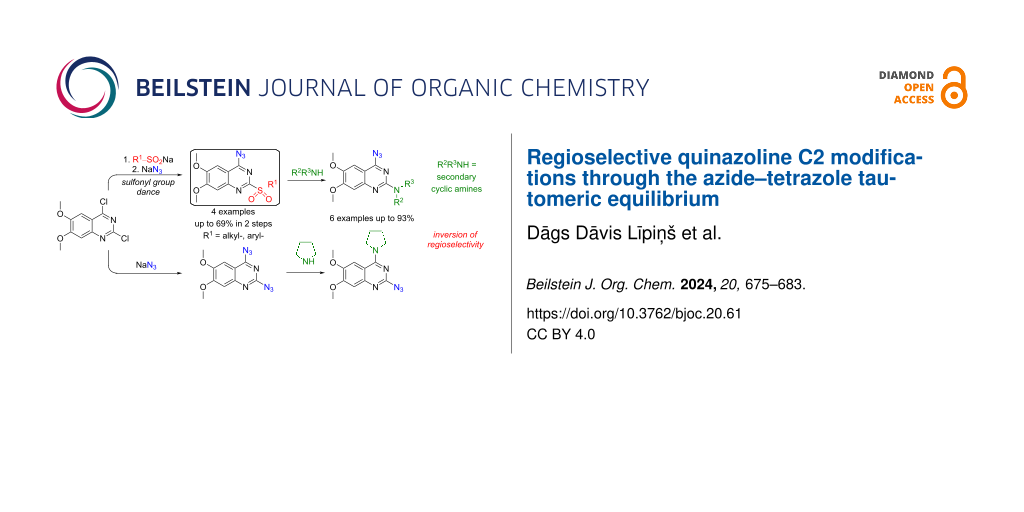
Abstract
2-Chloro-4-sulfonylquinazolines undergo functional group swap when treated with an azide nucleophile: 1) the azide replaces the sulfonyl group at the C4 position; 2) the intrinsic azide–tetrazole tautomeric equilibrium directs the nucleofugal sulfinate from the first step to replace chloride at the C2 position. This transformation is effective with quinazolines bearing electron-rich substituents. Therefore, the title transformations are demonstrated on the 6,7-dimethoxyquinazoline core, which is present in pharmaceutically active substances. The methodology application is showcased by transforming the obtained 4-azido-6,7-dimethoxy-2-sulfonylquinazolines into the α1-adrenoceptor blockers terazosin and prazosin by further C2-selective SNAr reaction and azide reduction.

Graphical Abstract
Introduction
The quinazoline core is a privileged structure with a wide range of applications. Quinazoline derivatives exhibit a broad spectrum of biological activities, finding use as anticancer, antimicrobial, antimalarial, and antiviral agents [1,2]. Furthermore, numerous 2-amino-6,7-dimethoxyquinazoline analogs are extensively employed as α1-adrenoceptor blockers [3,4]. In recent years quinazoline-based OLED materials have also gained attention showing great quantum efficiencies [5-7]. Consequently, ongoing efforts focus on advancing methodologies for synthesizing established quinazoline-based drugs and acquiring novel modified quinazoline derivatives for pharmaceutical or materials science purposes.
Aromatic nucleophilic substitution [8] or metal-catalyzed reactions [9,10] are commonly employed for quinazoline modification (Scheme 1). Existing literature underscores the reactivity of the C4 position in aromatic nucleophilic substitutions of quinazolines I while achieving regioselective replacement at the C2 position poses challenges [11]. Modification of the C2 position of quinazolines requires longer time, higher temperatures, and sometimes the use of expensive transition-metal catalysts [12]. A selective C2 modification can be achieved by using 2-chloroquinazolines IV, where the C4 position is blocked by an unreactive C–C or C–H bond (Scheme 1). Cyclization reactions of substituted anilines VI, VII or N-arylamidines VIII are frequently employed for synthesizing C2-substituted quinazolines (Scheme 1), thereby influencing the spatial arrangement of the desired substituents [13,14]. Moreover, there have been recent advancements in efficient C–H activation techniques employing transition-metal and photocatalysis [15,16]. These methods facilitate C–C bond formation, enabling the introduction of alkyl groups at the C2 position of quinazoline derivatives.
