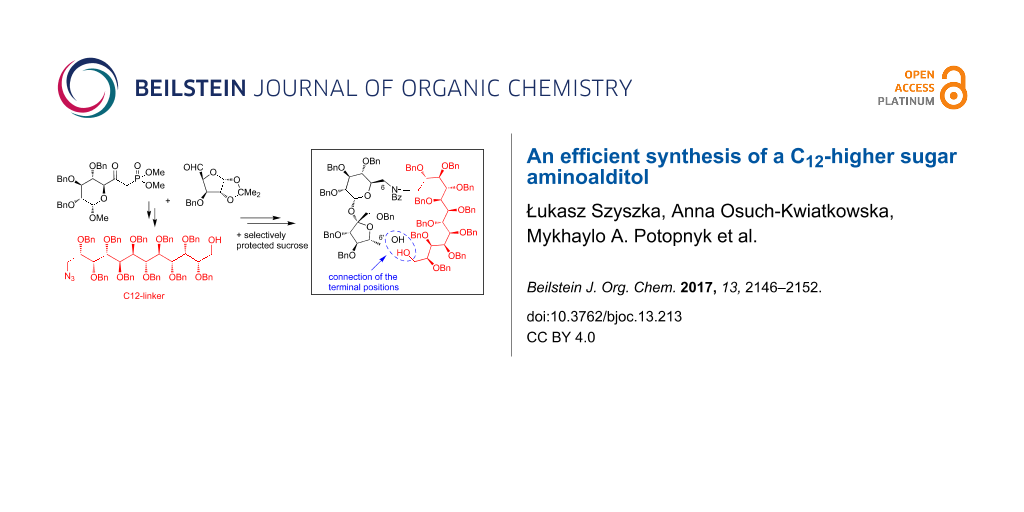
Abstract
The C12-aminoalditol H2NCH2–(CHOBn)10–CH2OH was prepared from two simple monosaccharide building blocks. The synthesis was realized by a regioselective introduction of the azide group and subsequent protection–deprotection transformations. The chemical reactivity of the aminoalditol was tested in the reductive amination reaction with a selectively protected sucrose monoaldehyde.

Graphical Abstract
Introduction
Carbohydrates, because of their availability in a variety of optical pure forms, are particularly useful in planning and executing the synthesis of chiral macrocyclic compounds [1-5]. In this context, polyhydroxylated derivatives with long chains are very interesting. However, the synthesis of this kind of molecules is a real challenge in carbohydrate chemistry. Very often reactions, which work well for ‘normal’ (C5–C7) sugars are not applicable for the elongated analogs [6,7]. Recently we have prepared such a derivative by the coupling of two simple monosaccharides and subsequent transformation of the resulting enone into the protected C12-sugar 1. The conversion of the latter into alditol 2 allowed us to prepare derivative 3 having a 18-membered ring (Figure 1) [8].
![[1860-5397-13-213-1]](/bjoc/content/figures/1860-5397-13-213-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Previous synthesis of a C12-higher sugar 1 and its application in the preparation of a polyhydroxylated macrocycle.
Figure 1: Previous synthesis of a C12-higher sugar 1 and its application in the preparation of a polyhydroxyl...
Higher sugar 1 has a substantial synthetic potential. Besides the approach shown in Figure 1, it could be used in the synthesis of other highly oxygenated targets if the terminal positions at this stage are differentiated. This would allow introducing various functions at either terminal position, thus opening a route to many interesting derivatives, including cyclic ones.
We decided to perform a model study on the efficient differentiation of the terminal positions in such higher sugars or alditols, which could open a route to complex polyhydroxylated derivatives.
Results and Discussion
A regioselective protection of one of the primary hydroxy groups in diol 2 is not possible. Thus we decided to differentiate the terminal positions in the stage of higher sugar 1.
The replacement of the hydroxy group at the C12-position with an azide, accomplished successfully under Mistunobu conditions, afforded azide 4 in 95% yield (Scheme 1).
![[1860-5397-13-213-i1]](/bjoc/content/inline/1860-5397-13-213-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Preparation of C12-aminoalditol 10.
Scheme 1: Preparation of C12-aminoalditol 10.
The crucial step was the liberation of the anomeric position (C1), which, as we noticed, can cause problems. We subjected azide 4 to acetolysis and, although this reaction is very capricious in higher sugar chemistry [7,8], succeeded in the preparation of the desired hemiacetal acetate 5 in very good yield (86%) as a mixture of two anomers (α:β = 2:1).
Treatment of azido ester 5 with NaBH4 gave predominantly azidodiol 6 (66%) and small amounts of aminodiol 7 (5%). Application of LiAlH4 as the reducing agent afforded only compound 7 (Scheme 1). The former result may open, eventually, a new possibility for the preparation of linear higher aminoalditol derivatives.
The subsequent protection of both primary positions in aminodiol 7 with the bulky trityl group (8), followed by a protection of the remaining secondary OH group as benzyl ether (9) and removal of the temporary trityl group provided aminoalditol 10 (Scheme 1). This compound can be modified selectively at either terminal position: C1 (OH group) or C12 (NH2 group).
Since we are engaged in the preparation of complex derivatives of sucrose (Figure 2) [9-16] we decided to prepare a sucrose derivative with this aminoalditol pendant which could eventually be used in the synthesis of macrocyclic derivatives. Sucrose, undoubtedly the most common disaccharide, has found applications in the synthesis of polymers [17] or surfactants [18]. There is also an increasing interest in the application of sucrose as a ‘normal’ chemical [16,19,20].
![[1860-5397-13-213-2]](/bjoc/content/figures/1860-5397-13-213-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Examples of highly functionalized sucrose derivatives from our laboratory.
