhang, W. Mar. Drugs 2014, 12, 4539–4577. doi:10.3390/md12084539
Return to citation in text: [1] -
Hanif, N.; Murni, A.; Tanaka, C.; Tanaka, J. Mar. Drugs 2019, 17, 364. doi:10.3390/md17060364
Return to citation in text: [1] -
Lin, W.; Brauers, G.; Ebel, R.; Wray, V.; Berg, A.; Sudarsono; Proksch, P. J. Nat. Prod. 2003, 66, 57–61. doi:10.1021/np020196b
Return to citation in text: [1] [2] -
Rotinsulu, H.; Yamazaki, H.; Sugai, S.; Iwakura, N.; Wewengkang, D. S.; Sumilat, D. A.; Namikoshi, M. J. Nat. Med. 2018, 72, 779–783. doi:10.1007/s11418-018-1193-y
Return to citation in text: [1] -
Sabdaningsih, A.; Cristianawati, O.; Sibero, M. T.; Nuryadi, H.; Radjasa, O. K.; Sabdono, A.; Trianto, A. IOP Conf. Ser. Earth Environ. Sci. 2017, 55, No. 012026. doi:10.1088/1755-1315/55/1/012026
Return to citation in text: [1] -
Sibero, M. T.; Radjasa, O. K.; Sabdono, A.; Trianto, A.; Triningsih, D. W.; Hutagaol, I. D. J. Appl. Pharm. Sci. 2018, 8, 88–94. doi:10.7324/japs.2018.8214
Return to citation in text: [1] -
Sibero, M. T.; Herdikiawan, D.; Radjasa, O. K.; Sabdono, A.; Trianto, A.; Triningsih, D. W. AACL Bioflux 2018, 11, 10–18.
Return to citation in text: [1] -
Betina, V.; Sedmera, P.; Vokoun, J.; Podojil, M. Experientia 1986, 42, 196–197. doi:10.1007/bf01952466
Return to citation in text: [1] [2] -
Khokhar, S.; Pierens, G. K.; Hooper, J. N. A.; Ekins, M. G.; Feng, Y.; Davis, R. A. J. Nat. Prod. 2016, 79, 946–953. doi:10.1021/acs.jnatprod.5b01029
Return to citation in text: [1] [2] [3] -
Li, S.; Shao, M.-W.; Lu, Y.-H.; Kong, L.-C.; Jiang, D.-H.; Zhang, Y.-L. J. Agric. Food Chem. 2014, 62, 8997–9001. doi:10.1021/jf502484n
Return to citation in text: [1] [2] -
Bashyal, B. P.; Gunatilaka, A. A. L. Nat. Prod. Res. 2010, 24, 349–356. doi:10.1080/14786410903125401
Return to citation in text: [1] [2] -
Nozoe, S.; Goi, M.; Morisaki, N. Tetrahedron Lett. 1970, 11, 1293–1296. doi:10.1016/s0040-4039(01)91612-0
Return to citation in text: [1] [2] -
Stodůlková, E.; Kolařík, M.; Křesinová, Z.; Kuzma, M.; Šulc, M.; Man, P.; Novák, P.; Maršík, P.; Landa, P.; Olšovská, J.; Chudíčková, M.; Pažoutová, S.; Černý, J.; Bella, J.; Flieger, M. Folia Microbiol. 2009, 54, 179–187. doi:10.1007/s12223-009-0028-3
Return to citation in text: [1] [2] -
Grimblat, N.; Zanardi, M. M.; Sarotti, A. M. J. Org. Chem. 2015, 80, 12526–12534. doi:10.1021/acs.joc.5b02396
Return to citation in text: [1] [2] -
