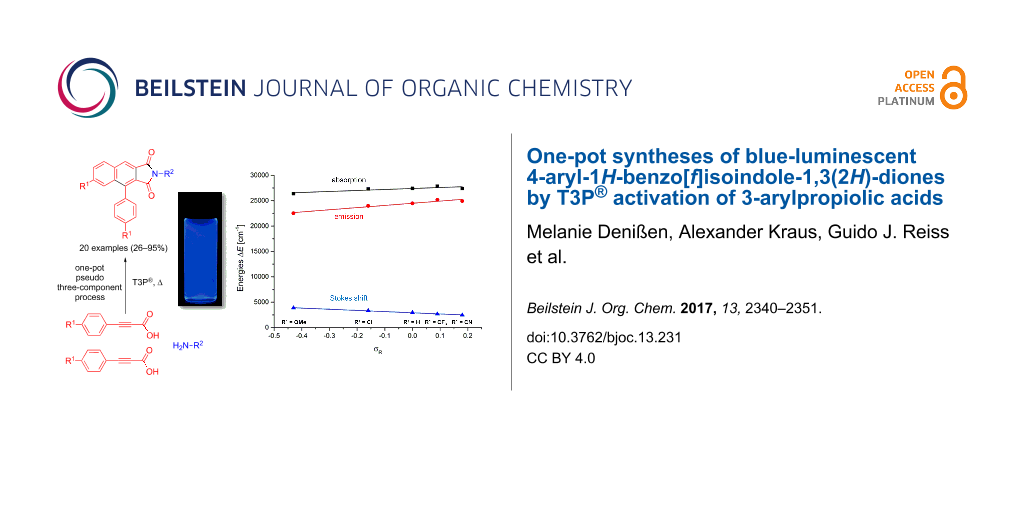
Abstract
In situ activation of 3-arylpropiolic acids with T3P® (n-propylphosphonic acid anhydride) initiates a domino reaction furnishing 4-arylnaphtho[2,3-c]furan-1,3-diones in excellent yields. Upon employing these anhydrides as reactive intermediates blue-luminescent 4-aryl-1H-benzo[f]isoindole-1,3(2H)-diones are formed by consecutive pseudo three-component syntheses in a one-pot fashion. The Stokes shifts correlate excellently with the Hammett–Taft σR parameter indicating an extended degree of resonance stabilization in the vibrationally relaxed excited singlet state.

Graphical Abstract
Introduction
Luminophores as functional π-electron systems [1] are crucial in modern illumination technologies, such as organic light-emitting diodes (OLEDs) [2-5]. As a consequence efficient and efficacious syntheses of fluorescent molecules can be most advantageously addressed by diversity-oriented syntheses [6-8], namely by multicomponent [9-21] and domino reactions [22-24] where fundamental organic reactions are combined in one-pot sequences [25-29]. Multicomponent reactions (MCR) take advantage of creating and transforming reactive functionalities in the same reaction vessel without intermediary work-up [30]. Syntheses of functional chromophores by MCR [31,32] have indeed become a powerful tool in synthetic chemistry for convergently approaching substance libraries of luminescent molecules. In particular, blue-emissive heterocyclic chromophores, intensively requested in illumination research, are equally accessible by MCR strategies [33].
Cyclic imides are often prepared by condensation of acid anhydrides and amines [34]. Their applications are widespread, ranging from pharmaceutically active compounds [35] to agrochemicals [36] and fluorophores [37]. The characteristic photophysical properties of 1,8- and 2,3-naphthalene imides render the substance class particularly attractive for the development of novel sensors and fluorescent dyes [38], for instance 6-chloro-2,3-naphthaleneimide derivatives were successfully used for labeling amino acids, and for studying peptide protein interactions [39]. Even the superficial attachment to a binding domain can be detected and monitored by absorption and emission spectroscopy. Furthermore, the investigation of a substance library of various 2,3- and 1,8-naphthalene imides has shown that the electronic nature of the ground and the excited state is decisively influenced by variation of the substitution pattern of the naphthalene scaffold. Even the smallest polarity change in the solvent system effects their absorption and emission behavior [34,40]. Very recent investigations on naphthaleneimide derivatives revealed enormous phosphorescence lifetimes that are particularly interesting for imaging, sensing and display applications [41].
4-Phenylnaphtho[2,3-c]furan-1,3-diones can well serve as reactive intermediates in multicomponent reactions, e.g., for synthesizing the corresponding imides. A particularly intriguing access to 4-phenylnaphtho[2,3-c]furan-1,3-diones is the intramolecular [4 + 2]-cycloaddition of phenylpropiolic acid anhydrides. Usually, this synthesis requires harsh reaction conditions starting from phenylpropiolic acids [42,43]. However, employing bis(2-oxooxazolidin-3-yl)phosphinic chloride as an activating agent represents a suitable access [44]. Alternative approaches include the use of phenylpropiolic acid chloride and phenylpropiolic acid as starting materials [45], and as well oxidative arene–alkyne cyclization with dichloro-5,6-dicyano-benzoquinone (DDQ) [46].
Based upon our experience in using propylphosphonic acid anhydride (T3P®) [47] as a condensation agent for in situ activation of benzyl alcohols in the synthesis of N-benzylphenothiazine derivatives [48], we reasoned that T3P® might be equally well suited for furnishing 4-phenylnaphtho[2,3-c]furan-1,3-diones, and thereby opening a straightforward entry to 4-aryl-1H-benzo[f]isoindole-1,3(2H)-diones in a diversity-oriented one-pot process. Here, we report the development of the one-pot synthesis of these title compounds by a consecutive pseudo three-component approach and the investigation of the luminescence behavior by absorption and emission spectroscopy.
Results and Discussion
Synthesis and structure of 4-arylnaphtho[2,3-c]furan-1,3-diones and 4-aryl-1H-benzo[f]isoindole-1,3(2H)-diones
Starting from phenylpropiolic acids 1 we first set out to optimize the reaction conditions for the domino synthesis of the 4-arylnaphtho[2,3-c]furan-1,3-diones 2a and 2b with T3P® as an activating agent, varying temperature and reaction time (Scheme 1, Table 1).
![[1860-5397-13-231-i1]](/bjoc/content/inline/1860-5397-13-231-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Mechanistic rationale and optimization of the domino synthesis of 4-arylnaphtho[2,3-c]furan-1,3-diones 2.
Scheme 1: Mechanistic rationale and optimization of the domino synthesis of 4-arylnaphtho[2,3-c]furan-1,3-dio...
Table 1: Optimization of the reaction conditions for the synthesis of 4-arylnaphtho[2,3-c]furan-1,3-diones 2.
| entry | arylpropiolic acid 1 | solvent | T [°C] | t [h] | 4-arylnaphtho[2,3-c]furan-1,3-dione 2 (isolated yield) [%] |
|---|---|---|---|---|---|
| 1 | R = H 1a | CH2Cl2 | 20 | 4 | 2a (81) |
| 2 | R = H 1a | CH2Cl2 | 20 | 20 | 2a (81) |
| 3 | R = H 1a | CH2Cl2 | 40 | 20 | 2a (21) |
| 4a | R = H 1a | CH2Cl2 | 20 | 20 | 2a (–) |
| 5 | R = OCH3 1b | CH2Cl2 | 20 | 20 | 2b (3) |
| 6 | R = OCH3 1b | THF | 20 | 20 | 2b (2) |
aWithout addition of T3P®.