Figure 2: Examples of highly functionalized sucrose derivatives from our laboratory.
Our synthesis towards a sucrose derivative with a long alditol pendant was initiated from aldehyde 12 (readily prepared from the known [15] alcohol 11 with the free 6-OH group) and C12-aminoalditol 10 (Scheme 2).
![[1860-5397-13-213-i2]](/bjoc/content/inline/1860-5397-13-213-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Preparation of a sucrose molecule with a higher aminoalditol pendant.
Scheme 2: Preparation of a sucrose molecule with a higher aminoalditol pendant.
Coupling of these two species under the reductive amination conditions provided amine 13 in 78% yield. For the transformation of compound 13 into the terminally functionalized derivative we decided to protect the amino function, located at the C6 position of the sucrose moiety, preferably as benzyl ether 15. Compound 13 was, therefore, treated with benzoyl chloride which installed the benzoyl group at the amino and the free hydroxy functions providing dibenzoyl derivative 14 in very good yield (94%, Scheme 2).
The reduction of derivative 14 with LiAlH4 removed the benzoyl protection from both hydroxy and amino groups and surprisingly, no reduction of the benzoyl located at the amino or the benzyl groups was noted.
Therefore, compound 14 was treated with 1 M KOH to remove the benzoyl groups only from the hydroxy group, which yielded compound 16, the starting material in the next step of our synthesis. Subsequent removal of the tert-butyldiphenylsilyl group from the C6’ position with TBAF afforded 17 in 93% yield (Scheme 2). Amide 16 with the free OH group at the C18 position and 17 having two hydroxy groups at the C18 and C6’ positions can be regarded as convenient starting materials for the preparation of more complex sucrose derivatives.
Conclusion
In summary, we have developed an efficient method for the synthesis of a C12-aminoalditol H2NCH2–(CHOBn)10–CH2OH 10. The chemical reactivity of this valuable synthon was demonstrated by its high yielding coupling with the C-6 position of a selectively protected sucrose by a reductive amination reaction.
Experimental
General
NMR spectra were recorded using Varian 600 MHz or Bruker 500 MHz spectrometers in CDCl3 at 25 °C unless otherwise stated. The structures were assigned, whenever necessary, with the help of 2D correlation experiments (COSY, HSQC, HMBC). Chemical shifts (in most cases only diagnostic signals were shown) were reported with reference to TMS. Optical rotations were measured with a Jasco P 1020 polarimeter (sodium light) in chloroform at 20 °C. The MS spectra were recorded with a Mariner PerSeptive Biosystems spectrometer. Thin-layer chromatography was performed on pre-coated plates (0.25 mm, silica gel 60 F254). Column chromatography was carried out with silica gel (230–400 mesh).
Synthesis of azide 4
This reaction was conducted under an argon atmosphere. To a cooled (to 0 °C) solution of alcohol 1 [8] (0.89 g, 0.75 mmol) and Ph3P (0.4 g, 1.5 mmol) in dry THF (12 mL), DIAD (0.3 mL, 1.5 mmol) was added and the mixture was stirred for 10 min. Diphenylphosphoryl azide (DPPA, 0.32 mL, 1.48 mmol) was then added dropwise within 10 min, the mixture was allowed to the reach 20 °C, and stirring was prolonged for another 1.5 h. Ethyl acetate (10 mL) was added and the mixture was washed with 1 M HCl (10 mL). The organic phase was separated and the aqueous phase extracted with ethyl acetate (2 × 10 mL). The combined organic solutions were washed with aqueous saturated NaHCO3 (15 mL) and brine (15 mL), dried, concentrated, and the product was isolated by column chromatography (hexanes/ethyl acetate 8:1) to afford pure compound 4 (0.87 g, 0.7 mmol, 95%) as an oil. [α]D +12.1 (c 0.4); 1H NMR (600 MHz) δ 7.41–7.11 (m, 35H, ArH), 4.91 (d, J = 11.0, 1H, benzylic H), 4.80 (d, J = 11.5 Hz, 1H, benzylic H), 4.79 (d, J = 11.2 Hz, 1H, benzylic H), 4.80–4.67 (m, 4H, benzylic H), 4.64 (d, J = 11.6 Hz, 2H, benzylic H), 4.62 (d, J = 11.8 Hz, 2H, benzylic H), 4.61 (d, J = 12.2 Hz, 1H, benzylic H), 4.59 (d, J1,2 = 3.5 Hz, 1H, H-1), 4.53 (d, J = 11.5 Hz, 1H , benzylic H), 4.49 (d, J = 11.5 Hz, 1H, benzylic H), 4.42 (s, 1H, benzylic H), 4.41 (s, 1H, benzylic H), 4.39 (d, J = 11.5, 1H, benzylic H), 4.28 (d, J = 11.7 Hz, 1H, benzylic H), 4.22 (d, J5,4 = 10.3 Hz, 1H, H-5), 4.18–4.13 (m, 2H, H-7, H-8), 4.05 (dd, J9,8 = 5.8 Hz, J9,10 = 4.3 Hz, 1H, H-9), 4.01 (d, J6,7 = 9.8 Hz, 1H, H-6), 3.97 (dd, J3,4 = 9.2 Hz, J3,2 = 9.3 Hz, 1H, H-3), 3.83 (dd, J4,5 = 10.1 Hz, 1H, H-4), 3.75 (m, 1H, H-10), 3.67 (m, 1H, H-11), 3.40 (dd, 1H, H-2), 3.29 (s, 3H, OCH3), 3.26–3.17 (m, 2H, H-12, H-12’) ppm; 13C NMR (150 MHz) δ 138.86, 138,86, 138.81, 138.75, 138.72, 138.69, 138.23, 138.12, 138.01 (9 × Cquat-Ph), 130.05 (C-Ph), 128.42–127.05 (m, 41 × C-Ph), 126.0 (C-Ph), 120.24 (C-Ph), 120.21 (C-Ph), 97.84 (C-1), 82.79 (C-3), 80.05 (C-2), 79.71 (C-10), 79.18 (C-11), 78.55 (C-4), 78.45 (C-6), 77.54 (C-7), 77.34 (C-8), 77.11 (C-9), 75.39, 74.65, 74.36, 73.33, 73.29, 73.01, 72.38, 72.08, 71.73 (9 × OCH2Ph), 69.64 (C-5), 55.01 (OCH3), 51.83 (C-12) ppm; HRMS (ESI) m/z: [M + Na]+ calcd for C76H79N3O11Na, 1232.5590; found, 1232.5612; Anal. calcd for C76H79N3O11: C, 75.41; H, 6.58; N, 3.47; found: C, 75.41; H, 6.47; N, 3.34.
