Abstract
The spliceostatins/thailanstatins are a family of linear peptides/polyketides that inhibit pre-mRNA splicing, and as such act as potent cytotoxic compounds. These compounds generally contain 9 stereocenters spread over a common (2Z,4S)-4-acetoxy-2-butenamide fragment, an (all-cis)-2,3,5,6-tetrasubstituted tetrahydropyran fragment and a terminal oxane ring joined by a dienyl chain. Due to the impressive antitumor properties of these compounds, along with their complex structure, a number of total syntheses have been reported. This review will compare the synthetic strategies reported through the end of 2019.

Graphical Abstract
Introduction
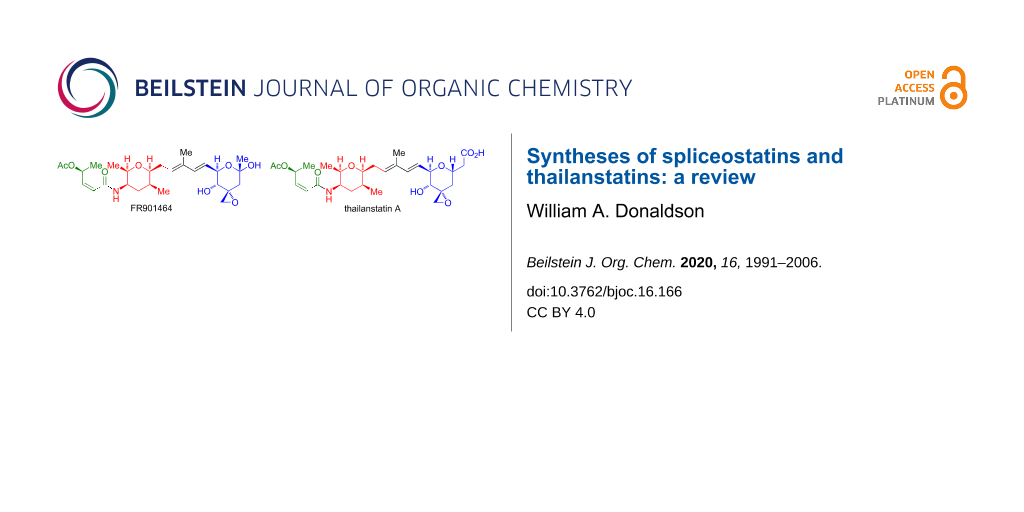
The spliceostatins/thailanstatins (Figure 1) are a family of linear peptide/polyketide natural products isolated from the bacteria Burkholderia sp. FERM BP-3421 [1-3] (originally identified as Pseudomonas sp. No 2663) and Burkholderia sp. MSMB 43 [4,5]. These compounds are of interest due to their ability to bind to a subunit of the human spliceosome, splicing factor 3b [6], which inhibits pre-mRNA splicing, and as such act as potent cytotoxic compounds. A review of the discovery, target identification, and biological applications of the compounds that exhibit these binding characteristics has been published [7]. These compounds all contain a common (2Z,4S)-4-acetoxy-2-butenamide fragment (in green, Figure 1), appended to an (all-cis)-2,3,5,6-tetrasubstituted tetrahydropyran fragment (in red, Figure 1). The members of this family primarily differ with respect to the terminal oxane ring (in blue, Figure 1) which is attached to the common fragments by a dienyl chain. The exciting antitumor properties of these compounds (Table 1), along with their complex structure, have led to a significant synthetic activity. The present review will cover the total syntheses of the spliceostatins/thailanstatins through the end of 2019.
![[1860-5397-16-166-1]](/bjoc/content/figures/1860-5397-16-166-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structures of spliceostatins/thailanstatins.
Figure 1: Structures of spliceostatins/thailanstatins.
Table 1: Biological activities of spliceostatins/thailanstatins (IC50 values in nM).
| tumor cell line | |||||
| compound | MCF-7 | A549 | HCT116 | SW480 | P388 |
| FR901464 (1)a | 0.91 | 0.66 | 0.31 | 0.51 | 1.69 |
| FR901465 (2)a | 0.59 | 0.44 | 0.34 | 0.53 | 0.48 |
| FR901463 (3)a | 0.46 | 0.35 | 0.22 | 0.40 | 0.82 |
| H1975 | N87 | BT474 | MDA-MB-DYT2 | MDA-MB-468 | |
| thailanstatin B (5)b | n.t.c | >100 | >100 | n.t. | >100 |
| spliceostatin F (6) b | n.t. | 0.641 | 1.85 | n.t. | 1.35 |
| thailanstatin A (7) b | 320 | 59 | 145 | 161 | 142 |
| spliceostatin B (8) b | 30 | n.t. | n.t. | n.t. | n.t. |
| spliceostatin D (9) b | 950 | n.t. | n.t. | n.t. | n.t. |
| spliceostatin E (10)b | n.t. | 3.67 | 3.72 | 4.16 | 1.56 |
| spliceostatin G (11)b | n.t. | >100 | >100 | >100 | n.t. |
Review
Synthesis of the (2Z,4S)-4-acetoxy- or protected (2Z,4S)-4-hydroxy-2-butenoic acid fragment
The (2Z,4S)-4-acetoxy-2-butenoic acid fragment is common to all of the spliceostatins/thailanstatins. The various routes to this building block (Scheme 1) serve as a tutorial on the methodology for the asymmetric synthesis of a (Z)-2-hydroxy-3-pentene unit, with most proceeding via the cis-reduction of 4-acetoxy-2-pentynoic acid (13). The synthesis by Kitahara et al. [8,9] is the exception, where the chiral center is derived from relatively inexpensive (S)-ethyl lactate (14). This was transformed into the silyl ether-protected 2-hydroxypropanals 15a and 15b via literature procedures [10], followed by the application of the Still–Gennari Z-selective Horner–Wadsworth–Emmons olefination [11]. Koide’s group [12,13] reported that the asymmetric addition of the enyne 16 to acetaldehyde in the presence of zinc triflate and the chiral additive N-methylephedrine [14] gave the (S)-propargyl alcohol 17 in 72% ee; the chiral purity could be improved to 96% ee by recrystallization. Unfortunately, attempts to catalyze this reaction with Zn(OTf)2/ephedrine were unsuccessful. More recently, Ghosh and co-workers [15] used the (R,R)-ProPhenol ligand [16] to accomplish a catalytic asymmetric addition of methyl propynoate to acetaldehyde to give 18 in high enantiopurity (98% ee). Jacobsen’s group [17,18] utilized the Noyori Ru-catalyzed transfer hydrogenation [19] of the 3-butyne-2-one 19, which gave 20 with 97% ee. The removal of the silyl protecting group, followed by a carboxylation and acylation gave 13. Koide’s group [13] reported a second-generation route to 13, which utilized the Corey–Bakshi–Shibata chiral oxazaborolidine catalyst 21 [20] for the asymmetric reduction of the THP-protected 5-hydroxy-3-pentyn-2-one 22 to generate the secondary alcohol 23. The acylation of 23, followed by the treatment with Jones’ reagent effected the THP deprotection as well as an overoxidation to give 13. The syn-selective reduction of 13 was accomplished with a balloon worth of pressure of H2 in the presence of the Lindlar catalyst. Of the asymmetric strategies, Jacobsen’s and Ghosh’s routes proceeded with the highest enantioselectivity; the catalyst loading was lower for the Jacobsen route (0.5 mol %) compared to the Ghosh route (20 mol %).
![[1860-5397-16-166-i1]](/bjoc/content/inline/1860-5397-16-166-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthetic routes to protected (2Z,4S)-4-hydroxy-2-butenoic acid fragments.
Scheme 1: Synthetic routes to protected (2Z,4S)-4-hydroxy-2-butenoic acid fragments.
Synthesis of the (all-cis)-2,3,4,5-tetrasubstituted tetrahydropyran fragment
Syntheses from ʟ-threonine-derived aldehyde
Three groups utilized 4-formyl-2,2,5-trimethyl-3-oxazolidine (24) [21], derived from relatively inexpensive ʟ-threonine (<$1/g in bulk) as a chiral pool precursor for the amine stereocenter of the (all-cis)-2,3,5,6-tetrasubstituted tetrahydropyran fragment. In Kitahara’s synthesis [8,9], the Wittig olefination of 24, followed by a catalytic hydrogenation, removal of the dimethylaminal protecting group, and lactonization gave 25 as a mixture of diastereomers (Scheme 2). The further transformation of 25 afforded the dihydropyran 26, which upon catalytic hydrogenation over Pt2O and then low-temperature DIBAL reduction afforded the all-cis tetrasubstituted tetrahydropyran 27.
![[1860-5397-16-166-i2]](/bjoc/content/inline/1860-5397-16-166-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Kitahara synthesis of the (all-cis)-2,3,5,6-tetrasubstituted tetrahydropyran.
Scheme 2: Kitahara synthesis of the (all-cis)-2,3,5,6-tetrasubstituted tetrahydropyran.
