Abstract
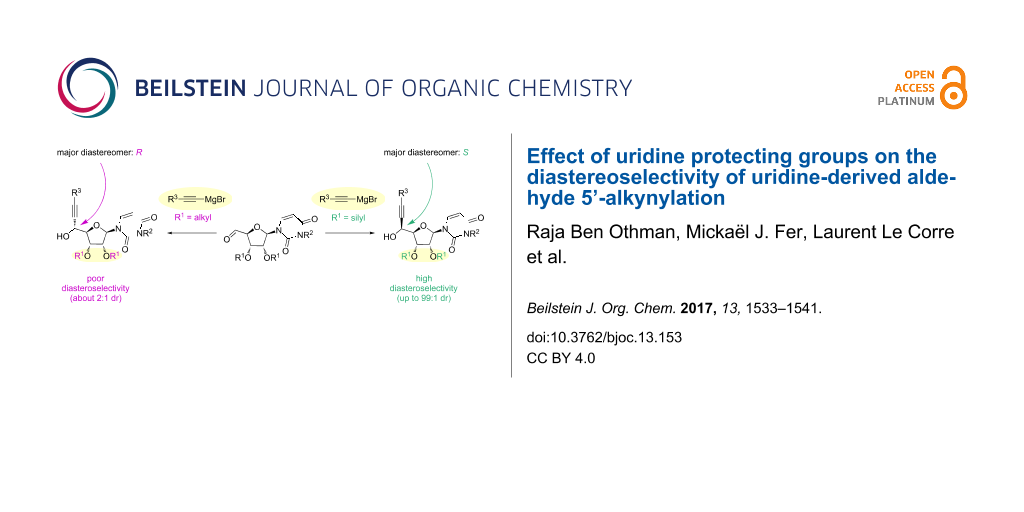
The 5’-alkynylation of uridine-derived aldehydes is described. The addition of alkynyl Grignard reagents on the carbonyl group is significantly influenced by the 2’,3’-di-O-protecting groups (R1): O-alkyl groups led to modest diastereoselectivities (65:35) in favor of the 5’R-isomer, whereas O-silyl groups promoted higher diastereoselectivities (up to 99:1) in favor of the 5’S-isomer. A study related to this protecting group effect on the diastereoselectivity is reported.

Graphical Abstract
Introduction
Nucleoside and nucleotide derivatives or analogues are biologically active compounds of major interest [1,2]. Their widespread applications span from therapeutic agents, such as antibacterial [3-5], antiviral [6] or antitumor [7,8] drugs, to epigenetic modulators [9] or chemical tools [10,11]. Furthermore, they represent central monomers for oligonucleos(t)ide synthesis [12-17]. In the past decades, chemical modifications of these crucial building blocks have been extensively studied, both on the nucleobase itself and on the sugar moiety [6]. Many of them concern the C-2’ and C-3’ sugar positions. However, functionalization at C-5’ has been much less reported, particularly the stereocontrolled formation of a stereogenic center at C-5’, in spite of its important role for biological activity (Figure 1) [16-19].
![[1860-5397-13-153-1]](/bjoc/content/figures/1860-5397-13-153-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: C-5’ configuration of nucleoside derivatives and related biological activity.
Figure 1: C-5’ configuration of nucleoside derivatives and related biological activity.
In numerous approaches, this 5′ stereogenic center comes from the chiral pool and the nucleobase is introduced under Vorbrüggen conditions [20-26]. It can also be created directly on the nucleoside, either by functionalization of an alkene at C-5’ [27-32] or by the diastereoselective addition of a nucleophile on a carbonyl group. The latter can involve either the reduction of a ketone at C-5’ [12,33-35], or the addition on an aldehyde of various nucleophiles such as enolates [15,36-38], allylborane [39], dialkyl phosphites [40], TMSCN [41] or Grignard reagents [12,17,35,42-47]. Aside from the use of chiral ligands promoting an excellent facial discrimination of the aldehyde [34], the addition of Grignard reagents usually proceed with moderate diastereoselectivity and yield (Table 1) [12,35,43-46,48].
Table 1: Reported C-5’ diastereoselectivity for the addition of an organometallic reagent on nucleoside aldehydes.
![[Graphic 1]](/bjoc/content/inline/1860-5397-13-153-i4.svg?max-width=637&scale=1.0)
|
||||||||
| Entry | Basea | Organometallic reagent | Solvent | Temp (°C) | dr | C-5’b | Yield (%)c | Ref. |
|---|---|---|---|---|---|---|---|---|
| 1 | A(Bz) | CH3MgBr | THF | −78 | 3:2 | R | n.d. | [35] |
| 2 | U | AllylMgBr | THF | 100 | 16:1 | R | n.d. | [42] |
| 3 | A | TMSC≡CMgBr | THF | −20 | 2:1 | R | 44d | [43] |
| 4 | U | TMSC≡CMgBr | THF | −15 | 2:1 | R | 21 | [12] |
| 5 | U | TESC≡CMgBr | THF | −15 | 2:1 | R | 42 | [12] |
| 6 | A(Bz) | TMSC≡CMgBr | THF | −15 | 2:1 | R | 40 | [44] |
| 7 | U | AllylMgBr | THF | −78 | 5:1 | R | n.d. | [45] |
| 8 | U | CH2=CHMgBr | THF | −78 | 5:1 | R | n.d. | [46] |
| 9 | U(MTPM)e | 4-phenyl-1-butyne, iPrMgCl, Zn(OTf2) | toluene | rt | 1:1.7 | S | 47 | [48] |
aThe nature of the nucleobase protecting group is mentioned in brackets; bC-5’ configuration of the major diastereomer; cisolated yield of the major diastereomer; dcontaminated with a small amount of the other isomer; eMTPM: monomethoxytetrachlorodiphenylmethoxymethyl.
In the course of our program devoted to the synthesis of new MraY inhibitors [49,50], we were interested in developing a more efficient access to 5’-ethynyluridine, a crucial building block for the further synthesis of triazole-containing compounds [49]. Intrigued by the moderate diastereomeric ratio reported for the addition or organometallic reagents onto nucleoside aldehyde (Table 1), we decided to investigate the influence of the protecting groups of the uridine aldehyde on the stereochemical outcome of the nucleophilic addition of a Grignard reagent and we wish to report herein the results of our study.
Results and Discussion
We first prepared primary alcohols 1–5 from uridine and differing by the nature of R1 protecting groups. The secondary alcohols at C-2’ and C-3’ were protected either as a cyclic ketal (isopropylidene (1a) [12], isopentylidene (2a), cyclohexylidene (3a)) or as acyclic silyl ethers (4a [15], R1 = TBDMS and 5a [51], R1 = TIPS). Some compounds were also N3-allylated (1b, 4b and 5b) to evaluate the possible influence of R2 on the diastereoselectivity of the nucleophilic addition (Scheme 1).
