Abstract
Isoxazolidines represent a very important class of N/O-containing heterocycles used as the key intermediates in the synthesis of more complex cyclic and acyclic compounds, including various biologically active molecules. Here, we present a fast and highly stereoselective approach towards both C-3/4-cis and C-3/4-trans isomers of 3-substituted isoxazolidin-4-ols. The strategy relies on a highly regio- and trans-stereoselective hydroboration–oxidation reaction of the 4,5-unsubstituted 2,3-dihydroisoxazoles with basic hydrogen peroxide. The consecutive oxidation/reduction route, sequentially employing Dess–Martin periodinane and ʟ-selectride, is used for the inversion of the C-3/4-trans relative configuration of the isoxazolidine ring. The significance of the method lies in its variability and applicability to a concise synthesis of various 4-hydroxyisoxazolidines, starting from the readily available C-alkyl/aryl-nitrones. The resemblance to 3-hydroxypyrrolidines certainly makes the 4-hydroxyisoxazolidines important and valuable structural fragments in drug discovery.

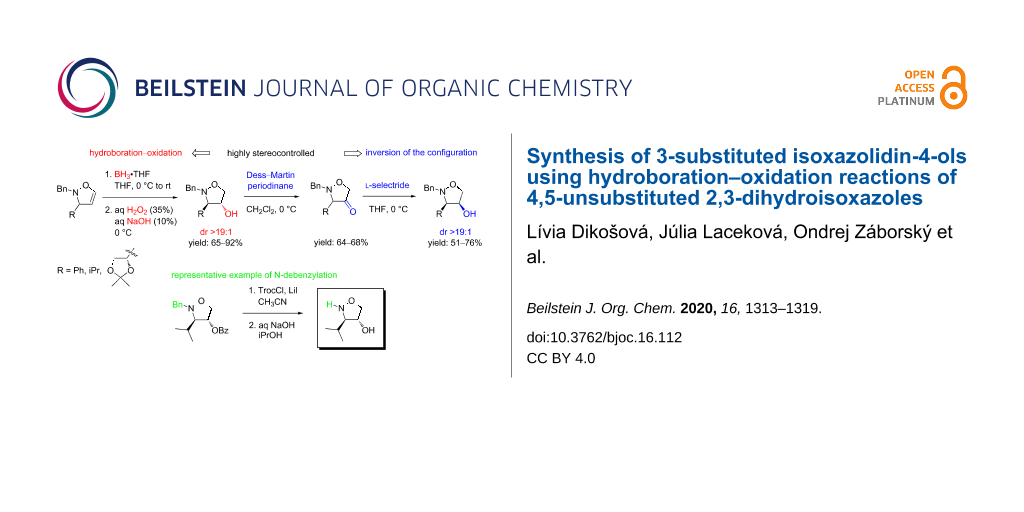
Graphical Abstract
Introduction
2,3-Dihydroisoxazoles (often referred to as 4-isoxazolines) represent a very important class of N/O-containing heterocycles used as the key intermediates in the synthesis of more complex cyclic and acyclic compounds, including various biologically active molecules [1-6].
In this regard, we have recently developed new methods for the preparation of 4,5-unsubstituted 2,3-dihydroisoxazoles in moderate to very good yields, starting from readily available 5-acetoxy- and 5-hydroxyisoxazolidines [7,8]. Their reactivity in electrophilic addition reactions allows for a straightforward introduction of a hydroxy group at the C-4 position of the resulting isoxazolidines by means of dihydroxylation [9,10] and epoxidation [11,12] reactions. Regarding the stereochemistry, almost all of the realized additions proceed with an excellent trans stereoselectivity relative to the substituent at C-3, giving isoxazolidine-4,5-diols and isoxazolidinyl epoxides with a C-3/4-trans relative configuration.
The resemblance between 3-hydroxypyrrolidines 1 and 4-hydroxyisoxazolidines 2 (Figure 1) makes the latter valuable structural fragments in drug discovery [13-18]. Up to now, this class of compounds was commonly prepared using intramolecular nucleophilic substitution reactions of properly substituted hydroxylamines [19,20] and N-hydroxyphthalimides [21-24] as well as by Tamao oxidations of 4-silylisoxazolidines [25,26]. Very recently, Tomkinson et al. described a simple and effective method for an intramolecular oxyamination of allylic N-tosyl hydroxylamines using malonoyl peroxide, providing N-tosylated 3-aryl-substituted 4-hydroxyisoxazolidines in a highly stereoselective manner in favor of the C-3/4-trans isomers [27]. We believe that the 3-substituted isoxazolidin-4-ols, represented by the general structures of the trans and the cis stereoisomer 3 and 4 (Figure 1), would be of particular interest in modern medicinal chemistry as eventual heterocyclic substructures.
![[1860-5397-16-112-1]](/bjoc/content/figures/1860-5397-16-112-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: 3-Substituted isoxazolidin-4-ols resembling 3-hydroxypyrrolidines.
Figure 1: 3-Substituted isoxazolidin-4-ols resembling 3-hydroxypyrrolidines.
Results and Discussion
At the beginning of our investigations, we envisaged that the above-mentioned 3-substituted 4-hydroxyisoxazolidines bearing no substituent at C5 could be readily obtained through the reductive cleavage of benzoylated isoxazolidines, employing the Lewis acid-catalyzed SN reaction with triethylsilane as the hydride source (Scheme 1). For this reason, the benzoates 6a and 6b were readily prepared from the corresponding 2,3-dihydroisoxazoles 5a and 5b, respectively, according to our procedure [7,10]. The compounds 5a and 5b were first converted into the isoxazolidine-4,5-diols by the treatment with potassium osmate/N-methylmorpholine N-oxide (NMO). The dihydroxylation reactions proceeded with an excellent trans selectivity with respect to the substituent at the C-3 carbon atom. The obtained products were benzoylated with benzoyl chloride/pyridine in the presence of DMAP to give the fully benzoylated isoxazolidine-4,5-diols 6a and 6b, which were subsequently treated with Et3SiH (3 equiv) and TMSOTf (2 equiv) in anhydrous CH2Cl2 at room temperature for 2 h [28]. To our delight, already the first attempts afforded the isoxazolidines 7a and 7b in very good 74% and 80% yield, respectively. Finally, their debenzoylation with K2CO3 in aqueous methanol gave the desired isoxazolidin-4-ols 8a (88% yield) and 8b (87% yield).