In Koide et al.’s synthesis, the unsaturated lactone 29 was prepared via the Wittig methenylation of 24, followed by aminal hydrolysis, methallylation of the 2° alcohol 28, and ring-closing metathesis using Grubbs’ 2nd generation catalyst (G-II, Scheme 3) [13]. Replacing the N-Boc protecting group with an N-tosyl group and allylic oxidation gave 30. The introduction of the allyl group at C-11 made use of the Kishi protocol [22] of the allyl-Grignard addition, followed by an ionic reduction. The N-tosyl group was removed and the resultant amine protected to give the N-Boc tetrahydropyran 31. The Koide group had originally attempted the reduction–allylation–ionic reduction sequence on the Boc-protected amine 32, however, this gave lower overall yields due to the competing formation of a pyrrolidine byproduct.
![[1860-5397-16-166-i3]](/bjoc/content/inline/1860-5397-16-166-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Koide synthesis of (all-cis)-2,3,5,6-tetrasubstituted tetrahydropyran.
Scheme 3: Koide synthesis of (all-cis)-2,3,5,6-tetrasubstituted tetrahydropyran.
Using the Stork–Zhao conditions [23], the Nicolaou group performed an α-methyliodomethylenation of 24 to give 33 with a good stereoselectivity (Z:E ≈ 95:5, Scheme 4) [24,25]. After the hydrolysis of the dimethylaminal and the reaction of the resultant amine with phthalic anhydride, a Pd-catalyzed Stille coupling [26] of 34 with 3-(tributylstannyl)-2-propen-1-ol gave 35. The oxidation of 35 with an excess of MnO2 gave the (2E,4Z)-dienal 36. Alternatively, the Stille coupling of 34 with 3-(tributylstannyl)acrolein also afforded 36. After much experimentation with up to eight amine catalysts, this group found that the intramolecular oxa-Michael addition using 20 mol % of the diarylprolinol organocatalyst 37 in the presence of benzoic acid gave 38. The use of the enantiomer of 37 gave the dihydropyran with the opposite configuration at C-11. The catalytic hydrogenation of 38 proceeded with a poor stereoselectivity, however, a similar reduction of the diethyl acetal of 38, followed by an acetal hydrolysis gave the aldehyde 39 with a good stereocontrol (>10:1).
![[1860-5397-16-166-i4]](/bjoc/content/inline/1860-5397-16-166-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Nicolaou synthesis of the (all-cis)-2,3,5,6-tetrasubstituted tetrahydropyran.
Scheme 4: Nicolaou synthesis of the (all-cis)-2,3,5,6-tetrasubstituted tetrahydropyran.
It is not possible to make a direct comparison of the efficiency of these three routes as they do not lead to an identical endpoint. However, Nicolaou’s route is the shortest (6 or 7 steps, 9.8–9.2% yield), while Kitahara’s synthesis is the highest-yielding and does not involve the use of expensive transition metals or organocatalysts.
Syntheses to generate the C-14 stereocenter via C–N bond formation
Two groups implemented strategies that rely on the generation of the C-14 stereocenter by stereoselective C–N bond formation. The Jacobsen group utilized an asymmetric Cr(III)-catalyzed cycloaddition reaction [27] between (2Z,4E)-3-(triethylsilyloxy)-2,4-hexadiene (40) and the aldehyde 41 to generate the 4-silyloxydihydropyran 43 in a high yield and enantioselectivity (Scheme 5) [17,18]. The Rubottom oxidation [28] of 43 gave a separable mixture of the desired 44 and its C-14 epimer (≈7:1 ratio). The reductive deoxygenation of 44 proceeded via the tosylhydrazone to afford 45, which upon desilylation and alkyne isomerization produced 46. The hydrozirconation of 46 with Schwartz’s reagent under equilibrating conditions, followed by the reaction with I2 gave the vinyl iodide 47. Finally, the activation of the C-14 hydroxy group and the SN2 displacement with azide gave the C-8–C-16 fragment 48.
![[1860-5397-16-166-i5]](/bjoc/content/inline/1860-5397-16-166-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Jacobsen synthesis of the (all-cis)-2,3,5,6-tetrasubstituted tetrahydropyran.
Scheme 5: Jacobsen synthesis of the (all-cis)-2,3,5,6-tetrasubstituted tetrahydropyran.
Ghosh relied on a reductive amination of the tetrahydropyranone 50 to generate the (all-cis)-tetrahydropyran fragment. This group reported multiple different routes to 50. In an abortive route, the addition of allyl-Grignard to 5-methylfurfural, followed by the resolution of the racemic homoallylic alcohol with Amano lipase gave (−)-51 (Scheme 6) [16]. An Achmatowicz oxidation [29] with tert-butyl hydroperoxide catalyzed by VO(acac)2 afforded the hemiketal 52, and the ionic reduction [22] of 52 gave the enone (−)-53. The enone transposition of 53 was effected by the addition of methyl-Grignard, followed by the oxidation with PCC. Unfortunately, all attempted 1,4-reduction conditions afforded an inseparable mixture of trans-54 as the major product along with minor amounts of the desired cis-55.
![[1860-5397-16-166-i6]](/bjoc/content/inline/1860-5397-16-166-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Unproductive attempt to generate the (all-cis)-tetrahydropyranone 50.
Scheme 6: Unproductive attempt to generate the (all-cis)-tetrahydropyranone 50.
Alternatively, the asymmetric reduction of the 2-acylfuran 56 using the Corey–Bakshi–Shibata reagent (21) [30] gave the alcohol 57 in a high yield and high enantioselectivity (Scheme 7) [15,16]. The Achmatowicz oxidation and ionic reduction generated the enone 58, which is regioisomeric with 53. The 1,4-addition of lithium dimethylcopper gave the desired cis-55 with high a diastereoselectivity (25:1). The cross-metathesis of 55 with 3-methyl-3-buten-1-yl tosylate in the presence of Grubbs’ 2nd generation catalyst yielded 59, which, upon elimination with potassium tert-butoxide led to the diene 50. The reductive amination of 50 afforded an inseparable mixture of the C-14 amines (6:1 ratio). However, the amidation of this mixture with 12c gave a separable mixture of the desired 49 (52%) along with the C-14 epimer (8%).
![[1860-5397-16-166-i7]](/bjoc/content/inline/1860-5397-16-166-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Ghosh synthesis of the C-7–C-14 (all-cis)-tetrahydropyran segment.
Scheme 7: Ghosh synthesis of the C-7–C-14 (all-cis)-tetrahydropyran segment.
More recently, the Ghosh group provided an alternative synthesis from the triacetyl ᴅ-glucal 60 (Scheme 8) [31]. The exhaustive hydrolysis and selective protection as the 4,6-O-di-tert-butylsilylene derivative 61 [32] was followed by a 3-O-vinylation. A thermal 3,3-sigmatropic Claisen rearrangement of 62 gave the cis-2,6-disubstituted dihydropyran 63, which upon sequential Wittig olefination with 2-(triphenylphosporanylidene)propanal and then methylenetriphenylphosphorane yielded the diene 64. The removal of the di-tert-butylsilylene protecting group and the selective arylsulfonylation of the primary alcohol was effected with the bulky 2,4,6-triisopropylsulfonyl chloride, which upon reduction with aluminum hydride, followed by the oxidation of the remaining alcohol group gave the dihydropyran-3-one 65. In a fashion similar to that of the dihydropyranone 58 [16], the 1,4-addition of dimethyl copper lithium to 65 gave the desired cis-50 as a single diastereomer [31], allowing for a convergence with the route in Scheme 7.
![[1860-5397-16-166-i8]](/bjoc/content/inline/1860-5397-16-166-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Ghosh’s alternative route to the (all-cis)-tetrahydropyranone 50.
Scheme 8: Ghosh’s alternative route to the (all-cis)-tetrahydropyranone 50.
Alternatively, the Wittig methenylation of 63, followed by a silyl ether cleavage gave 66 (Scheme 9) [33]. A three-step sequence similar to that from 64 to 65 allowed for the transformation of 66 to 58, and thus providing an additional pathway for a convergence.
![[1860-5397-16-166-i9]](/bjoc/content/inline/1860-5397-16-166-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Alternative synthesis of the dihydro-3-pyrone 58.
Scheme 9: Alternative synthesis of the dihydro-3-pyrone 58.
Synthesis of the C-1–C-6 tetrahydropyran fragment of FR901464 (1)/spliceostatins/thailanstatins
Syntheses of the C-1–C-6 segment of FR901464 (1) from chiral pool precursors
Kitahara’s group fashioned the C-1–C-6 tetrahydropyran fragment of FR901464 (1) from commercially available 2-deoxy-ᴅ-glucose. In the first-generation approach (66, Scheme 10) [8], a sequence of protection and oxidation steps generated the tetrahydropyrone 67 at which point an olefination with the Tebbe reagent and the addition of methanol afforded the cyclic ketal as a separable mixture of the diastereomers 68 and 69 (≈4.5:1 ratio). The desilylation of 68 gave the 2° alcohol, which after oxidation and Tebbe olefination afforded the exocyclic olefin 70. The manipulation of the C-4 and C-6 protecting groups gave the secondary allylic alcohol 71, which underwent an epoxidation with mCPBA to give 72. A second sequence of C-4/C-6 protection, manipulation, and oxidation gave the aldehyde 73. The disadvantages of this route include the overall length (13 or 14 steps), low yield (5.4%), and the relative expense of the starting material ($22.3/g).
