Abstract
Multiple fluorination of glycostructures has emerged as an attractive way of modulating their protein affinity, metabolic stability, and lipophilicity. Here we described the synthesis of a series of mono-, di- and trifluorinated N-acetyl-ᴅ-glucosamine and ᴅ-galactosamine analogs. The key intermediates are the corresponding multiply fluorinated glucosazide and galactosazide thioglycosides prepared from deoxyfluorinated 1,6-anhydro-2-azido-β-ᴅ-hexopyranose precursors by ring-opening reaction with phenyl trimethylsilyl sulfide. Nucleophilic deoxyfluorination at C4 and C6 by reaction with DAST, thioglycoside hydrolysis and azide/acetamide transformation completed the synthesis.

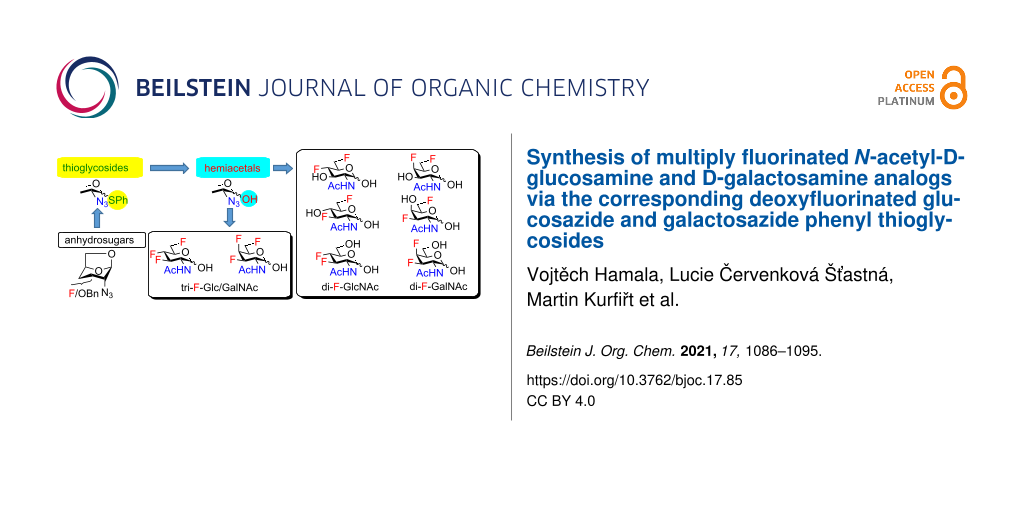
Graphical Abstract
Introduction
Fluorinated carbohydrates are versatile carbohydrate mimetics used to probe or manipulate the recognition of carbohydrates by carbohydrate-binding proteins or carbohydrate-processing enzymes [1-7]. The introduction of additional fluorine atoms into a monofluorinated carbohydrate is an attractive way of modulating the binding affinity and pharmacokinetic properties of fluorinated glycomimetics. Hydrophobic segments incorporating multiple C–F bonds could (1) reduce the desolvation penalty associated with binding of hydrophilic natural carbohydrates [8], and (2) create additional contacts with the binding cavity via electrostatic and dipolar interactions with C–F bonds [9,10], new intermolecular hydrogen bonds [11], or rearrangement of hydrogen bond-mediating water molecules [12]. The fluorination of sugars is also a promising strategy to improve unfavorable pharmacokinetic properties of natural carbohydrates such as low lipophilicity [13-16] and fast metabolic degradation [17-19]. Over the last few years, considerable effort has been expended on the synthesis of unprotected multiply-deoxyfluorinated monosaccharides, including a complete series of mono-, di-, and trifluorinated ᴅ-glucose [15], difluorinated [20] and tetrafluorinated [13] ᴅ-galactose, and 2,3,4-trifluoro analogs of ᴅ-mannose, ᴅ-galactose, ᴅ-allose, ᴅ-talose, and ᴅ-altrose [13,21].
Unprotected multiply-deoxyfluorinated N-acetyl-ᴅ-glucosamine (GlcNAc) and N-acetyl-ᴅ-galactosamine (GalNAc) have not yet been described except for a 4,6-difluoro-GalNAc analog [22], although GlcNAc is the most abundant monosaccharide component of mammalian glycans [23], and GalNAc occurs in important glycan structures including the TN and T antigen and their sialylated forms [24]. A complete series of O-protected monofluorinated [22,25-32] and several difluorinated [22,26,33,34] GlcNAc/GalNAc analogs have been prepared. Some acylated mono- and difluorinated analogs have potential in biomedical applications due to their ability to inhibit the glycan and glycosaminoglycan biosynthesis [34-37]. The fluorine substituent has typically been introduced into these GlcNAc and GalNAc analogues using nucleophilic fluorination. The primary position (C6 hydroxy group) was fluorinated by reaction with diethylamino sulfurtrifluoride (DAST) [27]. This reaction was greatly improved by microwave irradiation, especially in the GalNAc series [31,32]. The deoxyfluorination of the secondary hydroxy groups at the 3- and 4-positions was accomplished using a treatment of the C3/4 hydroxy groups with DAST [22,25,26,30,35], or reaction of C3/C4 methanesulfonate or trifluoromethanesulfonate esters with a source of nucleophilic fluorine, such as TBAF or KF [22,25,34]. Although these fluorinations usually proceeded with inversion of configuration, the acetylated 3-fluoro-GlcNAc analogue was most conveniently accessed using retentive DAST fluorination of 2-azido-4-O-benzyl-2-deoxy-1,6-anhydro-β-ᴅ-glucopyranose [26]. Preparation of glycostructures comprising multifluorinated GlcNAc and GalNAc will be greatly facilitated if synthetic routes to the corresponding glycosyl donors are developed. Here we describe the synthesis of a complete series of unprotected GlcNAc and GalNAc analogs systematically deoxyfluorinated at all non-anomeric hydroxy positions. The key synthetic intermediates are multifluorinated glucosazide and galactosazide thioglycosides and hemiacetals, which are also valuable glycosyl donors for the installation of a 1,2-cis-linked multifluorinated GlcNAc and GalNAc moiety.
Results and Discussion
Our approach to the synthesis is summarized in Scheme 1. Challenging regio- and stereoselective introduction of fluorine at C3 and C4 of the pyranose ring, together with azide installation at C2, can be accomplished by nucleophilic fluorination and azidolysis starting from dianhydro derivatives 1 and 2 as we described previously [26]. The resulting intermediates 3 can be transformed into 2-azidohexopyranosides 4 by cleavage of the internal acetal and protection of the anomeric position. Deoxyfluorination at C6 should then afford intermediates 6. Protecting-group manipulation of intermediates 4 and 6 should deliver the required fluoro analogs. The initially contemplated conversion of intermediates 3 into acetates 5 [26], followed by base-catalyzed O-deacetylation, led to substantial decomposition. These observations are consistent with the recently reported instability of O-acylated GlcNAc under basic conditions due to elimination reactions of transient hemiacetal intermediates [38]. This instability of amino sugar hemiacetals underscores the requirement to both protect the anomeric position with a robust protecting group and to conduct final deprotection under neutral conditions. After initial experimentation with benzyl glycosides (Scheme 1, PG = OBn), phenyl thioglycosides (Scheme 1, PG = SPh), readily available from 1,6-anhydropyranoses [39] as we described earlier [40] were found to fulfill this requirement satisfactorily.