![[1860-5397-16-112-i1]](/bjoc/content/inline/1860-5397-16-112-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthetic approach towards isoxazolidin-4-ols via the regioselective reductive cleavage of the C5–O bond.
Scheme 1: Synthetic approach towards isoxazolidin-4-ols via the regioselective reductive cleavage of the C5–O...
Although the obtained results clearly showed the applicability of the reductive cleavage of anomeric isoxazolidinyl carboxylates in the synthesis of the respective 5-unsubstituted 4-hydroxyisoxazolidines, this pioneering approach mainly suffered from a large number of synthetic steps starting from 2,3-dihydroisoxazoles, leading to the target alcohols. As a consequence, we assumed that the hydroboration–oxidation reaction of 2,3-dihydroisoxazoles would be an excellent way to prevent this obstacle. Recently, Kang et al. [29] reported the first hydroboration–oxidation reaction of the 5-substituted 4-isoxazolines even though the access to 4-hydroxyisoxazolidines by the treatment of boronic ester-substituted isoxazolidines with basic hydrogen peroxide has previously been described [30,31].
To start with, the phenyl-substituted 2,3-dihydroisoxazole 5a was chosen as the starting substrate. After optimizing Kang's reaction conditions in terms of the borane amount, reaction temperature, and reaction time, we were able to prepare the 4-hydroxyisoxazolidine 8a in the best yield of 76% (Scheme 2). Firstly, 2 equivalents of the BH3·THF complex were added at 0 °C, and the reaction was further conducted at room temperature for 12 h. Regarding the following oxidation step, after the disappearance of the starting material, 10% aqueous NaOH and 35% aqueous H2O2 were added at 0 °C dropwise as slowly as possible. It turned out that a patient addition of both NaOH (aq) and H2O2 (aq) accompanied by intense cooling is a crucial part of the procedure, preventing the formation of undesired byproducts, which can arise from an oxidation at the nitrogen atom, leading to the opening of the isoxazolidine ring. After stirring for 3 h at 0 °C, ethyl acetate was added to the reaction mixture, and stirring was continued at 0 °C for additional 10 min. Afterwards, the reaction was worked up and the product was isolated by flash column chromatography (FCC). Subsequently, the optimized reaction conditions were successfully applied for the 2,3-dihydroisoxazoles 5b and 5c (Scheme 2), the providing desired isoxazolidin-4-ols 8b and 8c in 92% and 65% yield, respectively.
![[1860-5397-16-112-i2]](/bjoc/content/inline/1860-5397-16-112-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Hydroboration-oxidation of 4,5-unsubstituted 2,3-dihydroisoxazoles.
Scheme 2: Hydroboration-oxidation of 4,5-unsubstituted 2,3-dihydroisoxazoles.
All hydroborations proceeded with the exclusive formation of isoxazolidines with a hydroxy group in the C-4 position. We assume that the excellent regioselectivity is unambiguously caused by electronic effects since the endocyclic oxygen atom donates electrons to the C=C double bond, developing a negative partial charge at C-4. The existence of 4-hydroxy regioisomers was established by comparison of the 1H NMR spectrum of 8a with the already reported values for the known 5-hydroxyisoxazolidine possessing the same substituents at the N2 and C-3 atom [32]. The main differences between both regioisomers can be observed in the chemical shifts of the H-4 and H-5 protons. The signals for the H-4 proton at 4.48 ppm and for the H-5 protons at 3.81 and 4.11 ppm clearly indicate the formation of the 4-hydroxy derivative, whereas in the 5-hydroxy regioisomer, the signals corresponding to the H-4 protons are shifted to lower values (2.08–3.30 ppm), and the signal for the H-5 proton can be found at 5.50 ppm due to the effect of the two oxygen atoms bound to the C5 carbon atom.
Regarding the stereochemistry, the borane attack on the C=C double bond of the 2,3-dihydroisoxazole occurs from the sterically less hindered side, which is opposite relatively to the phenyl substituent at the C-3 carbon. The proposed C-3/4-trans relative configuration was confirmed by NOESY 1D experiments for 8a (Figure 2), and the results obtained were later compared with those measured for the respective C-3/4-cis isomer. For 8a, the irradiation of the H-4 proton resulted in a stronger enhancement of the signal corresponding to the pseudoequatorial proton H-5b (1.5%), whereas a weak NOE was observed for H-3 (0.4%) and the pseudoaxial H-5a proton (0.5%). Furthermore, the protons of the phenyl ring at C-3 were also affected (0.8%). The stereochemical results are consistent with our previous findings on the direct dihydroxylation and epoxidation reactions of 4,5-unsubstituted 2,3-dihydroisoxazoles [9-12].
![[1860-5397-16-112-2]](/bjoc/content/figures/1860-5397-16-112-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Selected NOE enhancements observed in the isoxazolidin-4-ol trans-8a. The arrows show the NOESY correlations.
Figure 2: Selected NOE enhancements observed in the isoxazolidin-4-ol trans-8a. The arrows show the NOESY cor...
To invert the relative C-3/4-trans stereochemistry, the isoxazolidin-4-ols 5a–c were first oxidized to the corresponding ketones (Scheme 3). Dess–Martin periodinane was chosen as the oxidizing agent [33,34] as our primary choice, pyridinium dichromate, prove to be insufficiently effective even at an elevated temperature. The reaction in anhydrous dichloromethane at 0 °C led to the desired isoxazolidin-4-ones 9a–c in moderate isolated yields of 64–68%. The optimization of the reaction conditions showed that 2 equivalents of the oxidizing agent were necessary to bring the reaction to completion. Even though the ketones 9a–c were isolated by FCC on silica gel in a pure form, they decomposed gradually if kept for a longer time, even at a lower temperature.
![[1860-5397-16-112-i3]](/bjoc/content/inline/1860-5397-16-112-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Dess-Martin oxidation of isoxazolidin-4-ols to ketones.
Scheme 3: Dess-Martin oxidation of isoxazolidin-4-ols to ketones.
The structures of 9a–c were definitely confirmed on the basis of 13C NMR spectra. The chemical shifts in the range of 210.8–215.0 ppm for the carbonyl carbon atom clearly indicated the presence of a ketone.