![[1860-5397-20-61-i1]](/bjoc/content/inline/1860-5397-20-61-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Approaches for quinazoline modifications at the C2 and C4 positions.
Scheme 1: Approaches for quinazoline modifications at the C2 and C4 positions.
While arylsulfanyl group rearrangement reactions have been documented by us for modifying 2,4-substituted quinazolines [17,18], and sulfonyl group rearrangement has been applied to functionalize purines [19], the literature lacks information on sulfonyl group migration in quinazolines. Notably, this transformation has not been previously reported, despite its potential utility in the synthesis of drugs such as terazosin and prazosin [20].
Herein, we report the use of the sulfonyl group dance to synthesize novel 4-azido-2-sulfonylquinazolines and their C2-selective modification in SNAr reactions. In addition, we offer an approach for the synthesis of terazosin and prazosin, known medications against hypertension, using sulfonyl group dance products.
Results and Discussion
Synthesis of 4-azido-2-sulfonylquinazolines
We started our experiments with commercially available 2,4-dichloroquinazoline (1a). It was treated with sodium 4-methylphenylsulfinate in order to yield 4-sulfonylquinazoline 2a (Scheme 2), but the first attempts in iPrOH did not provide the starting material conversion. The reaction in THF resulted in the full conversion of the starting material, but the analysis of the crude product revealed the quantitative formation of hydrolysis product 5a. Assuming the instability of intermediate 2a, a one-pot reaction was performed by adding sodium 4-methylphenylsulfinate in the first step which was followed by NaN3. As the result, the formation of hydrolysis product 5a and 2,4-diazidoquinazoline (6a) was observed.
![[1860-5397-20-61-i2]](/bjoc/content/inline/1860-5397-20-61-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Attempts toward sulfonyl group dance using 2,4-dichloroquinazolines 1a‒c.
Scheme 2: Attempts toward sulfonyl group dance using 2,4-dichloroquinazolines 1a‒c.
Next, the reaction 1a → 4a in DMSO yielded diazidoquinazoline 6a as a major product and hydrolysis product 5a. In MeCN the conversion to derivative 2a was stopped at 50% and was not facilitated by an extra addition of sulfinate. To our delight, in MeOH we observed the formation of intermediate 2a over 5 hours, and after the subsequent addition of sodium azide product 4a was isolated in 31% yield over 2 steps. The full conversion was achieved by keeping the reaction mixture at a temperature of 0 °C and by the stepwise additions of the sulfinate and NaN3. Any deviation from these conditions facilitated the formation of byproducts.
In addition, the sulfonyl group dance reactions were carried out also with quinazoline derivatives 1b and 1c (Scheme 2), the structure features of which may slow-down the fast SNAr processes due to the substituents’ character. The desired products 4b and 4c were obtained in MeOH and isolated in 44 and 40% yields, respectively. Methanol is known to decrease reactivity in the SNAr reactions in comparison to polar solvents such as DMSO and DMF. This is explained by solvent hydrogen bond acidity and basicity descriptors α and β, for example, α(DMSO) = 0, β(DMSO) = 0.88, α(MeOH) = 0.43, β(MeOH) = 0.47. The rate constant of the SNAr process escalates with an increase of β parameters and diminishes with an increase of α parameters [21,22]. Therefore, it was possible to accomplish the sulfonyl group dance reactions of very reactive quinazolines 1a‒c in MeOH.