T3P®, employed as a 50 wt % solution in 1,2-dichloroethane (DCE), is added to a solution of phenylpropiolic acid (1a) in dichloromethane at 0 °C over the course of 30 min. After warming the reaction mixture to room temperature, stirring for 4 h and aqueous work-up the desired product 2a can be obtained in good yield (Table 1, entry 1). A prolonged reaction time indicates that product 2a is stable under the reaction conditions (Table 1, entry 2). However, a temperature increase to 40 °C already causes decomposition as indicated by a diminished isolated yield (Table 1, entry 3). The addition of T3P® as an activation agent is a prerequisite for achieving the transformation (Table 1, entry 4).
With these conditions in hand, 3-(4-methoxyphenyl)propiolic acid (1b) was also chosen as a model substrate (Table 1, entries 5 and 6). The optimized reaction conditions for the synthesis of 4-phenylnaphtho[2,3-c]furan-1,3-dione (2a) cannot directly be transferred to give the desired product 2b, which was only obtained in traces. This is probably caused by the poor solubility of starting material 1b. This problem can be circumvented by the addition of triethylamine as a deprotonating solubilizer [44]. Under these modified conditions four substituted 4-arylnaphtho[2,3-c]furan-1,3-dione derivatives 2 can be synthesized in very good to quantitative yields (Scheme 2, Table 2).
![[1860-5397-13-231-i2]](/bjoc/content/inline/1860-5397-13-231-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Domino synthesis of 4-arylnaphtho[2,3-c]furan-1,3-diones 2 via in situ activation of arylpropiolic acids 1.
Scheme 2: Domino synthesis of 4-arylnaphtho[2,3-c]furan-1,3-diones 2 via in situ activation of arylpropiolic ...
Interestingly, in this domino synthesis of 4-phenylnaphtho[2,3-c]furan-1,3-diones electron-rich and electro-neutral substrates are equally well tolerated (Table 2, entries 1–3), while the electron-poor derivative 1d results in a slightly decreased yield of 88% due to the increased formation of side products (Table 2, entry 4). Expectedly, the products 2 are not stable under acidic aqueous conditions (e.g., hydrochloric acid, silica gel and saturated aqueous solution of ammonium chloride) [49]. After 48 h in the presence of deuterated hydrochloric acid (36% in D2O), complete conversion of compound 2a to the free deuterated dicarboxylic acid was observed (20% conversion after 60 min) by NMR spectroscopy. For verification of the structure by NMR and IR spectroscopy, the hydrolysis was performed on a preparative scale with ammonium chloride in a mixture of water and acetone. After complete removal of water, the IR spectrum shows the characteristic OH-stretching vibrations of the free carboxylic acid (![[Graphic 1]](/bjoc/content/inline/1860-5397-13-231-i8.svg?max-width=637&scale=1.18182) 2901–3082 cm−1). The signals at δ 12.70 and 12.98 in the 1H NMR spectrum can be assigned to the two carboxylic acid functionalities (see Supporting Information File 1). The extraction of the crude anhydrides with a saturated aqueous solution of sodium bicarbonate can be achieved uneventfully, leaving the anhydride unimpaired.
2901–3082 cm−1). The signals at δ 12.70 and 12.98 in the 1H NMR spectrum can be assigned to the two carboxylic acid functionalities (see Supporting Information File 1). The extraction of the crude anhydrides with a saturated aqueous solution of sodium bicarbonate can be achieved uneventfully, leaving the anhydride unimpaired.
For employing 4-phenylnaphtho[2,3-c]furan-1,3-diones 2 as reactive intermediates for the en route conversion with primary amines 3 into 4-aryl-1H-benzo[f]isoindole-1,3(2H)-diones 4 the reaction conditions were optimized with 4-phenylnaphtho[2,3-c]furan-1,3-dione (2a) and aniline (3a) as model substrates (Scheme 3, Table 3).
![[1860-5397-13-231-i3]](/bjoc/content/inline/1860-5397-13-231-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Optimization of the synthesis of 2,4-diphenyl-1H-benzo[f]isoindole-1,3(2H)-dione (4a) by imidation of 4-phenylnaphtho[2,3-c]furan-1,3-dione (2a) with aniline (3a).
Scheme 3: Optimization of the synthesis of 2,4-diphenyl-1H-benzo[f]isoindole-1,3(2H)-dione (4a) by imidation ...
Table 3: Optimization of the reaction conditions for the synthesis of 2,4-diphenyl-1H-benzo[f]isoindole-1,3(2H)-dione (4a).
| entry |
equivalents
of aniline (3a) |
solvent c(3a) = 0.125 M | T [°C] | 2,4-diphenyl-1H-benzo[f]isoindole-1,3(2H)-dione (4a) (isolated yield [%]) |
|---|---|---|---|---|
| 1 | 1.0 | CH2Cl2 | 50 | – |
| 2 | 1.0 | CH2Cl2/DMF 1:1 (v/v) | 80 | 15 |
| 3 | 1.0 | CH2Cl2/DMF 1:1 (v/v) | 115 | 47 |
| 4 | 1.0 | CH2Cl2/DMF 1:1 (v/v) | 160 | 33 |
| 5 | 2.0 | CH2Cl2/DMF 1:1 (v/v) | 160 | 71 |
| 6 | 2.0 | CH2Cl2/DMF 1:1 (v/v) | 115 | 80 |
In dichloromethane at only slightly elevated temperatures imide 4a is not formed (Table 3, entry 1). Upon addition of N,N-dimethylformamide as a cosolvent at 80 °C the desired product 4a can be isolated in 15% yield (Table 3, entry 2). The yield of 4a can be increased to 47% upon raising the reaction temperature to 115 °C (Table 3, entry 3), however, higher temperatures, such as 160 °C, cause a significant drop in yield. Finally, at 115 °C with two equivalents of aniline the highest yield can be achieved (Table 3, entry 6).
These imidation conditions are well-suited for concatenating the arylpropiolic anhydride formation, intramolecular cycloaddition and imidation in a one-pot fashion in the sense of a consecutive pseudo three-component synthesis of 4-aryl-1H-benzo[f]isoindole-1,3(2H)-diones 4, which was first probed with an electroneutral (1a), an electron-rich (1b) and electron-poor (1c) propiolic acid substrate (Scheme 4, Table 4). Although the electroneutral and electron-rich substrates give good to excellent yields in the sequence (Table 4, entries 1–3), even higher (Table 4, entry 1) than for a stepwise synthesis furnishing an overall yield of 4a of 80%, the cyano-substituted substrate 1c furnishes a significantly lower yield of 27% (Table 4, entry 4), originating from the lower reactivity in the anhydride formation.
![[1860-5397-13-231-i4]](/bjoc/content/inline/1860-5397-13-231-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Pseudo three-component synthesis of 4-aryl-1H-benzo[f]isoindole-1,3(2H)-diones 4.
Scheme 4: Pseudo three-component synthesis of 4-aryl-1H-benzo[f]isoindole-1,3(2H)-diones 4.
Table 4: Pseudo three-component synthesis of 1H-benzo[f]isoindole-1,3(2H)-diones 4.
| entry | arylpropiolic acid 1 | amine 3 | 4-aryl-1H-benzo[f]isoindole-1,3(2H)-dione 4 (isolated yield [%]) |
|---|---|---|---|
| 1 | R1 = H (1a) | R2 = C6H5 (3a) | 4a (95) |
| 2 | R1 = OCH3 (1b) | 3a | 4b (79) |
| 3a | 1a |
R2 = CH2(10-methyl-10H-
phenothiazine-3-yl)∙HCl (3b) |
4c (85) |
| 4 | R1 = CN (1c) | 3a | 4d (27) |
aCompound 3b was employed as the corresponding hydrochloride with 202 mg (2.00 mmol) Et3N in CH2Cl2 (1.0 mL) at rt for 1 h.