![[1860-5397-16-166-i10]](/bjoc/content/inline/1860-5397-16-166-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Kitahara’s 1st-generation synthesis of the C-1–C-6 fragment of FR901464 (1).
Scheme 10: Kitahara’s 1st-generation synthesis of the C-1–C-6 fragment of FR901464 (1).
In their second-generation approach (Scheme 11) [9], the acetonide-protected dithiane was alkylated according to Horton’s procedure [34]. A deprotection and cyclic-ketal formation gave 70, allowing for a convergence with Scheme 10.
![[1860-5397-16-166-i11]](/bjoc/content/inline/1860-5397-16-166-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 11: Kitahara 1st-generation synthesis of the C-1–C-6 fragment of FR901464 (1).
Scheme 11: Kitahara 1st-generation synthesis of the C-1–C-6 fragment of FR901464 (1).
Very recently, the groups of Nimura and Arisawa reported the synthesis of a phenyl C-glucoside derivative of spliceostatin beginning from ᴅ-glucal (Scheme 12) [35]. A Heck coupling of the tris(trimethylsilyl) ether of 74 with phenylboronic acid in the presence of Pd(OAc)2 and benzoquinone (BQ) gave the C-phenyl glucoside 75 [36]. Notably, the use of oxidants other than BQ gave either the TMS enol ether or the 2,3-dihydro-6-phenyl-4H-pyran-4-one. The C-3 exocyclic methylene group was introduced by a Wittig olefination, and after the manipulation of the protecting groups, a VO(acac)2-catalyzed oxidation stereoselectively generated the spirocyclic epoxide 76. A second-protecting group shuffle afforded the primary alcohol 77. A Mitsunobu substitution of 77 with 2-mercaptobenzothiazole, followed by an oxidation with a large excess of mCPBA afforded the sulfone 78. While not producing the identical product, this route is shorter and higher-yielding (11 steps, 11.5%) than the Kitahara synthesis. However, it suffers from repeated protection/deprotection steps.
![[1860-5397-16-166-i12]](/bjoc/content/inline/1860-5397-16-166-i12.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 12: Nimura/Arisawa synthesis of the C-1-phenyl segment.
Scheme 12: Nimura/Arisawa synthesis of the C-1-phenyl segment.
The Ghosh group utilized (R)-glyceraldehyde acetonide (79, readily available from ᴅ-mannitol) as a chiral pool precursor for the introduction of the C-6 stereocenter (Scheme 13) [15,16]. The administration of the vinyllithium reagent, generated from the addition of the vinyl bromide 80 to 79, gave a separable 1:1 mixture of the diastereomeric alcohols 81a and 81b. The undesired stereoisomer 81b was converted into 81a by a Mitsunobu reaction/hydrolysis sequence. The use of the dithiane 80 protecting group was crucial for the following steps. Initial attempts of using a dioxolane protecting group for the C-1 ketone (instead of dithiane) led to an insurmountable difficulty of the selective hydrolysis of the dioxolane and acetonide ketals. The protection of the C-4 hydroxy group, hydrolysis of the acetonide, and selective tosylation of the 1° alcohol were prerequisites for the generation of the C-5–C-6 bond. To this end, the reaction of 82 with the ylide generated from trimethylsulfonium iodide gave the allylic alcohol 83. The removal of the dithiane protecting group and the cyclic-ketal formation gave 84. The oxidative hydrolysis of the PMB ether and the reaction with mCPBA afforded the epoxide 85. While relatively short (9 steps), the nonstereoselective formation of 81a/b led to a lower overall yield.
![[1860-5397-16-166-i13]](/bjoc/content/inline/1860-5397-16-166-i13.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 13: Ghosh synthesis of the C-1–C-6 fragment of FR901464 (1) from (R)-glyceraldehyde acetonide.
Scheme 13: Ghosh synthesis of the C-1–C-6 fragment of FR901464 (1) from (R)-glyceraldehyde acetonide.
Syntheses of the C-1–C-6 segment of FR901464 (1) via asymmetric catalysis
Similar to their preparation of the C-8–C-15 segment, Jacobsen’s group relied on a Cr(III)-catalyzed silyloxydiene/aldehyde cycloaddition for the C-1–C-7 segment (Scheme 14) [17,18]. The reaction of the protected glycolaldehyde 86 with (1E,3E)-2-triethylsilyloxy-1,3-hexadien-5-yne (87) in the presence of 42 gave the dihydropyran 88 with excellent enantioselectivity. A Rubottom oxidation, protection of the C-4 alcohol, and a Wittig methenylation afforded 89. The selective deprotection of the primary TBS ether, followed by an Appel iodination and the cleavage of the secondary triisopropylsilyl ether were prerequisites for a vanadium-catalyzed stereoselective epoxidation of the exocyclic double bond to give 90. The C-4 hydroxy group was eventually protected as a triethylsilyl ether. Through abortive attempts, Jacobsen found that the generation of the exocyclic epoxide prior to the formation of the C-6–C-9 conjugated diene was necessary in order to avoid the unwanted epoxidation of the C-6–C-7 olefin.
![[1860-5397-16-166-i14]](/bjoc/content/inline/1860-5397-16-166-i14.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 14: Jacobsen synthesis of the C-1–C-7 segment of FR901464 (1).
Scheme 14: Jacobsen synthesis of the C-1–C-7 segment of FR901464 (1).
Koide employed a unique strategy in which the exocyclic epoxide was generated as the initial stereocenter (Scheme 15) [12,13]. The Sharpless asymmetric epoxidation [37] of 5-methyl-2-methylene-4-penten-1-ol gave the epoxyalcohol 92 in 94% ee, which was oxidized to the aldehyde 93. While the addition of the lithium salt of methyl propynoate proceeded in a nondiastereoselective fashion, the use of a zirconium/silver-mediated alkynylation gave the alcohol 94 with a 6:1 diastereoselectivity. The Red-Al reduction of 94, protection of the 2° alcohol, and reduction of the enoate to an allylic alcohol were prerequisites for the stereoselective 2,3-sigmatropic selenoxide rearrangement to generate 95. Further protecting group manipulation and Johnson–Lemieux cleavage afforded the methyl ketone 96. The cyclic hemiketal 97 was unstable at an elevated temperature (37 °C), with t1/2 = 48 h. Both the Jacobsen (10 steps, 24.4% yield) and the Koide syntheses (11 steps, 22.3% yield) are relatively efficient in terms of the length and overall yield.
![[1860-5397-16-166-i15]](/bjoc/content/inline/1860-5397-16-166-i15.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 15: Koide synthesis of the C-1–C-7 segment of FR901464 (1).
Scheme 15: Koide synthesis of the C-1–C-7 segment of FR901464 (1).
Syntheses of the C-1–C-5 segment of thailanstatin methyl esters
Both syntheses of the C-1–C-5 tetrahydropyran segment of thailanstatin A (7) utilize sugar precursors. In the Ghosh synthesis [31], the triacetyl ᴅ-glucal 60 is converted into the trans C-1-allylated tetrahydropyran 98 according to literature procedures [38,39] (Scheme 16). After a protecting group manipulation, the C-1 allyl group was truncated via ozonolysis, and the C-3 hydroxy group was transformed into an exocyclic methylene group to give 99. The conversion of the C-6 alcohol moiety to a vinyl group by oxidation and Wittig methenylation to give 100 was relatively inefficient (31%) and was the main factor in diminishing the overall yield. The transformation of the silyl-protected 1° alcohol group of 100 into an ester afforded 101. Finally, the C-3 spirocyclic oxirane was introduced stereoselectively by a VO(acac)2-directed epoxidation. While highly stereoselective, this relatively lengthy route was somewhat inefficient (16 steps, 1.4% overall yield).
![[1860-5397-16-166-i16]](/bjoc/content/inline/1860-5397-16-166-i16.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 16: Ghosh synthesis of the C-1–C-5 segment 102 of thailanstatin A (7).
Scheme 16: Ghosh synthesis of the C-1–C-5 segment 102 of thailanstatin A (7).