Synthesis of 4-azido-6,7-dimethoxy-2-sulfonylquinazolines
Next, we aimed to explore the sulfonyl group dance process using a more electron-rich quinazoline. The commercially available 2,4-dichloro-6,7-dimethoxyquinazoline (7) was chosen for this purpose (Scheme 3). The common dimethoxy motif is also found in a variety of quinazoline-based pharmaceuticals [2,3,8,23].
![[1860-5397-20-61-i3]](/bjoc/content/inline/1860-5397-20-61-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of 2-chloro-6,7-dimethoxy-4-sulfonylquinazoline derivatives 8.
Scheme 3: Synthesis of 2-chloro-6,7-dimethoxy-4-sulfonylquinazoline derivatives 8.
We commenced our study with the preparation of 2-chloro-4-sulfonylquinazolines 8 (Scheme 3). The starting material 7 underwent SNAr reactions with sodium sulfinates and the C4-substituted products 8a,b were isolated [24]. The complete conversion was achieved in DMF or DMSO. In the case of sodium dodecylsulfinate, the reaction stopped at 70% conversion when 1 equivalent of sulfinate was used. Products 8 exhibited instability in the presence of water, leading to the formation of hydrolysis product 9 [25] in the reaction mixture. This instability caused issues during the reaction work-up, and attempts for purification using column chromatography resulted in full degradation of the formed product.
Consequently, an alternative pathway toward product 8 was explored (Scheme 4). 2-Chloro-4-thioquinazolines 10 were prepared from starting material 7 in an SNAr reaction with thiols in the presence of K2CO3 in good 75–93% yields. Next, thioquinazolines 10 were oxidized to the corresponding sulfonylquinazolines 8. Inspired by our previous work [19] a TFAA/H2O2 oxidizing system was tried first but yielded several side-products, such as the hydrolysis product and unwanted oxidation of the quinazoline N3 position. Changing the oxidant to mCPBA (with 96% purity) [26] provided a more selective reaction, no water-based work-up was needed and the pure product was obtained by simple recrystallization from ethanol in yields up to 88%. The oxidation step thiol → sulfoxide was fast and full conversion to the intermediate was achieved in one hour for most substrates, but the step sulfoxide → sulfone was entirely slower and required stirring overnight (except for 8d (R = iC3H7)).
![[1860-5397-20-61-i4]](/bjoc/content/inline/1860-5397-20-61-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Alternative synthesis pathway for 2-chloro-6,7-dimethoxy-4-sulfonylquinazoline derivatives 8.
Scheme 4: Alternative synthesis pathway for 2-chloro-6,7-dimethoxy-4-sulfonylquinazoline derivatives 8.
With 2-chloro-6,7-dimethoxy-4-sulfonylquinazolines 8 in hand, we started to explore the reactivity in SNAr reactions (Scheme 5). Sulfonyl group dance reactions did not work in anhydrous THF, MeCN, and dioxane, using such azide sources as NaN3, LiN3, and TMS-N3. Full conversion towards product 12a was observed by HPLC with NaN3 in anhydrous DMF. However, precipitation, direct, and reversed-phase column chromatography provided low yields (Scheme 5) due to the degradation of the product. Compounds 12 did not tolerate aqueous conditions or high temperatures and have also been observed to degrade under direct sunlight.
![[1860-5397-20-61-i5]](/bjoc/content/inline/1860-5397-20-61-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Sulfonyl group dance using 2-chloro-6,7-dimethoxy-4-sulfonylquinazolines 8.
Scheme 5: Sulfonyl group dance using 2-chloro-6,7-dimethoxy-4-sulfonylquinazolines 8.
Next, a stepwise one-pot approach was investigated to increase the overall yield (Scheme 6). The reaction in anhydrous DMF yielded a mixture of the desired product 12a, diazide 13, and hydrolysis product 9 [25] which were inseparable using common purification methods (Table 1).