![[1860-5397-17-85-i1]](/bjoc/content/inline/1860-5397-17-85-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Retrosynthetic analysis of the target fluoro analogs.
Scheme 1: Retrosynthetic analysis of the target fluoro analogs.
Accordingly, the synthesis started from known fluorinated 1,6-anhydro-2-azidohexopyranoses 7–13 (Scheme 2) [26,40]. Reaction of compounds 7–10 with phenyl trimethylsilyl sulfide (PhSTMS) and ZnI2 delivered phenyl thioglycosides 14–17 [40]. 1,6-Anhydropyranoses 11 and 12 under these conditions produced the expected thioglycosides 18 and 19, respectively. Difluorinated derivative 13 decomposed on reaction with the PhSTMS/ZnI2 system. The separation of the anomers of products 14–19 was attempted because of the risk of thiophenyl migration in the subsequent C6 deoxyfluorination, which would likely occur with the β-anomers of 14–19 [41]. The complete separation of the α-anomer by conventional silica gel column chromatography was possible for thioglycosides 14, 16, 17, and 19, while the products 15 and 18 were obtainable as enriched α-anomers (α/β ≥ 3.3:1). Cleavage of the internal acetal with PhSTMS was accompanied by the formation of low quantities of side-products detectable by TLC and separable by careful chromatography except for the cleavage of 12 where the side products co-eluted with the fraction containing the β-anomer of the product. In the case of the cleavage of 1,6-anhydro derivative 10, we were able to isolate one of the side-products in sufficient purity and quantity to assign the structure of C-furanoside 20 (Scheme 2). This compound resulted from pyranose ring contraction probably caused by intramolecular displacement of the C2 azide aided by coordination of ZnI2. When the α-anomer of thioglycoside 17 was separately subjected to the reaction conditions, the byproduct 20 started to form in trace amounts in accordance with the suggested mechanism. The ring contraction may involve formation of a transient oxiranium cation as suggested in Scheme 2 [42-45]. Analogous ring-contraction reactions have been described for substrates possessing a good C2 leaving group [42,46-50].
![[1860-5397-17-85-i2]](/bjoc/content/inline/1860-5397-17-85-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Conversion of 1,6-anhydro derivatives into thioglycosides, and a possible mechanism for the formation of C-furanosides by ring contraction.
Scheme 2: Conversion of 1,6-anhydro derivatives into thioglycosides, and a possible mechanism for the formati...
We initially considered converting thioglycosides 14–19 to benzyl glycosides because thioglycosides give glycosyl fluorides on reaction with diethylamino sulfurtrifluoride (DAST) [51], but our experiments revealed that DAST-mediated C6-deoxyfluorination of thioglycosides 14–17 and 19 proceeded satisfactorily under microwave irradiation, on condition that pure or substantially enriched α-anomers were subjected to reaction with DAST, yielding thioglycosides 22–26 (Scheme 3). Reaction of β-thiogalactosides possessing an unprotected C6 hydroxy group with DAST was accompanied by migration of the anomeric thio-aglycone to C6 [41,52] as shown for β-thiogalactoside β-17, which mostly delivered migration product 32 (Scheme 3, see also the synthesis of compound 24 in the Supporting Information File 1). However, β-thioglucoside β-14 yielded only 6% of migration product 33 together with the main C6-fluoro product β-22, suggesting that starting fluorinated 2-azido-thioglucosides were significantly less prone to thiophenyl migration than 2-azidothiogalactosides were. This was convenient because thioglucosides 15 and 18 (vide infra) were available for deoxyfluorination only as enriched anomeric mixtures α/β ≥ 3.3:1 and any traces of the migration products were removed in the subsequent thioaglycone hydrolysis. Thioglycosides 14–17 and 19 were also O6-benzylated [40] to thioglycosides 27–31 (Scheme 3).
![[1860-5397-17-85-i3]](/bjoc/content/inline/1860-5397-17-85-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Deoxyfluorination and O-benzylation of thioglycosides and thioaglycone migration.
Scheme 3: Deoxyfluorination and O-benzylation of thioglycosides and thioaglycone migration.
As the 3,4-difluorinated thiogalactoside could not be accessed from compound 13 by reaction with PhSTMS/ZnI2 (Scheme 2), it was necessary to obtain 3,4-difluoro and 3,4,6-trifluoro analogs of GalNAc from 3-fluoro-4,6-diol 18. According to precedents in the literature [53], deoxyfluorination of the C4-hydroxy group in compound 18 was expected to occur with inversion of configuration to give the desired galacto-configured 4-fluoro products. Accordingly, treatment of diol 18 with DAST resulted in deoxyfluorination of both hydroxy groups to yield trifluoro thiogalactosazide 34 (Scheme 3). 4,6-O-Benzylidenation of diol 18 followed by regioselective opening of the benzylidene acetal produced compound 35. Subsequent DAST deoxyfluorination delivered the desired thioglycoside 36 (Scheme 3). For both compounds 18 and 35, deoxyfluorination of the C4 hydroxy group occurred with inversion of configuration. Thioglycosides 22–31, 34 and 36 were then hydrolyzed into the corresponding hemiacetals 37–48 using treatment with NBS in acetone/water (Scheme 4).
![[1860-5397-17-85-i4]](/bjoc/content/inline/1860-5397-17-85-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
To obtain the target fluoro analogs, the hemiacetals 37–48 were debenzylated and their azide group converted to an acetamide. Although palladium-catalyzed hydrogenolysis in ethanol/acetic anhydride appeared to be a logical deprotection step [26], the desired fluoro sugars were contaminated with varying quantities of unidentified byproducts. However, clean debenzylation was achieved by first converting the azide to an acetamide on reaction with thioacetic acid [54,55]. Hence, the hemiacetals were reacted with thioacetic acid in pyridine to give acetamides 49–58 (Scheme 5) and the target trifluoro analogs 59 and 60. Reversing the order of hemiacetal and acetamide formation was not an option because NBS-promoted hydrolysis of 2-acetamido thioglycosides was sluggish and incomplete. Protecting the primary hydroxy group at C6 by O-benzylation (Scheme 3, compounds 27–31) was essential before treatment with thioacetic acid; otherwise, an O6-acetylated byproduct was formed. Acetylation of the anomeric hydroxy group occurred only to a very limited degree upon reaction with AcSH in pyridine and traces of O1 acetates were removed by chromatography or recrystallization. Palladium-catalyzed hydrogenolytic debenzylation of 49–58 then yielded the target fluoro analogs 61–70. To complete the series of fluorinated analogs for the purpose of comparing their NMR spectra, the known C6 monodeoxyfluorinated compounds 71 [27,28] and 72 [29] were prepared by published procedures [27,29].
![[1860-5397-17-85-i5]](/bjoc/content/inline/1860-5397-17-85-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Synthesis of the target compounds by azide/acetamide conversion and debenzylation.
Scheme 5: Synthesis of the target compounds by azide/acetamide conversion and debenzylation.