The Nicolaou synthesis [24,25] of the C-1–C-9 tetrahydropyranyl segment commenced with the protected dihydropyranone 103 (Scheme 17), readily prepared from ᴅ-glucal [38]. The application of an iodine-catalyzed Mukiyama–Michael addition of the ketene silyl acetal 104 to 103 afforded the trans-1,5-disubstituted tetrahydropyranone 105. After the generation of the C-3-exocyclic olefin and functional group manipulation, the Takai olefination [40] of the aldehyde 106 gave the trans-vinyl iodide 107. The removal of the silyl protecting group and hydrolysis of the methyl ester afforded the carboxylic acid 108. A subsequently attempted Suzuki–Miyaura coupling of 108 with vinyl boronates was described as “capricious”, and thus the acid was esterified with the tert-butyl donor reagent 109 to afford 110. In a fashion similar to that used by Ghosh’s group, the VO(acac)2-catalyzed epoxidation of 110 afforded the spirocyclic oxirane 111. A subsequent ring opening with LiCl gave 112, which was used in the synthesis of thailanstatin B (5). Nicolaou’s route to 111 (9 steps, 29.9% yield) is shorter and considerably more efficient than the Ghosh synthesis of 102.
![[1860-5397-16-166-i17]](/bjoc/content/inline/1860-5397-16-166-i17.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 17: Nicolaou synthesis of the C-1–C-9 segments of spliceostatin D (9) and thailanstatins A (7) and B (5).
Scheme 17: Nicolaou synthesis of the C-1–C-9 segments of spliceostatin D (9) and thailanstatins A (7) and B (5...
Syntheses of the C-1–C-6 segment of spliceostatin E (10)
The Ghosh group’s synthesis of the C-1–C-6 segment of spliceostatin E (10) relied on a Cu-catalyzed Grignard addition to tert-butyldiphenylsilyl-protected (R)-glycidol, followed by the generation of the mixed acetal 113 (Scheme 18) [33]. A ring-closing metathesis gave an inseparable mixture of the dihydropyranyl ethers 114a and 14b, which could be equilibrated under acidic conditions (114b/114a > 20:1). A standard functional group manipulation afforded the vinyldihydropyran-2-one (−)-115.
![[1860-5397-16-166-i18]](/bjoc/content/inline/1860-5397-16-166-i18.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 18: Ghosh synthesis of the C-1–C-6 segment 115 of spliceostatin E (10).
Scheme 18: Ghosh synthesis of the C-1–C-6 segment 115 of spliceostatin E (10).
Fragment coupling to complete the synthesis of FR901464 (1)/spliceostatins/thailanstatins
Fragment coupling via Wittig and Julia olefinations
Kitahara’s group utilized Wittig and modified/one-pot Julia olefination [41] reactions to fashion the dienyl segment, joining the two tetrahydropyran fragments. As their 2nd-generation synthesis was more efficient, this will be described (Scheme 19) [9]. To this end, the C-9–C-15 aldehyde 27 underwent an olefination with (carbethoxyethylidene)triphenylphosphorane to afford 116. The enoate 116 was elaborated into the 1,3-benzothiazolesulfone 117 by standard transformations, prior to the cleavage of the Boc amide protecting group. The free amine 118 underwent an amidation with the TBS-protected (2Z,4S)-4-hydroxy-2-butenoic acid 12b to give 119. The modified Julia olefination of the aldehyde 73 with the anion derived from 119 proceeded with a high E-selectivity to generate the mixed cyclic ketal 120. Finally, the removal of the C-4’ silyl protecting group, acylation of the resultant alcohol, removal of the C-4 silyl protecting group, and ketal hydrolysis generated FR901464 (1).
![[1860-5397-16-166-i19]](/bjoc/content/inline/1860-5397-16-166-i19.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 19: Fragment coupling via Wittig and modified Julia olefinations by Kitahara.
Scheme 19: Fragment coupling via Wittig and modified Julia olefinations by Kitahara.
Fragment coupling via olefin cross-metathesis
Koide’s group was the first to demonstrate the use of a Ru-catalyzed olefin cross-metathesis for the generation of the dienyl segment joining the two tetrahydropyranyl segments (Scheme 20) [12,13]. The cleavage of the Boc amide of 31 and the eventual amidation with (2Z,4S)-4-acetoxy-2-butenoic acid (12c) gave 121. The construction of the diene 49 involved cross-metathesis with an excess of methacrolein, catalyzed by 122, followed by a Wittig olefination. The union of the diene 49 with 1.8 equivalents of the vinyl tetrahydropyran 97 was achieved with the Ru catalyst 122. One recycle of the recovered starting material from this reaction gave FR901464 (1) in a combined yield of 40%. This reaction needed to be done at room temperature due to the lability of the hemiketal 97. The use of Grubbs’ 2nd generation catalyst or the Grubbs–Hoveyda catalyst also gave 1, albeit in a diminished yield (12–13%).
![[1860-5397-16-166-i20]](/bjoc/content/inline/1860-5397-16-166-i20.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 20: Fragment coupling via cross-metathesis by Koide.
Scheme 20: Fragment coupling via cross-metathesis by Koide.
The Ghosh synthesis of spliceostatin A (4), FR901464 (1) [15,16], spliceostatin E (10) [41], and thailanstatin A methyl ester (123) [31] used the cross-metathesis strategy for the coupling of the diene 49 with 85, 115, and 102, respectively (Scheme 21). In order to avoid the decomposition problems encountered by Koide, the mixed cyclic ketal 85 was used as a coupling partner for the preparation of spliceostatin A (4). Due to this change, Grubbs’ second-generation catalyst, at a lower loading and elevated reaction temperatures, could be used since 4 was not labile under these thermal conditions. Ketal 4 underwent a hydrolysis to 1 under acidic conditions. The yields for the cross-metathesis coupling were generally higher when using an excess of 85 or 115, as compared to the cross-metathesis of 102, which also used a relatively high catalyst loading (20 mol % for 46% yield). While these authors did not report on the hydrolysis of 123 to prepare thailanstatin A (7), they found that the methyl ester 123 (IC50 ≈ 0.4 μM) is nearly equipotent with 7 (IC50 ≈ 0.65 μM).
![[1860-5397-16-166-i21]](/bjoc/content/inline/1860-5397-16-166-i21.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 21: The Ghosh synthesis of spliceostatin A (4), FR901464 (1), spliceostatin E (10), and thailanstatin methyl ester (123).
Scheme 21: The Ghosh synthesis of spliceostatin A (4), FR901464 (1), spliceostatin E (10), and thailanstatin m...
Fragment coupling via Wittig olefination, cross-metathesis, and Julia olefination
In their recent synthesis of a 1-phenyl analog (124) of FR901464 (1), Arisawa’s group utilized a combination of previously reported methodologies (Scheme 22) [35]. Thus, the (all-cis)-2,3,5,6-tetrasubstituted aldehyde 27 was constructed according to Kitahara’s protocol [8,9]. The Wittig methenylation of 27, followed by the cleavage of the Boc protecting group and the amidation with 12b afforded the fragment 125. The cross-metathesis of 125 with an excess of methacrolein in the presence of the Ru catalyst 122 gave the aldehyde 126. This coupling is analogous to Koide’s cross-metathesis of 121 (cf. Scheme 20). A modified Julia olefination of 126 with the anion generated from the sulfone 78, followed by the cleavage of the 4’-TBS protecting group gave 127 in an acceptable yield (52%). A protecting-group adjustment finalized the synthesis of 124. Notably, these authors found that switching the sulfone and aldehyde functionalities, i.e., an olefination of the aldehyde generated by the oxidation of 77 with Kitahara’s sulfone 119, proceeded less efficiently (22% yield). In a subsequent assay for repressed cell proliferation against the human prostate cancer cell lines LNCaP, LNCaP95, and CWR22Rv, these authors reported IC50 values of 63, 175, and 93 nM, respectively, for 124. These can be compared to the IC50 values of 1.3, 1.0, and 0.36 nM, respectively, for spliceostatin A (4) against the same cell lines.
![[1860-5397-16-166-i22]](/bjoc/content/inline/1860-5397-16-166-i22.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 22: Arisawa synthesis of a C-1-phenyl analog of FR901464 (1).
Scheme 22: Arisawa synthesis of a C-1-phenyl analog of FR901464 (1).
Fragment union via Pd-catalyzed sp2–sp2 coupling
A Negishi coupling reaction [42] was used in the Jacobsen synthesis of FR901464 (1, Scheme 23) [17,18]. The hydrozirconation of 91, followed by a transmetalation provided a vinylzinc reagent that was coupled with 48 to afford 128, for which only the E-stereoisomer was observed. Notably, the Negishi conditions were tolerant to the azide present in 48 and the oxirane and 1° iodoalkane present in 91. The subsequent reduction of the azide and the amidation with 12c afforded 129. The synthesis was completed by the elimination of the 1° iodide, silyl ether cleavage, and hydration of the exocyclic enol ether. Of all the syntheses of FR901464 (1), the coupling of 91 with 48 is the most efficient sequence (6 steps, 45% yield).
![[1860-5397-16-166-i23]](/bjoc/content/inline/1860-5397-16-166-i23.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 23: Jacobsen fragment coupling by a Pd-catalyzed Negishi coupling.
Scheme 23: Jacobsen fragment coupling by a Pd-catalyzed Negishi coupling.