![[1860-5397-13-153-i1]](/bjoc/content/inline/1860-5397-13-153-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Primary alcohols 1–5 were submitted to an oxidation/Grignard addition sequence leading to the corresponding propargyl alcohols 11–15 (Scheme 2). The aldehydes 6–10 resulting from oxidation with IBX were isolated and directly submitted to Grignard addition without further purification. Grignard reagents were prepared from trimethylsilyl-, triethylsilyl- or triisopropylsilylacetylene and ethylmagnesium bromide in THF at 0 °C, in order to vary the steric hindrance of the silyl group (R3). To unambiguously determine the ratio of diastereomers and the C-5’ configuration of the major one for the synthesized propargylic alcohols 11–15, their partial or complete deprotection was carried out to take advantage of an unequivocal 1H NMR comparison with known compounds, or reliable derivatives.
![[1860-5397-13-153-i2]](/bjoc/content/inline/1860-5397-13-153-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of propargylic alcohols 11–15 and their partial or complete deprotection.
Scheme 2: Synthesis of propargylic alcohols 11–15 and their partial or complete deprotection.
Thus, we prepared compounds (5’R)-11ab [12] and (5’S)-11ab [12] from protected uridine 1a and triethylsilylethynylmagnesium chloride. Indeed, Vasella’s group devoted a huge amount of work to the synthesis and characterization of these compounds and notably reported that TES was better than TMS for efficient separation of both isomers, resulting in an improved 42% yield for the isolated major diastereomer (5’R)-11ab [12]. Then, subsequent acidic hydrolysis afforded (5’R)-16 and (5’S)-16 and alkyne desilylation under basic conditions provided (5’S)-17 and (5’R)-17 [12] (Scheme 3).
![[1860-5397-13-153-i3]](/bjoc/content/inline/1860-5397-13-153-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of reference compounds and strategy for assignment of C-5’ configuration.
Scheme 3: Synthesis of reference compounds and strategy for assignment of C-5’ configuration.
The differences between 1H NMR data of the related diastereomeric compounds (Figure 2 and Figure 3) provided a conclusive evidence to unequivocally attribute the absolute configuration at C-5’ of the synthesized compounds. Thus, for compounds 11–15 with R3 = TES, the configuration at C-5’ was determined according to the 1H NMR spectra of (5’R) or (5’S)-16 derivative (Figure 2). Indeed, in 1H NMR spectrum (500 MHz, CDCl3), H-1’ appears as a doublet at 6.02 ppm for (5’R)-16 (3JH-1’-H-2’ = 7.0 Hz) and as a doublet at 5.98 ppm for (5’S)-16 (3JH-1’-H-2’ = 6.0 Hz). Furthermore, H-4’ appears as a multiplet at 4.11–4.09 ppm for (5’R)-16 and as a triplet at 4.05 ppm for (5’S)-16 (3JH-4’-H-3’ = 3JH-4’-H-5’ = 3.0 Hz). For compounds 11–15 with R3 ≠ TES, the configuration at C-5’ was attributed after complete deprotection leading to compound (5’R) or (5’S)-17 (Figure 3). As observed for compound 16, the H-1’ chemical shift is more shielded for (5’S)-17 isomer (doublet (3JH-1’-H-2’ = 6.0 Hz) at 5.98 ppm) than for the (5’R)-17 diastereomer (doublet (3JH-1’-H-2’ = 7.0 Hz) at 6.04 ppm). Similarly, the H-4’ chemical shift is more shielded for (5’S)-17 isomer (triplet (3JH-4’-H-3’ = 3JH-4’-H-5’ = 3.5 Hz) at 4.03 ppm) than for the (5’R)-17 diastereomer (triplet (3JH-4’-H-3’ = 3JH-4’-H-5’ = 2.5 Hz) at 4.07 ppm). Moreover, chemical shifts for H-3’ (doublet of doublet (3JH3’-H2’ = 5.5 Hz, 3JH3’-H4’ = 2.5 Hz) at 4.30 ppm) and H-2’ (doublet of doublet (3JH2’-H1’ = 7.0 Hz, 3JH2’-H3’ = 5.5 Hz) at 4.22 ppm) for the (5’R)-17 isomer are also significantly different from that of the (5’S)-17 isomer (triplet (3JH2’-H1’ = 3JH2’-H3’ = 6.0 Hz) at 4.23 ppm for H-2’ and doublet of doublet (3JH3’-H2’ = 6.0 Hz, 3JH3’-H4’ = 3.5 Hz) at 4.20 ppm for H-3’.
![[1860-5397-13-153-2]](/bjoc/content/figures/1860-5397-13-153-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: 1H NMR of (5’R)-16 and (5’S)-16 and of a (5’R)/(5’S)-16 mixture.
Figure 2: 1H NMR of (5’R)-16 and (5’S)-16 and of a (5’R)/(5’S)-16 mixture.
![[1860-5397-13-153-3]](/bjoc/content/figures/1860-5397-13-153-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: 1H NMR of (5’R)-17 and (5’S)-17 and example of configuration determination for a pure isolated compound with unknown configuration.
Figure 3: 1H NMR of (5’R)-17 and (5’S)-17 and example of configuration determination for a pure isolated comp...
The results and conditions of assays involving the various synthesized substrates 1–5 and Grignard reagents are reported in Table 2. The diastereomeric ratio of 11–15 was determined by 1H NMR or HPLC analysis of the crude mixture and the C-5’ configuration of the major diastereomer was assigned as explained above. To evaluate the effect of the temperature on the diastereomeric ratio (Table 2, entries 1 and 2), we first tried to carry out the reaction at lower temperature but it remained close to 2:1 in favor of the (5’R)-11ab, either with conventional or inverse order of addition. We also tried to increase the reaction temperature [52] but, even at 0 °C, the components in the reaction mixture were largely degraded. The protection of uracil nitrogen at N-3 position by an allyl group did not significantly modify the 5’R/5’S ratio (Table 2, entry 3). Increasing the bulkiness of the ketal protecting group by introducing an isopentylidene 2a (Table 2, entry 4) or a cyclohexylidene 3a (Table 2, entry 5) did not modify the observed diastereoselectivity.
Table 2: Influence of the protecting groups and conditions on the diastereoselectivity of the alkynyl Grignard addition on uridine derived aldehydes.