The magnitudes of the vicinal 3JH-H, 3JH-F, 3JC-F, geminal 2JH-F, 2JC-F, and one-bond 1JC-F coupling constants confirmed the expected fluorination pattern and ᴅ-gluco or ᴅ-galacto configuration for all fluoro analogs 59–72. The values of the coupling constants correlated with the 4C1 conformation adopted by the target fluoro analogs in solution (Table 1 and Table S1 in Supporting Information File 1). For example, the magnitude of the germinal fluorine–carbon coupling 2JC5-F4 indicated an antiperiplanar (2JC5-F4 = 23.2–24.2 Hz, ᴅ-gluco configuration, F4 equatorial) or a gauche (2JC5-F4 = 17.5–18.1 Hz, ᴅ-galacto configuration, F4 axial) relationship between the C4‒F and C5‒O bonds [56]. Similarly, the values of 3JH3/H5-F4 coupling constants reflected an axial (3JH3/H5-F = 25.5–30.3 Hz) or equatorial (3JH3-F4 = 14.8–16.8 Hz, 3JH5-F4 = 2.5–4.8 Hz) position of the C4 fluorine substituent [57]. Moreover, evaluation of 3JH5-F6 coupling constants revealed that 6-fluoro ᴅ-gluco analogs 59, 62, 64, and 71 assumed preferentially gauche,gauche (gg) conformation of the exocyclic C5‒C6 bond in solution (3JH5-F6 = 24.6–27.1 Hz), whereas the corresponding ᴅ-galacto-configured analogs 60, 67, 69, and 72 adopted gauche,trans (gt) or trans,gauche (tg) conformations to a significant degree (3JH5-F6 = 12.7–14.6 Hz). These findings were in accordance with the previous reports by Giguѐre [13,15,58].
Table 1: The values [Hz] of selected coupling constants. Boldfaced values illustrate the trends discussed in the text.
| compound | 2JC3-F4 | 1JC4-F4 | 2JC5-F4 | 3JH3-F4 | 2JH4-F4 | 3JH5-F4 | ||
![[Graphic 1]](/bjoc/content/inline/1860-5397-17-85-i6.svg?max-width=637&scale=1.0)
β-63 |
18.6 | 180.9 | 24.2 | 15.6 | 50.8 | 2.5 | ||
![[Graphic 2]](/bjoc/content/inline/1860-5397-17-85-i7.svg?max-width=637&scale=1.0)
α-68 |
19.1 | 180.1 | 18.1 | 28.9 | 50.5 | 30.3 | ||
| 4JC3-F6 | 3JC4-F6 | 2JC5-F6 | 1JC6-F6 | 3JH5-F6 | 2JH6-F6 | 2JH6'-F6 | ||
![[Graphic 3]](/bjoc/content/inline/1860-5397-17-85-i8.svg?max-width=637&scale=1.0)
α-71 |
0.7 | 7 | 17.9 | 171.3 | 27.1 | 48.2 | 48.2 | |
![[Graphic 4]](/bjoc/content/inline/1860-5397-17-85-i9.svg?max-width=637&scale=1.0)
α-72 |
1.1 | 2.5 | 13.0 | 166.8 | 14.6 | 46.4 | 48.1 | |
| 3JC2-F4 | 2JC3-F4 | 1JC4-F4 | 2JC5-F4 | 3JC4-F6 | 2JC5-F6 | 1JC6-F6 | ||
![[Graphic 5]](/bjoc/content/inline/1860-5397-17-85-i10.svg?max-width=637&scale=1.0)
α-64 |
8.0 | 18.5 | 181.2 | 23.7 | 7.4 | 18.2 | 172.5 | |
![[Graphic 6]](/bjoc/content/inline/1860-5397-17-85-i11.svg?max-width=637&scale=1.0)
α-69 |
2.7 | 18.8 | 180.1 | 17.7 | 5.9 | 23.1 | 168.1 | |
| 5JH1-F4 | 3JH3-F4 | 2JH4-F4 | 3JH5-F4 | 3JH5-F6 | 2JH6-F6 | 2JH6'-F6 | ||
![[Graphic 7]](/bjoc/content/inline/1860-5397-17-85-i12.svg?max-width=637&scale=1.0)
α-64 |
3.3 | 14.8 | 50.6 | 4.1 | 26.6 | 48.1 | 47.5 | |
![[Graphic 8]](/bjoc/content/inline/1860-5397-17-85-i13.svg?max-width=637&scale=1.0)
α-69 |
n/a | 28.9 | 51.0 | 30.3 | 12.7 | 47.3 | 46.2 | |
Conclusion
In summary, we have demonstrated that multiply deoxyfluorinated GlcNAc and GalNAc are accessible via the corresponding multifluorinated 1-thiophenyl gluco- and galactosazides. Installation of the thiophenyl aglycone permits C6 deoxyfluorination and circumvents the problems resulting from the low stability of amino sugar hemiacetals. The prepared polyfluorinated thiodonors and hemiacetals are valuable intermediates in oligosaccharide synthesis and their utility in glycosylation is currently being studied in our group.
Funding
We thank the Czech Science Foundation (grant no. 17-18203S) and Ministry of Education, Youth and Sport (INTER COST, no. LTC20052) for supporting our research. This publication is partially based on research done within the framework of COST Action CA18103 (INNOGLY) supported by European Cooperation in Science and Technology.
References
-
Linclau, B.; Ardá, A.; Reichardt, N.-C.; Sollogoub, M.; Unione, L.; Vincent, S. P.; Jiménez-Barbero, J. Chem. Soc. Rev. 2020, 49, 3863–3888. doi:10.1039/c9cs00099b
Return to citation in text: [1] -
Williams, S. J.; Withers, S. G. Carbohydr. Res. 2000, 327, 27–46. doi:10.1016/s0008-6215(00)00041-0
Return to citation in text: [1] -
Namchuk, M.; Braun, C.; McCarter, J. D.; Withers, S. G. Fluorinated sugars as probes of glycosidase mechanisms. In Biomedical Frontiers of Fluorine Chemistry; Ojima, I.; Ojima, J. R.; Welch, J. T., Eds.; ACS Symposium Series, Vol. 639; American Chemical Society: Washington, DC, USA, 1996; pp 279–293. doi:10.1021/bk-1996-0639.ch020
Return to citation in text: [1] -
Williams, D. A.; Pradhan, K.; Paul, A.; Olin, I. R.; Tuck, O. T.; Moulton, K. D.; Kulkarni, S. S.; Dube, D. H. Chem. Sci. 2020, 11, 1761–1774. doi:10.1039/c9sc05955e
Return to citation in text: [1] -
Glaudemans, C. P. J.; Kováč, P.; Nashed, E. M. Methods Enzymol. 1994, 247, 305–322. doi:10.1016/s0076-6879(94)47023-5