Therefore, for accessing acceptor-substituted derivatives of 4, an intermediate extraction with bicarbonate after the anhydride formation–cycloaddition was attempted (Scheme 5). With this variation acceptor-substituted arylpropiolic acids 1c–e can be transformed to the corresponding 1H-benzo[f]isoindole-1,3(2H)-diones 4d–f in moderate yields.
![[1860-5397-13-231-i5]](/bjoc/content/inline/1860-5397-13-231-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Modified sequence for the synthesis of acceptor-substituted 4-aryl-1H-benzo[f]isoindole-1,3(2H)-diones 4.
Scheme 5: Modified sequence for the synthesis of acceptor-substituted 4-aryl-1H-benzo[f]isoindole-1,3(2H)-dio...
Upon lowering the amount of T3P® to 0.5 equiv, electron-neutral and electron-donating phenylpropiolic acids 1 can be employed with comparable efficiency applying various amines 3 and giving rise to a series of 4-aryl-1H-benzo[f]isoindole-1,3(2H)-diones 4 in moderate to good yield (Table 5). Variation of the amine nucleophile 3 allows for the introduction of functional groups for consecutive transformations. For instance, para-substituted halogenated anilines 3 (Table 5, entries 3–5) and ethyl 4-aminobenzoate (Table 5, entry 6) furnish the desired products in 51–65% yield. Interestingly, also sterically hindered anilines, such as 2,6-dimethylaniline can be obtained in 56% yield (Table 5, entry 8). In addition, aliphatic amines (Table 5, entries 10–13) are readily tolerated in the established reaction sequence.
Table 5: Variation of the amine 3 in pseudo three-component syntheses of 1H-benzo[f]isoindole-1,3(2H)-diones 4.
| entry | arylpropiolic acid 1 | amine 3 | 1H-benzo[f]isoindole-1,3(2H)-dione 4 (isolated yield [%]) |
|---|---|---|---|
| 1 | 1a | R2 = C6H5 (3a) | 4a (92) |
| 2 | R1 = CH3 (1f) | 3a | 4g (48) |
| 3 | R1 = 1a | R2 = p-FC6H4 (3c) | 4h (55) |
| 4 | 1a | R2 = p-ClC6H4 (3d) | 4i (65) |
| 5 | 1a | R2 = p-IC6H4 (3e) | 4j (51) |
| 6 | 1a | R2 = p-EtO2CC6H4 (3f) | 4k (57) |
| 7 | 1a | R2 = 3,5-Me2C6H3 (3g) | 4l (69) |
| 8 | 1a | R2 = 2,6-Me2C6H3 (3h) | 4m (56) |
| 9 | 1a | R2 = 3,5-(MeO)2C6H3 (3i) | 4n (64) |
| 10 | 1a | R2 = CH2Ph (3j) | 4o (41) |
| 11a | 1a | R2 = CH2CCH (3k) | 4p (26) |
| 12 | 1a | R2 = n-hexyl (3l) | 4q (53) |
| 13 | 1a | R2 = n-butyl (3m) | 4r (61) |
| 14 | 1b | 3h | 4s (57) |
Besides comprehensive NMR spectroscopic and mass spectrometric characterization the structures of the title compounds 4 were additionally corroborated by a crystal structure determination of compound 4b (Figure 1). The twist angle of the phenyl substituent (ring A) and the 1H-pyrrole-2,5-dionyl moiety is 51.37(7)°, whereas the p-anisyl substituent (ring B) is considerably twisted against the adjacent six-membered ring by 70.95(7)° (Figure 1) [50].
![[1860-5397-13-231-1]](/bjoc/content/figures/1860-5397-13-231-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: The ORTEP-style plot of crystal structure 4b (ellipsoids are draw at the 40% probability level).
Figure 1: The ORTEP-style plot of crystal structure 4b (ellipsoids are draw at the 40% probability level).
Upon slightly varying the reaction conditions the potential of the sequence can be extended. An increase of the T3P® concentration to 2.0 equiv leads to the regioselective formation of (E)-2,9-diphenyl-3-(phenylimino)-2,3-dihydro-1H-benzo[f]isoindol-1-one (5) in 15% yield in the sense of a pseudo four-component reaction (Scheme 6).
![[1860-5397-13-231-i6]](/bjoc/content/inline/1860-5397-13-231-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Pseudo four-component synthesis of (E)-2,9-diphenyl-3-(phenylimino)-2,3-dihydro-1H-benzo[f]isoindol-1-one (5).
Scheme 6: Pseudo four-component synthesis of (E)-2,9-diphenyl-3-(phenylimino)-2,3-dihydro-1H-benzo[f]isoindol...
The increased amount of the condensation agent T3P® obviously enables further activation of the initially formed 1H-benzo[f]isoindole-1,3(2H)-dione by electrophilic attack on the sterically easier accessible carbonyl group. As a consequence the imine condensation proceeds also with thermodynamic control giving exclusively the formation of the E-configured product 5. Interestingly the corresponding reaction with ortho-phenylenediamine (3n) gives rise to the regioselective formation of the pentacyclic condensation product 6, where the intramolecular imine formation formally occurred at the sterically more biased carbonyl group (Scheme 7).
![[1860-5397-13-231-i7]](/bjoc/content/inline/1860-5397-13-231-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Synthesis of 6-phenyl-12H-benzo[f]benzo[4,5]imidazo[2,1-a]isoindol-12-one (6).
Scheme 7: Synthesis of 6-phenyl-12H-benzo[f]benzo[4,5]imidazo[2,1-a]isoindol-12-one (6).
In addition to NMR spectroscopic and mass spectrometric characterization the crystal structures of the imine condensation products 5 and 6 were determined (Figure 2 and Figure 3) [50]. In similarity to 4b the twist angles of the phenyl substituents (rings A and B) are 61.1(1)° and 66.8(1)°, respectively (Figure 2, left part). Interestingly, the crystal structure of 5 shows centrosymmetric dimers, formed by a close π–π interaction of the planar naphthyl moieties (Figure 2, right part). The intermolecular distance of the naphthalene moieties of the two molecules accounts to 3.435(7) Å.
![[1860-5397-13-231-2]](/bjoc/content/figures/1860-5397-13-231-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: The ORTEP-type plot of the crystal structure 5 (left) and a centrosymmetric dimer formation by π–π interactions (right) (ellipsoids are drawn at the 40% probability level).
Figure 2: The ORTEP-type plot of the crystal structure 5 (left) and a centrosymmetric dimer formation by π–π ...
![[1860-5397-13-231-3]](/bjoc/content/figures/1860-5397-13-231-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: The ORTEP-type plot of the asymmetric unit of the crystal structure 6 (top) and π-stacking interactions (bottom) (ellipsoids are drawn at the 40% probability level).
Figure 3: The ORTEP-type plot of the asymmetric unit of the crystal structure 6 (top) and π-stacking interact...