With the isoxazolidin-4-ones 9a–c in hand, we were ready to examine the intended stereoselective reduction of cyclic ketones with the aim to obtain the 4-hydroxyisoxazolidines with a relative C-3/4-cis configuration. Whereas the initial attempts of the reduction of 9a with lithium borohydride resulted in a poor stereoselectivity (70:30 in favor of the desired cis isomer), the use of bulky, sterically hindered ʟ-selectride in anhydrous THF at 0 °C led to the formation of the cis product exclusively (Scheme 4) [35,36], and the C-3/4-cis 4-hydroxyisoxazolidine 10a was isolated in 76% yield. Its relative configuration was confirmed by means of NOESY 1D experiments (Figure 3). Not surprisingly, the NOE between the protons H-3 and H-4 was significantly stronger (2.4%) than in the respective trans isomer 8a.
![[1860-5397-16-112-i4]](/bjoc/content/inline/1860-5397-16-112-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Inversion of the relative configuration of the isoxazolidine ring.
Scheme 4: Inversion of the relative configuration of the isoxazolidine ring.
![[1860-5397-16-112-3]](/bjoc/content/figures/1860-5397-16-112-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Selected NOE enhancements observed in the isoxazolidin-4-ol cis-10a. The arrows show the NOESY correlations.
Figure 3: Selected NOE enhancements observed in the isoxazolidin-4-ol cis-10a. The arrows show the NOESY corr...
The C-3/4-cis isoxazolidin-4-ols 10b and 10c were obtained from the ketones 9b and 9c in a similar manner in satisfying (51%) and good yield (72%), respectively (Scheme 4).
According to the literature, the N-debenzylation by a Pd-catalyzed hydrogenolysis in methanol with formic acid as the hydrogen source [37,38] should allow the access to N-deprotected isoxazolidines. Unfortunately, all attempts to remove the benzyl group directly from the model isoxazolidine 8b resulted only in a reductive cleavage of the N–O bond.
A successful debenzylation was achieved in a two-step procedure using 2,2,2-trichloroethyl chloroformate (TrocCl). However, the protection of the hydroxy group was required first (for the reaction of 8b with benzoyl chloride, see Supporting Information File 1). The consecutive reaction of the O-benzoylated isoxazolidine 7b with TrocCl in the presence of lithium iodide [39,40] gave the protected N-Troc isoxazolidine 11 in 70% yield (Scheme 5). It is worth mentioning that considering suitable solvents, acetonitrile was superior to 1,2-dichloroethane. Finally, the subsequent basic hydrolysis with 6 M NaOH [39] in isopropyl alcohol [41] to remove the carbamate readily afforded the desired isoxazolidine 12 in good yield (72%). The progress of the reaction was easily followed by TLC (EtOAc), and the N-unsubstituted isoxazolidin-4-ol 12 was identified as a readily oxidizable polar component by permanganate detection even at room temperature. The prior attempts to cleave the Troc group by zinc dust in acetic acid [42] failed due to the reduction of the N–O bond.
![[1860-5397-16-112-i5]](/bjoc/content/inline/1860-5397-16-112-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: N-debenzylation via N-Troc-protected isoxazolidines.
Scheme 5: N-debenzylation via N-Troc-protected isoxazolidines.
Conclusion
In summary, we have developed a new synthetic approach towards 3-substituted isoxazolidin-4-ols employing the hydroboration–oxidation reaction of 4,5-unsubstituted 2,3-dihydroisoxazoles with basic hydrogen peroxide. A patient addition of both NaOH (aq) and H2O2 (aq) accompanied by intense cooling was a crucial part of the procedure in terms of product yield. All hydroborations were highly regioselective, leading only to the formation of isoxazolidines with the hydroxy group in the C-4 position. From a stereochemical point of view, the additions took place with an excellent trans selectivity with respect to the substituent at C-3.
A sequential oxidation/reduction route was successfully used for the inversion of the C-3/4-trans relative configuration of the isoxazolidine ring. The bulkier ʟ-selectride showed an excellent selectivity toward the C-3/4-cis isomer compared to lithium borohydride. The final N-debenzylation by a two-step procedure involving a basic hydrolysis of the corresponding N-Troc intermediate was successfully exemplified on the 3-isopropyl-substituted derivative. We believe that the presented method will provide a fast and highly stereoselective approach towards both C-3/4-cis and C-3/4-trans isomers of 3-substituted isoxazolidin-4-ols as valuable structural fragments in drug discovery due to their resemblance with 3-hydroxypyrrolidines.
Experimental
Typical procedure for the hydroboration of 2,3-dihydroisoxazoles
(±)-2-Benzyl-3-phenylisoxazolidin-4-ol (8a): A round-bottom flask was charged with the 2,3-dihydroisoxazole 5a (590 mg, 2.49 mmol), evacuated, and flushed with argon. Afterwards, dry THF was added (25 mL), the mixture was cooled to 0 °C, and BH3∙THF (5 mL, 5 mmol, 1 M solution in THF) was added dropwise. The reaction mixture was stirred at rt for 12 h. After the disappearance of the starting material (TLC, hexanes/EtOAc, 4:1), a 10% solution of NaOH (7.5 mL) was added dropwise as slowly as possible at 0 °C, followed by a 35% solution of H2O2 (15 mL) added in a likewise manner. After 3 h of stirring at 0 °C (TLC, hexanes/EtOAc, 1:1), the reaction was diluted with EtOAc (30 mL). The organic layer was separated, washed with brine (2 × 40 mL), dried over MgSO4, and concentrated under reduced pressure. The product was purified by FCC (hexanes/EtOAc, 7:3) to give the isoxazolidinol 8a (485 mg, 1.90 mmol, 76%) as colorless oil. Rf 0.43 (n-hexane/EtOAc, 1:1); IR (ATR) νmax: 3392, 3030, 2862, 1495, 1454, 1095, 993, 753, 695, 635, 527 cm−1; 1H NMR (600 MHz, CDCl3) δ 2.44 (bs, 1H, OH), 3.66 (d, J = 4.7 Hz, 1H, H-3), 3.81 (dd, J = 2.6, 9.3 Hz, 1H, H-5a), 3.82 (d, J = 14.2 Hz, 1H, PhCH2), 3.98 (d, J = 14.2 Hz, 1H, PhCH2), 4.11 (dd, J = 6.1, 9.3 Hz, 1H, H-5b), 4.48 (ddd, J = 2.6, 4.8, 6.1 Hz, 1H, H-4), 7.21–7.43 (m, 10H, H-Ph); 13C NMR (150 MHz, CDCl3) δ 60.3 (PhCH2), 73.8 (C-5), 79.5 (C-3), 83.5 (C-4), 127.4 (CH-Ph), 127.9 (CH-Ph), 128.2 (CH-Ph), 128.3 (CH-Ph), 2×128.9 (CH-Ph), 137.4 (C-Ph), 138.2 (C-Ph); HRMS (ESI) (m/z): [M + H]+ calcd for C16H18NO2, 256.1333; found 256.1329.