Fouillaud, M.; Venkatachalam, M.; Girard-Valenciennes, E.; Caro, Y.; Dufossé, L. Mar. Drugs 2016, 14, 64. doi:10.3390/md14040064
Return to citation in text: [1] -
Cao, Q.-X.; Wei, J.-H.; Deng, R.; Feng, G.-K.; Zhu, X.-F.; Lan, W.-J.; Li, H.-J. Chem. Biodiversity 2017, 14, e1600298. doi:10.1002/cbdv.201600298
Return to citation in text: [1] -
Liu, S.-Z.; Yan, X.; Tang, X.-X.; Lin, J.-G.; Qiu, Y.-K. Mar. Drugs 2018, 16, No. 483. doi:10.3390/md16120483
Return to citation in text: [1] -
Veitch, N. C.; Simmonds, M. S. J.; Blaney, W. M.; Reynolds, T. Phytochemistry 1994, 35, 1163–1166. doi:10.1016/s0031-9422(00)94814-3
Return to citation in text: [1] -
Kjer, J.; Debbab, A.; Aly, A. H.; Proksch, P. Nat. Protoc. 2010, 5, 479–490. doi:10.1038/nprot.2009.233
Return to citation in text: [1] -
Kim, Y.; Ogura, H.; Akasaka, K.; Oikawa, T.; Matsuura, N.; Imada, C.; Yasuda, H.; Igarashi, Y. Mar. Drugs 2014, 12, 4110–4125. doi:10.3390/md12074110
Return to citation in text: [1] -
MacroModel, Version 12.1; Schrödinger, LLC: New York, NY, 2018.
Return to citation in text: [1] -
Maestro, Version 11.7; Schrödinger, LLC: New York, NY, 2018.
Return to citation in text: [1] -
Gaussian 16, Revision B.01; Gaussian, Inc.: Wallingford, CT, 2016.
Return to citation in text: [1]
| 1. | Blunt, J. W.; Carroll, A. R.; Copp, B. R.; Davis, R. A.; Keyzers, R. A.; Prinsep, M. R. Nat. Prod. Rep. 2018, 35, 8–53. doi:10.1039/c7np00052a |
| 2. | Carroll, A. R.; Copp, B. R.; Davis, R. A.; Keyzers, R. A.; Prinsep, M. R. Nat. Prod. Rep. 2019, 36, 122–173. doi:10.1039/c8np00092a |
| 5. | Indraningrat, A. A. G.; Smidt, H.; Sipkema, D. Mar. Drugs 2016, 14, No. 87. doi:10.3390/md14050087 |
| 10. | Zhang, J.; Yuan, B.; Liu, D.; Gao, S.; Proksch, P.; Lin, W. Front. Chem. (Lausanne, Switz.) 2018, 6, No. 314. doi:10.3389/fchem.2018.00314 |
| 11. | Kumla, D.; Dethoup, T.; Gales, L.; Pereira, J. A.; Freitas-Silva, J.; Costa, P. M.; Silva, A. M. S.; Pinto, M. M. M.; Kijjoa, A. Molecules 2019, 24, No. 208. doi:10.3390/molecules24010208 |
| 24. | Stodůlková, E.; Kolařík, M.; Křesinová, Z.; Kuzma, M.; Šulc, M.; Man, P.; Novák, P.; Maršík, P.; Landa, P.; Olšovská, J.; Chudíčková, M.; Pažoutová, S.; Černý, J.; Bella, J.; Flieger, M. Folia Microbiol. 2009, 54, 179–187. doi:10.1007/s12223-009-0028-3 |
| 8. | Blockley, A.; Elliott, D. R.; Roberts, A. P.; Sweet, M. Diversity 2017, 9, 49. doi:10.3390/d9040049 |
| 9. | Romano, G.; Costantini, M.; Sansone, C.; Lauritano, C.; Ruocco, N.; Ianora, A. Mar. Environ. Res. 2017, 128, 58–69. doi:10.1016/j.marenvres.2016.05.002 |