Nicolaou utilized the sequential application of a cross-metathesis and a Suzuki–Miyaura coupling in the syntheses of thailanstatin A and B (7 and 5) and spliceostatin D (9, Scheme 24) [24,25]. To this end, the methylhydrazinolysis of the phthalimide 39 and the amide formation with 12c yielded 121. The cross-metathesis of 121 with a five-fold excess of isopropenylboronic acid pinacol ester afforded the lynchpin vinylborane 130. A Pd-catalyzed Suzuki–Miyaura coupling of 130 with the vinyl iodide 110, 111, or 112 gave the tert-butyl ester 131, 132, or 133 in a moderate yield (42–63%). The hydrolysis of the tert-butyl esters with formic acid gave the carboxylic acid 9, 7, or 5. Additionally, the treatment of the thailanstatin A tert-butyl ester with lithium chloride generated the thailanstatin B tert-butyl ester.
![[1860-5397-16-166-i24]](/bjoc/content/inline/1860-5397-16-166-i24.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 24: Nicolaou syntheses of thailanstatin A and B (7 and 5) and spliceostatin D (9) via a Pd-catalyzed Suzuki–Miyaura coupling.
Scheme 24: Nicolaou syntheses of thailanstatin A and B (7 and 5) and spliceostatin D (9) via a Pd-catalyzed Su...
Ghosh used a similar cross-metathesis/Suzuki–Miyaura coupling sequence for the preparation of spliceostatin G (11, Scheme 25) [43]. The catalyst loading for the coupling of 130 with methyl (E)-3-iodoacrylate (20 mol %) was twice the amount used by Nicolaou (10 mol %).
![[1860-5397-16-166-i25]](/bjoc/content/inline/1860-5397-16-166-i25.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 25: The Ghosh synthesis of spliceostatin G (11) via Suzuki–Miyaura coupling.
Scheme 25: The Ghosh synthesis of spliceostatin G (11) via Suzuki–Miyaura coupling.
Conclusion
As delineated above, a wide variety of synthetic strategies has been employed to either introduce the stereocenters present in the spliceostatins/thailanstatins or to couple the subfragments of these molecules (Table 2). The most common route to the (2Z,4S)-4-acetoxy-2-butenoic acid fragment relies on the cis-reduction of 4-acetoxy-2-pentynoic acid; the most efficient of these routes utilizes a Noyori asymmetric reduction of an alkynyl ketone to produce the required stereocenter. Of the syntheses to date, Jacobsen’s use of an asymmetric Cr(III)-catalyzed cycloaddition stands as the most efficient route, in terms of synthetic steps and low catalyst loading, to the (all-cis)-2,3,5,6-tetrasubstituted tetrahydropyran and the C-1–C-6 tetrahydropyran fragments of FR901464 (1). The preparation of the C-1–C-5 tetrahydropyran fragment of the thailanstatins has, so far, utilized sugar-derived precursors. A variety of reactions have been utilized for union of these fragments via a diene segment. While a Ru-catalyzed cross-metathesis reaction was employed in numerous syntheses, the drawbacks of this strategy include the high catalyst loading and the excess of the olefin coupling partners. To date, the most efficient strategy for the union of the fragments relies on Pd-catalyzed sp2–sp2 couplings.
Table 2: Summary of the total syntheses.
| target | PI | year | Reference | Scheme | no. of steps (LLS) | overall yield |
| FR901464 (1) | Jacobsen | 2000 | [17,18] | 5, 14, 23 | 16 | 8.46% |
| FR901464 (1) 1st generation | Kitahara | 2001 | [8] | 2, 10, 19 | 22 | 1.63% |
| FR901464 (1) 2nd generation | Kitahara | 2006 | [9] | 2, 11, 19 | 23 | 3.18% |
| FR901464 (1) | Koide | 2006 | [12,13] | 3, 15, 20 | 15 | 3.77% |
| spliceostatin A (4) | Ghosh | 2013 | [15,16] | 3, 15, 20 | 11 | 6.61% |
| FR901464 (1) | Ghosh | 2013 | [15,16] | 7, 13, 21 | 12 | 5.22% |
| spliceostatin E (10) | Ghosh | 2014 | [14] | 7, 18, 21 | 9 | 8.24% |
| thailanstatin A (7) | Nicolaou | 2016 | [24] | 4, 17, 24 | 12 | 2.23% |
| thailanstatin A methyl ester (123) | Ghosh | 2018 | [31] | 8, 16, 21 | 15 | 0.54% |
| spliceostatin G (11) | Ghosh | 2018 | [43] | 9, 25 | 15 | 2.31% |
| spliceostatin D (9) | Nicolaou | 2018 | [25] | 4, 17, 24 | 12 | 2.24% |
| thailanstatin B (5) | Nicolaou | 2018 | [25] | 4, 17, 24 | 12 | 1.87% |
| spliceostatin A analog | Arisawa | 2019 | [34] | 2, 12, 22 | 18 | 3.99% |
References
-
Nakajima, H.; Sato, B.; Fujita, T.; Takase, S.; Terano, H.; Okuhara, M. J. Antibiot. 1996, 49, 1196–1203. doi:10.7164/antibiotics.49.1196
Return to citation in text: [1] [2] -
Nakajima, H.; Hori, Y.; Terano, H.; Okuhara, M.; Manda, T.; Matsumoto, S.; Shimomura, K. J. Antibiot. 1996, 49, 1204–1211. doi:10.7164/antibiotics.49.1204
Return to citation in text: [1] -
Liu, X.; Biswas, S.; Tang, G.-L.; Cheng, Y.-Q. J. Antibiot. 2013, 66, 555–558. doi:10.1038/ja.2013.38
Return to citation in text: [1] -
Liu, X.; Biswas, S.; Berg, M. G.; Antapli, C. M.; Xie, F.; Wang, Q.; Tang, M.-C.; Tang, G.-L.; Zhang, L.; Dreyfuss, G.; Cheng, Y.-Q. J. Nat. Prod. 2013, 76, 685–693. doi:10.1021/np300913h
Return to citation in text: [1] -
He, H.; Ratnayake, A. S.; Janso, J. E.; He, M.; Yang, H. Y.; Loganzo, F.; Shor, B.; O’Donnell, C. J.; Koehn, F. E. J. Nat. Prod. 2014, 77, 1864–1870. doi:10.1021/np500342m
Return to citation in text: [1] [2] -
Kaida, D.; Motoyoshi, H.; Tashiro, E.; Nojima, T.; Hagiwara, M.; Ishigami, K.; Watanabe, H.; Kitahara, T.; Yoshida, T.; Nakajima, H.; Tani, T.; Horinouchi, S.; Yoshida, M. Nat. Chem. Biol. 2007, 3, 576–583. doi:10.1038/nchembio.2007.18
Return to citation in text: [1] -
Pham, D.; Koide, K. Nat. Prod. Rep. 2016, 33, 637–647. doi:10.1039/c5np00110b
Return to citation in text: [1] -
Horigome, M.; Motoyoshi, H.; Watanabe, H.; Kitahara, T. Tetrahedron Lett. 2001, 42, 8207–8210. doi:10.1016/s0040-4039(01)01763-4
Return to citation in text: [1] [2] [3] [4] [5] -
Motoyoshi, H.; Horigome, M.; Watanabe, H.; Kitahara, T. Tetrahedron 2006, 62, 1378–1389. doi:10.1016/j.tet.2005.11.031
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Massad, S. K.; Hawkins, L. D.; Baker, D. C. J. Org. Chem. 1983, 48, 5180–5182. doi:10.1021/jo00174a006
Return to citation in text: [1] -
Still, W. C.; Gennari, C. Tetrahedron Lett. 1983, 24, 4405–4408. doi:10.1016/s0040-4039(00)85909-2