Acetolysis of glycoside 4; synthesis of 5
Azide 4 (0.36 g, 0.3 mmol) was dissolved in ethyl acetate (2.3 mL) to which acetic anhydride (4.6 mL) and sulfuric acid (0.72 mL of the solution: conc. H2SO4 (0.05 mL) in ethyl acetate (100 mL)) were added. The mixture was stirred at 20 °C for 46 h and then diluted with ether (15 mL). It was then neutralized with saturated aq NaHCO3 (15 mL), the organic phase was separated, and the aqueous phase extracted with ether (2 × 10 mL). The combined organic solutions were washed with water (40 mL) and brine (40 mL), dried, concentrated, and the crude product was purified by column chromatography (hexanes/ethyl acetate 9:1) to yield 5 (0.32 g, 0.26 mmol, 86%) as a 2:1 mixture of α/β anomers (calculated by integration of the anomeric signals at δ: 6.34 and 5.63). 1H NMR (600 MHz) δ 7.32–7.13 (m, 35H, ArH), 6.34 (d, J1,2 = 3.6 Hz, 1H, H-1), 4.88 (d, J = 11.0 Hz, 1H, benzylic H), 4.85–4.37 (m, 43H, benzylic H), 4.34 (dd, J5,4 = 10 Hz, J5,6 = 2.4 Hz, 1H, H-5), 4.27 (d, J = 11.9 Hz, 1H, benzylic H), 4.16–4.05 (m, 3H, H-7, H-8, H-9), 3.93–3.86 (m, 2H, H-3, H-4), 3.73 (dd, 1H, H-10), 3.69–3.62 (m, 2H, H-6, H-11), 3.47 (dd, J2,3 = 9.0 Hz, 1H, H-2), 3.23–3.22 (d, 2H, H-12, H-12’), 1.97 (s, 3H, CH3) ppm; 13C NMR (150 MHz) δ 169.50 (C=O), 138.95, 138.65, 138.62, 138.60, 138.55, 138.46, 138.13, 137.99, 137.61 (9 × Cquat-Ph), 128.44–127.08 (m, 45 × C-Ph), 89.58 (C-1), 85.26 (C-6), 82.26 (C-3), 79.62 (C-10), 79.06 (C-2), 78.90 (C-11), 77.79 (C-4), 77.63, 77.39, 77.34 (C-7, C-8, C-9), 75.32, 74.70, 74.54, 73.16, 73.10, 72.84, 72.61, 72.22, 72.14 (9 × OCH2Ph), 72.61 (C-5), 51.78 (C-12), 20.94 (CH3) ppm; HRMS (ESI) m/z: [M + Na]+ calcd for C77H79N3O12Na: 1260.5537; found, 1260.5561; Anal. calcd for C77H79N3O12: C, 74.61; H, 6.51; N, 3.39; found: C, 74.70; H, 6.58; N, 3.38.
Preparation of compounds 6 and 7
Method a: To a solution of 5 (0.23 g, 0.18 mmol) in THF/methanol (3:1 v/v, 30 mL), NaBH4 (3 g, 79 mmol) was added in few portions and the mixture was stirred for 3 h at 20 °C. Then it was partitioned between ether (30 mL) and water (15 mL), the organic phase was separated, and the aqueous phase extracted with ether (2 × 15 mL). The combined organic solutions were washed with water (10 mL), dried, concentrated, and the residue was subjected to column chromatography (hexanes/ethyl acetate 5:1 to 3:1) to give two products: 6 (0.15 g, 0.12 mmol, 66%) and 7 (12 mg, 5%). Products 6 and 7 were isolated by preparative TLC (for 6 hexanes/ethyl acetate 2:1, for 7 CH2Cl2/MeOH 10:1).
Product 6: [α]D −6.5 (c 0.5); 1H NMR (600 MHz) δ 7.29–7.15 (m, 45H, 45 × H-Ph), 4.77 (d, J = 11.5 Hz 1H,), 4.70–4.44 (m, 19H), 4.35 (m, 1H, H-5), 4.23–4.16 (m, 2H), 4.02–3.94 (m, 3H), 3.87–3.82 (m, 2H), 3.80 (dd, J = 3.7 Hz, J = 5.9 Hz, 1H), 3.71 (dd, J = 4.3 Hz, J = 10.3 Hz, 1H), 3.48 (dd, J1,2 = 4.5 Hz, J1,1’ = 12.0 Hz, 1H, H-1), 3.39 (dd, J12,11 = 3.1 Hz, J12,12’ = 12.8 Hz, 1H, H-12), 3.33–3.26 (m, 2H, H-1’, H-12’) ppm; 13C NMR (150 MHz) δ 128.42–127.25 (m, 45 × C-Ph), 79.78, 79.74, 79.35, 79.15, 79.12, 78.87, 78.68, 77.76, 77.67, 74.69, 74.03, 73.74, 73.47, 73.16, 72.82, 72.73, 72.04, 72.04 (9 × OCH2Ph), 70.74 (C-5), 61.60 (C-1), 51.88 (C-12) ppm; HRMS (ESI) m/z: [M + Na]+ calcd for C75H79N3O11Na, 1220.5627; found, 1220.5612; Anal. calcd for C75H79N3O11: C, 75.16; H, 6.64; N, 3.51; found: C, 75.13; H, 6.82; N, 3.40.