![[1860-5397-20-61-i6]](/bjoc/content/inline/1860-5397-20-61-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: One-pot synthesis of 4-azido-6,7-dimethoxy-2-sulfonylquinazolines 12. The crystallographic information for 12a has been submitted to the Cambridge Crystallographic Data Centre and is available as supplementary publication No. CCDC-2312750.
Scheme 6: One-pot synthesis of 4-azido-6,7-dimethoxy-2-sulfonylquinazolines 12. The crystallographic informat...
Table 1: Conditions for one-pot synthesis of 4-azido-6,7-dimethoxy-2-sulfonylquinazolines 12.
| Entry | Solvent | Azide source | Time, h | R | Yield, % |
| 1 | anh. DMF | 1.0 equiv NaN3a | 1 | 4-CH3C6H4 | –b |
| 2 | anh. DMSO | 0.6 equiv NaN3a | 1 | 4-CH3C6H4 | 12a, 39 |
| 3 | anh. DMSO | 0.8 equiv NaN3a | 4 | 4-CH3C6H4 | 12a, 69 |
| 4 | anh. DMSO | 0.8 equiv NaN3c | 2 | 4-CH3C6H4 | 12a, 66d |
| 5 | anh. DMSO | 0.8 equiv NaN3c | 12 | n-C12H25 | 12b, 28 |
| 6 | anh. DMSO | 0.8 equiv NaN3c | 12 | iC3H7 | 12d, 63e |
| 7 | anh. DMSO | 0.8 equiv NaN3c | 2 | C6H5 | 12c, 50e |
aAdded in portions; ba mixture of products 12 and 13 (Scheme 6); c0.5 M solution of NaN3 in anh. DMSO added over 2 hours; d5.8 mmol scale; eqNMR yield.
The pivotal advancement occurred when attempting the reaction in DMSO (Table 1). In the case of 8a (R = 4-CH3C6H4), the product precipitated out when full conversion was reached. Filtration of this precipitate yielded the pure desired product 12a in 39% yield (Table 1, entry 2). Incremental additions of NaN3, coupled with HPLC analysis following each addition, facilitated the achievement of full conversion of the starting material after 0.7–0.8 equivalents of NaN3. This approach limited the formation of diazide 13 and significantly elevated the yield of the desired product to 69% over two steps. When other sufinates were employed, the product failed to precipitate, necessitating isolation through preparative HPLC. Quantitative nuclear magnetic resonance (qNMR) yields were consequently reported.
To reduce the formation of diazide 13, an overnight addition of the azide solution via a dispenser was employed at a rate of 0.1 equivalents of NaN3 per hour. This strategy improved the ratio of product 12 to diazide 13. For arylsulfinates, the addition time was finally reduced to 2 hours without compromising selectivity. Although tetrabutylammonium azide (TBAA) is better soluble in DMSO than NaN3, practical challenges associated with its use led to the preference for NaN3.
Confirmation of regioselectivity for the sulfonyl group dance products
The regioselectivity and the structure of 4-azido-6,7-dimethoxy-2-sulfonylquinazoline derivatives 12 were proven by chemical synthesis of the regioisomers 15 (Scheme 7) and X-ray analysis of 12a (Scheme 6). 6,7-Dimethoxy-2,4-diazidoquinazoline (13) was synthesized from commercially available dichloroquinazoline 7 in 93% yield. Further, thioether substituents were installed in the presence of K2CO3. For alkylthiols, DMF was used, but arylthiols required milder conditions with MeOH and cooling to acquire regioselectivity to the C4 position which resulted in yields up to 91%. Oxidation with purified mCPBA (commercial mCPBA with 68% purity was washed with pH 7.4 phosphate buffer to reach 96% purity [26]) yielded the regioisomers 15 of the sulfonyl group dance products at a lower yield than the previously mentioned oxidation step, which was most likely caused by the high reactivity of product 14, but the reaction conditions were not further optimized since the products were only needed for analytical purposes.
![[1860-5397-20-61-i7]](/bjoc/content/inline/1860-5397-20-61-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Synthesis of 2-azido-4-sulfonyl-6,7-dimethoxyquinazolines 15.