Return to citation in text: [1] -
Albert, B. J.; Sivaramakrishnan, A.; Naka, T.; Koide, K. J. Am. Chem. Soc. 2006, 128, 2792–2793. doi:10.1021/ja058216u
Return to citation in text: [1] [2] [3] [4] -
Albert, B. J.; Sivaramakrishnan, A.; Naka, T.; Czaicki, N. L.; Koide, K. J. Am. Chem. Soc. 2007, 129, 2648–2659. doi:10.1021/ja067870m
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Frantz, D. E.; Fässler, R.; Carreira, E. M. J. Am. Chem. Soc. 2000, 122, 1806–1807. doi:10.1021/ja993838z
Return to citation in text: [1] [2] -
Ghosh, A. K.; Chen, Z.-H. Org. Lett. 2013, 15, 5088–5091. doi:10.1021/ol4024634
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Ghosh, A. K.; Chen, Z.-H.; Effenberger, K. A.; Jurica, M. S. J. Org. Chem. 2014, 79, 5697–5709. doi:10.1021/jo500800k
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] -
Thompson, C. F.; Jamison, T. F.; Jacobsen, E. N. J. Am. Chem. Soc. 2000, 122, 10482–10483. doi:10.1021/ja0055357
Return to citation in text: [1] [2] [3] [4] [5] -
Thompson, C. F.; Jamison, T. F.; Jacobsen, E. N. J. Am. Chem. Soc. 2001, 123, 9974–9983. doi:10.1021/ja016615t
Return to citation in text: [1] [2] [3] [4] [5] -
Matsumura, K.; Hashiguchi, S.; Ikariya, T.; Noyori, R. J. Am. Chem. Soc. 1997, 119, 8738–8739. doi:10.1021/ja971570a
Return to citation in text: [1] -
Corey, E. J.; Bakshi, R. K.; Shibata, S.; Chen, C. P.; Singh, V. K. J. Am. Chem. Soc. 1987, 109, 7925–7926. doi:10.1021/ja00259a075
Return to citation in text: [1] -
Garner, P.; Ramakanth, S. J. Org. Chem. 1987, 52, 2629–2631. doi:10.1021/jo00388a063
Return to citation in text: [1] -
Lewis, M. D.; Cha, J. K.; Kishi, Y. J. Am. Chem. Soc. 1982, 104, 4976–4978. doi:10.1021/ja00382a053
Return to citation in text: [1] [2] -
Stork, G.; Zhao, K. Tetrahedron Lett. 1989, 30, 2173–2174. doi:10.1016/s0040-4039(00)99640-0
Return to citation in text: [1] -
Nicolaou, K. C.; Rhoades, D.; Lamani, M.; Pattanayak, M. R.; Kumar, S. M. J. Am. Chem. Soc. 2016, 138, 7532–7535. doi:10.1021/jacs.6b04781
Return to citation in text: [1] [2] [3] [4] -
Nicolaou, K. C.; Rhoades, D.; Kumar, S. M. J. Am. Chem. Soc. 2018, 140, 8303–8320. doi:10.1021/jacs.8b04634
Return to citation in text: [1] [2] [3] [4] [5] -
Espinet, P.; Echavarren, A. M. Angew. Chem., Int. Ed. 2004, 43, 4704–4734. doi:10.1002/anie.200300638
Return to citation in text: [1] -
Dossetter, A. G.; Jamison, T. F.; Jacobsen, E. N. Angew. Chem., Int. Ed. 1999, 38, 2398–2400. doi:10.1002/(sici)1521-3773(19990816)38:16<2398::aid-anie2398>3.0.co;2-e
Return to citation in text: [1] -
Rubottom, G. M.; Vazquez, M. A.; Pelegrina, D. R. Tetrahedron Lett. 1974, 15, 4319–4322. doi:10.1016/s0040-4039(01)92153-7
Return to citation in text: [1] -
Deska, J.; Thiel, D.; Gianolio, E. Synthesis 2015, 47, 3435–3450. doi:10.1055/s-0035-1560345
Return to citation in text: [1] -
Corey, E. J.; Bakshi, R. K.; Shibata, S. J. Am. Chem. Soc. 1987, 109, 5551–5553. doi:10.1021/ja00252a056
Return to citation in text: [1] -
Ghosh, A. K.; Veitschegger, A. M.; Nie, S.; Relitti, N.; MacRae, A. J.; Jurica, M. S. J. Org. Chem. 2018, 83, 5187–5198. doi:10.1021/acs.joc.8b00593
Return to citation in text: [1] [2] [3] [4] [5] -
Pazos, G.; Pérez, M.; Gándara, Z.; Gómez, G.; Fall, Y. Tetrahedron Lett. 2009, 50, 5285–5287. doi:10.1016/j.tetlet.2009.07.041
Return to citation in text: [1] -
Ghosh, A. K.; Veitschegger, A. M.; Sheri, V. R.; Effenberger, K. A.; Prichard, B. E.; Jurica, M. S. Org. Lett. 2014, 16, 6200–6203. doi:10.1021/ol503127r
Return to citation in text: [1] [2] -
Horton, D.; Markovs, R. A. Carbohydr. Res. 1980, 78, 295–303. doi:10.1016/0008-6215(80)90010-5
Return to citation in text: [1] [2] -
Yoshikawa, Y.; Ishibashi, A.; Murai, K.; Kaneda, Y.; Nimura, K.; Arisawa, M. Tetrahedron Lett. 2019, 60, 151313. doi:10.1016/j.tetlet.2019.151313
Return to citation in text: [1] [2] -
Xiong, D.-C.; Zhang, L.-H.; Ye, X.-S. Org. Lett. 2009, 11, 1709–1712. doi:10.1021/ol900273d
Return to citation in text: [1] -
Pfenninger, A. Synthesis 1986, 89–116. doi:10.1055/s-1986-31489
Return to citation in text: [1] -
Yadav, J. S.; Reddy, B. V. S.; Reddy, K. B.; Satyanarayana, M. Tetrahedron Lett. 2002, 43, 7009–7012. doi:10.1016/s0040-4039(02)01584-8
Return to citation in text: [1] [2] -
Fujiwara, K.; Sato, D.; Watanabe, M.; Morishita, H.; Murai, A.; Kawai, H.; Suzuki, T. Tetrahedron Lett. 2004, 45, 5243–5246. doi:10.1016/j.tetlet.2004.05.020
Return to citation in text: [1] -
Takai, K.; Nitta, K.; Utimoto, K. J. Am. Chem. Soc. 1986, 108, 7408–7410. doi:10.1021/ja00283a046
Return to citation in text: [1] -
Blakemore, P. R. J. Chem. Soc., Perkin Trans. 1 2002, 2563–2585. doi:10.1039/b208078h
Return to citation in text: [1] [2] -
Negishi, E.-i. Angew. Chem., Int. Ed. 2011, 50, 6738–6764. doi:10.1002/anie.201101380
Return to citation in text: [1] -
Ghosh, A. K.; Reddy, G. C.; MacRae, A. J.; Jurica, M. S. Org. Lett. 2018, 20, 96–99. doi:10.1021/acs.orglett.7b03456
Return to citation in text: [1] [2]
| 16. | Ghosh, A. K.; Chen, Z.-H.; Effenberger, K. A.; Jurica, M. S. J. Org. Chem. 2014, 79, 5697–5709. doi:10.1021/jo500800k |
| 29. | Deska, J.; Thiel, D.; Gianolio, E. Synthesis 2015, 47, 3435–3450. doi:10.1055/s-0035-1560345 |
| 31. | Ghosh, A. K.; Veitschegger, A. M.; Nie, S.; Relitti, N.; MacRae, A. J.; Jurica, M. S. J. Org. Chem. 2018, 83, 5187–5198. doi:10.1021/acs.joc.8b00593 |
| 22. | Lewis, M. D.; Cha, J. K.; Kishi, Y. J. Am. Chem. Soc. 1982, 104, 4976–4978. doi:10.1021/ja00382a053 |
| 43. | Ghosh, A. K.; Reddy, G. C.; MacRae, A. J.; Jurica, M. S. Org. Lett. 2018, 20, 96–99. doi:10.1021/acs.orglett.7b03456 |
| 14. | Frantz, D. E.; Fässler, R.; Carreira, E. M. J. Am. Chem. Soc. 2000, 122, 1806–1807. doi:10.1021/ja993838z |
| 24. | Nicolaou, K. C.; Rhoades, D.; Lamani, M.; Pattanayak, M. R.; Kumar, S. M. J. Am. Chem. Soc. 2016, 138, 7532–7535. doi:10.1021/jacs.6b04781 |
| 15. | Ghosh, A. K.; Chen, Z.-H. Org. Lett. 2013, 15, 5088–5091. doi:10.1021/ol4024634 |