This intermolecular plane distance of the two adjacent naphthyl moieties (C2–C11) must be understood as a π–π interaction of two fused aromatic systems [51]. The twist angle of the attached phenyl substituent (ring B) of the asymmetric unit of the crystal structure 6 were determined as 62.28(5)° (Figure 3). The significant difference of the twist angles in 4b, 5, and 6 could be a consequence of packing in the individual crystal structure. The crystal structure of the pentacyclic molecules 6 shows stacks with an antiparallel arrangement of the monomers. The found interplanar distance is 3.437(4) Å. Consequently, π–π interactions must be considered for this structure [51,52]. Bond lengths and angles of the reported crystal structures 4b, 5, and 6 are in the expected range. The tricyclic 1H-benzo[f]isoindole-1,3(2H)-dionyl moiety in 4b, 5 and the corresponding 3-imino-1-oxo-2,3-dihydro-1H-benzo[f]isoindolyl moiety in 6 are absolutely planar.
Photophysical properties
The pseudo three-component synthesis of 1H-benzo[f]isoindole-1,3(2H)-diones 4 furnishes a substance library with electronically diverse substitution patterns and already upon eyesight several derivatives are intensively blue and greenish luminescent in solution at low concentration (Figure 4). Therefore, the 1H-benzo[f]isoindole-1,3(2H)-diones 4 and 5 and the pentacyclic compound 6 were investigated with absorption and static fluorescence spectroscopy (Table 6).
![[1860-5397-13-231-4]](/bjoc/content/figures/1860-5397-13-231-4.jpg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Emission properties of compounds 4a,b,d–f, 5, and 6 under handheld UV-lamp (λexc ≈ 350 nm).
Figure 4: Emission properties of compounds 4a,b,d–f, 5, and 6 under handheld UV-lamp (λexc ≈ 350 nm).
Table 6:
Selected photophysical properties (absorption and emission maxima,a,b fluorescence quantum yields (Φf [53]),c and Stokes shifts Δ![[Graphic 4]](/bjoc/content/inline/1860-5397-13-231-i11.svg?max-width=637&scale=1.18182) d) of compounds 4a–g,m,n,s, 5, and 6.
d) of compounds 4a–g,m,n,s, 5, and 6.
| entry | compound | λmax,abs (ε) [nm] ([L∙(mol∙cm)−1]) | λmax,em [nm] (Φf) |
Stokes shift Δ![[Graphic 3]](/bjoc/content/inline/1860-5397-13-231-i10.svg?max-width=637&scale=1.18182) [cm−1] [cm−1]
|
|---|---|---|---|---|
| 1 | 4a | 364.5 (3900), 264.0 (49500) | 468.0 sh, 408.5 (0.01) | 3000 |
| 2 | 4b | 379.0 (4000), 273.5 (55300) | 444.0 (0.14) | 3900 |
| 3 | 4c | 363.0 (5080), 300.5 (13510), 262.5 (77840) | 525.0 sh, 450.0 | 5300 |
| 4 | 4d | 365.0 (3600), 266.5 (57300) | 512.5, 401.0 sh | 2500 |
| 5 | 4e | 366.0 (3000), 268.5 (52600) | 499.5, 417.0 sh | 3300 |
| 6 | 4f | 359.0 (3100), 343.0 (2600), 258.5 (50700) | 500.0, 397.0 sh | 2700 |
| 7 | 4g | 369.5 (4200), 270.5 (53000) | 415.0 | 3000 |
| 8 | 4m | 364.0 (5400), 263.5 (58000) | 391.0 | 2000 |
| 9 | 4n | 364.0 (5600), 264.5 (67000) | 520.0 | 8200 |
| 10 | 4s | 371.5 (4700), 271.5 (56300) | 437.0 (0.17) | 4000 |
| 11 | 5 | 362.5 (5900), 350.0 (4800) 265.5 (52900), 261.0 (52500) | – | – |
| 12 | 6 | 388.5 sh (21600), 360.0 (54900) 296.0 (53000), 255.5 (21500) | 500.0 (0.10)d | 5700 |
aRecorded in CH2Cl2 UVASOL® at T = 293 K. bRecorded in CH2Cl2 UVASOL® at T = 293 K with λexc = 360.0/380.0 nm. cQuantum yields Φf were determined with coumarine 1 in ethanol as a standard, Φf = 0.73, at T = 293 K with λexc = 360.0 nm. dΔ = 1/λmax,abs – 1/λmax,em [cm−1]. eQuantum yields Φf were determined with coumarine 30 in acetonitrile as a standard, Φf = 0.67, at T = 293 K with λexc = 380.0 nm.
All 1-aryl-2,3-naphthaleneimides 4 possess two characteristic absorption maxima λmax,abs between 258.5 and 273.5 nm with molar extinction coefficients ε of 55000 L∙mol−1∙cm−1 and between 359.0 and 379.0 nm with molar extinction coefficients ε of about 3500 L∙mol−1∙cm−1. While electron-withdrawing substituents R1 in tendency shift the absorption maxima slightly hypsochromically (Table 6, entries 1, and 3–6) the electron-donating methoxy group (Table 6, entry 2) clearly causes a red shift. The absorption spectra of the imine condensation products 5 and 6 essentially display a similar appearance; however, the pentacycle 6 possesses a considerably more intense longest wavelength absorption, which appears in the spectrum at 388.5 nm as a shoulder with a molar extinction coefficients ε of 21600 L∙mol−1∙cm−1 (Table 6, entry 12).
Although most of the 1H-benzo[f]isoindole-1,3(2H)-diones 4 fluorescence upon excitation with UV light, a closer inspection, by comparing the relative intensities of the emission maxima at identical concentrations, reveals that only the methoxy derivative 4b (Table 6, entry 2) is substantially fluorescent (Figure 5). With this exception all qualitatively determined emission spectra reveal a broad unstructured maximum followed by a shoulder. This appearance might result from the free rotation of the N-phenyl substituents, furnishing emissive conformers that arise from a coplanar (λmax,em at 468.0 nm as a shoulder for compound 4a) and torsional orientation (λmax,em at 408.5 nm as a maximum for compound 4a) of the N-phenyl substituent in the corresponding excited Franck–Condon states [54]. Qualitatively, also a red shift of the emission maximum can be detected upon increasing the electron-withdrawing character of the substituent R1. In the consanguineous series of 1H-benzo[f]isoindole-1,3(2H)-diones 4a,b,d–f the emission maxima λmax,em are found in a range from 408.5 to 512.5 nm with Stokes shifts lying between 3900 and 2500 cm−1 (Table 6, entries 1, 2 and 4–6, Figure 6).
![[1860-5397-13-231-5]](/bjoc/content/figures/1860-5397-13-231-5.png?scale=2.5&max-width=1024&background=FFFFFF)
Figure 5: Relative emission intensities of compounds 4a,b,d–f (recorded in CH2Cl2 UVASOL® at T = 293 K; λexc = 350 nm.
Figure 5: Relative emission intensities of compounds 4a,b,d–f (recorded in CH2Cl2 UVASOL® at T = 293 K; λexc ...
![[1860-5397-13-231-6]](/bjoc/content/figures/1860-5397-13-231-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Absorption and emission properties of selected imides 4 measured in CH2Cl2 UVASOL® at 293 K with λexc = 360 nm.
Figure 6: Absorption and emission properties of selected imides 4 measured in CH2Cl2 UVASOL® at 293 K with λe...