Method b: To a solution of 5 (0.3 g, 0.24 mmol) in dry THF (20 mL), LiAlH4 (55 mg, 1.45 mmol) was slowly added and the mixture was stirred for 1 h at 20 °C. Excess hydride was carefully decomposed with aqueous saturated Na2SO4 (5 mL). Celite (0.5 g) was added, the mixture was stirred for 5 min, and then filtered through a short pad of silica which was subsequently washed with ethyl acetate (3 × 20 mL). The organic solution was dried and concentrated to give 7 (0.27 g) as a single product, which was used in the next step without purification. The structure of 7 was confirmed by MS. MS (ESI) m/z: [M + H]+ calcd for C75H81NO11, 1172.6; found, 1172.8.
Synthesis of aminoalcohol 10
Crude compound 7 was dissolved in dichloromethane (20 mL) containing triethylamine (0.4 mL), DMAP (5 mg) and trityl chloride (0.4 g, 1.4 mmol). The mixture was boiled under reflux for 24 h, then cooled to 20 °C, and partitioned between ether (20 mL) and water (20 mL). The layers were separated and the aqueous layer extracted with ether (2 × 20 mL). The combined organic solutions were washed with water (20 mL) and brine (20 mL), dried, concentrated, and the residue was purified by column chromatography (hexanes/ethyl acetate 100:0 to 90:10) to yield alcohol 8 (0.19 g, 0.11 mmol, 47% after two steps). [α]D +1.0 (c 0.2); 1H NMR (600 MHz) δ 7.4–6.90 (m, 75H, 75 × H-Ph), 4.74–4.58 (m, 7H, 7 × benzylic H), 4.56 (d, J = 11.7 Hz, 1H, benzylic H), 4.50 (d, J = 11.2 Hz, 1H, benzylic H), 4.49 (d, J = 11.3 Hz, 1H, benzylic H), 4.48 (d, J = 11.2 Hz, 1H, benzylic H), 4.45 (d, J = 11.4 Hz, 1H, benzylic H), 4.41–4.32 (m, 4H, 4 × benzylic H), 4.31–4.21 (m, 4H, H-6, H-5, H-9, benzylic H), 4.16 (d, J = 11.6 Hz, 1H, benzylic H), 4.06–4.03 (m, 2H, H-8, H-7), 4.00–3.98 (m, 2H, H-4, H-10), 3.94–3.89 (m, 2H, H-2, H-11), 3.78 (m, 1H, H-3), 3.46 (br s, 1H, NH), 3.35 (dd J1,1’ = 10.0 Hz, J1,2 = 6.3 Hz, 1H, H-1), 3.25 (dd, 1H, H-1’), 2.44 (dd, J12,12’ = 12.4 Hz, J12,11 = 3.9 Hz, 1H, H-12), 2.37 (dd, 1H, H-12’) ppm; 13C NMR (150 MHz) δ 145.92 (3C, 3 × Cquat-Ph), 143.95 (3C, 3 × Cquat-Ph), 139.34, 139,10, 139.01, 138.97, 138.57, 138.54, 138.24, 138.16, 137.83 (9 × Cquat-Ph), 128.71–126.07 (m, 75 × C-Ph), 86.96 (OCPh3), 80.36 (C-11), 80.21 (C-4), 80.01 (C-8), 78.24 (C-3), 78.11 (C-9), 77.83 (C-6), 77.72 (C-7), 77.69 (C-2), 76.7 (C-10), 74.52, 73.06, 72.97, 72.86, 72.61, 72.42, 72.34, 72.34, 71.69 (9 × OCH2Ph), 71.58 (C-5), 70.67 (NCPh3), 63.64 (C-1), 43.62 (C-12) ppm; HRMS (ESI) m/z: [M + H]+ calcd for C113H110NO11: 1656.8102; found: 1656.8079; Anal. calcd for C113H109NO11: C, 81.90; H, 6.63; N, 0.85; found: C, 81.92; H, 6.72; N, 0.88.
To a stirred solution of 8 (0.35 g, 0.21 mmol) in DMF (20 mL) containing imidazole (1.5 mg), sodium hydride (60% dispersion in mineral oil, 80.6 mg, 2.1 mmol) was added, and the mixture was stirred at 20 °C for 30 min. Benzyl bromide (0.2 mL, 1.68 mmol) was added dropwise and the mixture was stirred for 24 h at 20 °C. Excess of hydride was decomposed by careful addition of water (5 mL) and the mixture was partitioned between water (30 mL) and ether (40 mL). The layers were separated and the aqueous one extracted with ethyl acetate (3 × 20 mL). The combined organic solutions were washed with water (40 mL) and brine (40 mL), dried, concentrated, and the crude 9 (0.4 g) was used to the next step without purification.