| 16. | Ghosh, A. K.; Chen, Z.-H.; Effenberger, K. A.; Jurica, M. S. J. Org. Chem. 2014, 79, 5697–5709. doi:10.1021/jo500800k |
| 33. | Ghosh, A. K.; Veitschegger, A. M.; Sheri, V. R.; Effenberger, K. A.; Prichard, B. E.; Jurica, M. S. Org. Lett. 2014, 16, 6200–6203. doi:10.1021/ol503127r |
| 8. | Horigome, M.; Motoyoshi, H.; Watanabe, H.; Kitahara, T. Tetrahedron Lett. 2001, 42, 8207–8210. doi:10.1016/s0040-4039(01)01763-4 |
| 16. | Ghosh, A. K.; Chen, Z.-H.; Effenberger, K. A.; Jurica, M. S. J. Org. Chem. 2014, 79, 5697–5709. doi:10.1021/jo500800k |
| 31. | Ghosh, A. K.; Veitschegger, A. M.; Nie, S.; Relitti, N.; MacRae, A. J.; Jurica, M. S. J. Org. Chem. 2018, 83, 5187–5198. doi:10.1021/acs.joc.8b00593 |
| 31. | Ghosh, A. K.; Veitschegger, A. M.; Nie, S.; Relitti, N.; MacRae, A. J.; Jurica, M. S. J. Org. Chem. 2018, 83, 5187–5198. doi:10.1021/acs.joc.8b00593 |
| 34. | Horton, D.; Markovs, R. A. Carbohydr. Res. 1980, 78, 295–303. doi:10.1016/0008-6215(80)90010-5 |
| 32. | Pazos, G.; Pérez, M.; Gándara, Z.; Gómez, G.; Fall, Y. Tetrahedron Lett. 2009, 50, 5285–5287. doi:10.1016/j.tetlet.2009.07.041 |
| 30. | Corey, E. J.; Bakshi, R. K.; Shibata, S. J. Am. Chem. Soc. 1987, 109, 5551–5553. doi:10.1021/ja00252a056 |
| 25. | Nicolaou, K. C.; Rhoades, D.; Kumar, S. M. J. Am. Chem. Soc. 2018, 140, 8303–8320. doi:10.1021/jacs.8b04634 |
| 15. | Ghosh, A. K.; Chen, Z.-H. Org. Lett. 2013, 15, 5088–5091. doi:10.1021/ol4024634 |
| 16. | Ghosh, A. K.; Chen, Z.-H.; Effenberger, K. A.; Jurica, M. S. J. Org. Chem. 2014, 79, 5697–5709. doi:10.1021/jo500800k |
| 25. | Nicolaou, K. C.; Rhoades, D.; Kumar, S. M. J. Am. Chem. Soc. 2018, 140, 8303–8320. doi:10.1021/jacs.8b04634 |
| 9. | Motoyoshi, H.; Horigome, M.; Watanabe, H.; Kitahara, T. Tetrahedron 2006, 62, 1378–1389. doi:10.1016/j.tet.2005.11.031 |
| 34. | Horton, D.; Markovs, R. A. Carbohydr. Res. 1980, 78, 295–303. doi:10.1016/0008-6215(80)90010-5 |
| 35. | Yoshikawa, Y.; Ishibashi, A.; Murai, K.; Kaneda, Y.; Nimura, K.; Arisawa, M. Tetrahedron Lett. 2019, 60, 151313. doi:10.1016/j.tetlet.2019.151313 |
| 38. | Yadav, J. S.; Reddy, B. V. S.; Reddy, K. B.; Satyanarayana, M. Tetrahedron Lett. 2002, 43, 7009–7012. doi:10.1016/s0040-4039(02)01584-8 |
| 39. | Fujiwara, K.; Sato, D.; Watanabe, M.; Morishita, H.; Murai, A.; Kawai, H.; Suzuki, T. Tetrahedron Lett. 2004, 45, 5243–5246. doi:10.1016/j.tetlet.2004.05.020 |
| 24. | Nicolaou, K. C.; Rhoades, D.; Lamani, M.; Pattanayak, M. R.; Kumar, S. M. J. Am. Chem. Soc. 2016, 138, 7532–7535. doi:10.1021/jacs.6b04781 |
| 25. | Nicolaou, K. C.; Rhoades, D.; Kumar, S. M. J. Am. Chem. Soc. 2018, 140, 8303–8320. doi:10.1021/jacs.8b04634 |
| 31. | Ghosh, A. K.; Veitschegger, A. M.; Nie, S.; Relitti, N.; MacRae, A. J.; Jurica, M. S. J. Org. Chem. 2018, 83, 5187–5198. doi:10.1021/acs.joc.8b00593 |
| 17. | Thompson, C. F.; Jamison, T. F.; Jacobsen, E. N. J. Am. Chem. Soc. 2000, 122, 10482–10483. doi:10.1021/ja0055357 |
| 18. | Thompson, C. F.; Jamison, T. F.; Jacobsen, E. N. J. Am. Chem. Soc. 2001, 123, 9974–9983. doi:10.1021/ja016615t |
| 12. | Albert, B. J.; Sivaramakrishnan, A.; Naka, T.; Koide, K. J. Am. Chem. Soc. 2006, 128, 2792–2793. doi:10.1021/ja058216u |
| 13. | Albert, B. J.; Sivaramakrishnan, A.; Naka, T.; Czaicki, N. L.; Koide, K. J. Am. Chem. Soc. 2007, 129, 2648–2659. doi:10.1021/ja067870m |
| 36. | Xiong, D.-C.; Zhang, L.-H.; Ye, X.-S. Org. Lett. 2009, 11, 1709–1712. doi:10.1021/ol900273d |
| 15. | Ghosh, A. K.; Chen, Z.-H. Org. Lett. 2013, 15, 5088–5091. doi:10.1021/ol4024634 |
| 16. | Ghosh, A. K.; Chen, Z.-H.; Effenberger, K. A.; Jurica, M. S. J. Org. Chem. 2014, 79, 5697–5709. doi:10.1021/jo500800k |
| 40. | Takai, K.; Nitta, K.; Utimoto, K. J. Am. Chem. Soc. 1986, 108, 7408–7410. doi:10.1021/ja00283a046 |
| 33. | Ghosh, A. K.; Veitschegger, A. M.; Sheri, V. R.; Effenberger, K. A.; Prichard, B. E.; Jurica, M. S. Org. Lett. 2014, 16, 6200–6203. doi:10.1021/ol503127r |
| 38. | Yadav, J. S.; Reddy, B. V. S.; Reddy, K. B.; Satyanarayana, M. Tetrahedron Lett. 2002, 43, 7009–7012. doi:10.1016/s0040-4039(02)01584-8 |
| 1. | Nakajima, H.; Sato, B.; Fujita, T.; Takase, S.; Terano, H.; Okuhara, M. J. Antibiot. 1996, 49, 1196–1203. doi:10.7164/antibiotics.49.1196 |
| 2. | Nakajima, H.; Hori, Y.; Terano, H.; Okuhara, M.; Manda, T.; Matsumoto, S.; Shimomura, K. J. Antibiot. 1996, 49, 1204–1211. doi:10.7164/antibiotics.49.1204 |
| 3. | Liu, X.; Biswas, S.; Tang, G.-L.; Cheng, Y.-Q. J. Antibiot. 2013, 66, 555–558. doi:10.1038/ja.2013.38 |
| 1. | Nakajima, H.; Sato, B.; Fujita, T.; Takase, S.; Terano, H.; Okuhara, M. J. Antibiot. 1996, 49, 1196–1203. doi:10.7164/antibiotics.49.1196 |
| 19. | Matsumura, K.; Hashiguchi, S.; Ikariya, T.; Noyori, R. J. Am. Chem. Soc. 1997, 119, 8738–8739. doi:10.1021/ja971570a |
| 35. | Yoshikawa, Y.; Ishibashi, A.; Murai, K.; Kaneda, Y.; Nimura, K.; Arisawa, M. Tetrahedron Lett. 2019, 60, 151313. doi:10.1016/j.tetlet.2019.151313 |
| 13. | Albert, B. J.; Sivaramakrishnan, A.; Naka, T.; Czaicki, N. L.; Koide, K. J. Am. Chem. Soc. 2007, 129, 2648–2659. doi:10.1021/ja067870m |
| 6. | Kaida, D.; Motoyoshi, H.; Tashiro, E.; Nojima, T.; Hagiwara, M.; Ishigami, K.; Watanabe, H.; Kitahara, T.; Yoshida, T.; Nakajima, H.; Tani, T.; Horinouchi, S.; Yoshida, M. Nat. Chem. Biol. 2007, 3, 576–583. doi:10.1038/nchembio.2007.18 |
| 16. | Ghosh, A. K.; Chen, Z.-H.; Effenberger, K. A.; Jurica, M. S. J. Org. Chem. 2014, 79, 5697–5709. doi:10.1021/jo500800k |
| 41. | Blakemore, P. R. J. Chem. Soc., Perkin Trans. 1 2002, 2563–2585. doi:10.1039/b208078h |
| 4. | Liu, X.; Biswas, S.; Berg, M. G.; Antapli, C. M.; Xie, F.; Wang, Q.; Tang, M.-C.; Tang, G.-L.; Zhang, L.; Dreyfuss, G.; Cheng, Y.-Q. J. Nat. Prod. 2013, 76, 685–693. doi:10.1021/np300913h |
| 5. | He, H.; Ratnayake, A. S.; Janso, J. E.; He, M.; Yang, H. Y.; Loganzo, F.; Shor, B.; O’Donnell, C. J.; Koehn, F. E. J. Nat. Prod. 2014, 77, 1864–1870. doi:10.1021/np500342m |
| 17. | Thompson, C. F.; Jamison, T. F.; Jacobsen, E. N. J. Am. Chem. Soc. 2000, 122, 10482–10483. doi:10.1021/ja0055357 |