Typical procedure for the oxidation of isoxazolidin-4-ols with Dess–Martin periodinane
(±)-2-Benzyl-3-phenylisoxazolidin-4-one (9a): A Schlenk flask was charged with the isoxazolidinol 8a (450 mg, 1.76 mmol), evacuated, and filled with argon. The starting material was dissolved in anhydrous CH2Cl2 (18 mL), the reaction mixture was cooled down to 0 °C, and solid Dess–Martin periodinane (1.5 g, 3.54 mmol) was slowly added under a stream of argon. The reaction was stirred at 0 °C for 12 h, and after the complete conversion of the starting material (TLC analysis: hexanes/EtOAc, 1:1), a saturated aq NaHCO3 (20 mL) and a saturated aq Na2S2O3∙5H2O (20 mL) solution were added. The mixture was stirred for 15 min at 0 °C and then, the solution was allowed to warm to rt. The organic layer was separated and washed with water (2 × 20 mL), dried over MgSO4, and evaporated in vacuo. The residue was purified by FCC (hexanes/EtOAc, 9:1) to give the isoxazolidin-4-one 9a (305 mg, 1.20 mmol, 68%) as yellowish waxy solid that gradually decomposed over time. mp 35–38 °C; Rf 0.61 (n-hexane/EtOAc, 1:1); IR (ATR) νmax: 3032, 2871, 2814, 1770, 1495, 1454, 1123, 1048, 737, 695, 615, 545, 470 cm−1; 1H NMR (300 MHz, CDCl3) δ 3.97 (s, 1H, H-3), 3.98 (d, J = 14.3 Hz, 1H, PhCH2), 4.14 (d, J = 15.7 Hz, 1H, H-5a), 4.24 (d, J = 14.3 Hz, 1H, PhCH2), 4.29 (d, J = 15.7, 1H, H-5b), 7.28–7.41 (m, 10H, H-Ph); 13C NMR (150 MHz, CDCl3) δ 60.7 (PhCH2), 70.7 (C-5), 75.1 (C-3), 127.9 (CH-Ph), 128.5 (CH-Ph), 128.7 (CH-Ph), 128.8 (CH-Ph), 129.0 (CH-Ph), 129.3 (CH-Ph), 133.6 (C-Ph), 136.0 (C-Ph), 210.8 (C=O); HRMS (APCI) (m/z): [M + H]+ calcd for C16H16NO2, 254.1176; found, 254.1174.
Typical procedure for the reduction of isoxazolidin-4-one with ʟ-selectride
(±)-2-Benzyl-3-phenylisoxazolidin-4-ol (10a): ʟ-Selectride (1.4 mL, 1.4 mmol, 1 M solution in THF) was added dropwise to a solution of the isoxazolidin-4-one 9a (280 mg, 1.11 mmol) in anhydrous THF (11 mL) under an argon atmosphere at 0 °C, and the reaction mixture was stirred for 30 min. When TLC analysis showed that the starting isoxazolidinone had disappeared (hexanes/EtOAc, 1:1), a saturated aq NH4Cl solution was added slowly (20 mL), and the mixture was stirred for additional 10 min. Afterwards, the mixture was extracted with CH2Cl2 (3×20 mL). The combined organic layers were washed with water (50 mL), dried over MgSO4, and concentrated under reduced pressure. The product was isolated by FCC (hexanes/EtOAc, 7:3) to give the isoxazolidinol 10a (215 mg, 0.84 mmol, 76%) as colorless oil. Rf 0.41 (n-hexane/EtOAc, 1:1); IR (ATR) νmax: 3421, 3028, 2924, 2868, 1495, 1454, 1107, 1028, 749, 696, 599, 531 cm−1; 1H NMR (600 MHz, CDCl3) δ 1.65 (bs, 1H, OH), 3.70 (d, J = 14.4 Hz, 1H, PhCH2), 3.79 (d, J = 5.5 Hz, 1H, H-3), 3.85 (dd, J = 3.5, 9.2 Hz, 1H, H-5a), 4.07 (d, J = 14.4 Hz, 1H, PhCH2), 4.38 (dd, J = 6.1, 9.2 Hz, 1H, H-5b), 4.59 (td, J = 3.5, 5.8 Hz, 1H, H-4), 7.24–7.47 (m, 10H, H-Ph); 13C NMR (150 MHz, CDCl3) δ 60.4 (PhCH2), 74.5 (C-5), 75.3 (C-3), 76.8 (C-4), 127.4 (CH-Ph), 128.3 (CH-Ph), 128.4 (CH-Ph), 128.9 (CH-Ph), 129.0 (CH-Ph), 129.2 (CH-Ph), 134.4 (C-Ph), 137.3 (C-Ph); HRMS (ESI) (m/z): [M + H]+ calcd for C16H18NO2, 256.1333; found 256.1329.