Return to citation in text: [1] -
Valverde, P.; Quintana, J. I.; Santos, J. I.; Ardá, A.; Jiménez-Barbero, J. ACS Omega 2019, 4, 13618–13630. doi:10.1021/acsomega.9b01901
Return to citation in text: [1] -
Gimeno, A.; Valverde, P.; Ardá, A.; Jiménez-Barbero, J. Curr. Opin. Struct. Biol. 2020, 62, 22–30. doi:10.1016/j.sbi.2019.11.004
Return to citation in text: [1] -
Biffinger, J. C.; Kim, H. W.; DiMagno, S. G. ChemBioChem 2004, 5, 622–627. doi:10.1002/cbic.200300910
Return to citation in text: [1] -
van Straaten, K. E.; Kuttiyatveetil, J. R. A.; Sevrain, C. M.; Villaume, S. A.; Jiménez-Barbero, J.; Linclau, B.; Vincent, S. P.; Sanders, D. A. R. J. Am. Chem. Soc. 2015, 137, 1230–1244. doi:10.1021/ja511204p
Return to citation in text: [1] -
Dohi, H.; Périon, R.; Durka, M.; Bosco, M.; Roué, Y.; Moreau, F.; Grizot, S.; Ducruix, A.; Escaich, S.; Vincent, S. P. Chem. – Eur. J. 2008, 14, 9530–9539. doi:10.1002/chem.200801279
Return to citation in text: [1] -
Thanna, S.; Lindenberger, J. J.; Gaitonde, V. V.; Ronning, D. R.; Sucheck, S. J. Org. Biomol. Chem. 2015, 13, 7542–7550. doi:10.1039/c5ob00867k
Return to citation in text: [1] -
Vermersch, P. S.; Tesmer, J. J. G.; Quiocho, F. A. J. Mol. Biol. 1992, 226, 923–929. doi:10.1016/0022-2836(92)91041-m
Return to citation in text: [1] -
Denavit, V.; Lainé, D.; St-Gelais, J.; Johnson, P. A.; Giguère, D. Nat. Commun. 2018, 9, 4721. doi:10.1038/s41467-018-06901-y
Return to citation in text: [1] [2] [3] [4] -
St-Gelais, J.; Bouchard, M.; Denavit, V.; Giguère, D. J. Org. Chem. 2019, 84, 8509–8522. doi:10.1021/acs.joc.9b00795
Return to citation in text: [1] -
St‐Gelais, J.; Côté, É.; Lainé, D.; Johnson, P. A.; Giguère, D. Chem. – Eur. J. 2020, 26, 13499–13506. doi:10.1002/chem.202002825
Return to citation in text: [1] [2] [3] -
Linclau, B.; Wang, Z.; Compain, G.; Paumelle, V.; Fontenelle, C. Q.; Wells, N.; Weymouth-Wilson, A. Angew. Chem., Int. Ed. 2016, 55, 674–678. doi:10.1002/anie.201509460
Return to citation in text: [1] -
Johannes, M.; Reindl, M.; Gerlitzki, B.; Schmitt, E.; Hoffmann-Röder, A. Beilstein J. Org. Chem. 2015, 11, 155–161. doi:10.3762/bjoc.11.15
Return to citation in text: [1] -
Sun, X.-L.; Kanie, Y.; Guo, C.-T.; Kanie, O.; Suzuki, Y.; Wong, C.-H. Eur. J. Org. Chem. 2000, 2643–2653. doi:10.1002/1099-0690(200007)2000:14<2643::aid-ejoc2643>3.0.co;2-1
Return to citation in text: [1] -
Axer, A.; Jumde, R. P.; Adam, S.; Faust, A.; Schäfers, M.; Fobker, M.; Koehnke, J.; Hirsch, A. K. H.; Gilmour, R. Chem. Sci. 2021, 12, 1286–1294. doi:10.1039/d0sc04297h
Return to citation in text: [1] -
Malassis, J.; Vendeville, J.-B.; Nguyen, Q.-H.; Boujon, M.; Gaignard-Gaillard, Q.; Light, M.; Linclau, B. Org. Biomol. Chem. 2019, 17, 5331–5340. doi:10.1039/c9ob00707e
Return to citation in text: [1] -
Bresciani, S.; Lebl, T.; Slawin, A. M. Z.; O'Hagan, D. Chem. Commun. 2010, 46, 5434–5436. doi:10.1039/c0cc01128b
Return to citation in text: [1] -
Sharma, M.; Bernacki, R. J.; Paul, B.; Korytnyk, W. Carbohydr. Res. 1990, 198, 205–221. doi:10.1016/0008-6215(90)84293-4
Return to citation in text: [1] [2] [3] [4] [5] -
Werz, D. B.; Ranzinger, R.; Herget, S.; Adibekian, A.; von der Lieth, C.-W.; Seeberger, P. H. ACS Chem. Biol. 2007, 2, 685–691. doi:10.1021/cb700178s
Return to citation in text: [1] -
Fu, C.; Zhao, H.; Wang, Y.; Cai, H.; Xiao, Y.; Zeng, Y.; Chen, H. HLA 2016, 88, 275–286. doi:10.1111/tan.12900
Return to citation in text: [1] -
Berkin, A.; Szarek, W. A.; Kisilevsky, R. Carbohydr. Res. 2000, 326, 250–263. doi:10.1016/s0008-6215(00)00049-5
Return to citation in text: [1] [2] [3] -
Horník, Š.; Červenková Šťastná, L.; Cuřínová, P.; Sýkora, J.; Káňová, K.; Hrstka, R.; Císařová, I.; Dračínský, M.; Karban, J. Beilstein J. Org. Chem. 2016, 12, 750–759. doi:10.3762/bjoc.12.75
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] -
Morrison, Z. A.; Nitz, M. Carbohydr. Res. 2020, 495, 108071. doi:10.1016/j.carres.2020.108071
Return to citation in text: [1] [2] [3] [4] -
Hough, L.; Penglis, A. A. E.; Richardson, A. C. Can. J. Chem. 1981, 59, 396–405. doi:10.1139/v81-061
Return to citation in text: [1] [2] -
Sharma, M.; Bernacki, R. J.; Hillman, M. J.; Korytnyk, W. Carbohydr. Res. 1993, 240, 85–93. doi:10.1016/0008-6215(93)84174-5
Return to citation in text: [1] [2] [3] -
Wasonga, G.; Tatara, Y.; Kakizaki, I.; Huang, X. J. Carbohydr. Chem. 2013, 32, 392–409. doi:10.1080/07328303.2013.815196
Return to citation in text: [1] [2] -
Wagner, S.; Mersch, C.; Hoffmann-Röder, A. Chem. – Eur. J. 2010, 16, 7319–7330. doi:10.1002/chem.200903294
Return to citation in text: [1] [2] -
Mersch, C.; Wagner, S.; Hoffmann-Röder, A. Synlett 2009, 2167–2171. doi:10.1055/s-0029-1217566
Return to citation in text: [1] [2] -
Johannes, M.; Oberbillig, T.; Hoffmann-Röder, A. Org. Biomol. Chem. 2011, 9, 5541–5546. doi:10.1039/c1ob05373f
Return to citation in text: [1] -