This electronic substituent effect of R1 on the Stokes shifts was further corroborated by linear structure–property relationships based upon Hammett–Taft correlations with the consanguineous series 4a,b,d–f. Correlation studies of the longest wavelength absorption maxima λmax,abs, the shortest wavelength emission maxima λmax,em, and the Stokes shifts Δ![[Graphic 2]](/bjoc/content/inline/1860-5397-13-231-i9.svg?max-width=637&scale=1.18182) with the Hammett–Taft parameters σp, σR, σp+, and σp− [55] disclose an interesting insight on electronic substituent effects in the electronic ground and excited states (see Table S6 in Supporting Information File 1). Although the linear correlations of λmax,abs with all σ parameters are relatively poor, the correlations of λmax,em with σR and σp+ indicate a strong influence of resonance stabilization in the vibrationally relaxed excited state. This is even more the case in the nearly perfect linear correlation of the Stokes shift with σR (r2 = 0.989) and can be interpreted as a significant structural change upon photonic excitation and excited state relaxation resulting from a considerable charge transfer character, as supported by the influence of the remote polar substitution (Figure 7).
with the Hammett–Taft parameters σp, σR, σp+, and σp− [55] disclose an interesting insight on electronic substituent effects in the electronic ground and excited states (see Table S6 in Supporting Information File 1). Although the linear correlations of λmax,abs with all σ parameters are relatively poor, the correlations of λmax,em with σR and σp+ indicate a strong influence of resonance stabilization in the vibrationally relaxed excited state. This is even more the case in the nearly perfect linear correlation of the Stokes shift with σR (r2 = 0.989) and can be interpreted as a significant structural change upon photonic excitation and excited state relaxation resulting from a considerable charge transfer character, as supported by the influence of the remote polar substitution (Figure 7).
![[1860-5397-13-231-7]](/bjoc/content/figures/1860-5397-13-231-7.png?scale=2.5&max-width=1024&background=FFFFFF)
Figure 7: Hammett–Taft correlations of the emission maxima (red circles, lmax,em = 4274 · sR + 24495 [cm−1], R2 = 0.925) and Stokes shifts (blue triangles, Δ = −2319 · sR + 2909 [cm−1], R2 = 0.989) of compounds 4a,b,d–f with sR (black squares are the corresponding absorption maxima).
Figure 7: Hammett–Taft correlations of the emission maxima (red circles, lmax,em = 4274 · sR + 24495 [cm−1], R...
Interestingly, compound 4a, which has a fluorescence quantum yield Φf of less than 0.01 in dichloromethane solution, experiences an over eightfold increase to 0.08 in the solid state emission as determined from the powder by an integrating sphere. The emission maximum appears at 468.5 nm, i.e., at the same longest wavelength band as in solution. Therefore, 1H-benzo[f]isoindole-1,3(2H)-diones 4 can be considered as AIE (aggregation induced emission) chromophores [56-58].
In comparison to the 1-phenyl-[2,3-c]-naphthaleneimide 5, which is only weakly luminescent, the pentacycle 6 displays a relative enhancement of the greenish emission at 500 nm by a factor of 340 (relative to compound 4a, Figure 8) and can be quantified with a fluorescence quantum yield of 0.10 (Table 6, entry 12).
![[1860-5397-13-231-8]](/bjoc/content/figures/1860-5397-13-231-8.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: Relative emission intensities of the 1-phenyl-2,3-naphthaleneimide 4a (blue) and the pentacyclus 6 (green, normalized on the absorptivity at the excitation wavelength λexc = 350 nm).
Figure 8: Relative emission intensities of the 1-phenyl-2,3-naphthaleneimide 4a (blue) and the pentacyclus 6 ...
Conclusion
3-Arylpropiolic acids can be readily activated with T3P® (n-propylphosphonic acid anhydride) to initiate a domino reaction furnishing 4-arylnaphtho[2,3-c]furan-1,3-diones in excellent yields. These anhydrides can be considered as reactive intermediates for a subsequent imidation with primary amines and, therefore, a one-pot reaction in the sense of a consecutive pseudo three-component process evolved. The resulting 4-aryl-1H-benzo[f]isoindole-1,3(2H)-diones are interestingly blue to greenish-blue emissive upon excitation of the longest wavelength absorption bands. The photophysical characterization by absorption and emission spectroscopy revealed that the Stokes shifts are excellently correlated with Hammett–Taft's σR parameters indicating an extended degree of resonance stabilization as a result of a charge transfer character in the vibrationally relaxed S1-state. The fluorescence can be redshifted by employing 1,2-phenylenediamine as a reaction partner in the terminal step of the sequence furnishing a rigidified planar pentacyclic condensation product. The interesting emission properties and the straightforward diversity-oriented synthetic approach are therefore well-suited for the design of covalently ligated, conjugated and non-conjugated bichromophores in a rapid fashion. Studies directed towards the one-pot synthesis of more complex polycyclic emitters are currently underway.
Supporting Information
The Supporting Information contains all experimental procedures, spectroscopic and analytical data as well as copies of NMR spectra of compounds 2, 4, 5, and 6. X-ray structure analyses of compounds 4b, 5, and 6, and Hammett–Taft correlations of compounds 4a,b,d–f are also given.
| Supporting Information File 1: Experimental part. | ||
| Format: PDF | Size: 3.3 MB | Download |
Acknowledgements
The authors cordially thank the Fonds der Chemischen Industrie and the Deutsche Forschungsgemeinschaft (Mu 1088/9-1) for financial support. The work primarily originates from the doctoral thesis of Melanie Denißen (inaugural dissertation, University of Düsseldorf, 2015) and the master thesis of Alexander Kraus (master thesis, University of Düsseldorf, 2015), who are both first and second authors of this paper.
References
-
Müller, T. J. J.; Bunz, U. H. F., Eds. Functional Organic Materials. Syntheses, Strategies, and Applications; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2007. doi:10.1002/9783527610266
Return to citation in text: [1] -
Li, Z. R., Ed. Organic Light-Emitting Materials and Devices, 2nd ed.; CRC Press: Boca Raton, 2015. doi:10.1201/b18540
Return to citation in text: [1] -
Thejo Kalayani, N.; Dhoble, S. J. Renewable Sustainable Energy Rev. 2012, 16, 2696–2723. doi:10.1016/j.rser.2012.02.021
Return to citation in text: [1] -
Müllen, K.; Scherf, U., Eds. Organic Light-Emitting Diodes: Synthesis, Properties, and Applications; Wiley-VCH: Weinheim, Germany, 2006. doi:10.1002/3527607986
Return to citation in text: [1] -
Park, J.-S.; Chae, H.; Chung, H. K.; Lee, S. I. Semicond. Sci. Technol. 2011, 26, 034001–034008. doi:10.1088/0268-1242/26/3/034001
See for a review on flexible AM-OLED.
Return to citation in text: [1] -
Briehn, C. A.; Bäuerle, P. Chem. Commun. 2002, 1015–1023. doi:10.1039/B108846G
Return to citation in text: [1] -
Müller, T. J. J.; D’Souza, D. M. Pure Appl. Chem. 2008, 80, 609–620. doi:10.1351/pac200880030609
Return to citation in text: [1] -
Müller, T. J. J. Diversity-oriented Synthesis of Chromophores by Combinatorial Strategies and Multi-component Reactions. In Functional Organic Materials. Syntheses, Strategies, and Applications; Müller, T. J. J.; Bunz, U. H. F., Eds.; Wiley-VCH: Weinheim, 2007; pp 179–223. doi:10.1002/9783527610266.ch5
Return to citation in text: [1] -
Zhu, J.; Wang, Q.; Wang, M.-X. Multicomponent Reactions in Organic Synthesis; Wiley-VHC: Weinheim, Germany, 2015. doi:10.1002/9783527678174
Return to citation in text: [1] -
Müller, T. J. J. Multicomponent Reactions; Science of Synthesis Series; Georg Thieme Verlag KG: Stuttgart, 2014.