Crude compound 9 was dissolved in ether/methanol (v/v 1:1; 40 mL) to which p-TsOH∙H2O (3.5 g, 18 mmol) was added, and the mixture was stirred and boiled under reflux for 24 h. Then it was cooled to 20 °C and partitioned between ethyl acetate (50 mL) and water (50 mL). The organic phase was separated and the aqueous layer was extracted with ethyl acetate (2 × 30 mL). The combined organic solutions were washed with water (50 mL), aq saturated Na2CO3 (30 mL), and brine (30 mL), dried, concentrated, and the product was isolated by column chromatography (hexanes/ethyl acetate 5:1 to 1:2) to afford 10 (0.24 g, 0.19 mmol, 87% after two steps). [α]D −15.7 (c 0.2); 1H NMR (600 MHz) δ 7.35–7.10 (m, 50H, 50 × H-Ph), 4.91 (d, J = 11.0 Hz, 1H), 4.90 (d, J = 11.9 Hz, 1H), 4.78 (d, J = 11.0 Hz, 1H), 4.74 (d, J = 11.9 Hz, 1H), 4.67 (d, J = 11.0 Hz, 1H), 4.67 (d, J = 11.0 Hz, 1H), 4.63 (d, J = 11.9 Hz, 1H), 4.54–4.42 (m, 13H), 4.36 (d, J = 11.2 Hz, 1H), 4.26–4.18 (m, 7H), 4.02 (m, 1H), 3.91 (dd, J = 5.0 Hz, J = 5.3 Hz, 1H), 3.75 (dd, J = 5.6 Hz, J = 5.2 Hz, 1H), 3.59 (m, 2H), 3.52 (dd, J = 4.7 Hz, J = 11.9 Hz, 1H), 3.30 (dd, J = 3.6 Hz, J = 11.9 Hz, 1H), 2.74 (dd, J = 4.5 Hz, J = 13.7 Hz, 1H), 2.64 (dd, J = 5.6 Hz, J = 13.7 Hz, 1H) ppm; 13C NMR (150 MHz) δ 139.42, 139.05, 139.01, 138.68, 138.59, 138.59, 138.40, 138.32, 138.32, 138.18 (10 × Cquat-Ph), 128.34–127.17 (m, 50 × C-Ph), 81.66, 81.01, 80.52, 79.99, 79.11, 78.97, 79.97, 78.80, 78.19, 78.19, 75.24, 74.98, 74.74, 74.63, 73.71, 72.52, 72.44, 72.23, 71.86, 71.75 (10 × OCH2Ph), 61.46 (C-1), 42.09 (C-12) ppm; HRMS (ESI) m/z: [M + H]+ calcd for C82H88NO11, 1262.6356; found, 1262.6357.
Synthesis of sucrose with a long chain at the C6-position; preparation of 17
This reaction was conducted under an argon atmosphere. Sucrose alcohol 11 [10] (110 mg, 0.1 mmol) was oxidized under Swern conditions [21] to afford aldehyde 12. To a solution of this crude aldehyde in CH2Cl2 (5 mL), freshly dried molecular sieves (150 mg), acetic acid (6 μL, 0.1mmol), and aminoalcohol 10 (130 mg, 0.1 mmol in 5 mL CH2Cl2) were added, and the mixture was stirred at 20 °C for 2 h. NaBH3CN (7.5 mg, 0.12 mmol) was added and stirring was continued overnight at rt. Water (20 mL), 25% NH3 aq (0.5 mL), and CH2Cl2 (10 mL) were added, the phases were separated, and the aqueous layer was extracted with CH2Cl2 (3 × 20 mL). The combined organic solutions were washed with water (30 mL) and brine (30 mL), dried, concentrated, and the crude material was purified by flash chromatography (hexanes/ethyl acetate 5:1 to 2:1) to afford 13 (0.19 g, 0.08 mmol, 78%). [α]D −5.3 (c 0.2); 1H NMR (500 MHz) δ 7.59–7.61, 7.28–7.04 (m, 90H, 90 × H-Ph), 5.68 (d, J = 3.5 Hz, 1H, H-1), 4.80 (d, J = 11.3 Hz, 1H), 4.78 (d, J = 11.0 Hz, 1H), 4.73 (d, J = 11.0 Hz, 1H), 4.69 (d, J = 11.5 Hz, 1H), 4.67–4.15 (m, 34H), 4.09–4.02 (m, 2H), 4.03 (d, J = 11.6 Hz, 1H), 3.95 (d, J = 10.7 Hz, 1H), 3.94 (d, J = 10.7 Hz, 1H), 3.93–3.85 (m, 2H), 3.83 (d, J = 10.4 Hz, 1H), 3.82 (d, J = 10.5 Hz, 1H), 3.81–3.74 (m, 2H), 3.56–3.48 (m, 3H), 3.42 (d, J = 11.7 Hz, 1H), 3.30 (dd, J = 3.5 Hz, J = 9.6 Hz, 1H), 3.20 (d, J = 9.7 Hz, 1H), 2.78 (dd, J = 3.3 Hz, J = 12.5 Hz, 1H), 2.65 (dd, J = 6.7 Hz, J = 12.6 Hz, 1H), 2.61–2.51 (m, 2H), 1.90 (s, 1H), 1.00 (s, 9H, t-Bu) ppm; 13C NMR (125 MHz) δ 139. 37, 139.16, 139.12, 138.95, 138.89, 138.89, 138.85, 138.82, 138.60, 138.54, 138.37, 138.35, 138.34, 138.29, 138.29, 137.94, 133.41, 133.22 (18 × Cquat-Ph), 135.57, 135.49, 129.64, 129.61, 128.30–127.08 (m) (90 × C-Ph), 105.13 (C-2’), 90.47 (C-1), 84.25, 83.67, 81.77, 81.68, 81.19, 81.17, 80.68, 80.33, 80.23, 80.23, 79.14, 79.01, 79.01, 78.43, 78.14, 78.03, 70.61, (C-2, C-3, C-3’ C-4, C-4’ C-5, C-5’, C-8, C-9, C-10, C-11, C-12, C-13, C-14, C-15, C-16, C-17), 75.36, 74.83, 74.76, 74.65, 74.38, 74.33, 73.46, 73.35, 72.97, 72.57, 72.47, 72.30, 72.13, 72.04, 71.84, 71.62, 70.44 (16 × OCH2Ph, C-1’), 65.37, 61.57 (C-6’, C-18), 51.41 (NCH2), 49.78 (NCH2), 26.92 (triple intensity, 3C – t-Bu), 19.24 (Cquat, t-Bu) ppm; HRMS (ESI) m/z: [M + Na]+ calcd for C152H161NO21SiNa, 2388.1334; found, 2388.1295; Anal. calcd for C152H161NO21Si: C, 77.16; H, 6.86; N, 0.59; found: C, 77.13; H, 6.78; N, 0.57.