![[Graphic 2]](/bjoc/content/inline/1860-5397-13-153-i5.svg?max-width=637&scale=1.0)
|
||||||||||
| Entry | Alcohol | Aldehydea |
Grignard
reagent |
Temperature
(°C) |
Propargyl alcohol | |||||
| 1–5 | R1 | R2 | 6–10 | R3 | equiv | 11–15 | 5’R/5’S ratiob | Yield (%)c | ||
| 1 | 1a | CMe2 | H | 6a | TES | 2.5 | −50 | 11ab | 65:35d | conv: 65% |
| 2 | 1a | CMe2 | H | 6a | TES | 2.5 | −78 | 11ab | 65:35d | conv: 65% |
| 3 | 1b | CMe2 | allyl | 6b | TES | 2.5 | −15 | 11bb | 65:35e | 52 (24) |
| 4 | 2a | CEt2 | H | 7a | TES | 2.5 | −15 | 12ab | 65:35 | 54 (15) |
| 5 | 3a | CcHex | H | 8a | TES | 2.5 | −15 | 13ab | 70:30 | 65 (18) |
| 6 | 4a | TBDMS | H | 9a | TES | 2.5 | −15 | 14ab | 15:85 | 64 (48) |
| 7 | 4a | TBDMS | H | 9a | TES | 3.5 | −78 | 14ab | 15:85 | 51 (38) |
| 8 | 4b | TBDMS | allyl | 9b | TES | 3.5 | −78 | 14bb | 9:91e | 59 (39) |
| 9 | 5a | TIPS | H | 10a | TES | 5 | −15 | 15ab | 5:95 | 64 (53) |
| 10 | 5a | TIPS | H | 10a | TES | 5 | −78 | 15ab | 5:95 | 65 (32) |
| 11 | 5a | TIPS | H | 10a | TMS | 5 | −78 | 15aa | 10:90 | 66 (36) |
| 12 | 5a | TIPS | H | 10a | TIPS | 5 | −78 | 15ac | 1:99 | 61 |
| 13 | 5a | TIPS | H | 10a | TIPS | 5 | −15 | 15ac | 2:98 | 55 |
| 14 | 5a | TIPS | H | 10a | TIPS | 2.5 | −15 | 15ac | 2:98 | conv: 85% |
| 15 | 5b | TIPS | allyl | 10b | TIPS | 5 | −78 | 15bc | 10:90e | 43 (27) |
aNormal addition procedure; bdetermined by 1H NMR and/or HPLC of the crude mixture; cyield of the mixture of diastereomers over two steps from the corresponding primary alcohol, isolated yield of the major diastereomer is shown in brackets; dan “inverse” order of addition led to the same diastereomeric ratio; e5’-configuration of the major diastereomer was not determined.
We next turned to the use of acyclic protecting groups. tert-Butyldimethylsilyl ether 4a (Table 2, entry 6) led to an improved 85:15 ratio. Furthermore, we were delighted to discover that, contrary to that which was observed with ketal groups, the major diastereomer 4a obtained using tert-butyldimethylsilyl ether displayed the 5’S configuration. As was noted for isopropylidene as a protecting group, decreasing the temperature (Table 2, entry 7) or protecting the N-3 nitrogen of uracil (Table 2, entry 8) did not significantly change the 5’R/5’S ratio. In both cases, using 3.5 equivalents of Grignard reagent was required to complete the reaction.
In order to improve this reverse diastereoselectivity, we envisaged the use of a more bulky protecting group, such as the triisopropylsilyl group. The addition of triethylsilylacetylide magnesium bromide gave a 5’R /5’S ratio of 5:95 (Table 2, entries 9 and 10). To determine if R3 was able to influence this ratio, we also tested trimethylsilylacetylmagnesium bromide (Table 2, entry 11) and triisopropylsilylacetylmagnesium bromide (Table 2, entry 12). When R3 = TMS, the 5’R/5’S ratio was only 10:90, whereas for R3 = TIPS, an excellent 5’R/5’S 1:99 ratio was reached, allowing the direct synthesis of (5’S)-15ac with a substantial 61% isolated yield over two steps. Running this reaction at −15 °C (Table 2, entry 13) slightly diminished the yield to 55%. The bulkiness of the TIPS groups required the use of 5 equivalents of Grignard reagent to get a clean reaction and complete conversion of the starting material (Table 2, entry 14). The protection of the N-3 nitrogen with an allyl group was unfavorable and led to a 5’R/5’S 10:90 ratio (Table 2, entry 15). The three attempts of C-5’ alkynylation of N-3-allylated uridine aldehydes, did not revealed marked influence on the diastereoselective ratio (Table 2, entries 3, 8, 15).
To explain the diastereoselective outcome of the reaction, we first considered Cram chelated models (Figure 4).
![[1860-5397-13-153-4]](/bjoc/content/figures/1860-5397-13-153-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Hypothetical Cram chelated models.
Figure 4: Hypothetical Cram chelated models.
A 1,3-chelation of the metal with the carbonyl group and the protecting group oxygen atom at C-3’ would promote an attack of the nucleophile on the Re face of the aldehyde and thus the formation of the 5’R diastereoisomers, whereas a 1,2-metal chelation with the carbonyl group and the endocyclic oxygen atom of uridine at C-4’ would explain the formation of the 5’S diastereoisomer (attack of the nucleophile on the Si face of the aldehyde, Figure 4). In such a case, the poor chelating ability of silyl ether groups, along with their bulkiness, would strongly disfavor the 1,3-chelate and the 5’R-isomer formation, justifying the high selectivity observed for the silyl protected compounds in favor of the 5’S-isomer. However, when ketal groups are used, the 5’R-isomer is predominantly formed with a modest diastereoselectivity, meaning that the most stable transition state would involve a 1,3-metal chelation with the carbonyl group and the protecting group oxygen atom at C-3’ of uridine. Even if the later 1,3-chelation can be envisioned, we found this hypothesis very unlikely due to the large distance between the two oxygen atoms.
Therefore, the following models would be more consistent with our results (Figure 5).
![[1860-5397-13-153-5]](/bjoc/content/figures/1860-5397-13-153-5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Proposed stereochemical models.
Figure 5: Proposed stereochemical models.
When R1 is a ketal group (R1 = alkyl, Figure 5 top), on the one hand, the major formation of the 5’R-isomer would be explained by the Felkin Anh model (Figure 5A top, left) with an attack of the nucleophile on the Re face of the aldehyde. On the other hand, the minor formation of the 5’S diastereomer would result from an attack of the nucleophile on the Si face of the aldehyde on the 1,2-chelated model (Figure 5B top, right).
When R1 is a trialkylsilyl protecting group (R1 = silyl, Figure 5 bottom), on the one hand, the 5’S diastereomer formation is in agreement with an attack on the least hindered face of a 1,2-chelated conformer (Figure 5C bottom, right). On the other hand, the minute formation of the 5’R-isomer that would be explained by the Felkin–Ahn model (Figure 5D bottom, left) is strongly disfavored due to the the bulkiness of the silyl groups hampering an approach of the nucleophile on the Re face: the bulkier the silyl group, the stronger the effect.
Conclusion
In summary, we report a study on the diastereoselective 5’-alkynylation of uridine aldehydes displaying various protecting groups on the secondary alcohols at C-2’ and C-3’ and at the N-3 position of the uracile. Our results show that while the N-3 protection has little influence on the alkynylation diastereoselectivity, the nature of the diol protecting group strongly impact the diastereoselective outcome of the reaction. Indeed, whatever the ketal group used, the major 5’R-isomer is obtained with a 2:1 ratio whereas the protection as silyl ethers leads to an inverse diastereoselectivity in favor of the 5’S-isomer. We propose stereochemical models to rationalize the observed diastereoselectivity. Furthermore by increasing the bulkiness of the silyl group both on the diol and the Grignard reagent, we manage to obtain an excellent diastereoselectivity. Indeed, by using the most bulky 2’,3’-O-TIPS protecting groups and TIPS-ethynylmagnesium bromide, the 5’-ethynylation was achieved in a 99:1 ratio in favor of the 5’S-isomer. The resulting building block with a broad potential in nucleos(t)ide derivative syntheses is obtained in a very satisfactory 61% yield over two steps.