Scheme 7: Synthesis of 2-azido-4-sulfonyl-6,7-dimethoxyquinazolines 15.
Two different pyrrolidine-substituted derivatives were additionally synthesized to prove the regioselectivity of the sulfonyl group dance products (Scheme 8). Compound 16 was obtained in the C4-selective SNAr reaction between diazidoquinazoline 13 and pyrrolidine in 93% yield. A cross peak for the H–C5 position of quinazoline and CH2 groups of pyrrolidine at the second position was observed in the NOESY spectrum and unequivocally proved the structure 16. Selective C2 substitution was achieved between sulfonylquinazoline 12 and pyrrolidine in CHCl3 yielding product 17a. No NOESY signals were seen between the quinazoline core and the pyrrolidine moiety. Interestingly, the C4 substitution was achieved when DMF was used as a solvent in the transformation 12 → 18, resulting in product 18.
![[1860-5397-20-61-i8]](/bjoc/content/inline/1860-5397-20-61-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Synthesis of 6,7-dimethoxyquinazoline derivatives 16, 17a and 18.
Scheme 8: Synthesis of 6,7-dimethoxyquinazoline derivatives 16, 17a and 18.
In addition, the reaction 12a + pyrrolidine in MeCN and THF gave only product 17a, but in DMSO resulted in the mixture of 17a/18/6,7-dimethoxy-2,4-di(pyrrolidin-1-yl)quinazoline = 36:15:48% (HPLC analysis). The selectivity of 17a/18 was interesting but was not further developed in the scope of this study.
Consequently, an investigation into the azide–tetrazole equilibrium of product 12a was initiated, revealing a singular form present in all solvents. Despite attempts to increase the amount of the azide form with the increase of the temperature in NMR experiments [27], no observable alteration in the tautomeric equilibrium was observed. FTIR analysis of 12a in CHCl3 and DMSO solutions revealed the absence of the azide form (see Supporting Information File 1), precluding an explanation of the reactivity of 12a through the tautomeric equilibrium. The presence of electron-donating methoxy groups in the structure was proposed as a plausible explanation for the present tetrazole form in the solutions. Surprisingly, FTIR and X-ray analyses of 12a in the solid state indicated the existence of 12a in the azide form.
In subsequent experiments it was discovered that for less nucleophilic N-nucleophiles (piperidine, morpholine, N-methylpiperazine) C2 selectivity was reached only in polar solvents such as DMF, DMSO, and MeCN. In other solvents, no reactivity was observed at the C2 or C4 positions.
Selective modification of the C2 position of 6,7-dimethoxyquinazoline
Products 12 are useful intermediates to achieve selective modification at the C2 position of quinazolines. A scope of 2-amino-4-azido-6,7-dimetoxyquinazolines 17 was synthesized. For pyrrolidine, selective C2 substitution was achieved in a non-polar solvent such as CHCl3. Less nucleophilic amines gave C2-selective SNAr in MeCN.
To apply the developed technique to the synthesis of pharmaceutically active substances such as terazosin and prazosin, nucleophilic substitution at the C2 position was carried out with the corresponding amines – piperazin-1-yl(tetrahydrofuran-2-yl)methanone and furan-2-yl(piperazin-1-yl)methanone to give products 17e and 17f. Products 17e,f can be obtained through the aromatic nucleophilic substitution of 2-azido-4-sulfonylquinazoline 12a or by performing three subsequent SNAr reactions starting from 2,4-dichloroquinazoline 7 in a one-pot procedure [28] (Scheme 9, Table 2).
![[1860-5397-20-61-i9]](/bjoc/content/inline/1860-5397-20-61-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Synthesis of 2-amino-4-azido-6,7-dimethoxyquinazolines 17.
Scheme 9: Synthesis of 2-amino-4-azido-6,7-dimethoxyquinazolines 17.