N-Debenzylation with 2,2,2-trichloroethyl chloroformate
(±)-2,2,2-Trichloroethyl 4-(benzoyloxy)-3-isopropylisoxazolidine-2-carboxylate (11): 2,2,2-Trichloroethyl chloroformate (0.33 mL, 2.4 mmol) was added dropwise to a stirred solution of the N-benzylisoxazolidine 7b (260 mg, 0.8 mmol) and lithium iodide (160 mg, 1.2 mmol) in anhydrous acetonitrile (4 mL) under argon. The reaction mixture was stirred at 60 °C for 8 h. When the TLC analysis showed that the starting isoxazolidine had disappeared (hexanes/EtOAc, 9:1), a saturated aq NaHCO3 (10 mL) solution and CH2Cl2 (20 mL) were added. After vigorous stirring for additional 5 min, the organic layer was separated, and the aqueous phase was extracted with CH2Cl2 (10 mL). The combined organic layers were washed with water (20 mL), dried over MgSO4, and concentrated under reduced pressure. The product was purified by FCC (hexanes/EtOAc, 9:1) to give the N-Troc-isoxazolidine 11 (230 mg, 0.56 mmol, 70%) as colorless sticky oil. Rf 0.20 (n-hexane/EtOAc, 9:1); IR (ATR) νmax: 3067, 2963, 2881, 1755, 1717, 1374, 1315, 1265, 1107, 1052, 805, 708, 572 cm−1; 1H NMR (600 MHz, CDCl3): δ 1.09 (d, J = 6.7 Hz, 3H, CH3), 1.14 (d, J = 6.7 Hz, 3H, CH3), 1.89–1.97 (m, 1H, CH(CH3)2), 4.12 (dd, J = 3.4, 9.5 Hz, 1H, H-5a), 4.22 (d, J = 8.4 Hz, 1H, H-3), 4.51 (dd, J = 5.9, 9.6 Hz, 1H, H-5b), 4.72 (d, J = 11.9 Hz, 1H, Cl3CCH2O), 4.86 (d, J = 11.9 Hz, 1H, Cl3CCH2O), 5.64 (ddd, J = 1.1, 3.5, 5.9 Hz, 1H, H-4), 7.43–7.46 (m, 2H, H-Ph), 7.57–7.60 (m, 1H, H-Ph), 7.95–7.98 (m, 2H, H-Ph); 13C NMR (150 MHz, CDCl3) δ 19.1 (CH3), 19.2 (CH3), 29.7 (CH(CH3)2), 72.1 (C-3), 74.4, 75.3 (C-5, CO2CH2), 78.9 (C-4), 94.8 (CCl3), 128.6 (CH-Ph), 128.9 (C-Ph), 129.7 (CH-Ph), 133.7 (CH-Ph), 157.1 (CO2CH2), 165.9 (COPh); HRMS (ESI) (m/z): [M + H]+ calcd for C16H19Cl3NO5, 410.0324; found 410.0329.
Hydrolysis of trichloroethyl carbamate
(±)-3-Isopropylisoxazolidin-4-ol (12): The NaOH solution (0.6 mL, 3.6 mmol, 6 M) was added to a solution of the N-Troc-isoxazolidine 11 (120 mg, 0.29 mmol) in isopropyl alcohol (1.2 mL), and the mixture was stirred at rt for 1 h. After the disappearance of the starting material (TLC, hexanes/EtOAc, 5:1), the solution was neutralized with HCl (6 M). Afterwards, a saturated aq NaHCO3 solution (2 mL) and solid NaCl were added, and the resulting slurry was vigorously extracted with EtOAc (3×5 mL). The combined organic layers were dried over MgSO4 and concentrated under reduced pressure. The product was purified by FCC (EtOAc) to give the isoxazolidinol 12 (28 mg, 0.21 mmol, 72%) as colorless sticky oil. Rf 0.25 (EtOAc); IR (ATR): νmax = 3160, 2971, 2899, 2874, 1473, 1093, 1036, 1013, 935, 886, 754, 718, 647 cm−1; 1H NMR (600 MHz, CDCl3) δ 1.02 (d, J = 6.7 Hz, 3H, CH3), 1.04 (d, J = 6.7 Hz, 3H, CH3), 1.58–1.66 (m, 1H, CH(CH3)2), 2.90 (dd, J = 1.8, 8.8 Hz, 1H, H-3), 3.83 (dd, J = 1.9, 9.4 Hz, 1H, H-5a), 3.89 (dd, J = 5.0, 9.4 Hz, 1H, H-5b), 4.51 (dt, J = 2.0, 5.0 Hz, 1H, H-4); 13C NMR (150 MHz, CDCl3) δ 19.8 (CH3), 20.0 (CH3), 29.1 (CH(CH3)2), 75.7 (C-3), 78.0 (C-5), 78.4 (C-4); HRMS (ESI) (m/z): [M + H]+ calcd for C6H14NO2, 132.1020; found 132.1020.
Supporting Information
| Supporting Information File 1: Detailed experimental procedures, analytical data, and NMR spectra of all compounds. | ||
| Format: PDF | Size: 2.1 MB | Download |
Funding
This work was supported by the Slovak Grant Agency for Science VEGA (project no. 1/0552/18) and the Research and Development Operational Programmes funded by the ERDF (ITMS project nos. 26240120001 and 26240120025). This article was also created with the support of the MŠVVaŠ of the Slovak Republic within the Research and Development Operational Programme for the project “University Science Park of STU Bratislava” (ITMS project no. 26240220084), cofunded by the European Regional Development Fund.
References
-
Freeman, J. P. Chem. Rev. 1983, 83, 241–261. doi:10.1021/cr00055a002
Return to citation in text: [1] -
Chukanov, N. V.; Reznikov, V. A. Russ. Chem. Bull. 2011, 60, 379–399. doi:10.1007/s11172-011-0062-6
Return to citation in text: [1] -
Pinho e Melo, T. M. V. D. Eur. J. Org. Chem. 2010, 3363–3376. doi:10.1002/ejoc.201000321
Return to citation in text: [1] -
Cordero, F. M.; Giomi, D.; Lascialfari, L. Prog. Heterocycl. Chem. 2017, 29, 353–382. doi:10.1016/b978-0-08-102310-5.00011-4
Return to citation in text: [1] -
Cordero, F. M.; Giomi, D.; Lascialfari, L. Prog. Heterocycl. Chem. 2018, 30, 279–309. doi:10.1016/b978-0-08-102788-2.00011-8
Return to citation in text: [1] -
Ram, V.; Sethi, A.; Nath, M.; Pratap, R. Five-Membered Heterocycles. In The Chemistry of Heterocycles; Ram, V.; Sethi, A.; Nath, M.; Pratap, R., Eds.; Elsevier: Amsterdam, 2019; pp 149–478. doi:10.1016/b978-0-08-101033-4.00005-x.