Experimental
General procedure for N3-allylation of protected uridine derivatives. To a solution of protected uridine derivative (1 equiv) in DMF/acetone (1:1, final concentration 0.4 M) was added K2CO3 (1.8 equiv) and allyl bromide (1.5 equiv). The suspension was stirred at 50 °C for 12 h, filtered and concentrated in vacuo. The resulting residue was submitted to flash chromatography (elution conditions mentioned below) to afford the corresponding N-allyl compound.
General procedure for the oxidation of uridine derivatives 1–5. To a suspension of protected uridine derivative 1–5 (1 equiv) in acetonitrile (5 × 10−2 M) was added IBX (3 equiv). The suspension was refluxed for 45 min–1.5 h until complete conversion of starting material (TLC). The suspension was cooled to rt, filtered on a celite® pad and the cake was washed with EtOAc. The filtrate was then concentrated in vacuo to afford crude aldehydes 6–10 which were used without other purification (quantitative yield). Aldehydes have been characterized by 1H and 13C NMR. Since they are quite unstable, crude aldehydes were used without any further purification and directly engaged in the alkynylation reaction.
General procedure for the 5'-alkynylation of uridine-derived aldehydes 6–10. At 0 °C, under Ar, to a solution of trialkylsilylacetylene (1.5–2.45 equiv), in THF (0.25 M) was added dropwise a solution of ethylmagnesium bromide (3 M in Et2O, 1.5–2.45 equiv). The solution was stirred at 0 °C for 10 min and then at rt for 1 h. Crude aldehydes 6–10 were dissolved in THF (0.1 M), transferred into a dropping funnel and slowly added to the solution of the Grignard reagent at −15 °C. The mixture was stirred at −15 °C for 1 h 30 and was allowed to slowly warm to rt for 24 h. The reaction was then quenched by addition of a saturated aqueous solution of NH4Cl (40 mL) and THF was removed in vacuo. The aqueous phase was extracted with EtOAc and the combined organic layers were dried over Na2SO4, filtered and concentrated in vacuo. The crude foam was purified by flash chromatography.
Supporting Information
| Supporting Information File 1: Description of the materials and methods, and the preparation and characterization of new compounds. | ||
| Format: PDF | Size: 448.1 KB | Download |
| Supporting Information File 2: Copies of spectra for final compounds and NMR studies. | ||
| Format: PDF | Size: 4.1 MB | Download |
Acknowledgements
We thank the “Centre National de la Recherche Scientifique” and the “Ministère de l′Enseignement Supérieur et de la Recherche” for financial support of this work and for a Ph.D. grant to M. J. Fer. Assia Hessani (Université Paris Descartes) is gratefully acknowledged for mass spectra analyzes and Serge Turcaud for his expertise and advice in HPLC analysis.
References
-
Peters, H. L.; Ku, T. C.; Seley-Radtke, K. L. Curr. Med. Chem. 2015, 22, 3910–3921. doi:10.2174/0929867322666150818103624
Thematic Issue: Nucleoside/tide Analogues in Modern Drug Design.
Return to citation in text: [1] -
Burke, M. P.; Borland, K. M.; Litosh, V. A. Curr. Top. Med. Chem. 2016, 16, 1231–1241. doi:10.2174/1568026615666150915111933
Return to citation in text: [1] -
Bugg, T. D. H.; Lloyd, A. J.; Roper, D. I. Infect. Disord.: Drug Targets 2006, 6, 85–106. doi:10.2174/187152606784112128
Return to citation in text: [1] -
Shmalenyuk, E. R.; Chernousova, L. N.; Karpenko, I. L.; Kochetkov, S. N.; Smirnova, T. G.; Andreevskaya, S. N.; Chizhov, A. O.; Efremenkova, O. V.; Alexandrova, L. A. Bioorg. Med. Chem. 2013, 21, 4874–4884. doi:10.1016/j.bmc.2013.07.003
Return to citation in text: [1] -
Ichikawa, S.; Yamaguchi, M.; Matsuda, A. Curr. Med. Chem. 2015, 22, 3951–3979. doi:10.2174/0929867322666150818103502
Return to citation in text: [1] -
Jordheim, L. P.; Durantel, D.; Zoulim, F.; Dumontet, C. Nat. Rev. Drug Discovery 2013, 12, 447–464. doi:10.1038/nrd4010
Return to citation in text: [1] [2] -
Ewald, B.; Sampath, D.; Plunkett, W. Oncogene 2008, 27, 6522–6537. doi:10.1038/onc.2008.316
Return to citation in text: [1] -
Lesnikowski, Z.; Olejniczak, A.; Kilianska, Z.; Zolnierczyk, J.; Robak, T.; Mieczkowski, A. A nucleoside derivative for use as a drug, particularly for the treatment of chronic lymphocytic leukemia. PCT Int. Appl. WO2016020858 A2, Feb 11, 2016.
Return to citation in text: [1] -
Ivanov, M.; Barragan, I.; Ingelman-Sundberg, M. Trends Pharmacol. Sci. 2014, 35, 384–396. doi:10.1016/j.tips.2014.05.004
Return to citation in text: [1] -
Muthu, P.; Chen, H. X.; Lutz, S. ACS Chem. Biol. 2014, 9, 2326–2333. doi:10.1021/cb500463f
Return to citation in text: [1] -
Granqvist, L.; Virta, P. J. Org. Chem. 2015, 80, 7961–7970. doi:10.1021/acs.joc.5b00973
Return to citation in text: [1] -
Eppacher, S.; Solladié, N.; Bernet, B.; Vasella, A. Helv. Chim. Acta 2000, 83, 1311–1330. doi:10.1002/1522-2675(20000705)83:7<1311::AID-HLCA1311>3.0.CO;2-2
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11] -
Eppacher, S.; Christen, M.; Vasella, A. Helv. Chim. Acta 2004, 87, 3004–3020. doi:10.1002/hlca.200490271
Return to citation in text: [1] -
Rozners, E.; Katkevica, D.; Bizdena, E.; Strömberg, R. J. Am. Chem. Soc. 2003, 125, 12125–12136. doi:10.1021/ja0360900
Return to citation in text: [1] -
Maturano, M.; Catana, D.-A.; Lavedan, P.; Tarrat, N.; Saffon, N.; Payrastre, C.; Escudier, J.-M. Eur. J. Org. Chem. 2012, 721–730. doi:10.1002/ejoc.201101353
Return to citation in text: [1] [2] [3] -
Prakash, T. P.; Lima, W. F.; Murray, H. M.; Li, W.; Kinberger, G. A.; Chappell, A. E.; Gaus, H.; Seth, P. P.; Bhat, B.; Crooke, S. T.; Swayze, E. E. Nucleic Acids Res. 2015, 43, 2993–3011. doi:10.1093/nar/gkv162
Return to citation in text: [1] [2] -
Kel'in, A. V.; Zlatev, I.; Harp, J.; Jayaraman, M.; Bisbe, A.; O'Shea, J.; Taneja, N.; Manoharan, R. M.; Khan, S.; Charisse, K.; Maier, M. A.; Egli, M.; Rajeev, K. G.; Manoharan, M. J. Org. Chem. 2016, 81, 2261–2279. doi:10.1021/acs.joc.5b02375
Return to citation in text: [1] [2] [3] -
Meurillon, M.; Marton, Z.; Hospital, A.; Jordheim, L. P.; Béjaud, J.; Corinne Lionne, C.; Dumontet, C.; Périgaud, C.; Chaloin, L.; Peyrottes, S. Eur. J. Med. Chem. 2014, 77, 18–37. doi:10.1016/j.ejmech.2014.02.055
Return to citation in text: [1] -
Dini, C.; Collette, P.; Drochon, N.; Guillot, J. C.; Lemoine, G.; Mauvais, P.; Aszodi, J. Bioorg. Med. Chem. Lett. 2000, 10, 1839–1843. doi:10.1016/S0960-894X(00)00349-8
Return to citation in text: [1] -
Vorbrüggen, H.; Krolikiewicz, K. Angew. Chem., Int. Ed. Engl. 1975, 14, 421–422. doi:10.1002/anie.197504211
Return to citation in text: [1] -
Vorbrüggen, H.; Krolikiewicz, K.; Bennua, B. Chem. Ber. 1981, 114, 1234–1255. doi:10.1002/cber.19811140404
Return to citation in text: [1] -
Vorbrüggen, H.; Ruh-Pohlenz, C. Org. React. 2000, 55, 1–630. doi:10.1002/0471264180.or055.01
See for a review on nucleoside synthesis.