Table 2: Diversity and yields for 2-amino-4-azido-6,7-dimethoxyquinazolines 17.
| Entry | R1R2NH | Solvent | Yield |
| 1 |
![[Graphic 1]](/bjoc/content/inline/1860-5397-20-61-i12.svg?max-width=637&scale=1.0)
|
CHCl3 | 17a, 93a |
| 2 |
![[Graphic 2]](/bjoc/content/inline/1860-5397-20-61-i13.svg?max-width=637&scale=1.0)
|
MeCN | 17b, 73a |
| 3 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-20-61-i14.svg?max-width=637&scale=1.0)
|
MeCN | 17c, 75a |
| 4 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-20-61-i15.svg?max-width=637&scale=1.0)
|
MeCN | 17d, 77a |
| 5 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-20-61-i16.svg?max-width=637&scale=1.0)
|
DMSO | 17e, 80a, 41b |
| 6 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-20-61-i17.svg?max-width=637&scale=1.0)
|
DMSO | 17f, 75a, 49b |
aYield 12a→17, %; byield 7→17, % (over 3 steps).
The resulting products exist in an azide–tetrazole equilibrium in solution, but in solid form can be in either the azide (17e, 17f) or tetrazole form (17a–d).
With derivatives 17e,f in hand, the reduction of the azido group in the C4 position was carried out by bubbling hydrogen through the solution in the presence of palladium on charcoal. In the last step, the product was acidified with a 4 M HCl solution in iPrOH, forming the respective hydrochlorides of terazosin [29,30] and prazosin [31,32] (Scheme 10).
![[1860-5397-20-61-i10]](/bjoc/content/inline/1860-5397-20-61-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Synthesis of terazosin and prazosin hydrochlorides 19a and 19b.
Scheme 10: Synthesis of terazosin and prazosin hydrochlorides 19a and 19b.
In addition, we explored some other reactions of the azido group, and derivatives 17 were used in CuAAC and Staudinger reactions, yielding products 20 and 21 (Scheme 11).
![[1860-5397-20-61-i11]](/bjoc/content/inline/1860-5397-20-61-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 11: Modifications of derivatives 17.
Scheme 11: Modifications of derivatives 17.
For CuAAC reactions no conversion towards the desired triazolyl product 20 was observed in systems such as CuSO4·5H2O/sodium ascorbate/t-BuOH/H2O, CuSO4·5H2O/sodium ascorbate/THF/H2O, CuI/DIPEA/DCM. Instead, triazolyl derivatives 20 were synthesized using [Cu(MeCN)4]PF6/TBTA (tris(benzyltriazolylmethyl)amine) [33] in toluene.
Conclusion
To summarize, an approach toward 4-azido-6,7-dimethoxy-2-alkyl/arylsulfonylquinazolines was developed employing a sulfonyl group dance caused by the azide–tetrazole equilibrium in quinazolines. 4-Azido-6,7-dimethoxy-2-alkyl/arylsulfonylquinazolines were obtained using two pathways: 1) SNAr reaction between 2-chloro-6,7-dimethoxy-4-sulfonylquinazoline derivatives and NaN3; 2) SNAr reaction between 2,4-dichloro-6,7-dimethoxyquinazoline and alkyl/arylsulfinates, followed by substitution with NaN3. 4-Azido-6,7-dimethoxy-2-alkyl/arylsulfonylquinazolines serve as valuable precursors for the C2-regioselective modification in quinazolines. Furthermore, the developed methodology was valorized by successfully employing it in the synthesis of adrenoblockers terazosin and prazosin.
Supporting Information
| Supporting Information File 1: Experimental, copies of spectra and crystal data, data collection and structure refinement details for compound 12a. | ||
| Format: PDF | Size: 7.4 MB | Download |
| Supporting Information File 2: Checkcif for compound 12a. | ||
| Format: PDF | Size: 129.5 KB | Download |
| Supporting Information File 3: Crystallographic information file (CIF) for compound 12a. | ||
| Format: CIF | Size: 485.0 KB | Download |
Data Availability Statement
All data that supports the findings of this study is available in the published article and/or the supporting information to this article.