Stephenson, E. L.; Zhang, P.; Ghorbani, S.; Wang, A.; Gu, J.; Keough, M. B.; Rawji, K. S.; Silva, C.; Yong, V. W.; Ling, C.-C. ACS Cent. Sci. 2019, 5, 1223–1234. doi:10.1021/acscentsci.9b00327
Return to citation in text: [1] [2] [3] -
Nishimura, S.-I.; Hato, M.; Hyugaji, S.; Feng, F.; Amano, M. Angew. Chem., Int. Ed. 2012, 51, 3386–3390. doi:10.1002/anie.201108742
Return to citation in text: [1] [2] -
van Wijk, X. M.; Lawrence, R.; Thijssen, V. L.; van den Broek, S. A.; Troost, R.; van Scherpenzeel, M.; Naidu, N.; Oosterhof, A.; Griffioen, A. W.; Lefeber, D. J.; van Delft, F. L.; van Kuppevelt, T. H. FASEB J. 2015, 29, 2993–3002. doi:10.1096/fj.14-264226
Return to citation in text: [1] -
Keough, M. B.; Rogers, J. A.; Zhang, P.; Jensen, S. K.; Stephenson, E. L.; Chen, T.; Hurlbert, M. G.; Lau, L. W.; Rawji, K. S.; Plemel, J. R.; Koch, M.; Ling, C.-C.; Yong, V. W. Nat. Commun. 2016, 7, 11312. doi:10.1038/ncomms11312
Return to citation in text: [1] -
Qin, K.; Zhang, H.; Zhao, Z.; Chen, X. J. Am. Chem. Soc. 2020, 142, 9382–9388. doi:10.1021/jacs.0c02110
Return to citation in text: [1] -
Wang, L.-X.; Sakairi, N.; Kuzuhara, H. J. Chem. Soc., Perkin Trans. 1 1990, 1677–1682. doi:10.1039/p19900001677
Return to citation in text: [1] -
Kurfiřt, M.; Červenková Št’astná, L.; Dračínský, M.; Müllerová, M.; Hamala, V.; Cuřínová, P.; Karban, J. J. Org. Chem. 2019, 84, 6405–6431. doi:10.1021/acs.joc.9b00705
Return to citation in text: [1] [2] [3] [4] -
Lin, P.-C.; Adak, A. K.; Ueng, S.-H.; Huang, L.-D.; Huang, K.-T.; Ho, J.-a. A.; Lin, C.-C. J. Org. Chem. 2009, 74, 4041–4048. doi:10.1021/jo900516r
Return to citation in text: [1] [2] -
Dax, K.; Albert, M.; Hammond, D.; Illaszewicz, C.; Purkarthofer, T.; Tscherner, M.; Weber, H. Rearrangements in the Course of Fluorination by Diethylaminosulfur Trifluoride at C-2 of Glycopyranosides: Some New Parameters. In Timely Research Perspectives in Carbohydrate Chemistry; Schmid, W.; Stütz, A. E., Eds.; Springer: Vienna, Austria, 2002; pp 77–98. doi:10.1007/978-3-7091-6130-2_5
Return to citation in text: [1] [2] -
Karban, J.; Císařová, I.; Strašák, T.; Šťastná, L. Č.; Sýkora, J. Org. Biomol. Chem. 2012, 10, 394–403. doi:10.1039/c1ob06336g
Return to citation in text: [1] -
Lainé, D.; Denavit, V.; Lessard, O.; Carrier, L.; Fecteau, C.-É.; Johnson, P. A.; Giguère, D. Beilstein J. Org. Chem. 2020, 16, 2880–2887. doi:10.3762/bjoc.16.237
Return to citation in text: [1] -
Quiquempoix, L.; Wang, Z.; Graton, J.; Latchem, P. G.; Light, M.; Le Questel, J.-Y.; Linclau, B. J. Org. Chem. 2019, 84, 5899–5906. doi:10.1021/acs.joc.9b00310
Return to citation in text: [1] -
Baer, H. H.; Mateo, F. H.; Siemsen, L. Carbohydr. Res. 1989, 187, 67–92. doi:10.1016/0008-6215(89)80056-4
Return to citation in text: [1] -
Popsavin, V.; Benedeković, G.; Popsavin, M.; Divjaković, V.; Armbruster, T. Tetrahedron 2004, 60, 5225–5235. doi:10.1016/j.tet.2004.04.040
Return to citation in text: [1] -
Chen, S. Y.; Joullie, M. M. J. Org. Chem. 1984, 49, 1769–1772. doi:10.1021/jo00184a020
Return to citation in text: [1] -
Vera-Ayoso, Y.; Borrachero, P.; Cabrera-Escribano, F.; Gómez-Guillén, M. Tetrahedron: Asymmetry 2005, 16, 889–897. doi:10.1016/j.tetasy.2004.12.024
Return to citation in text: [1] -
Vera-Ayoso, Y.; Borrachero, P.; Cabrera-Escribano, F.; Carmona, A. T.; Gómez-Guillén, M. Tetrahedron: Asymmetry 2004, 15, 429–444. doi:10.1016/j.tetasy.2003.11.034
Return to citation in text: [1] -
Suzuki, K.; Ito, Y.; Kanie, O. Carbohydr. Res. 2012, 359, 81–91. doi:10.1016/j.carres.2012.07.003
Return to citation in text: [1] -
Herczeg, M.; Mező, E.; Eszenyi, D.; Lázár, L.; Csávás, M.; Bereczki, I.; Antus, S.; Borbás, A. Eur. J. Org. Chem. 2013, 5570–5573. doi:10.1002/ejoc.201300681
Return to citation in text: [1] -
Card, P. J.; Reddy, G. S. J. Org. Chem. 1983, 48, 4734–4743. doi:10.1021/jo00172a054
Return to citation in text: [1] -
Kolakowski, R. V.; Shangguan, N.; Sauers, R. R.; Williams, L. J. J. Am. Chem. Soc. 2006, 128, 5695–5702. doi:10.1021/ja057533y
Return to citation in text: [1] -
Dhakal, B.; Crich, D. J. Am. Chem. Soc. 2018, 140, 15008–15015. doi:10.1021/jacs.8b09654
Return to citation in text: [1] -
Wray, V. J. Chem. Soc., Perkin Trans. 2 1976, 1598–1605. doi:10.1039/p29760001598
Return to citation in text: [1] -
Phillips, L.; Wray, V. J. Chem. Soc. B 1971, 1618–1624. doi:10.1039/j29710001618
Return to citation in text: [1] -
Denavit, V.; Lainé, D.; Bouzriba, C.; Shanina, E.; Gillon, É.; Fortin, S.; Rademacher, C.; Imberty, A.; Giguère, D. Chem. – Eur. J. 2019, 25, 4478–4490. doi:10.1002/chem.201806197
Return to citation in text: [1]
| 26. | Horník, Š.; Červenková Šťastná, L.; Cuřínová, P.; Sýkora, J.; Káňová, K.; Hrstka, R.; Císařová, I.; Dračínský, M.; Karban, J. Beilstein J. Org. Chem. 2016, 12, 750–759. doi:10.3762/bjoc.12.75 |