To a solution of 13 (150 mg, 0.063 mmol) in dry dichloromethane (5 mL), containing DMAP (1 mg), and triethylamine (53 μL, 0.38 mmol), benzoyl chloride (30 μL, 0.25 mmol) was added dropwise, and the mixture was stirred for 5 h at 20 °C. It was then partitioned between water (5 mL) and saturated aqueous NaHCO3 (2 mL), the layers were separated, and the aqueous one extracted with dichloromethane (2 × 15 mL). The combined organic solutions were dried, concentrated, and the residue was purified by column chromatography (hexanes/ethyl acetate 5:1 to 2:1) to afford 14 (0.15 g, 0.06 mmol, 94%). [α]D −11.0 (c 0.2); 1H NMR (500 MHz, C7D8, 80 °C) δ 7.75–7.72 (m, 2H, H-Ph), 7.55–7.46 (m, 4H, H-Ph), 7.24–7.16 (m, 3H, H-Ph), 7.12–6.64 (m, 91H, 91 × H-Ph), 5.60 (d, J = 2.3 Hz, 1H, H-1), 4.81–3.59 (m, 58H), 3.52 (d, J = 11.1 Hz, 1H), 0.88 (s, 9H, t-Bu) ppm; 13C NMR (125 MHz, C7D8, 80 °C) δ 135.68, 135.56, 132.37, 132.32, 129.63, 129.56, 128.77, 128.47–126.93 (m), 125.00 (100 × C-Ph), 90.54 (C-1), 84.76, 84.16, 83.14, 82.69, 81.32, 80.59, 80.44, 80.34, 80.16, 80.02, 79.79, 79.55, 79.15, 78.93, 78.61, 78.38, 78.25 (C-2, C-3, C-3’ C-4, C-4’ C-5, C-5’, C-8, C-9, C-10, C-11, C-12, C-13, C-14, C-15, C-16, C-17), 75.07, 74.72, 74.58, 74.54, 74.18, 73.92, 73.45, 73.39, 73.25, 72.93, 72.69, 72.58, 72.40, 71.99, 71.86, 71.53, 71.29 (16 × OCH2Ph, C-1’), 70.07, 69.91 (C-6’, C-18), 67.05, 65.43 (2 × NCH2), 26.82 (triple intensity, 3C – t-Bu) ppm; HRMS (ESI) m/z: [M + 2Na]+ calcd for C166H169NO23SiNa2, 2618.1662; found, 2618.1639; Anal. calcd for C166H169NO23Si: C, 77.45; H, 6.62; N, 0.54; found: C, 77.22; H, 6.70; N, 0.51.
To a solution of 14 (30 mg, 0.01 mmol) in dry THF (4 mL), LiAlH4 (2 mg, 0.044 mmol) was added, the mixture was stirred for 5 h at 20 °C and then boiled under reflux for additional 5 h. After cooling to rt, the excess of hydride was decomposed with aq saturated Na2SO4 (0.5 mL). Celite (100 mg) was added, the mixture was stirred for 5 min and then was filtered through a short pad of silica which was subsequently washed with ethyl acetate (3 × 10 mL). The organic solution was dried and concentrated to give 13 (15 mg) as a single product. HRMS (ESI) m/z: [M + Na]+ calcd for C152H161NO21SiNa, 2388.1334; found, 2388.1295.
Compound 14 (108 mg, 0.042 mmol) was dissolved in dry THF (2 mL) to which methanol (2 mL) and 1 M KOH (0.5 mL) were added, and the mixture was stirred at 20 °C overnight. The mixture was neutralized with 1 M HCl and partitioned between ethyl acetate (10 mL) and water (10 mL). The aqueous layer was extracted with ethyl acetate (2 × 10 mL) and the combined organic solutions were washed with water (10 mL) and brine (10 mL), dried, and concentrated. The crude material was purified by column chromatography (hexanes/ethyl acetate 6:1 to 3:1) to give product 16 (86.3 mg) detected by MS (HRMS (ESI) m/z: [M + 2Na]+ calcd for C159H165NO22SiNa2, 2514.1377; found, 2514.1384) and traces of secondary amine 13.