Return to citation in text: [1] -
Knapp, S.; Morriello, G. J.; Doss, G. A. Org. Lett. 2002, 4, 603–606. doi:10.1021/ol0102904
Return to citation in text: [1] -
Chun, M. W.; Kim, M. J.; Kim, H. O.; Kim, H.-D.; Kim, J. H.; Moon, H. R.; Jeong, L. S. Nucleosides, Nucleotides Nucleic Acids 2003, 22, 915–917. doi:10.1081/NCN-120022685
Return to citation in text: [1] -
Besada, P.; Costas, T.; Teijeira, M.; Terán, C. Eur. J. Med. Chem. 2010, 45, 6114–6119. doi:10.1016/j.ejmech.2010.09.046
Return to citation in text: [1] -
Hospital, A.; Meurillon, M.; Peyrottes, S.; Périgaud, C. Org. Lett. 2013, 15, 4778–4781. doi:10.1021/ol402143y
Return to citation in text: [1] -
Evina, C. M.; Guillerm, G. Tetrahedron Lett. 1996, 37, 163–166. doi:10.1016/0040-4039(95)02116-7
Return to citation in text: [1] -
Jung, K.-Y.; Hohl, R. J.; Wiemer, A. J.; Wiemer, D. F. Bioorg. Med. Chem. 2000, 8, 2501–2509. doi:10.1016/S0968-0896(00)00183-8
Return to citation in text: [1] -
Hirano, S.; Ichikawa, S.; Matsuda, A. Angew. Chem., Int. Ed. 2005, 44, 1854–1856. doi:10.1002/anie.200462439
Return to citation in text: [1] -
Gallier, F.; Alexandre, J. A. C.; El Amri, C.; Deville-Bonne, D.; Peyrottes, S.; Périgaud, C. ChemMedChem 2011, 6, 1094–1106. doi:10.1002/cmdc.201100068
Return to citation in text: [1] -
Sarabia, F.; Vivar-García, C.; García-Ruiz, C.; Martin-Ortiz, L.; Romero-Carasco, A. J. Org. Chem. 2012, 77, 1328–1339. doi:10.1021/jo202061t
Return to citation in text: [1] -
Fer, M. J.; Doan, P.; Prangé, T.; Calvet-Vitale, S.; Gravier-Pelletier, C. J. Org. Chem. 2014, 79, 7758–7765. doi:10.1021/jo501410m
Return to citation in text: [1] -
Sarabia, F.; Martín-Ortiz, L.; López-Herrera, F. J. Org. Biomol. Chem. 2003, 1, 3716–3725. doi:10.1039/B307674A
Return to citation in text: [1] -
Bligh, C. M.; Anzalone, L.; Jung, Y. C.; Zhang, Y.; Nugent, W. A. J. Org. Chem. 2014, 79, 3238–3243. doi:10.1021/jo500089t
Return to citation in text: [1] [2] -
Ranganathan, R.; Jones, G. H.; Moffatt, J. G. J. Org. Chem. 1974, 39, 290–298. doi:10.1021/jo00917a003
Return to citation in text: [1] [2] [3] [4] -
Ichikawa, S.; Matsuda, A. Nucleosides, Nucleotides Nucleic Acids 2004, 23, 239–253. doi:10.1081/NCN-120027831
Return to citation in text: [1] -
Kim, K. S.; Ahn, Y. H. Tetrahedron: Asymmetry 1998, 9, 3601–3605. doi:10.1016/S0957-4166(98)00366-8
Return to citation in text: [1] -
Gopinath, P.; Wang, L.; Abe, H.; Ravi, G.; Masuda, T.; Watanabe, T.; Shibasaki, M. Org. Lett. 2014, 16, 3364–3367. doi:10.1021/ol501397b
Return to citation in text: [1] -
Chen, X.; Wiemer, D. F. J. Org. Chem. 2003, 68, 6597–6604. doi:10.1021/jo0300136
Return to citation in text: [1] -
Králíková, Š.; Buděšínký, M.; Masojídková, M.; Rosenberg, I. Tetrahedron 2006, 62, 4917–4932. doi:10.1016/j.tet.2006.03.008
Return to citation in text: [1] -
Kurosu, M.; Li, K.; Crick, D. C. Org. Lett. 2009, 11, 2393–2396. doi:10.1021/ol900458w
Return to citation in text: [1] -
Hanessian, S.; Kloss, J.; Sugawara, T. J. Am. Chem. Soc. 1986, 108, 2758–2759. doi:10.1021/ja00270a047
Return to citation in text: [1] [2] -
Matsuda, A.; Kosaki, H.; Saitoh, Y.; Yoshimura, Y.; Minakawa, N.; Nakata, H. J. Med. Chem. 1998, 41, 2676–2678. doi:10.1021/jm9802822
Return to citation in text: [1] [2] [3] -
Eppacher, S.; Bhardwaj, P. K.; Bernet, B.; Gala, J. L. B.; Knöpfel, T.; Vasella, A. Helv. Chim. Acta 2004, 87, 2969–2986. doi:10.1002/hlca.200490269
Return to citation in text: [1] [2] [3] -
Murata, S.; Ichikawa, S.; Matsuda, A. Tetrahedron 2005, 61, 5837–5842. doi:10.1016/j.tet.2005.04.019
Return to citation in text: [1] [2] [3] -
Nakaya, T.; Matsuda, A.; Ichikawa, S. Org. Biomol. Chem. 2015, 13, 7720–7735. doi:10.1039/C5OB01037C
Return to citation in text: [1] [2] [3] -
Lolk, L.; Pøhlsgaard, J.; Jepsen, A. S.; Hansen, L. H.; Nielsen, H.; Steffansen, S. I.; Sparving, L.; Nielsen, A. B.; Vester, B.; Nielsen, P. J. Med. Chem. 2008, 51, 4957–4967. doi:10.1021/jm800261u
Return to citation in text: [1] -
Mitachi, K.; Aleiwi, B. A.; Schneider, C. M.; Siricilla, S.; Kurosu, M. J. Am. Chem. Soc. 2016, 138, 12975–12980. doi:10.1021/jacs.6b07395
Return to citation in text: [1] [2] -
Fer, M. J.; Olatunji, S.; Bouhss, A.; Calvet-Vitale, S.; Gravier-Pelletier, C. J. Org. Chem. 2013, 78, 10088–10105. doi:10.1021/jo4014035
Return to citation in text: [1] [2] -