| 40. | Kurfiřt, M.; Červenková Št’astná, L.; Dračínský, M.; Müllerová, M.; Hamala, V.; Cuřínová, P.; Karban, J. J. Org. Chem. 2019, 84, 6405–6431. doi:10.1021/acs.joc.9b00705 |
| 40. | Kurfiřt, M.; Červenková Št’astná, L.; Dračínský, M.; Müllerová, M.; Hamala, V.; Cuřínová, P.; Karban, J. J. Org. Chem. 2019, 84, 6405–6431. doi:10.1021/acs.joc.9b00705 |
| 41. | Lin, P.-C.; Adak, A. K.; Ueng, S.-H.; Huang, L.-D.; Huang, K.-T.; Ho, J.-a. A.; Lin, C.-C. J. Org. Chem. 2009, 74, 4041–4048. doi:10.1021/jo900516r |
| 1. | Linclau, B.; Ardá, A.; Reichardt, N.-C.; Sollogoub, M.; Unione, L.; Vincent, S. P.; Jiménez-Barbero, J. Chem. Soc. Rev. 2020, 49, 3863–3888. doi:10.1039/c9cs00099b |
| 2. | Williams, S. J.; Withers, S. G. Carbohydr. Res. 2000, 327, 27–46. doi:10.1016/s0008-6215(00)00041-0 |
| 3. | Namchuk, M.; Braun, C.; McCarter, J. D.; Withers, S. G. Fluorinated sugars as probes of glycosidase mechanisms. In Biomedical Frontiers of Fluorine Chemistry; Ojima, I.; Ojima, J. R.; Welch, J. T., Eds.; ACS Symposium Series, Vol. 639; American Chemical Society: Washington, DC, USA, 1996; pp 279–293. doi:10.1021/bk-1996-0639.ch020 |
| 4. | Williams, D. A.; Pradhan, K.; Paul, A.; Olin, I. R.; Tuck, O. T.; Moulton, K. D.; Kulkarni, S. S.; Dube, D. H. Chem. Sci. 2020, 11, 1761–1774. doi:10.1039/c9sc05955e |
| 5. | Glaudemans, C. P. J.; Kováč, P.; Nashed, E. M. Methods Enzymol. 1994, 247, 305–322. doi:10.1016/s0076-6879(94)47023-5 |
| 6. | Valverde, P.; Quintana, J. I.; Santos, J. I.; Ardá, A.; Jiménez-Barbero, J. ACS Omega 2019, 4, 13618–13630. doi:10.1021/acsomega.9b01901 |
| 7. | Gimeno, A.; Valverde, P.; Ardá, A.; Jiménez-Barbero, J. Curr. Opin. Struct. Biol. 2020, 62, 22–30. doi:10.1016/j.sbi.2019.11.004 |
| 12. | Vermersch, P. S.; Tesmer, J. J. G.; Quiocho, F. A. J. Mol. Biol. 1992, 226, 923–929. doi:10.1016/0022-2836(92)91041-m |
| 22. | Sharma, M.; Bernacki, R. J.; Paul, B.; Korytnyk, W. Carbohydr. Res. 1990, 198, 205–221. doi:10.1016/0008-6215(90)84293-4 |
| 25. | Berkin, A.; Szarek, W. A.; Kisilevsky, R. Carbohydr. Res. 2000, 326, 250–263. doi:10.1016/s0008-6215(00)00049-5 |
| 26. | Horník, Š.; Červenková Šťastná, L.; Cuřínová, P.; Sýkora, J.; Káňová, K.; Hrstka, R.; Císařová, I.; Dračínský, M.; Karban, J. Beilstein J. Org. Chem. 2016, 12, 750–759. doi:10.3762/bjoc.12.75 |
| 27. | Morrison, Z. A.; Nitz, M. Carbohydr. Res. 2020, 495, 108071. doi:10.1016/j.carres.2020.108071 |
| 28. | Hough, L.; Penglis, A. A. E.; Richardson, A. C. Can. J. Chem. 1981, 59, 396–405. doi:10.1139/v81-061 |
| 29. | Sharma, M.; Bernacki, R. J.; Hillman, M. J.; Korytnyk, W. Carbohydr. Res. 1993, 240, 85–93. doi:10.1016/0008-6215(93)84174-5 |
| 30. | Wasonga, G.; Tatara, Y.; Kakizaki, I.; Huang, X. J. Carbohydr. Chem. 2013, 32, 392–409. doi:10.1080/07328303.2013.815196 |
| 31. | Wagner, S.; Mersch, C.; Hoffmann-Röder, A. Chem. – Eur. J. 2010, 16, 7319–7330. doi:10.1002/chem.200903294 |
| 32. | Mersch, C.; Wagner, S.; Hoffmann-Röder, A. Synlett 2009, 2167–2171. doi:10.1055/s-0029-1217566 |
| 26. | Horník, Š.; Červenková Šťastná, L.; Cuřínová, P.; Sýkora, J.; Káňová, K.; Hrstka, R.; Císařová, I.; Dračínský, M.; Karban, J. Beilstein J. Org. Chem. 2016, 12, 750–759. doi:10.3762/bjoc.12.75 |
| 11. | Thanna, S.; Lindenberger, J. J.; Gaitonde, V. V.; Ronning, D. R.; Sucheck, S. J. Org. Biomol. Chem. 2015, 13, 7542–7550. doi:10.1039/c5ob00867k |
| 22. | Sharma, M.; Bernacki, R. J.; Paul, B.; Korytnyk, W. Carbohydr. Res. 1990, 198, 205–221. doi:10.1016/0008-6215(90)84293-4 |
| 26. | Horník, Š.; Červenková Šťastná, L.; Cuřínová, P.; Sýkora, J.; Káňová, K.; Hrstka, R.; Císařová, I.; Dračínský, M.; Karban, J. Beilstein J. Org. Chem. 2016, 12, 750–759. doi:10.3762/bjoc.12.75 |
| 33. | Johannes, M.; Oberbillig, T.; Hoffmann-Röder, A. Org. Biomol. Chem. 2011, 9, 5541–5546. doi:10.1039/c1ob05373f |
| 34. | Stephenson, E. L.; Zhang, P.; Ghorbani, S.; Wang, A.; Gu, J.; Keough, M. B.; Rawji, K. S.; Silva, C.; Yong, V. W.; Ling, C.-C. ACS Cent. Sci. 2019, 5, 1223–1234. doi:10.1021/acscentsci.9b00327 |
| 54. | Kolakowski, R. V.; Shangguan, N.; Sauers, R. R.; Williams, L. J. J. Am. Chem. Soc. 2006, 128, 5695–5702. doi:10.1021/ja057533y |
| 55. | Dhakal, B.; Crich, D. J. Am. Chem. Soc. 2018, 140, 15008–15015. doi:10.1021/jacs.8b09654 |
| 9. | van Straaten, K. E.; Kuttiyatveetil, J. R. A.; Sevrain, C. M.; Villaume, S. A.; Jiménez-Barbero, J.; Linclau, B.; Vincent, S. P.; Sanders, D. A. R. J. Am. Chem. Soc. 2015, 137, 1230–1244. doi:10.1021/ja511204p |
| 10. | Dohi, H.; Périon, R.; Durka, M.; Bosco, M.; Roué, Y.; Moreau, F.; Grizot, S.; Ducruix, A.; Escaich, S.; Vincent, S. P. Chem. – Eur. J. 2008, 14, 9530–9539. doi:10.1002/chem.200801279 |
| 23. | Werz, D. B.; Ranzinger, R.; Herget, S.; Adibekian, A.; von der Lieth, C.-W.; Seeberger, P. H. ACS Chem. Biol. 2007, 2, 685–691. doi:10.1021/cb700178s |