To a solution of the above crude mixture (43.6 mg) in THF (4 mL) tetrabutylammonium fluoride trihydrate (35 mg) was added and the mixture was stirred overnight at 20 °C. Then it was concentrated and the residue was purified by preparative thin-layer chromatography (hexanes/acetone 1:1) to yield pure compound 17 (36.7 mg, 0.016 mmol, 93%). [α]D −9.5 (c 0.5); 1H NMR (500 MHz, C7D8, 80 °C) δ 7.54–7.43 (m, 4H, H-Ph), 7.40–6.97 (m, 81H, H-Ph), 5.65 (d, J = 3.4 Hz, H-1, 1H), 5.12–3.55 (m, 57H) ppm; 13C NMR (125 MHz, C7D8, 80 °C) δ 128.63, 128.48, 128.20–126.80, 124.86 (85 × C-Ph), 91.04 (C-1), 84.95, 84.78, 83.01, 82.97, 82.41, 82.15, 81.31, 81.17, 80.99, 80.93, 80.66, 80.37, 80.25, 80.06, 79.92, 79.79, 79.22 (C-2, C-3, C-3’ C-4, C-4’ C-5, C-5’, C-8, C-9, C-10, C-11, C-12, C-13, C-14, C-15, C-16, C-17), 75.40, 74.90, 74.58, 74.53, 74.24, 73.96, 73.83, 73.75, 73.51, 72.83, 72.59, 72.42, 72.35, 71.97, 71.54, 71.01, 70.96 (16 × OCH2Ph, C-1’), 66.32, 61.92 (C-6’, C-18) ppm; HRMS (ESI) m/z: [M + 2Na]+ calcd for C143H147NO22Na2, 2276.0198; found, 2276.0199; Anal. calcd for C143H147NO22: C, 76.96; H, 6.64; N, 0.63; found: C, 76.85; H, 6.59; N, 0.64.
Supporting Information
| Supporting Information File 1: Copies of NMR spectra. | ||
| Format: PDF | Size: 2.0 MB | Download |
References
-
Potopnyk, M. A.; Jarosz, S. Adv. Carbohydr. Chem. Biochem. 2014, 71, 227–295. doi:10.1016/B978-0-12-800128-8.00003-0
Return to citation in text: [1] -
Desmond, R. T.; Magpusao, A. N.; Lorenc, C.; Alverson, J. B.; Priestley, N.; Peczuh, M. W. Beilstein J. Org. Chem. 2014, 10, 2215–2221. doi:10.3762/bjoc.10.229
Return to citation in text: [1] -
Pintal, M.; Kryczka, B.; Marsura, A.; Porwański, S. Carbohydr. Res. 2014, 386, 18–22. doi:10.1016/j.carres.2013.12.017
Return to citation in text: [1] -
Murphy, P. V. Eur. J. Org. Chem. 2007, 4177–4187. doi:10.1002/ejoc.200700248
Return to citation in text: [1] -
Ågren, J. K. M.; Billing, J. F.; Grundberg, H. E.; Nilsson, U. J. Synthesis 2006, 3141–3145. doi:10.1055/s-2006-942503
Return to citation in text: [1] -
Jarosz, S.; Mach, M. J. Chem. Soc., Perkin Trans. 1 1998, 3943–3948. doi:10.1039/a807190j
Return to citation in text: [1] -
Jarosz, S.; Gajewska, A.; Luboradzki, R. Tetrahedron: Asymmetry 2008, 19, 1385–1391. doi:10.1016/j.tetasy.2008.05.010
Return to citation in text: [1] [2] -
Osuch-Kwiatkowska, A.; Jarosz, S. Tetrahedron: Asymmetry 2013, 24, 468–473. doi:10.1016/j.tetasy.2013.03.009
Return to citation in text: [1] [2] [3] -
Lewandowski, B.; Jarosz, S. Chem. Commun. 2008, 6399–6401. doi:10.1039/b816476b
Return to citation in text: [1] -
Potopnyk, M. A.; Cmoch, P.; Jarosz, S. Org. Lett. 2012, 14, 4258–4261. doi:10.1021/ol301993d
Return to citation in text: [1] [2] -
Potopnyk, M. A.; Lewandowski, B.; Jarosz, S. Tetrahedron: Asymmetry 2012, 23, 1474–1479. doi:10.1016/j.tetasy.2012.10.003
Return to citation in text: [1] -
Potopnyk, M. A.; Jarosz, S. Monatsh. Chem. 2013, 144, 437–443. doi:10.1007/s00706-012-0894-2
Return to citation in text: [1] -
Potopnyk, M. A.; Jarosz, S. Eur. J. Org. Chem. 2013, 5117–5126. doi:10.1002/ejoc.201300427
Return to citation in text: [1] -
Pakulski, Z.; Gajda, N.; Jawiczuk, M.; Frelek, J.; Cmoch, P.; Jarosz, S. Beilstein J. Org. Chem. 2014, 10, 1246–1254. doi:10.3762/bjoc.10.124
Return to citation in text: [1] -
Gaweł, A.; Jarosz, S. J. Carbohydr. Chem. 2010, 29, 332–347. doi:10.1080/07328303.2010.524958
Return to citation in text: [1] [2] -
Jarosz, S. J. Carbohydr. Chem. 2015, 34, 365–387. doi:10.1080/07328303.2015.1074243
Return to citation in text: [1] [2] -
Petrova, K. T.; Correia-da-Silva, P.; Crucho, C. I. C.; Barros, M. T. Curr. Org. Chem. 2014, 18, 1788–1802. doi:10.2174/1385272819666140527231535
Return to citation in text: [1] -
Plat, T.; Linhardt, R. J. J. Surfactants Deterg. 2001, 4, 415–421. doi:10.1007/s11743-001-0196-y
Return to citation in text: [1] -
Eggleston, G. Sucrose and Related Oligosaccharides. In Glycoscience; Fraser-Reid, B.; Tatsuta, K.; Thiem, J., Eds.; Springer: Berlin, 2008; pp 1164–1183.