Fer, M. J.; Bouhss, A.; Patrão, M.; Le Corre, L.; Pietrancosta, N.; Amoroso, A.; Joris, B.; Mengin-Lecreulx, D.; Calvet-Vitale, S.; Gravier-Pelletier, C. Org. Biomol. Chem. 2015, 13, 7193–7222. doi:10.1039/C5OB00707K
Return to citation in text: [1] -
Hwu, J. R.; Jain, M. L.; Tsai, F.-Y.; Tsay, S.-C.; Balakumar, A.; Hakimelahi, G. H. J. Org. Chem. 2000, 65, 5077–5088. doi:10.1021/jo000024o
Return to citation in text: [1] -
Mowat, J.; Kang, B.; Fonovic, B.; Dudding, T.; Britton, R. Org. Lett. 2009, 11, 2057–2060. doi:10.1021/ol900324s
Return to citation in text: [1]
| 48. | Mitachi, K.; Aleiwi, B. A.; Schneider, C. M.; Siricilla, S.; Kurosu, M. J. Am. Chem. Soc. 2016, 138, 12975–12980. doi:10.1021/jacs.6b07395 |
| 49. | Fer, M. J.; Olatunji, S.; Bouhss, A.; Calvet-Vitale, S.; Gravier-Pelletier, C. J. Org. Chem. 2013, 78, 10088–10105. doi:10.1021/jo4014035 |
| 50. | Fer, M. J.; Bouhss, A.; Patrão, M.; Le Corre, L.; Pietrancosta, N.; Amoroso, A.; Joris, B.; Mengin-Lecreulx, D.; Calvet-Vitale, S.; Gravier-Pelletier, C. Org. Biomol. Chem. 2015, 13, 7193–7222. doi:10.1039/C5OB00707K |
| 49. | Fer, M. J.; Olatunji, S.; Bouhss, A.; Calvet-Vitale, S.; Gravier-Pelletier, C. J. Org. Chem. 2013, 78, 10088–10105. doi:10.1021/jo4014035 |
| 1. |
Peters, H. L.; Ku, T. C.; Seley-Radtke, K. L. Curr. Med. Chem. 2015, 22, 3910–3921. doi:10.2174/0929867322666150818103624
Thematic Issue: Nucleoside/tide Analogues in Modern Drug Design. |
| 2. | Burke, M. P.; Borland, K. M.; Litosh, V. A. Curr. Top. Med. Chem. 2016, 16, 1231–1241. doi:10.2174/1568026615666150915111933 |
| 9. | Ivanov, M.; Barragan, I.; Ingelman-Sundberg, M. Trends Pharmacol. Sci. 2014, 35, 384–396. doi:10.1016/j.tips.2014.05.004 |
| 40. | Králíková, Š.; Buděšínký, M.; Masojídková, M.; Rosenberg, I. Tetrahedron 2006, 62, 4917–4932. doi:10.1016/j.tet.2006.03.008 |
| 12. | Eppacher, S.; Solladié, N.; Bernet, B.; Vasella, A. Helv. Chim. Acta 2000, 83, 1311–1330. doi:10.1002/1522-2675(20000705)83:7<1311::AID-HLCA1311>3.0.CO;2-2 |
| 7. | Ewald, B.; Sampath, D.; Plunkett, W. Oncogene 2008, 27, 6522–6537. doi:10.1038/onc.2008.316 |
| 8. | Lesnikowski, Z.; Olejniczak, A.; Kilianska, Z.; Zolnierczyk, J.; Robak, T.; Mieczkowski, A. A nucleoside derivative for use as a drug, particularly for the treatment of chronic lymphocytic leukemia. PCT Int. Appl. WO2016020858 A2, Feb 11, 2016. |
| 41. | Kurosu, M.; Li, K.; Crick, D. C. Org. Lett. 2009, 11, 2393–2396. doi:10.1021/ol900458w |
| 52. | Mowat, J.; Kang, B.; Fonovic, B.; Dudding, T.; Britton, R. Org. Lett. 2009, 11, 2057–2060. doi:10.1021/ol900324s |
| 6. | Jordheim, L. P.; Durantel, D.; Zoulim, F.; Dumontet, C. Nat. Rev. Drug Discovery 2013, 12, 447–464. doi:10.1038/nrd4010 |
| 15. | Maturano, M.; Catana, D.-A.; Lavedan, P.; Tarrat, N.; Saffon, N.; Payrastre, C.; Escudier, J.-M. Eur. J. Org. Chem. 2012, 721–730. doi:10.1002/ejoc.201101353 |
| 36. | Ichikawa, S.; Matsuda, A. Nucleosides, Nucleotides Nucleic Acids 2004, 23, 239–253. doi:10.1081/NCN-120027831 |
| 37. | Kim, K. S.; Ahn, Y. H. Tetrahedron: Asymmetry 1998, 9, 3601–3605. doi:10.1016/S0957-4166(98)00366-8 |
| 38. | Gopinath, P.; Wang, L.; Abe, H.; Ravi, G.; Masuda, T.; Watanabe, T.; Shibasaki, M. Org. Lett. 2014, 16, 3364–3367. doi:10.1021/ol501397b |
| 12. | Eppacher, S.; Solladié, N.; Bernet, B.; Vasella, A. Helv. Chim. Acta 2000, 83, 1311–1330. doi:10.1002/1522-2675(20000705)83:7<1311::AID-HLCA1311>3.0.CO;2-2 |
| 3. | Bugg, T. D. H.; Lloyd, A. J.; Roper, D. I. Infect. Disord.: Drug Targets 2006, 6, 85–106. doi:10.2174/187152606784112128 |
| 4. | Shmalenyuk, E. R.; Chernousova, L. N.; Karpenko, I. L.; Kochetkov, S. N.; Smirnova, T. G.; Andreevskaya, S. N.; Chizhov, A. O.; Efremenkova, O. V.; Alexandrova, L. A. Bioorg. Med. Chem. 2013, 21, 4874–4884. doi:10.1016/j.bmc.2013.07.003 |
| 5. | Ichikawa, S.; Yamaguchi, M.; Matsuda, A. Curr. Med. Chem. 2015, 22, 3951–3979. doi:10.2174/0929867322666150818103502 |
| 39. | Chen, X.; Wiemer, D. F. J. Org. Chem. 2003, 68, 6597–6604. doi:10.1021/jo0300136 |
| 12. | Eppacher, S.; Solladié, N.; Bernet, B.; Vasella, A. Helv. Chim. Acta 2000, 83, 1311–1330. doi:10.1002/1522-2675(20000705)83:7<1311::AID-HLCA1311>3.0.CO;2-2 |
| 16. | Prakash, T. P.; Lima, W. F.; Murray, H. M.; Li, W.; Kinberger, G. A.; Chappell, A. E.; Gaus, H.; Seth, P. P.; Bhat, B.; Crooke, S. T.; Swayze, E. E. Nucleic Acids Res. 2015, 43, 2993–3011. doi:10.1093/nar/gkv162 |