References
-
Karan, R.; Agarwal, P.; Sinha, M.; Mahato, N. ChemEngineering 2021, 5, 73. doi:10.3390/chemengineering5040073
Return to citation in text: [1] -
Gomaa, H. A. M. Chem. Biol. Drug Des. 2022, 100, 639–655. doi:10.1111/cbdd.14129
Return to citation in text: [1] [2] -
Minarini, A.; Bolognesi, M. L.; Tumiatti, V.; Melchiorre, C. Expert Opin. Drug Discovery 2006, 1, 395–407. doi:10.1517/17460441.1.5.395
Return to citation in text: [1] [2] -
Leonardi, A.; Motta, G.; Boi, C.; Testa, R. Quinazolinyl-amino derivatives having α-antagonist activity. WO Patent WO95/25726, Sept 28, 1995.
Return to citation in text: [1] -
Li, B.; Wang, Z.; Su, S.-J.; Guo, F.; Cao, Y.; Zhang, Y. Adv. Opt. Mater. 2019, 7, 1801496. doi:10.1002/adom.201801496
Return to citation in text: [1] -
Li, P.; Xiang, Y.; Gong, S.; Lee, W.-K.; Huang, Y.-H.; Wang, C.-Y.; Yang, C.; Wu, C.-C. J. Mater. Chem. C 2021, 9, 12633–12641. doi:10.1039/d1tc02633j
Return to citation in text: [1] -
Li, B.; Li, Z.; Guo, F.; Song, J.; Jiang, X.; Wang, Y.; Gao, S.; Wang, J.; Pang, X.; Zhao, L.; Zhang, Y. ACS Appl. Mater. Interfaces 2020, 12, 14233–14243. doi:10.1021/acsami.9b20162
Return to citation in text: [1] -
Khan, I.; Ibrar, A.; Ahmed, W.; Saeed, A. Eur. J. Med. Chem. 2015, 90, 124–169. doi:10.1016/j.ejmech.2014.10.084
Return to citation in text: [1] [2] -
Tamatam, R.; Kim, S.-H.; Shin, D. Front. Chem. (Lausanne, Switz.) 2023, 11, 1140562. doi:10.3389/fchem.2023.1140562
Return to citation in text: [1] -
Shui, H.; Zhong, Y.; Ouyang, L.; Luo, N.; Luo, R. Synthesis 2022, 54, 2876–2884. doi:10.1055/a-1755-4700
Return to citation in text: [1] -
Connolly, D. J.; Cusack, D.; O'Sullivan, T. P.; Guiry, P. J. Tetrahedron 2005, 61, 10153–10202. doi:10.1016/j.tet.2005.07.010
Return to citation in text: [1] -
Gheidari, D.; Mehrdad, M.; Maleki, S. Sustainable Chem. Pharm. 2022, 27, 100696. doi:10.1016/j.scp.2022.100696
Return to citation in text: [1] -
Tamatam, R.; Shin, D. Molecules 2023, 28, 3227. doi:10.3390/molecules28073227
Return to citation in text: [1] -
Faisal, M.; Saeed, A. Front. Chem. (Lausanne, Switz.) 2021, 8, 594717. doi:10.3389/fchem.2020.594717
Return to citation in text: [1] -
Chen, X.; Luo, X.; Wang, K.; Liang, F.; Wang, P. Synlett 2021, 32, 733–737. doi:10.1055/a-1294-0158
Return to citation in text: [1] -
Xie, D.; Liu, Y.; Liu, X.; Yang, Q.; Peng, Y. Eur. J. Org. Chem. 2024, 27, e202300993. doi:10.1002/ejoc.202300993
Return to citation in text: [1] -
Jeminejs, A.; Goliškina, S. M.; Novosjolova, I.; Stepanovs, D.; Bizdēna, Ē.; Turks, M. Synthesis 2021, 53, 1543–1556. doi:10.1055/s-0040-1706568