Return to citation in text: [1] -
Goodby, J. W.; Görtz, V.; Cowling, S. J.; Mackenzie, G.; Martin, P.; Plusquellec, D.; Benvegnu, T.; Boullanger, P.; Lafont, D.; Queneau, Y.; Chambert, S.; Fitremann, J. Chem. Soc. Rev. 2007, 36, 1971–2032. doi:10.1039/b708458g
Return to citation in text: [1] -
Mancuso, A. J.; Huang, S.-L.; Swern, D. J. Org. Chem. 1978, 43, 2480–2482. doi:10.1021/jo00406a041
Return to citation in text: [1]
| 1. | Potopnyk, M. A.; Jarosz, S. Adv. Carbohydr. Chem. Biochem. 2014, 71, 227–295. doi:10.1016/B978-0-12-800128-8.00003-0 |
| 2. | Desmond, R. T.; Magpusao, A. N.; Lorenc, C.; Alverson, J. B.; Priestley, N.; Peczuh, M. W. Beilstein J. Org. Chem. 2014, 10, 2215–2221. doi:10.3762/bjoc.10.229 |
| 3. | Pintal, M.; Kryczka, B.; Marsura, A.; Porwański, S. Carbohydr. Res. 2014, 386, 18–22. doi:10.1016/j.carres.2013.12.017 |
| 4. | Murphy, P. V. Eur. J. Org. Chem. 2007, 4177–4187. doi:10.1002/ejoc.200700248 |
| 5. | Ågren, J. K. M.; Billing, J. F.; Grundberg, H. E.; Nilsson, U. J. Synthesis 2006, 3141–3145. doi:10.1055/s-2006-942503 |
| 9. | Lewandowski, B.; Jarosz, S. Chem. Commun. 2008, 6399–6401. doi:10.1039/b816476b |
| 10. | Potopnyk, M. A.; Cmoch, P.; Jarosz, S. Org. Lett. 2012, 14, 4258–4261. doi:10.1021/ol301993d |
| 11. | Potopnyk, M. A.; Lewandowski, B.; Jarosz, S. Tetrahedron: Asymmetry 2012, 23, 1474–1479. doi:10.1016/j.tetasy.2012.10.003 |
| 12. | Potopnyk, M. A.; Jarosz, S. Monatsh. Chem. 2013, 144, 437–443. doi:10.1007/s00706-012-0894-2 |
| 13. | Potopnyk, M. A.; Jarosz, S. Eur. J. Org. Chem. 2013, 5117–5126. doi:10.1002/ejoc.201300427 |
| 14. | Pakulski, Z.; Gajda, N.; Jawiczuk, M.; Frelek, J.; Cmoch, P.; Jarosz, S. Beilstein J. Org. Chem. 2014, 10, 1246–1254. doi:10.3762/bjoc.10.124 |
| 15. | Gaweł, A.; Jarosz, S. J. Carbohydr. Chem. 2010, 29, 332–347. doi:10.1080/07328303.2010.524958 |
| 16. | Jarosz, S. J. Carbohydr. Chem. 2015, 34, 365–387. doi:10.1080/07328303.2015.1074243 |
| 7. | Jarosz, S.; Gajewska, A.; Luboradzki, R. Tetrahedron: Asymmetry 2008, 19, 1385–1391. doi:10.1016/j.tetasy.2008.05.010 |
| 8. | Osuch-Kwiatkowska, A.; Jarosz, S. Tetrahedron: Asymmetry 2013, 24, 468–473. doi:10.1016/j.tetasy.2013.03.009 |
| 8. | Osuch-Kwiatkowska, A.; Jarosz, S. Tetrahedron: Asymmetry 2013, 24, 468–473. doi:10.1016/j.tetasy.2013.03.009 |
| 6. | Jarosz, S.; Mach, M. J. Chem. Soc., Perkin Trans. 1 1998, 3943–3948. doi:10.1039/a807190j |
| 7. | Jarosz, S.; Gajewska, A.; Luboradzki, R. Tetrahedron: Asymmetry 2008, 19, 1385–1391. doi:10.1016/j.tetasy.2008.05.010 |
| 15. | Gaweł, A.; Jarosz, S. J. Carbohydr. Chem. 2010, 29, 332–347. doi:10.1080/07328303.2010.524958 |
| 10. | Potopnyk, M. A.; Cmoch, P.; Jarosz, S. Org. Lett. 2012, 14, 4258–4261. doi:10.1021/ol301993d |
| 16. | Jarosz, S. J. Carbohydr. Chem. 2015, 34, 365–387. doi:10.1080/07328303.2015.1074243 |
| 19. | Eggleston, G. Sucrose and Related Oligosaccharides. In Glycoscience; Fraser-Reid, B.; Tatsuta, K.; Thiem, J., Eds.; Springer: Berlin, 2008; pp 1164–1183. |
| 20. | Goodby, J. W.; Görtz, V.; Cowling, S. J.; Mackenzie, G.; Martin, P.; Plusquellec, D.; Benvegnu, T.; Boullanger, P.; Lafont, D.; Queneau, Y.; Chambert, S.; Fitremann, J. Chem. Soc. Rev. 2007, 36, 1971–2032. doi:10.1039/b708458g |
| 21. | Mancuso, A. J.; Huang, S.-L.; Swern, D. J. Org. Chem. 1978, 43, 2480–2482. doi:10.1021/jo00406a041 |
| 18. | Plat, T.; Linhardt, R. J. J. Surfactants Deterg. 2001, 4, 415–421. doi:10.1007/s11743-001-0196-y |
| 17. | Petrova, K. T.; Correia-da-Silva, P.; Crucho, C. I. C.; Barros, M. T. Curr. Org. Chem. 2014, 18, 1788–1802. doi:10.2174/1385272819666140527231535 |
| 8. | Osuch-Kwiatkowska, A.; Jarosz, S. Tetrahedron: Asymmetry 2013, 24, 468–473. doi:10.1016/j.tetasy.2013.03.009 |
© 2017 Szyszka et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)