| 17. | Kel'in, A. V.; Zlatev, I.; Harp, J.; Jayaraman, M.; Bisbe, A.; O'Shea, J.; Taneja, N.; Manoharan, R. M.; Khan, S.; Charisse, K.; Maier, M. A.; Egli, M.; Rajeev, K. G.; Manoharan, M. J. Org. Chem. 2016, 81, 2261–2279. doi:10.1021/acs.joc.5b02375 |
| 18. | Meurillon, M.; Marton, Z.; Hospital, A.; Jordheim, L. P.; Béjaud, J.; Corinne Lionne, C.; Dumontet, C.; Périgaud, C.; Chaloin, L.; Peyrottes, S. Eur. J. Med. Chem. 2014, 77, 18–37. doi:10.1016/j.ejmech.2014.02.055 |
| 19. | Dini, C.; Collette, P.; Drochon, N.; Guillot, J. C.; Lemoine, G.; Mauvais, P.; Aszodi, J. Bioorg. Med. Chem. Lett. 2000, 10, 1839–1843. doi:10.1016/S0960-894X(00)00349-8 |
| 27. | Evina, C. M.; Guillerm, G. Tetrahedron Lett. 1996, 37, 163–166. doi:10.1016/0040-4039(95)02116-7 |
| 28. | Jung, K.-Y.; Hohl, R. J.; Wiemer, A. J.; Wiemer, D. F. Bioorg. Med. Chem. 2000, 8, 2501–2509. doi:10.1016/S0968-0896(00)00183-8 |
| 29. | Hirano, S.; Ichikawa, S.; Matsuda, A. Angew. Chem., Int. Ed. 2005, 44, 1854–1856. doi:10.1002/anie.200462439 |
| 30. | Gallier, F.; Alexandre, J. A. C.; El Amri, C.; Deville-Bonne, D.; Peyrottes, S.; Périgaud, C. ChemMedChem 2011, 6, 1094–1106. doi:10.1002/cmdc.201100068 |
| 31. | Sarabia, F.; Vivar-García, C.; García-Ruiz, C.; Martin-Ortiz, L.; Romero-Carasco, A. J. Org. Chem. 2012, 77, 1328–1339. doi:10.1021/jo202061t |
| 32. | Fer, M. J.; Doan, P.; Prangé, T.; Calvet-Vitale, S.; Gravier-Pelletier, C. J. Org. Chem. 2014, 79, 7758–7765. doi:10.1021/jo501410m |
| 51. | Hwu, J. R.; Jain, M. L.; Tsai, F.-Y.; Tsay, S.-C.; Balakumar, A.; Hakimelahi, G. H. J. Org. Chem. 2000, 65, 5077–5088. doi:10.1021/jo000024o |
| 6. | Jordheim, L. P.; Durantel, D.; Zoulim, F.; Dumontet, C. Nat. Rev. Drug Discovery 2013, 12, 447–464. doi:10.1038/nrd4010 |
| 12. | Eppacher, S.; Solladié, N.; Bernet, B.; Vasella, A. Helv. Chim. Acta 2000, 83, 1311–1330. doi:10.1002/1522-2675(20000705)83:7<1311::AID-HLCA1311>3.0.CO;2-2 |
| 33. | Sarabia, F.; Martín-Ortiz, L.; López-Herrera, F. J. Org. Biomol. Chem. 2003, 1, 3716–3725. doi:10.1039/B307674A |
| 34. | Bligh, C. M.; Anzalone, L.; Jung, Y. C.; Zhang, Y.; Nugent, W. A. J. Org. Chem. 2014, 79, 3238–3243. doi:10.1021/jo500089t |
| 35. | Ranganathan, R.; Jones, G. H.; Moffatt, J. G. J. Org. Chem. 1974, 39, 290–298. doi:10.1021/jo00917a003 |
| 12. | Eppacher, S.; Solladié, N.; Bernet, B.; Vasella, A. Helv. Chim. Acta 2000, 83, 1311–1330. doi:10.1002/1522-2675(20000705)83:7<1311::AID-HLCA1311>3.0.CO;2-2 |
| 12. | Eppacher, S.; Solladié, N.; Bernet, B.; Vasella, A. Helv. Chim. Acta 2000, 83, 1311–1330. doi:10.1002/1522-2675(20000705)83:7<1311::AID-HLCA1311>3.0.CO;2-2 |
| 13. | Eppacher, S.; Christen, M.; Vasella, A. Helv. Chim. Acta 2004, 87, 3004–3020. doi:10.1002/hlca.200490271 |
| 14. | Rozners, E.; Katkevica, D.; Bizdena, E.; Strömberg, R. J. Am. Chem. Soc. 2003, 125, 12125–12136. doi:10.1021/ja0360900 |
| 15. | Maturano, M.; Catana, D.-A.; Lavedan, P.; Tarrat, N.; Saffon, N.; Payrastre, C.; Escudier, J.-M. Eur. J. Org. Chem. 2012, 721–730. doi:10.1002/ejoc.201101353 |
| 16. | Prakash, T. P.; Lima, W. F.; Murray, H. M.; Li, W.; Kinberger, G. A.; Chappell, A. E.; Gaus, H.; Seth, P. P.; Bhat, B.; Crooke, S. T.; Swayze, E. E. Nucleic Acids Res. 2015, 43, 2993–3011. doi:10.1093/nar/gkv162 |
| 17. | Kel'in, A. V.; Zlatev, I.; Harp, J.; Jayaraman, M.; Bisbe, A.; O'Shea, J.; Taneja, N.; Manoharan, R. M.; Khan, S.; Charisse, K.; Maier, M. A.; Egli, M.; Rajeev, K. G.; Manoharan, M. J. Org. Chem. 2016, 81, 2261–2279. doi:10.1021/acs.joc.5b02375 |
| 12. | Eppacher, S.; Solladié, N.; Bernet, B.; Vasella, A. Helv. Chim. Acta 2000, 83, 1311–1330. doi:10.1002/1522-2675(20000705)83:7<1311::AID-HLCA1311>3.0.CO;2-2 |
| 10. | Muthu, P.; Chen, H. X.; Lutz, S. ACS Chem. Biol. 2014, 9, 2326–2333. doi:10.1021/cb500463f |
| 11. | Granqvist, L.; Virta, P. J. Org. Chem. 2015, 80, 7961–7970. doi:10.1021/acs.joc.5b00973 |
| 20. | Vorbrüggen, H.; Krolikiewicz, K. Angew. Chem., Int. Ed. Engl. 1975, 14, 421–422. doi:10.1002/anie.197504211 |
| 21. | Vorbrüggen, H.; Krolikiewicz, K.; Bennua, B. Chem. Ber. 1981, 114, 1234–1255. doi:10.1002/cber.19811140404 |
| 22. |
Vorbrüggen, H.; Ruh-Pohlenz, C. Org. React. 2000, 55, 1–630. doi:10.1002/0471264180.or055.01
See for a review on nucleoside synthesis. |