| 25. | Grimblat, N.; Zanardi, M. M.; Sarotti, A. M. J. Org. Chem. 2015, 80, 12526–12534. doi:10.1021/acs.joc.5b02396 |
| 5. | Indraningrat, A. A. G.; Smidt, H.; Sipkema, D. Mar. Drugs 2016, 14, No. 87. doi:10.3390/md14050087 |
| 6. | Tian, R.-M.; Sun, J.; Cai, L.; Zhang, W.-P.; Zhou, G.-W.; Qiu, J.-W.; Qian, P.-Y. Environ. Microbiol. 2016, 18, 2481–2494. doi:10.1111/1462-2920.13161 |
| 7. | Kiran, G. S.; Sekar, S.; Ramasamy, P.; Thinesh, T.; Hassan, S.; Lipton, A. N.; Ninawe, A. S.; Selvin, J. Mar. Environ. Res. 2018, 140, 169–179. doi:10.1016/j.marenvres.2018.04.017 |
| 22. | Bashyal, B. P.; Gunatilaka, A. A. L. Nat. Prod. Res. 2010, 24, 349–356. doi:10.1080/14786410903125401 |
| 3. | Hunt, B.; Vincent, A. C. J. Ambio 2006, 35, 57–64. doi:10.1579/0044-7447(2006)35[57:sasomb]2.0.co;2 |
| 4. | Bell, J. J. Estuarine, Coastal Shelf Sci. 2008, 79, 341–353. doi:10.1016/j.ecss.2008.05.002 |
| 23. | Nozoe, S.; Goi, M.; Morisaki, N. Tetrahedron Lett. 1970, 11, 1293–1296. doi:10.1016/s0040-4039(01)91612-0 |
| 16. | Sabdaningsih, A.; Cristianawati, O.; Sibero, M. T.; Nuryadi, H.; Radjasa, O. K.; Sabdono, A.; Trianto, A. IOP Conf. Ser. Earth Environ. Sci. 2017, 55, No. 012026. doi:10.1088/1755-1315/55/1/012026 |
| 17. | Sibero, M. T.; Radjasa, O. K.; Sabdono, A.; Trianto, A.; Triningsih, D. W.; Hutagaol, I. D. J. Appl. Pharm. Sci. 2018, 8, 88–94. doi:10.7324/japs.2018.8214 |
| 18. | Sibero, M. T.; Herdikiawan, D.; Radjasa, O. K.; Sabdono, A.; Trianto, A.; Triningsih, D. W. AACL Bioflux 2018, 11, 10–18. |
| 20. | Khokhar, S.; Pierens, G. K.; Hooper, J. N. A.; Ekins, M. G.; Feng, Y.; Davis, R. A. J. Nat. Prod. 2016, 79, 946–953. doi:10.1021/acs.jnatprod.5b01029 |
| 14. | Lin, W.; Brauers, G.; Ebel, R.; Wray, V.; Berg, A.; Sudarsono; Proksch, P. J. Nat. Prod. 2003, 66, 57–61. doi:10.1021/np020196b |
| 15. | Rotinsulu, H.; Yamazaki, H.; Sugai, S.; Iwakura, N.; Wewengkang, D. S.; Sumilat, D. A.; Namikoshi, M. J. Nat. Med. 2018, 72, 779–783. doi:10.1007/s11418-018-1193-y |
| 21. | Li, S.; Shao, M.-W.; Lu, Y.-H.; Kong, L.-C.; Jiang, D.-H.; Zhang, Y.-L. J. Agric. Food Chem. 2014, 62, 8997–9001. doi:10.1021/jf502484n |
| 13. | Hanif, N.; Murni, A.; Tanaka, C.; Tanaka, J. Mar. Drugs 2019, 17, 364. doi:10.3390/md17060364 |
| 12. | Mehbub, M. F.; Lei, J.; Franco, C.; Zhang, W. Mar. Drugs 2014, 12, 4539–4577. doi:10.3390/md12084539 |