| 40. | Kurfiřt, M.; Červenková Št’astná, L.; Dračínský, M.; Müllerová, M.; Hamala, V.; Cuřínová, P.; Karban, J. J. Org. Chem. 2019, 84, 6405–6431. doi:10.1021/acs.joc.9b00705 |
| 8. | Biffinger, J. C.; Kim, H. W.; DiMagno, S. G. ChemBioChem 2004, 5, 622–627. doi:10.1002/cbic.200300910 |
| 24. | Fu, C.; Zhao, H.; Wang, Y.; Cai, H.; Xiao, Y.; Zeng, Y.; Chen, H. HLA 2016, 88, 275–286. doi:10.1111/tan.12900 |
| 53. | Card, P. J.; Reddy, G. S. J. Org. Chem. 1983, 48, 4734–4743. doi:10.1021/jo00172a054 |
| 20. | Malassis, J.; Vendeville, J.-B.; Nguyen, Q.-H.; Boujon, M.; Gaignard-Gaillard, Q.; Light, M.; Linclau, B. Org. Biomol. Chem. 2019, 17, 5331–5340. doi:10.1039/c9ob00707e |
| 13. | Denavit, V.; Lainé, D.; St-Gelais, J.; Johnson, P. A.; Giguère, D. Nat. Commun. 2018, 9, 4721. doi:10.1038/s41467-018-06901-y |
| 21. | Bresciani, S.; Lebl, T.; Slawin, A. M. Z.; O'Hagan, D. Chem. Commun. 2010, 46, 5434–5436. doi:10.1039/c0cc01128b |
| 51. | Suzuki, K.; Ito, Y.; Kanie, O. Carbohydr. Res. 2012, 359, 81–91. doi:10.1016/j.carres.2012.07.003 |
| 15. | St‐Gelais, J.; Côté, É.; Lainé, D.; Johnson, P. A.; Giguère, D. Chem. – Eur. J. 2020, 26, 13499–13506. doi:10.1002/chem.202002825 |
| 22. | Sharma, M.; Bernacki, R. J.; Paul, B.; Korytnyk, W. Carbohydr. Res. 1990, 198, 205–221. doi:10.1016/0008-6215(90)84293-4 |
| 41. | Lin, P.-C.; Adak, A. K.; Ueng, S.-H.; Huang, L.-D.; Huang, K.-T.; Ho, J.-a. A.; Lin, C.-C. J. Org. Chem. 2009, 74, 4041–4048. doi:10.1021/jo900516r |
| 52. | Herczeg, M.; Mező, E.; Eszenyi, D.; Lázár, L.; Csávás, M.; Bereczki, I.; Antus, S.; Borbás, A. Eur. J. Org. Chem. 2013, 5570–5573. doi:10.1002/ejoc.201300681 |
| 17. | Johannes, M.; Reindl, M.; Gerlitzki, B.; Schmitt, E.; Hoffmann-Röder, A. Beilstein J. Org. Chem. 2015, 11, 155–161. doi:10.3762/bjoc.11.15 |
| 18. | Sun, X.-L.; Kanie, Y.; Guo, C.-T.; Kanie, O.; Suzuki, Y.; Wong, C.-H. Eur. J. Org. Chem. 2000, 2643–2653. doi:10.1002/1099-0690(200007)2000:14<2643::aid-ejoc2643>3.0.co;2-1 |
| 19. | Axer, A.; Jumde, R. P.; Adam, S.; Faust, A.; Schäfers, M.; Fobker, M.; Koehnke, J.; Hirsch, A. K. H.; Gilmour, R. Chem. Sci. 2021, 12, 1286–1294. doi:10.1039/d0sc04297h |
| 42. | Dax, K.; Albert, M.; Hammond, D.; Illaszewicz, C.; Purkarthofer, T.; Tscherner, M.; Weber, H. Rearrangements in the Course of Fluorination by Diethylaminosulfur Trifluoride at C-2 of Glycopyranosides: Some New Parameters. In Timely Research Perspectives in Carbohydrate Chemistry; Schmid, W.; Stütz, A. E., Eds.; Springer: Vienna, Austria, 2002; pp 77–98. doi:10.1007/978-3-7091-6130-2_5 |
| 43. | Karban, J.; Císařová, I.; Strašák, T.; Šťastná, L. Č.; Sýkora, J. Org. Biomol. Chem. 2012, 10, 394–403. doi:10.1039/c1ob06336g |
| 44. | Lainé, D.; Denavit, V.; Lessard, O.; Carrier, L.; Fecteau, C.-É.; Johnson, P. A.; Giguère, D. Beilstein J. Org. Chem. 2020, 16, 2880–2887. doi:10.3762/bjoc.16.237 |
| 45. | Quiquempoix, L.; Wang, Z.; Graton, J.; Latchem, P. G.; Light, M.; Le Questel, J.-Y.; Linclau, B. J. Org. Chem. 2019, 84, 5899–5906. doi:10.1021/acs.joc.9b00310 |
| 13. | Denavit, V.; Lainé, D.; St-Gelais, J.; Johnson, P. A.; Giguère, D. Nat. Commun. 2018, 9, 4721. doi:10.1038/s41467-018-06901-y |
| 14. | St-Gelais, J.; Bouchard, M.; Denavit, V.; Giguère, D. J. Org. Chem. 2019, 84, 8509–8522. doi:10.1021/acs.joc.9b00795 |
| 15. | St‐Gelais, J.; Côté, É.; Lainé, D.; Johnson, P. A.; Giguère, D. Chem. – Eur. J. 2020, 26, 13499–13506. doi:10.1002/chem.202002825 |
| 16. | Linclau, B.; Wang, Z.; Compain, G.; Paumelle, V.; Fontenelle, C. Q.; Wells, N.; Weymouth-Wilson, A. Angew. Chem., Int. Ed. 2016, 55, 674–678. doi:10.1002/anie.201509460 |
| 13. | Denavit, V.; Lainé, D.; St-Gelais, J.; Johnson, P. A.; Giguère, D. Nat. Commun. 2018, 9, 4721. doi:10.1038/s41467-018-06901-y |
| 42. | Dax, K.; Albert, M.; Hammond, D.; Illaszewicz, C.; Purkarthofer, T.; Tscherner, M.; Weber, H. Rearrangements in the Course of Fluorination by Diethylaminosulfur Trifluoride at C-2 of Glycopyranosides: Some New Parameters. In Timely Research Perspectives in Carbohydrate Chemistry; Schmid, W.; Stütz, A. E., Eds.; Springer: Vienna, Austria, 2002; pp 77–98. doi:10.1007/978-3-7091-6130-2_5 |
| 46. | Baer, H. H.; Mateo, F. H.; Siemsen, L. Carbohydr. Res. 1989, 187, 67–92. doi:10.1016/0008-6215(89)80056-4 |
| 47. | Popsavin, V.; Benedeković, G.; Popsavin, M.; Divjaković, V.; Armbruster, T. Tetrahedron 2004, 60, 5225–5235. doi:10.1016/j.tet.2004.04.040 |
| 48. | Chen, S. Y.; Joullie, M. M. J. Org. Chem. 1984, 49, 1769–1772. doi:10.1021/jo00184a020 |
| 49. | Vera-Ayoso, Y.; Borrachero, P.; Cabrera-Escribano, F.; Gómez-Guillén, M. Tetrahedron: Asymmetry 2005, 16, 889–897. doi:10.1016/j.tetasy.2004.12.024 |
| 50. | Vera-Ayoso, Y.; Borrachero, P.; Cabrera-Escribano, F.; Carmona, A. T.; Gómez-Guillén, M. Tetrahedron: Asymmetry 2004, 15, 429–444. doi:10.1016/j.tetasy.2003.11.034 |