Return to citation in text: [1] -
Sunderhaus, J. D.; Martin, S. F. Chem. – Eur. J. 2009, 15, 1300–1308. doi:10.1002/chem.200802140
Return to citation in text: [1] -
Isambert, N.; Lavilla, R. Chem. – Eur. J. 2008, 14, 8444–8454. doi:10.1002/chem.200800473
Return to citation in text: [1] -
Dömling, A. Chem. Rev. 2006, 106, 17–89. doi:10.1021/cr0505728
Return to citation in text: [1] -
Zhu, J.; Bienaymé, H., Eds. Multicomponent Reactions; Wiley-VHC: Weinheim, Germany, 2005. doi:10.1002/3527605118
Return to citation in text: [1] -
Orru, R. V. A.; de Greef, M. Synthesis 2003, 1471–1499. doi:10.1055/s-2003-40507
Return to citation in text: [1] -
Bienaymé, H.; Hulme, C.; Oddon, G.; Schmitt, P. Chem. – Eur. J. 2000, 6, 3321–3329. doi:10.1002/1521-3765(20000915)6:18<3321::AID-CHEM3321>3.0.CO;2-A
Return to citation in text: [1] -
Dömling, A.; Ugi, I. Angew. Chem., Int. Ed. 2000, 39, 3168–3210. doi:10.1002/1521-3773(20000915)39:18<3168::AID-ANIE3168>3.0.CO;2-U
Return to citation in text: [1] -
Ugi, I.; Dömling, A.; Werner, B. J. Heterocycl. Chem. 2000, 37, 647–658. doi:10.1002/jhet.5570370322
Return to citation in text: [1] -
Weber, L.; Illgen, K.; Almstetter, M. Synlett 1999, 366–374. doi:10.1055/s-1999-2612
Return to citation in text: [1] -
Armstrong, R. W.; Combs, A. P.; Tempest, P. A.; Brown, S. D.; Keating, T. A. Acc. Chem. Res. 1996, 29, 123–131. doi:10.1021/ar9502083
Return to citation in text: [1] -
Ugi, I.; Dömling, A.; Hörl, W. Endeavour 1994, 18, 115–122. doi:10.1016/S0160-9327(05)80086-9
Return to citation in text: [1] -
Tietze, L. F.; Brasche, G.; Gericke, K. M. Domino Reactions in Organic Synthesis; Wiley-VCH: Weinheim, Germany, 2006. doi:10.1002/9783527609925
Return to citation in text: [1] -
Tietze, L. F., Ed. Domino Reactions: Concepts for Efficient Organic Synthesis; Wiley-VCH: Weinheim, Germany, 2014. doi:10.1002/9783527671304
Return to citation in text: [1] -
Tietze, L. F. J. Heterocycl. Chem. 1990, 27, 47–69. doi:10.1002/jhet.5570270105
Return to citation in text: [1] -
Posner, G. H. Chem. Rev. 1986, 86, 831–844. doi:10.1021/cr00075a007
Return to citation in text: [1] -
Tietze, L. F.; Beifuss, U. Angew. Chem., Int. Ed. Engl. 1993, 32, 131–163. doi:10.1002/anie.199301313
Return to citation in text: [1] -
Tietze, L. F. Chem. Rev. 1996, 96, 115–136. doi:10.1021/cr950027e
Return to citation in text: [1] -
Ganem, B. Acc. Chem. Res. 2009, 42, 463–472. doi:10.1021/ar800214s
Return to citation in text: [1] -
Brauch, S.; van Berkel, S. S.; Westermann, B. Chem. Soc. Rev. 2013, 42, 4948–4962. doi:10.1039/C3CS35505E
Return to citation in text: [1] -
Müller, T. J. J. General Discussion and Reactions Involving a Carbonyl Compound as Electrophilic Component. In Multicomponent Reactions 1; Müller, T. J. J., Ed.; Science of Synthesis Series; Georg Thieme Verlag KG: Stuttgart, 2014; pp 5–27.
Return to citation in text: [1] -
Levi, L.; Müller, T. J. J. Chem. Soc. Rev. 2016, 45, 2825–2846. doi:10.1039/C5CS00805K
Return to citation in text: [1] -
Levi, L.; Müller, T. J. J. Eur. J. Org. Chem. 2016, 2907–2918. doi:10.1002/ejoc.201600409
Return to citation in text: [1] -
Riva, R.; Moni, L.; Müller, T. J. J. Targets Heterocycl. Syst. 2016, 20, 85–112.
Return to citation in text: [1] -
Baathulaa, K.; Xu, Y.; Quian, X. J. Photochem. Photobiol., A 2010, 216, 24–34. doi:10.1016/j.jphotochem.2010.09.002
Return to citation in text: [1] [2] -
Miyachi, H.; Azuma, A.; Ogasawara, A.; Uchimura, E.; Watabe, N.; Kobayashi, Y.; Kato, F.; Kato, M.; Washimoto, Y. J. Med. Chem. 1997, 40, 2858–2865. doi:10.1021/jm970109q
Return to citation in text: [1] -
Hargeaves, M. K.; Pritchard, J. G.; Dave, H. R. Chem. Rev. 1970, 70, 439–469. doi:10.1021/cr60266a001
Return to citation in text: [1] -
Banerjee, S.; Veale, E. B.; Phelan, C. M.; Murphy, S. A.; Tocci, G. M.; Gillespie, L. J.; Frimannsson, D. O.; Kelly, J. M.; Gunnlaugsson, T. Chem. Soc. Rev. 2013, 42, 1601–1618. doi:10.1039/C2CS35467E
Return to citation in text: [1] -
Katritzky, A. R.; Ozcan, S.; Todadze, E. Org. Biomol. Chem. 2010, 8, 1296–1300. doi:10.1039/c000684j
Return to citation in text: [1] -
Vázquez, M. E.; Blanco, J. B.; Imperiali, B. J. Am. Chem. Soc. 2005, 127, 1300–1306. doi:10.1021/ja0449168
Return to citation in text: [1] -
Alexiou, M. S.; Tychopoubs, V.; Ghorbanian, S.; Tyman, J. H. P.; Brown, R. G.; Brittain, P. I. J. Chem. Soc., Perkin Trans. 2 1990, 5, 837–842. doi:10.1039/P29900000837
Return to citation in text: [1] -
Chen, X.; Xu, C.; Wang, T.; Zhou, C.; Du, J.; Wang, Z.; Xu, H.; Xie, T.; Bi, G.; Jiang, J.; Zhang, X.; Demas, J. N.; Trindle, C. O.; Luo, Y.; Zhang, G. Angew. Chem., Int. Ed. 2016, 55, 9872–9875. doi:10.1002/anie.201601252
Return to citation in text: [1] -
Michael, A.; Bucher, J. E. Ber. Dtsch. Chem. Ges. 1908, 41, 70–73. doi:10.1002/cber.19080410119
Return to citation in text: [1] -
Lanser, T. Ber. Dtsch. Chem. Ges. 1899, 32, 2477–2478. doi:10.1002/cber.189903202194
Return to citation in text: [1] -
Cabré-Castellvi, J.; Palomo-Coll, A.; Palomo-Coll, A. L. Synthesis 1981, 616–620. doi:10.1055/s-1981-29544
Return to citation in text: [1] [2] -
Ruhemann, S.; Merriman, R. W. J. Chem. Soc., Trans. 1905, 87, 1383–1395. doi:10.1039/CT9058701383
Return to citation in text: [1] -
Kim, K. H.; Lim, C. H.; Lim, J. W.; Kim, J. N. Adv. Synth. Catal. 2014, 356, 697–704. doi:10.1002/adsc.201301169
Return to citation in text: [1] -
Wissmann, H.; Kleiner, H.-J. Angew. Chem., Int. Ed. 1980, 19, 133–134. doi:10.1002/anie.198001331
Return to citation in text: [1] -
Levi, L.; Scheuren, S.; Müller, T. J. J. Synthesis 2014, 46, 3059–3066. doi:10.1055/s-0034-1378541
Return to citation in text: [1] -
Barros, T. C.; Yunes, S.; Menegon, G.; Nome, F.; Chaimovich, H.; Politi, M. J.; Dias, L. G.; Cuccovia, I. M. J. Chem. Soc., Perkin Trans. 2 2001, 2342–2350. doi:10.1039/B104148G
Return to citation in text: [1] -
Deposited with the Cambridge Crystallographic Data Centre as supplementary publication nos. CCDC 1563985 (4b), 1563988 (5), and 1564617 (6).