Return to citation in text: [1] -
Fischer, R.; Lackovičová, D.; Fišera, L. Synthesis 2012, 44, 3783–3788. doi:10.1055/s-0032-1317682
Return to citation in text: [1] [2] -
Záborský, O.; Šoral, M.; Fischer, R. Tetrahedron Lett. 2015, 56, 2155–2158. doi:10.1016/j.tetlet.2015.03.048
Return to citation in text: [1] -
Fischer, R.; Stanko, B.; Prónayová, N. Synlett 2013, 24, 2132–2136. doi:10.1055/s-0033-1339674
Return to citation in text: [1] [2] -
Beňadiková, D.; Čurillová, J.; Lacek, T.; Rakovský, E.; Moncoľ, J.; Doháňošová, J.; Fischer, R. Tetrahedron 2014, 70, 5585–5593. doi:10.1016/j.tet.2014.06.093
Return to citation in text: [1] [2] [3] -
Záborský, O.; Malatinský, T.; Marek, J.; Moncol, J.; Fischer, R. Eur. J. Org. Chem. 2016, 3993–4002. doi:10.1002/ejoc.201600471
Return to citation in text: [1] [2] -
Záborský, O.; Štadániová, R.; Doháňošová, J.; Moncol, J.; Fischer, R. Synthesis 2017, 49, 4942–4954. doi:10.1055/s-0036-1590924
Return to citation in text: [1] [2] -
Guile, S. D.; Bantick, J. R.; Cooper, M. E.; Donald, D. K.; Eyssade, C.; Ingall, A. H.; Lewis, R. J.; Martin, B. P.; Mohammed, R. T.; Potter, T. J.; Reynolds, R. H.; St-Gallay, S. A.; Wright, A. D. J. Med. Chem. 2007, 50, 254–263. doi:10.1021/jm060995h
Return to citation in text: [1] -
Xu, L.; Liao, Y.; Liu, C.; Lei, H.-S.; Deng, J.; Zou, Y.; Lei, W.; Zhou, C.; He, Z.; Wu, K.; Yuan, Q.; Fan, B. Pyrimidinedione derivative capable of inhibiting monocarboxylate transporter. Chinese Patent CN109422749 A, March 5, 2019.
Return to citation in text: [1] -
Dorsch, D.; Muzerelle, M.; Burgdorf, L.; Wucherer-Plietker, M.; Czodrowski, P.; Esdar, C. Quinolin-2-one derivatives. World Patent WO2017121444 A1, July 20, 2017.
Return to citation in text: [1] -
Hartman, G. D.; Kuduk, S. Derivatives and methods of treating hepatitis B infections. U.S. Patent US2016185777 A1, June 30, 2016.
Return to citation in text: [1] -
Brookings, D. C.; Hutchings, M. C.; Langham, B. J. Thieno-pyridine derivatives as MEK inhibitors. World Patent WO2009093008 A1, July 30, 2009.
Return to citation in text: [1] -
Guile, S. D.; Ingall, A. H. Thienopyridazinones and their use in modulation of autoimmune disease. World Patent WO2004065395 A1, Aug 5, 2004.
Return to citation in text: [1] -
Ishikawa, T.; Nagai, K.; Senzaki, M.; Tatsukawa, A.; Saito, S. Tetrahedron 1998, 54, 2433–2448. doi:10.1016/s0040-4020(98)00008-8
Return to citation in text: [1] -
Pusterla, I.; Bode, J. W. Nat. Chem. 2015, 7, 668–672. doi:10.1038/nchem.2282
Return to citation in text: [1] -
Amlaiky, N.; Leclerc, G. Synthesis 1982, 426–428. doi:10.1055/s-1982-29825
Return to citation in text: [1] -
Amlaiky, N.; Leclerc, G.; Carpy, A. J. Org. Chem. 1982, 47, 517–523. doi:10.1021/jo00342a029
Return to citation in text: [1] -
Buchalska, E.; Plenkiewicz, J. Synth. Commun. 2000, 30, 1467–1477. doi:10.1080/00397910008087175
Return to citation in text: [1] -
Martin, B. P.; Cooper, M. E.; Donald, D. K.; Guile, S. D. Tetrahedron Lett. 2006, 47, 7635–7639. doi:10.1016/j.tetlet.2006.08.054
Return to citation in text: [1] -
Ishikawa, T.; Kudo, T.; Shigemori, K.; Saito, S. J. Am. Chem. Soc. 2000, 122, 7633–7637. doi:10.1021/ja000248o
Return to citation in text: [1] -
Kudoh, T.; Ishikawa, T.; Shimizu, Y.; Saito, S. Org. Lett. 2003, 5, 3875–3878. doi:10.1021/ol035423v
Return to citation in text: [1] -
Alamillo-Ferrer, C.; Curle, J. M.; Davidson, S. C.; Lucas, S. C. C.; Atkinson, S. J.; Campbell, M.; Kennedy, A. R.; Tomkinson, N. C. O. J. Org. Chem. 2018, 83, 6728–6740. doi:10.1021/acs.joc.8b00392
Return to citation in text: [1] -
Jeffery, A.; Nair, V. Tetrahedron Lett. 1995, 36, 3627–3630. doi:10.1016/0040-4039(95)00618-m
Return to citation in text: [1] -
Xiao, Z.-F.; Ding, T.-H.; Mao, S.-W.; Ning, X.-S.; Kang, Y.-B. Adv. Synth. Catal. 2016, 358, 1859–1863. doi:10.1002/adsc.201600044
Return to citation in text: [1] -
Carboni, B.; Ollivault, M.; Le Bouguenec, F.; Carrié, R.; Jazouli, M. Tetrahedron Lett. 1997, 38, 6665–6668. doi:10.1016/s0040-4039(97)01560-8
Return to citation in text: [1] -
Davies, C. D.; Marsden, S. P.; Stokes, E. S. E. Tetrahedron Lett. 2000, 41, 4229–4233. doi:10.1016/s0040-4039(00)00571-2
Return to citation in text: [1] -