| 31. | Wagner, S.; Mersch, C.; Hoffmann-Röder, A. Chem. – Eur. J. 2010, 16, 7319–7330. doi:10.1002/chem.200903294 |
| 32. | Mersch, C.; Wagner, S.; Hoffmann-Röder, A. Synlett 2009, 2167–2171. doi:10.1055/s-0029-1217566 |
| 34. | Stephenson, E. L.; Zhang, P.; Ghorbani, S.; Wang, A.; Gu, J.; Keough, M. B.; Rawji, K. S.; Silva, C.; Yong, V. W.; Ling, C.-C. ACS Cent. Sci. 2019, 5, 1223–1234. doi:10.1021/acscentsci.9b00327 |
| 35. | Nishimura, S.-I.; Hato, M.; Hyugaji, S.; Feng, F.; Amano, M. Angew. Chem., Int. Ed. 2012, 51, 3386–3390. doi:10.1002/anie.201108742 |
| 36. | van Wijk, X. M.; Lawrence, R.; Thijssen, V. L.; van den Broek, S. A.; Troost, R.; van Scherpenzeel, M.; Naidu, N.; Oosterhof, A.; Griffioen, A. W.; Lefeber, D. J.; van Delft, F. L.; van Kuppevelt, T. H. FASEB J. 2015, 29, 2993–3002. doi:10.1096/fj.14-264226 |
| 37. | Keough, M. B.; Rogers, J. A.; Zhang, P.; Jensen, S. K.; Stephenson, E. L.; Chen, T.; Hurlbert, M. G.; Lau, L. W.; Rawji, K. S.; Plemel, J. R.; Koch, M.; Ling, C.-C.; Yong, V. W. Nat. Commun. 2016, 7, 11312. doi:10.1038/ncomms11312 |
| 27. | Morrison, Z. A.; Nitz, M. Carbohydr. Res. 2020, 495, 108071. doi:10.1016/j.carres.2020.108071 |
| 28. | Hough, L.; Penglis, A. A. E.; Richardson, A. C. Can. J. Chem. 1981, 59, 396–405. doi:10.1139/v81-061 |
| 27. | Morrison, Z. A.; Nitz, M. Carbohydr. Res. 2020, 495, 108071. doi:10.1016/j.carres.2020.108071 |
| 29. | Sharma, M.; Bernacki, R. J.; Hillman, M. J.; Korytnyk, W. Carbohydr. Res. 1993, 240, 85–93. doi:10.1016/0008-6215(93)84174-5 |
| 27. | Morrison, Z. A.; Nitz, M. Carbohydr. Res. 2020, 495, 108071. doi:10.1016/j.carres.2020.108071 |
| 29. | Sharma, M.; Bernacki, R. J.; Hillman, M. J.; Korytnyk, W. Carbohydr. Res. 1993, 240, 85–93. doi:10.1016/0008-6215(93)84174-5 |
| 39. | Wang, L.-X.; Sakairi, N.; Kuzuhara, H. J. Chem. Soc., Perkin Trans. 1 1990, 1677–1682. doi:10.1039/p19900001677 |
| 40. | Kurfiřt, M.; Červenková Št’astná, L.; Dračínský, M.; Müllerová, M.; Hamala, V.; Cuřínová, P.; Karban, J. J. Org. Chem. 2019, 84, 6405–6431. doi:10.1021/acs.joc.9b00705 |
| 26. | Horník, Š.; Červenková Šťastná, L.; Cuřínová, P.; Sýkora, J.; Káňová, K.; Hrstka, R.; Císařová, I.; Dračínský, M.; Karban, J. Beilstein J. Org. Chem. 2016, 12, 750–759. doi:10.3762/bjoc.12.75 |
| 38. | Qin, K.; Zhang, H.; Zhao, Z.; Chen, X. J. Am. Chem. Soc. 2020, 142, 9382–9388. doi:10.1021/jacs.0c02110 |
| 26. | Horník, Š.; Červenková Šťastná, L.; Cuřínová, P.; Sýkora, J.; Káňová, K.; Hrstka, R.; Císařová, I.; Dračínský, M.; Karban, J. Beilstein J. Org. Chem. 2016, 12, 750–759. doi:10.3762/bjoc.12.75 |
| 13. | Denavit, V.; Lainé, D.; St-Gelais, J.; Johnson, P. A.; Giguère, D. Nat. Commun. 2018, 9, 4721. doi:10.1038/s41467-018-06901-y |
| 15. | St‐Gelais, J.; Côté, É.; Lainé, D.; Johnson, P. A.; Giguère, D. Chem. – Eur. J. 2020, 26, 13499–13506. doi:10.1002/chem.202002825 |
| 58. | Denavit, V.; Lainé, D.; Bouzriba, C.; Shanina, E.; Gillon, É.; Fortin, S.; Rademacher, C.; Imberty, A.; Giguère, D. Chem. – Eur. J. 2019, 25, 4478–4490. doi:10.1002/chem.201806197 |
| 26. | Horník, Š.; Červenková Šťastná, L.; Cuřínová, P.; Sýkora, J.; Káňová, K.; Hrstka, R.; Císařová, I.; Dračínský, M.; Karban, J. Beilstein J. Org. Chem. 2016, 12, 750–759. doi:10.3762/bjoc.12.75 |
| 22. | Sharma, M.; Bernacki, R. J.; Paul, B.; Korytnyk, W. Carbohydr. Res. 1990, 198, 205–221. doi:10.1016/0008-6215(90)84293-4 |
| 25. | Berkin, A.; Szarek, W. A.; Kisilevsky, R. Carbohydr. Res. 2000, 326, 250–263. doi:10.1016/s0008-6215(00)00049-5 |
| 26. | Horník, Š.; Červenková Šťastná, L.; Cuřínová, P.; Sýkora, J.; Káňová, K.; Hrstka, R.; Císařová, I.; Dračínský, M.; Karban, J. Beilstein J. Org. Chem. 2016, 12, 750–759. doi:10.3762/bjoc.12.75 |
| 30. | Wasonga, G.; Tatara, Y.; Kakizaki, I.; Huang, X. J. Carbohydr. Chem. 2013, 32, 392–409. doi:10.1080/07328303.2013.815196 |
| 35. | Nishimura, S.-I.; Hato, M.; Hyugaji, S.; Feng, F.; Amano, M. Angew. Chem., Int. Ed. 2012, 51, 3386–3390. doi:10.1002/anie.201108742 |
| 56. | Wray, V. J. Chem. Soc., Perkin Trans. 2 1976, 1598–1605. doi:10.1039/p29760001598 |
| 22. | Sharma, M.; Bernacki, R. J.; Paul, B.; Korytnyk, W. Carbohydr. Res. 1990, 198, 205–221. doi:10.1016/0008-6215(90)84293-4 |
| 25. | Berkin, A.; Szarek, W. A.; Kisilevsky, R. Carbohydr. Res. 2000, 326, 250–263. doi:10.1016/s0008-6215(00)00049-5 |
| 34. | Stephenson, E. L.; Zhang, P.; Ghorbani, S.; Wang, A.; Gu, J.; Keough, M. B.; Rawji, K. S.; Silva, C.; Yong, V. W.; Ling, C.-C. ACS Cent. Sci. 2019, 5, 1223–1234. doi:10.1021/acscentsci.9b00327 |
| 57. | Phillips, L.; Wray, V. J. Chem. Soc. B 1971, 1618–1624. doi:10.1039/j29710001618 |
© 2021 Hamala et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the author(s) and source are credited and that individual graphics may be subject to special legal provisions.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc/terms)