Return to citation in text: [1] [2] -
Grimme, S. Angew. Chem., Int. Ed. 2008, 47, 3430–3434. doi:10.1002/anie.200705157
Return to citation in text: [1] [2] -
The dashed lines in Figure 3, right part are shown to support a better unterstanding of the geometric orientation of the molecules 6 in its crystal structure. They do not represent descrete bonds.
Return to citation in text: [1] -
Fery-Forgues, S.; Lavabre, D. J. Chem. Educ. 1999, 76, 1260–1264. doi:10.1021/ed076p1260
Return to citation in text: [1] -
Demeter, A.; Bérces, T.; Biczók, L.; Wintgens, V.; Valat, P.; Kossanyi, J. J. Phys. Chem. 1996, 100, 2001–2011. doi:10.1021/jp951133n
Return to citation in text: [1] -
Hansch, C.; Leo, A.; Taft, R. W. Chem. Rev. 1991, 91, 165–195. doi:10.1021/cr00002a004
Return to citation in text: [1] -
Hong, Y.; Lam, J. W. Y.; Tang, B. Z. Chem. Commun. 2009, 4332–4353. doi:10.1039/B904665H
Return to citation in text: [1] -
Hong, Y.; Lam, J. W. Y.; Tang, B. Z. Chem. Soc. Rev. 2011, 40, 5361–5388. doi:10.1039/C1CS15113D
Return to citation in text: [1] -
Hu, R.; Leung, N. L. C.; Tang, B. Z. Chem. Soc. Rev. 2014, 43, 4494–4562. doi:10.1039/C4CS00044G
Return to citation in text: [1]
| 51. | Grimme, S. Angew. Chem., Int. Ed. 2008, 47, 3430–3434. doi:10.1002/anie.200705157 |
| 51. | Grimme, S. Angew. Chem., Int. Ed. 2008, 47, 3430–3434. doi:10.1002/anie.200705157 |
| 52. | The dashed lines in Figure 3, right part are shown to support a better unterstanding of the geometric orientation of the molecules 6 in its crystal structure. They do not represent descrete bonds. |
| 53. | Fery-Forgues, S.; Lavabre, D. J. Chem. Educ. 1999, 76, 1260–1264. doi:10.1021/ed076p1260 |
| 1. | Müller, T. J. J.; Bunz, U. H. F., Eds. Functional Organic Materials. Syntheses, Strategies, and Applications; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2007. doi:10.1002/9783527610266 |
| 22. | Tietze, L. F.; Brasche, G.; Gericke, K. M. Domino Reactions in Organic Synthesis; Wiley-VCH: Weinheim, Germany, 2006. doi:10.1002/9783527609925 |
| 23. | Tietze, L. F., Ed. Domino Reactions: Concepts for Efficient Organic Synthesis; Wiley-VCH: Weinheim, Germany, 2014. doi:10.1002/9783527671304 |
| 24. | Tietze, L. F. J. Heterocycl. Chem. 1990, 27, 47–69. doi:10.1002/jhet.5570270105 |
| 39. | Vázquez, M. E.; Blanco, J. B.; Imperiali, B. J. Am. Chem. Soc. 2005, 127, 1300–1306. doi:10.1021/ja0449168 |
| 9. | Zhu, J.; Wang, Q.; Wang, M.-X. Multicomponent Reactions in Organic Synthesis; Wiley-VHC: Weinheim, Germany, 2015. doi:10.1002/9783527678174 |
| 10. | Müller, T. J. J. Multicomponent Reactions; Science of Synthesis Series; Georg Thieme Verlag KG: Stuttgart, 2014. |
| 11. | Sunderhaus, J. D.; Martin, S. F. Chem. – Eur. J. 2009, 15, 1300–1308. doi:10.1002/chem.200802140 |
| 12. | Isambert, N.; Lavilla, R. Chem. – Eur. J. 2008, 14, 8444–8454. doi:10.1002/chem.200800473 |
| 13. | Dömling, A. Chem. Rev. 2006, 106, 17–89. doi:10.1021/cr0505728 |
| 14. | Zhu, J.; Bienaymé, H., Eds. Multicomponent Reactions; Wiley-VHC: Weinheim, Germany, 2005. doi:10.1002/3527605118 |
| 15. | Orru, R. V. A.; de Greef, M. Synthesis 2003, 1471–1499. doi:10.1055/s-2003-40507 |
| 16. | Bienaymé, H.; Hulme, C.; Oddon, G.; Schmitt, P. Chem. – Eur. J. 2000, 6, 3321–3329. doi:10.1002/1521-3765(20000915)6:18<3321::AID-CHEM3321>3.0.CO;2-A |
| 17. | Dömling, A.; Ugi, I. Angew. Chem., Int. Ed. 2000, 39, 3168–3210. doi:10.1002/1521-3773(20000915)39:18<3168::AID-ANIE3168>3.0.CO;2-U |
| 18. | Ugi, I.; Dömling, A.; Werner, B. J. Heterocycl. Chem. 2000, 37, 647–658. doi:10.1002/jhet.5570370322 |
| 19. | Weber, L.; Illgen, K.; Almstetter, M. Synlett 1999, 366–374. doi:10.1055/s-1999-2612 |
| 20. | Armstrong, R. W.; Combs, A. P.; Tempest, P. A.; Brown, S. D.; Keating, T. A. Acc. Chem. Res. 1996, 29, 123–131. doi:10.1021/ar9502083 |
| 21. | Ugi, I.; Dömling, A.; Hörl, W. Endeavour 1994, 18, 115–122. doi:10.1016/S0160-9327(05)80086-9 |
| 34. | Baathulaa, K.; Xu, Y.; Quian, X. J. Photochem. Photobiol., A 2010, 216, 24–34. doi:10.1016/j.jphotochem.2010.09.002 |
| 40. | Alexiou, M. S.; Tychopoubs, V.; Ghorbanian, S.; Tyman, J. H. P.; Brown, R. G.; Brittain, P. I. J. Chem. Soc., Perkin Trans. 2 1990, 5, 837–842. doi:10.1039/P29900000837 |
| 6. | Briehn, C. A.; Bäuerle, P. Chem. Commun. 2002, 1015–1023. doi:10.1039/B108846G |
| 7. | Müller, T. J. J.; D’Souza, D. M. Pure Appl. Chem. 2008, 80, 609–620. doi:10.1351/pac200880030609 |
| 8. | Müller, T. J. J. Diversity-oriented Synthesis of Chromophores by Combinatorial Strategies and Multi-component Reactions. In Functional Organic Materials. Syntheses, Strategies, and Applications; Müller, T. J. J.; Bunz, U. H. F., Eds.; Wiley-VCH: Weinheim, 2007; pp 179–223. doi:10.1002/9783527610266.ch5 |