Keirs, D.; Moffat, D.; Overton, K.; Tomanek, R. J. Chem. Soc., Perkin Trans. 1 1991, 1041–1051. doi:10.1039/p19910001041
Return to citation in text: [1] -
Dess, D. B.; Martin, J. C. J. Am. Chem. Soc. 1991, 113, 7277–7287. doi:10.1021/ja00019a027
Return to citation in text: [1] -
Cook, G. P.; Greenberg, M. M. J. Org. Chem. 1994, 59, 4704–4706. doi:10.1021/jo00095a060
Return to citation in text: [1] -
Brown, H. C.; Krishnamurthy, S. J. Am. Chem. Soc. 1972, 94, 7159–7161. doi:10.1021/ja00775a053
Return to citation in text: [1] -
Crich, D.; Hu, T.; Cai, F. J. Org. Chem. 2008, 73, 8942–8953. doi:10.1021/jo801630m
Return to citation in text: [1] -
Nguyen, T. B.; Beauseigneur, A.; Martel, A.; Dhal, R.; Laurent, M.; Dujardin, G. J. Org. Chem. 2010, 75, 611–620. doi:10.1021/jo902107j
Return to citation in text: [1] -
Chiacchio, U.; Corsaro, A.; Mates, J.; Merino, P.; Piperno, A.; Rescifina, A.; Romeo, G.; Romeo, R.; Tejero, T. Tetrahedron 2003, 59, 4733–4738. doi:10.1016/s0040-4020(03)00689-6
Return to citation in text: [1] -
Baumgartner, H.; O'Sullivan, A. C.; Schneider, J. Heterocycles 1997, 45, 1537–1549. doi:10.3987/com-97-7836
Return to citation in text: [1] [2] -
Arora, J.; Bordeleau, M.; Dube, L.; Jarvie, K.; Mazzocco, L.; Peragine, J.; Tehim, A.; Egle, I. Bioorg. Med. Chem. Lett. 2005, 15, 5253–5256. doi:10.1016/j.bmcl.2005.08.051
Return to citation in text: [1] -
Gangula, S.; Kumar Kolla, N.; Elati, C.; Dongamanti, A.; Bandichhor, R. Synth. Commun. 2012, 42, 3344–3360. doi:10.1080/00397911.2011.582216
Return to citation in text: [1] -
Whitby, L. R.; Ando, Y.; Setola, V.; Vogt, P. K.; Roth, B. L.; Boger, D. L. J. Am. Chem. Soc. 2011, 133, 10184–10194. doi:10.1021/ja201878v
Return to citation in text: [1]
| 39. | Baumgartner, H.; O'Sullivan, A. C.; Schneider, J. Heterocycles 1997, 45, 1537–1549. doi:10.3987/com-97-7836 |
| 40. | Arora, J.; Bordeleau, M.; Dube, L.; Jarvie, K.; Mazzocco, L.; Peragine, J.; Tehim, A.; Egle, I. Bioorg. Med. Chem. Lett. 2005, 15, 5253–5256. doi:10.1016/j.bmcl.2005.08.051 |
| 35. | Brown, H. C.; Krishnamurthy, S. J. Am. Chem. Soc. 1972, 94, 7159–7161. doi:10.1021/ja00775a053 |
| 36. | Crich, D.; Hu, T.; Cai, F. J. Org. Chem. 2008, 73, 8942–8953. doi:10.1021/jo801630m |
| 37. | Nguyen, T. B.; Beauseigneur, A.; Martel, A.; Dhal, R.; Laurent, M.; Dujardin, G. J. Org. Chem. 2010, 75, 611–620. doi:10.1021/jo902107j |
| 38. | Chiacchio, U.; Corsaro, A.; Mates, J.; Merino, P.; Piperno, A.; Rescifina, A.; Romeo, G.; Romeo, R.; Tejero, T. Tetrahedron 2003, 59, 4733–4738. doi:10.1016/s0040-4020(03)00689-6 |
| 1. | Freeman, J. P. Chem. Rev. 1983, 83, 241–261. doi:10.1021/cr00055a002 |
| 2. | Chukanov, N. V.; Reznikov, V. A. Russ. Chem. Bull. 2011, 60, 379–399. doi:10.1007/s11172-011-0062-6 |
| 3. | Pinho e Melo, T. M. V. D. Eur. J. Org. Chem. 2010, 3363–3376. doi:10.1002/ejoc.201000321 |
| 4. | Cordero, F. M.; Giomi, D.; Lascialfari, L. Prog. Heterocycl. Chem. 2017, 29, 353–382. doi:10.1016/b978-0-08-102310-5.00011-4 |
| 5. | Cordero, F. M.; Giomi, D.; Lascialfari, L. Prog. Heterocycl. Chem. 2018, 30, 279–309. doi:10.1016/b978-0-08-102788-2.00011-8 |
| 6. | Ram, V.; Sethi, A.; Nath, M.; Pratap, R. Five-Membered Heterocycles. In The Chemistry of Heterocycles; Ram, V.; Sethi, A.; Nath, M.; Pratap, R., Eds.; Elsevier: Amsterdam, 2019; pp 149–478. doi:10.1016/b978-0-08-101033-4.00005-x. |
| 13. | Guile, S. D.; Bantick, J. R.; Cooper, M. E.; Donald, D. K.; Eyssade, C.; Ingall, A. H.; Lewis, R. J.; Martin, B. P.; Mohammed, R. T.; Potter, T. J.; Reynolds, R. H.; St-Gallay, S. A.; Wright, A. D. J. Med. Chem. 2007, 50, 254–263. doi:10.1021/jm060995h |
| 14. | Xu, L.; Liao, Y.; Liu, C.; Lei, H.-S.; Deng, J.; Zou, Y.; Lei, W.; Zhou, C.; He, Z.; Wu, K.; Yuan, Q.; Fan, B. Pyrimidinedione derivative capable of inhibiting monocarboxylate transporter. Chinese Patent CN109422749 A, March 5, 2019. |
| 15. | Dorsch, D.; Muzerelle, M.; Burgdorf, L.; Wucherer-Plietker, M.; Czodrowski, P.; Esdar, C. Quinolin-2-one derivatives. World Patent WO2017121444 A1, July 20, 2017. |
| 16. | Hartman, G. D.; Kuduk, S. Derivatives and methods of treating hepatitis B infections. U.S. Patent US2016185777 A1, June 30, 2016. |
| 17. | Brookings, D. C.; Hutchings, M. C.; Langham, B. J. Thieno-pyridine derivatives as MEK inhibitors. World Patent WO2009093008 A1, July 30, 2009. |