| 19. | Betina, V.; Sedmera, P.; Vokoun, J.; Podojil, M. Experientia 1986, 42, 196–197. doi:10.1007/bf01952466 |
| 24. | Stodůlková, E.; Kolařík, M.; Křesinová, Z.; Kuzma, M.; Šulc, M.; Man, P.; Novák, P.; Maršík, P.; Landa, P.; Olšovská, J.; Chudíčková, M.; Pažoutová, S.; Černý, J.; Bella, J.; Flieger, M. Folia Microbiol. 2009, 54, 179–187. doi:10.1007/s12223-009-0028-3 |
| 26. | Fouillaud, M.; Venkatachalam, M.; Girard-Valenciennes, E.; Caro, Y.; Dufossé, L. Mar. Drugs 2016, 14, 64. doi:10.3390/md14040064 |
| 27. | Cao, Q.-X.; Wei, J.-H.; Deng, R.; Feng, G.-K.; Zhu, X.-F.; Lan, W.-J.; Li, H.-J. Chem. Biodiversity 2017, 14, e1600298. doi:10.1002/cbdv.201600298 |
| 28. | Liu, S.-Z.; Yan, X.; Tang, X.-X.; Lin, J.-G.; Qiu, Y.-K. Mar. Drugs 2018, 16, No. 483. doi:10.3390/md16120483 |
| 20. | Khokhar, S.; Pierens, G. K.; Hooper, J. N. A.; Ekins, M. G.; Feng, Y.; Davis, R. A. J. Nat. Prod. 2016, 79, 946–953. doi:10.1021/acs.jnatprod.5b01029 |
| 25. | Grimblat, N.; Zanardi, M. M.; Sarotti, A. M. J. Org. Chem. 2015, 80, 12526–12534. doi:10.1021/acs.joc.5b02396 |
| 32. | MacroModel, Version 12.1; Schrödinger, LLC: New York, NY, 2018. |
| 33. | Maestro, Version 11.7; Schrödinger, LLC: New York, NY, 2018. |
| 31. | Kim, Y.; Ogura, H.; Akasaka, K.; Oikawa, T.; Matsuura, N.; Imada, C.; Yasuda, H.; Igarashi, Y. Mar. Drugs 2014, 12, 4110–4125. doi:10.3390/md12074110 |
| 19. | Betina, V.; Sedmera, P.; Vokoun, J.; Podojil, M. Experientia 1986, 42, 196–197. doi:10.1007/bf01952466 |
| 20. | Khokhar, S.; Pierens, G. K.; Hooper, J. N. A.; Ekins, M. G.; Feng, Y.; Davis, R. A. J. Nat. Prod. 2016, 79, 946–953. doi:10.1021/acs.jnatprod.5b01029 |
| 21. | Li, S.; Shao, M.-W.; Lu, Y.-H.; Kong, L.-C.; Jiang, D.-H.; Zhang, Y.-L. J. Agric. Food Chem. 2014, 62, 8997–9001. doi:10.1021/jf502484n |
| 22. | Bashyal, B. P.; Gunatilaka, A. A. L. Nat. Prod. Res. 2010, 24, 349–356. doi:10.1080/14786410903125401 |
| 23. | Nozoe, S.; Goi, M.; Morisaki, N. Tetrahedron Lett. 1970, 11, 1293–1296. doi:10.1016/s0040-4039(01)91612-0 |
| 29. | Veitch, N. C.; Simmonds, M. S. J.; Blaney, W. M.; Reynolds, T. Phytochemistry 1994, 35, 1163–1166. doi:10.1016/s0031-9422(00)94814-3 |
| 14. | Lin, W.; Brauers, G.; Ebel, R.; Wray, V.; Berg, A.; Sudarsono; Proksch, P. J. Nat. Prod. 2003, 66, 57–61. doi:10.1021/np020196b |
| 30. | Kjer, J.; Debbab, A.; Aly, A. H.; Proksch, P. Nat. Protoc. 2010, 5, 479–490. doi:10.1038/nprot.2009.233 |
© 2019 Sibero et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)