| 37. | Banerjee, S.; Veale, E. B.; Phelan, C. M.; Murphy, S. A.; Tocci, G. M.; Gillespie, L. J.; Frimannsson, D. O.; Kelly, J. M.; Gunnlaugsson, T. Chem. Soc. Rev. 2013, 42, 1601–1618. doi:10.1039/C2CS35467E |
| 2. | Li, Z. R., Ed. Organic Light-Emitting Materials and Devices, 2nd ed.; CRC Press: Boca Raton, 2015. doi:10.1201/b18540 |
| 3. | Thejo Kalayani, N.; Dhoble, S. J. Renewable Sustainable Energy Rev. 2012, 16, 2696–2723. doi:10.1016/j.rser.2012.02.021 |
| 4. | Müllen, K.; Scherf, U., Eds. Organic Light-Emitting Diodes: Synthesis, Properties, and Applications; Wiley-VCH: Weinheim, Germany, 2006. doi:10.1002/3527607986 |
| 5. |
Park, J.-S.; Chae, H.; Chung, H. K.; Lee, S. I. Semicond. Sci. Technol. 2011, 26, 034001–034008. doi:10.1088/0268-1242/26/3/034001
See for a review on flexible AM-OLED. |
| 38. | Katritzky, A. R.; Ozcan, S.; Todadze, E. Org. Biomol. Chem. 2010, 8, 1296–1300. doi:10.1039/c000684j |
| 33. | Riva, R.; Moni, L.; Müller, T. J. J. Targets Heterocycl. Syst. 2016, 20, 85–112. |
| 35. | Miyachi, H.; Azuma, A.; Ogasawara, A.; Uchimura, E.; Watabe, N.; Kobayashi, Y.; Kato, F.; Kato, M.; Washimoto, Y. J. Med. Chem. 1997, 40, 2858–2865. doi:10.1021/jm970109q |
| 56. | Hong, Y.; Lam, J. W. Y.; Tang, B. Z. Chem. Commun. 2009, 4332–4353. doi:10.1039/B904665H |
| 57. | Hong, Y.; Lam, J. W. Y.; Tang, B. Z. Chem. Soc. Rev. 2011, 40, 5361–5388. doi:10.1039/C1CS15113D |
| 58. | Hu, R.; Leung, N. L. C.; Tang, B. Z. Chem. Soc. Rev. 2014, 43, 4494–4562. doi:10.1039/C4CS00044G |
| 31. | Levi, L.; Müller, T. J. J. Chem. Soc. Rev. 2016, 45, 2825–2846. doi:10.1039/C5CS00805K |
| 32. | Levi, L.; Müller, T. J. J. Eur. J. Org. Chem. 2016, 2907–2918. doi:10.1002/ejoc.201600409 |
| 36. | Hargeaves, M. K.; Pritchard, J. G.; Dave, H. R. Chem. Rev. 1970, 70, 439–469. doi:10.1021/cr60266a001 |
| 30. | Müller, T. J. J. General Discussion and Reactions Involving a Carbonyl Compound as Electrophilic Component. In Multicomponent Reactions 1; Müller, T. J. J., Ed.; Science of Synthesis Series; Georg Thieme Verlag KG: Stuttgart, 2014; pp 5–27. |
| 54. | Demeter, A.; Bérces, T.; Biczók, L.; Wintgens, V.; Valat, P.; Kossanyi, J. J. Phys. Chem. 1996, 100, 2001–2011. doi:10.1021/jp951133n |
| 25. | Posner, G. H. Chem. Rev. 1986, 86, 831–844. doi:10.1021/cr00075a007 |
| 26. | Tietze, L. F.; Beifuss, U. Angew. Chem., Int. Ed. Engl. 1993, 32, 131–163. doi:10.1002/anie.199301313 |
| 27. | Tietze, L. F. Chem. Rev. 1996, 96, 115–136. doi:10.1021/cr950027e |
| 28. | Ganem, B. Acc. Chem. Res. 2009, 42, 463–472. doi:10.1021/ar800214s |
| 29. | Brauch, S.; van Berkel, S. S.; Westermann, B. Chem. Soc. Rev. 2013, 42, 4948–4962. doi:10.1039/C3CS35505E |
| 34. | Baathulaa, K.; Xu, Y.; Quian, X. J. Photochem. Photobiol., A 2010, 216, 24–34. doi:10.1016/j.jphotochem.2010.09.002 |
| 55. | Hansch, C.; Leo, A.; Taft, R. W. Chem. Rev. 1991, 91, 165–195. doi:10.1021/cr00002a004 |
| 44. | Cabré-Castellvi, J.; Palomo-Coll, A.; Palomo-Coll, A. L. Synthesis 1981, 616–620. doi:10.1055/s-1981-29544 |
| 41. | Chen, X.; Xu, C.; Wang, T.; Zhou, C.; Du, J.; Wang, Z.; Xu, H.; Xie, T.; Bi, G.; Jiang, J.; Zhang, X.; Demas, J. N.; Trindle, C. O.; Luo, Y.; Zhang, G. Angew. Chem., Int. Ed. 2016, 55, 9872–9875. doi:10.1002/anie.201601252 |
| 42. | Michael, A.; Bucher, J. E. Ber. Dtsch. Chem. Ges. 1908, 41, 70–73. doi:10.1002/cber.19080410119 |
| 43. | Lanser, T. Ber. Dtsch. Chem. Ges. 1899, 32, 2477–2478. doi:10.1002/cber.189903202194 |
| 50. | Deposited with the Cambridge Crystallographic Data Centre as supplementary publication nos. CCDC 1563985 (4b), 1563988 (5), and 1564617 (6). |
| 50. | Deposited with the Cambridge Crystallographic Data Centre as supplementary publication nos. CCDC 1563985 (4b), 1563988 (5), and 1564617 (6). |
| 44. | Cabré-Castellvi, J.; Palomo-Coll, A.; Palomo-Coll, A. L. Synthesis 1981, 616–620. doi:10.1055/s-1981-29544 |
| 49. | Barros, T. C.; Yunes, S.; Menegon, G.; Nome, F.; Chaimovich, H.; Politi, M. J.; Dias, L. G.; Cuccovia, I. M. J. Chem. Soc., Perkin Trans. 2 2001, 2342–2350. doi:10.1039/B104148G |
| 47. | Wissmann, H.; Kleiner, H.-J. Angew. Chem., Int. Ed. 1980, 19, 133–134. doi:10.1002/anie.198001331 |
| 48. | Levi, L.; Scheuren, S.; Müller, T. J. J. Synthesis 2014, 46, 3059–3066. doi:10.1055/s-0034-1378541 |
| 45. | Ruhemann, S.; Merriman, R. W. J. Chem. Soc., Trans. 1905, 87, 1383–1395. doi:10.1039/CT9058701383 |
| 46. | Kim, K. H.; Lim, C. H.; Lim, J. W.; Kim, J. N. Adv. Synth. Catal. 2014, 356, 697–704. doi:10.1002/adsc.201301169 |
© 2017 Denißen et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)