| 18. | Guile, S. D.; Ingall, A. H. Thienopyridazinones and their use in modulation of autoimmune disease. World Patent WO2004065395 A1, Aug 5, 2004. |
| 9. | Fischer, R.; Stanko, B.; Prónayová, N. Synlett 2013, 24, 2132–2136. doi:10.1055/s-0033-1339674 |
| 10. | Beňadiková, D.; Čurillová, J.; Lacek, T.; Rakovský, E.; Moncoľ, J.; Doháňošová, J.; Fischer, R. Tetrahedron 2014, 70, 5585–5593. doi:10.1016/j.tet.2014.06.093 |
| 11. | Záborský, O.; Malatinský, T.; Marek, J.; Moncol, J.; Fischer, R. Eur. J. Org. Chem. 2016, 3993–4002. doi:10.1002/ejoc.201600471 |
| 12. | Záborský, O.; Štadániová, R.; Doháňošová, J.; Moncol, J.; Fischer, R. Synthesis 2017, 49, 4942–4954. doi:10.1055/s-0036-1590924 |
| 11. | Záborský, O.; Malatinský, T.; Marek, J.; Moncol, J.; Fischer, R. Eur. J. Org. Chem. 2016, 3993–4002. doi:10.1002/ejoc.201600471 |
| 12. | Záborský, O.; Štadániová, R.; Doháňošová, J.; Moncol, J.; Fischer, R. Synthesis 2017, 49, 4942–4954. doi:10.1055/s-0036-1590924 |
| 33. | Dess, D. B.; Martin, J. C. J. Am. Chem. Soc. 1991, 113, 7277–7287. doi:10.1021/ja00019a027 |
| 34. | Cook, G. P.; Greenberg, M. M. J. Org. Chem. 1994, 59, 4704–4706. doi:10.1021/jo00095a060 |
| 9. | Fischer, R.; Stanko, B.; Prónayová, N. Synlett 2013, 24, 2132–2136. doi:10.1055/s-0033-1339674 |
| 10. | Beňadiková, D.; Čurillová, J.; Lacek, T.; Rakovský, E.; Moncoľ, J.; Doháňošová, J.; Fischer, R. Tetrahedron 2014, 70, 5585–5593. doi:10.1016/j.tet.2014.06.093 |
| 30. | Carboni, B.; Ollivault, M.; Le Bouguenec, F.; Carrié, R.; Jazouli, M. Tetrahedron Lett. 1997, 38, 6665–6668. doi:10.1016/s0040-4039(97)01560-8 |
| 31. | Davies, C. D.; Marsden, S. P.; Stokes, E. S. E. Tetrahedron Lett. 2000, 41, 4229–4233. doi:10.1016/s0040-4039(00)00571-2 |
| 7. | Fischer, R.; Lackovičová, D.; Fišera, L. Synthesis 2012, 44, 3783–3788. doi:10.1055/s-0032-1317682 |
| 8. | Záborský, O.; Šoral, M.; Fischer, R. Tetrahedron Lett. 2015, 56, 2155–2158. doi:10.1016/j.tetlet.2015.03.048 |
| 32. | Keirs, D.; Moffat, D.; Overton, K.; Tomanek, R. J. Chem. Soc., Perkin Trans. 1 1991, 1041–1051. doi:10.1039/p19910001041 |
| 27. | Alamillo-Ferrer, C.; Curle, J. M.; Davidson, S. C.; Lucas, S. C. C.; Atkinson, S. J.; Campbell, M.; Kennedy, A. R.; Tomkinson, N. C. O. J. Org. Chem. 2018, 83, 6728–6740. doi:10.1021/acs.joc.8b00392 |
| 28. | Jeffery, A.; Nair, V. Tetrahedron Lett. 1995, 36, 3627–3630. doi:10.1016/0040-4039(95)00618-m |
| 42. | Whitby, L. R.; Ando, Y.; Setola, V.; Vogt, P. K.; Roth, B. L.; Boger, D. L. J. Am. Chem. Soc. 2011, 133, 10184–10194. doi:10.1021/ja201878v |
| 25. | Ishikawa, T.; Kudo, T.; Shigemori, K.; Saito, S. J. Am. Chem. Soc. 2000, 122, 7633–7637. doi:10.1021/ja000248o |
| 26. | Kudoh, T.; Ishikawa, T.; Shimizu, Y.; Saito, S. Org. Lett. 2003, 5, 3875–3878. doi:10.1021/ol035423v |
| 29. | Xiao, Z.-F.; Ding, T.-H.; Mao, S.-W.; Ning, X.-S.; Kang, Y.-B. Adv. Synth. Catal. 2016, 358, 1859–1863. doi:10.1002/adsc.201600044 |
| 21. | Amlaiky, N.; Leclerc, G. Synthesis 1982, 426–428. doi:10.1055/s-1982-29825 |
| 22. | Amlaiky, N.; Leclerc, G.; Carpy, A. J. Org. Chem. 1982, 47, 517–523. doi:10.1021/jo00342a029 |
| 23. | Buchalska, E.; Plenkiewicz, J. Synth. Commun. 2000, 30, 1467–1477. doi:10.1080/00397910008087175 |
| 24. | Martin, B. P.; Cooper, M. E.; Donald, D. K.; Guile, S. D. Tetrahedron Lett. 2006, 47, 7635–7639. doi:10.1016/j.tetlet.2006.08.054 |
| 39. | Baumgartner, H.; O'Sullivan, A. C.; Schneider, J. Heterocycles 1997, 45, 1537–1549. doi:10.3987/com-97-7836 |
| 19. | Ishikawa, T.; Nagai, K.; Senzaki, M.; Tatsukawa, A.; Saito, S. Tetrahedron 1998, 54, 2433–2448. doi:10.1016/s0040-4020(98)00008-8 |
| 20. | Pusterla, I.; Bode, J. W. Nat. Chem. 2015, 7, 668–672. doi:10.1038/nchem.2282 |
| 7. | Fischer, R.; Lackovičová, D.; Fišera, L. Synthesis 2012, 44, 3783–3788. doi:10.1055/s-0032-1317682 |
| 10. | Beňadiková, D.; Čurillová, J.; Lacek, T.; Rakovský, E.; Moncoľ, J.; Doháňošová, J.; Fischer, R. Tetrahedron 2014, 70, 5585–5593. doi:10.1016/j.tet.2014.06.093 |
| 41. | Gangula, S.; Kumar Kolla, N.; Elati, C.; Dongamanti, A.; Bandichhor, R. Synth. Commun. 2012, 42, 3344–3360. doi:10.1080/00397911.2011.582216 |
© 2020 Dikošová et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)