| 23. | Knapp, S.; Morriello, G. J.; Doss, G. A. Org. Lett. 2002, 4, 603–606. doi:10.1021/ol0102904 |
| 24. | Chun, M. W.; Kim, M. J.; Kim, H. O.; Kim, H.-D.; Kim, J. H.; Moon, H. R.; Jeong, L. S. Nucleosides, Nucleotides Nucleic Acids 2003, 22, 915–917. doi:10.1081/NCN-120022685 |
| 25. | Besada, P.; Costas, T.; Teijeira, M.; Terán, C. Eur. J. Med. Chem. 2010, 45, 6114–6119. doi:10.1016/j.ejmech.2010.09.046 |
| 26. | Hospital, A.; Meurillon, M.; Peyrottes, S.; Périgaud, C. Org. Lett. 2013, 15, 4778–4781. doi:10.1021/ol402143y |
| 15. | Maturano, M.; Catana, D.-A.; Lavedan, P.; Tarrat, N.; Saffon, N.; Payrastre, C.; Escudier, J.-M. Eur. J. Org. Chem. 2012, 721–730. doi:10.1002/ejoc.201101353 |
| 12. | Eppacher, S.; Solladié, N.; Bernet, B.; Vasella, A. Helv. Chim. Acta 2000, 83, 1311–1330. doi:10.1002/1522-2675(20000705)83:7<1311::AID-HLCA1311>3.0.CO;2-2 |
| 35. | Ranganathan, R.; Jones, G. H.; Moffatt, J. G. J. Org. Chem. 1974, 39, 290–298. doi:10.1021/jo00917a003 |
| 43. | Matsuda, A.; Kosaki, H.; Saitoh, Y.; Yoshimura, Y.; Minakawa, N.; Nakata, H. J. Med. Chem. 1998, 41, 2676–2678. doi:10.1021/jm9802822 |
| 44. | Eppacher, S.; Bhardwaj, P. K.; Bernet, B.; Gala, J. L. B.; Knöpfel, T.; Vasella, A. Helv. Chim. Acta 2004, 87, 2969–2986. doi:10.1002/hlca.200490269 |
| 45. | Murata, S.; Ichikawa, S.; Matsuda, A. Tetrahedron 2005, 61, 5837–5842. doi:10.1016/j.tet.2005.04.019 |
| 46. | Nakaya, T.; Matsuda, A.; Ichikawa, S. Org. Biomol. Chem. 2015, 13, 7720–7735. doi:10.1039/C5OB01037C |
| 48. | Mitachi, K.; Aleiwi, B. A.; Schneider, C. M.; Siricilla, S.; Kurosu, M. J. Am. Chem. Soc. 2016, 138, 12975–12980. doi:10.1021/jacs.6b07395 |
| 12. | Eppacher, S.; Solladié, N.; Bernet, B.; Vasella, A. Helv. Chim. Acta 2000, 83, 1311–1330. doi:10.1002/1522-2675(20000705)83:7<1311::AID-HLCA1311>3.0.CO;2-2 |
| 17. | Kel'in, A. V.; Zlatev, I.; Harp, J.; Jayaraman, M.; Bisbe, A.; O'Shea, J.; Taneja, N.; Manoharan, R. M.; Khan, S.; Charisse, K.; Maier, M. A.; Egli, M.; Rajeev, K. G.; Manoharan, M. J. Org. Chem. 2016, 81, 2261–2279. doi:10.1021/acs.joc.5b02375 |
| 35. | Ranganathan, R.; Jones, G. H.; Moffatt, J. G. J. Org. Chem. 1974, 39, 290–298. doi:10.1021/jo00917a003 |
| 42. | Hanessian, S.; Kloss, J.; Sugawara, T. J. Am. Chem. Soc. 1986, 108, 2758–2759. doi:10.1021/ja00270a047 |
| 43. | Matsuda, A.; Kosaki, H.; Saitoh, Y.; Yoshimura, Y.; Minakawa, N.; Nakata, H. J. Med. Chem. 1998, 41, 2676–2678. doi:10.1021/jm9802822 |
| 44. | Eppacher, S.; Bhardwaj, P. K.; Bernet, B.; Gala, J. L. B.; Knöpfel, T.; Vasella, A. Helv. Chim. Acta 2004, 87, 2969–2986. doi:10.1002/hlca.200490269 |
| 45. | Murata, S.; Ichikawa, S.; Matsuda, A. Tetrahedron 2005, 61, 5837–5842. doi:10.1016/j.tet.2005.04.019 |
| 46. | Nakaya, T.; Matsuda, A.; Ichikawa, S. Org. Biomol. Chem. 2015, 13, 7720–7735. doi:10.1039/C5OB01037C |
| 47. | Lolk, L.; Pøhlsgaard, J.; Jepsen, A. S.; Hansen, L. H.; Nielsen, H.; Steffansen, S. I.; Sparving, L.; Nielsen, A. B.; Vester, B.; Nielsen, P. J. Med. Chem. 2008, 51, 4957–4967. doi:10.1021/jm800261u |
| 34. | Bligh, C. M.; Anzalone, L.; Jung, Y. C.; Zhang, Y.; Nugent, W. A. J. Org. Chem. 2014, 79, 3238–3243. doi:10.1021/jo500089t |
| 45. | Murata, S.; Ichikawa, S.; Matsuda, A. Tetrahedron 2005, 61, 5837–5842. doi:10.1016/j.tet.2005.04.019 |
| 46. | Nakaya, T.; Matsuda, A.; Ichikawa, S. Org. Biomol. Chem. 2015, 13, 7720–7735. doi:10.1039/C5OB01037C |
| 12. | Eppacher, S.; Solladié, N.; Bernet, B.; Vasella, A. Helv. Chim. Acta 2000, 83, 1311–1330. doi:10.1002/1522-2675(20000705)83:7<1311::AID-HLCA1311>3.0.CO;2-2 |
| 44. | Eppacher, S.; Bhardwaj, P. K.; Bernet, B.; Gala, J. L. B.; Knöpfel, T.; Vasella, A. Helv. Chim. Acta 2004, 87, 2969–2986. doi:10.1002/hlca.200490269 |
| 43. | Matsuda, A.; Kosaki, H.; Saitoh, Y.; Yoshimura, Y.; Minakawa, N.; Nakata, H. J. Med. Chem. 1998, 41, 2676–2678. doi:10.1021/jm9802822 |
| 12. | Eppacher, S.; Solladié, N.; Bernet, B.; Vasella, A. Helv. Chim. Acta 2000, 83, 1311–1330. doi:10.1002/1522-2675(20000705)83:7<1311::AID-HLCA1311>3.0.CO;2-2 |
| 35. | Ranganathan, R.; Jones, G. H.; Moffatt, J. G. J. Org. Chem. 1974, 39, 290–298. doi:10.1021/jo00917a003 |
| 42. | Hanessian, S.; Kloss, J.; Sugawara, T. J. Am. Chem. Soc. 1986, 108, 2758–2759. doi:10.1021/ja00270a047 |
© 2017 Ben Othman et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)