| 18. | Thompson, C. F.; Jamison, T. F.; Jacobsen, E. N. J. Am. Chem. Soc. 2001, 123, 9974–9983. doi:10.1021/ja016615t |
| 31. | Ghosh, A. K.; Veitschegger, A. M.; Nie, S.; Relitti, N.; MacRae, A. J.; Jurica, M. S. J. Org. Chem. 2018, 83, 5187–5198. doi:10.1021/acs.joc.8b00593 |
| 11. | Still, W. C.; Gennari, C. Tetrahedron Lett. 1983, 24, 4405–4408. doi:10.1016/s0040-4039(00)85909-2 |
| 14. | Frantz, D. E.; Fässler, R.; Carreira, E. M. J. Am. Chem. Soc. 2000, 122, 1806–1807. doi:10.1021/ja993838z |
| 12. | Albert, B. J.; Sivaramakrishnan, A.; Naka, T.; Koide, K. J. Am. Chem. Soc. 2006, 128, 2792–2793. doi:10.1021/ja058216u |
| 13. | Albert, B. J.; Sivaramakrishnan, A.; Naka, T.; Czaicki, N. L.; Koide, K. J. Am. Chem. Soc. 2007, 129, 2648–2659. doi:10.1021/ja067870m |
| 10. | Massad, S. K.; Hawkins, L. D.; Baker, D. C. J. Org. Chem. 1983, 48, 5180–5182. doi:10.1021/jo00174a006 |
| 15. | Ghosh, A. K.; Chen, Z.-H. Org. Lett. 2013, 15, 5088–5091. doi:10.1021/ol4024634 |
| 15. | Ghosh, A. K.; Chen, Z.-H. Org. Lett. 2013, 15, 5088–5091. doi:10.1021/ol4024634 |
| 16. | Ghosh, A. K.; Chen, Z.-H.; Effenberger, K. A.; Jurica, M. S. J. Org. Chem. 2014, 79, 5697–5709. doi:10.1021/jo500800k |
| 8. | Horigome, M.; Motoyoshi, H.; Watanabe, H.; Kitahara, T. Tetrahedron Lett. 2001, 42, 8207–8210. doi:10.1016/s0040-4039(01)01763-4 |
| 9. | Motoyoshi, H.; Horigome, M.; Watanabe, H.; Kitahara, T. Tetrahedron 2006, 62, 1378–1389. doi:10.1016/j.tet.2005.11.031 |
| 41. | Blakemore, P. R. J. Chem. Soc., Perkin Trans. 1 2002, 2563–2585. doi:10.1039/b208078h |
| 5. | He, H.; Ratnayake, A. S.; Janso, J. E.; He, M.; Yang, H. Y.; Loganzo, F.; Shor, B.; O’Donnell, C. J.; Koehn, F. E. J. Nat. Prod. 2014, 77, 1864–1870. doi:10.1021/np500342m |
| 12. | Albert, B. J.; Sivaramakrishnan, A.; Naka, T.; Koide, K. J. Am. Chem. Soc. 2006, 128, 2792–2793. doi:10.1021/ja058216u |
| 13. | Albert, B. J.; Sivaramakrishnan, A.; Naka, T.; Czaicki, N. L.; Koide, K. J. Am. Chem. Soc. 2007, 129, 2648–2659. doi:10.1021/ja067870m |
| 9. | Motoyoshi, H.; Horigome, M.; Watanabe, H.; Kitahara, T. Tetrahedron 2006, 62, 1378–1389. doi:10.1016/j.tet.2005.11.031 |
| 8. | Horigome, M.; Motoyoshi, H.; Watanabe, H.; Kitahara, T. Tetrahedron Lett. 2001, 42, 8207–8210. doi:10.1016/s0040-4039(01)01763-4 |
| 9. | Motoyoshi, H.; Horigome, M.; Watanabe, H.; Kitahara, T. Tetrahedron 2006, 62, 1378–1389. doi:10.1016/j.tet.2005.11.031 |
| 20. | Corey, E. J.; Bakshi, R. K.; Shibata, S.; Chen, C. P.; Singh, V. K. J. Am. Chem. Soc. 1987, 109, 7925–7926. doi:10.1021/ja00259a075 |
| 21. | Garner, P.; Ramakanth, S. J. Org. Chem. 1987, 52, 2629–2631. doi:10.1021/jo00388a063 |
| 17. | Thompson, C. F.; Jamison, T. F.; Jacobsen, E. N. J. Am. Chem. Soc. 2000, 122, 10482–10483. doi:10.1021/ja0055357 |
| 18. | Thompson, C. F.; Jamison, T. F.; Jacobsen, E. N. J. Am. Chem. Soc. 2001, 123, 9974–9983. doi:10.1021/ja016615t |
| 24. | Nicolaou, K. C.; Rhoades, D.; Lamani, M.; Pattanayak, M. R.; Kumar, S. M. J. Am. Chem. Soc. 2016, 138, 7532–7535. doi:10.1021/jacs.6b04781 |
| 25. | Nicolaou, K. C.; Rhoades, D.; Kumar, S. M. J. Am. Chem. Soc. 2018, 140, 8303–8320. doi:10.1021/jacs.8b04634 |
| 8. | Horigome, M.; Motoyoshi, H.; Watanabe, H.; Kitahara, T. Tetrahedron Lett. 2001, 42, 8207–8210. doi:10.1016/s0040-4039(01)01763-4 |
| 9. | Motoyoshi, H.; Horigome, M.; Watanabe, H.; Kitahara, T. Tetrahedron 2006, 62, 1378–1389. doi:10.1016/j.tet.2005.11.031 |
| 42. | Negishi, E.-i. Angew. Chem., Int. Ed. 2011, 50, 6738–6764. doi:10.1002/anie.201101380 |
| 17. | Thompson, C. F.; Jamison, T. F.; Jacobsen, E. N. J. Am. Chem. Soc. 2000, 122, 10482–10483. doi:10.1021/ja0055357 |
| 18. | Thompson, C. F.; Jamison, T. F.; Jacobsen, E. N. J. Am. Chem. Soc. 2001, 123, 9974–9983. doi:10.1021/ja016615t |
| 28. | Rubottom, G. M.; Vazquez, M. A.; Pelegrina, D. R. Tetrahedron Lett. 1974, 15, 4319–4322. doi:10.1016/s0040-4039(01)92153-7 |
| 26. | Espinet, P.; Echavarren, A. M. Angew. Chem., Int. Ed. 2004, 43, 4704–4734. doi:10.1002/anie.200300638 |
| 12. | Albert, B. J.; Sivaramakrishnan, A.; Naka, T.; Koide, K. J. Am. Chem. Soc. 2006, 128, 2792–2793. doi:10.1021/ja058216u |
| 13. | Albert, B. J.; Sivaramakrishnan, A.; Naka, T.; Czaicki, N. L.; Koide, K. J. Am. Chem. Soc. 2007, 129, 2648–2659. doi:10.1021/ja067870m |
| 27. | Dossetter, A. G.; Jamison, T. F.; Jacobsen, E. N. Angew. Chem., Int. Ed. 1999, 38, 2398–2400. doi:10.1002/(sici)1521-3773(19990816)38:16<2398::aid-anie2398>3.0.co;2-e |
| 15. | Ghosh, A. K.; Chen, Z.-H. Org. Lett. 2013, 15, 5088–5091. doi:10.1021/ol4024634 |
| 16. | Ghosh, A. K.; Chen, Z.-H.; Effenberger, K. A.; Jurica, M. S. J. Org. Chem. 2014, 79, 5697–5709. doi:10.1021/jo500800k |
| 23. | Stork, G.; Zhao, K. Tetrahedron Lett. 1989, 30, 2173–2174. doi:10.1016/s0040-4039(00)99640-0 |
| 8. | Horigome, M.; Motoyoshi, H.; Watanabe, H.; Kitahara, T. Tetrahedron Lett. 2001, 42, 8207–8210. doi:10.1016/s0040-4039(01)01763-4 |
| 24. | Nicolaou, K. C.; Rhoades, D.; Lamani, M.; Pattanayak, M. R.; Kumar, S. M. J. Am. Chem. Soc. 2016, 138, 7532–7535. doi:10.1021/jacs.6b04781 |
| 25. | Nicolaou, K. C.; Rhoades, D.; Kumar, S. M. J. Am. Chem. Soc. 2018, 140, 8303–8320. doi:10.1021/jacs.8b04634 |
| 9. | Motoyoshi, H.; Horigome, M.; Watanabe, H.; Kitahara, T. Tetrahedron 2006, 62, 1378–1389. doi:10.1016/j.tet.2005.11.031 |
| 13. | Albert, B. J.; Sivaramakrishnan, A.; Naka, T.; Czaicki, N. L.; Koide, K. J. Am. Chem. Soc. 2007, 129, 2648–2659. doi:10.1021/ja067870m |
| 43. | Ghosh, A. K.; Reddy, G. C.; MacRae, A. J.; Jurica, M. S. Org. Lett. 2018, 20, 96–99. doi:10.1021/acs.orglett.7b03456 |
| 22. | Lewis, M. D.; Cha, J. K.; Kishi, Y. J. Am. Chem. Soc. 1982, 104, 4976–4978. doi:10.1021/ja00382a053 |
| 17. | Thompson, C. F.; Jamison, T. F.; Jacobsen, E. N. J. Am. Chem. Soc. 2000, 122, 10482–10483. doi:10.1021/ja0055357 |
| 18. | Thompson, C. F.; Jamison, T. F.; Jacobsen, E. N. J. Am. Chem. Soc. 2001, 123, 9974–9983. doi:10.1021/ja016615t |
© 2020 Donaldson; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)