Return to citation in text: [1] -
Jeminejs, A.; Novosjolova, I.; Bizdēna, Ē.; Turks, M. Org. Biomol. Chem. 2021, 19, 7706–7723. doi:10.1039/d1ob01315g
Return to citation in text: [1] -
Zaķis, J. M.; Ozols, K.; Novosjolova, I.; Vilšķe̅rsts, R.; Mishnev, A.; Turks, M. J. Org. Chem. 2020, 85, 4753–4771. doi:10.1021/acs.joc.9b03518
Return to citation in text: [1] [2] -
Bottini, A.; De, S. K.; Wu, B.; Tang, C.; Varani, G.; Pellecchia, M. Chem. Biol. Drug Des. 2015, 86, 663–673. doi:10.1111/cbdd.12534
Return to citation in text: [1] -
El Guesmi, N.; Berionni, G.; Asghar, B. H. Monatsh. Chem. 2013, 144, 1537–1545. doi:10.1007/s00706-013-1030-7
Return to citation in text: [1] -
Marenich, A. V.; Cramer, C. J.; Truhlar, D. G. J. Phys. Chem. B 2009, 113, 6378–6396. doi:10.1021/jp810292n
Return to citation in text: [1] -
Mizukawa, Y.; Ikegami-Kawai, M.; Horiuchi, M.; Kaiser, M.; Kojima, M.; Sakanoue, S.; Miyagi, S.; Nanga Chick, C.; Togashi, H.; Tsubuki, M.; Ihara, M.; Usuki, T.; Itoh, I. Bioorg. Med. Chem. 2021, 33, 116018. doi:10.1016/j.bmc.2021.116018
Return to citation in text: [1] -
Nguyen, V. D.; Nguyen, V. T.; Haug, G. C.; Dang, H. T.; Arman, H. D.; Ermler, W. C.; Larionov, O. V. ACS Catal. 2019, 9, 4015–4024. doi:10.1021/acscatal.9b00464
Return to citation in text: [1] -
Feng, J.; Zhang, Z.; Wallace, M. B.; Stafford, J. A.; Kaldor, S. W.; Kassel, D. B.; Navre, M.; Shi, L.; Skene, R. J.; Asakawa, T.; Takeuchi, K.; Xu, R.; Webb, D. R.; Gwaltney, S. L., II. J. Med. Chem. 2008, 51, 4357. doi:10.1021/jm8006799
Return to citation in text: [1] [2] -
Horn, A.; Kazmaier, U. Eur. J. Org. Chem. 2018, 2531–2536. doi:10.1002/ejoc.201701645
Return to citation in text: [1] [2] -
Sebris, A.; Turks, M. Chem. Heterocycl. Compd. 2019, 55, 1041–1043. doi:10.1007/s10593-019-02574-7
Return to citation in text: [1] -
Līpiņš, D. D.; Jeminejs, A.; Novosjolova, I.; Turks, M. The use of the sulfonyl group dance in quinazolines for the synthesis of 2-amino substituted 6,7-dimetoxyquinazolines. Latvian Patent LVP2023/000121, Nov 27, 2023.
Return to citation in text: [1] -
Joshi, S. V.; Soni, M. N.; Fulwala, K. M.; Jalani, H. B.; Prajapati, K. K. Process for preparing anhydrous terazosin hydrochloride. Indian Patent 2004MU01090, April 27, 2007.
Return to citation in text: [1] -
Schwartz, E.; Mendelovici, M.; Gershon, N. Process for the preparation of terazosin hydrochloride dihydrate. U.S. Patent US6248888B1, June 19, 2001.
Return to citation in text: [1] -
Honkanen, E.; Pippuri, A.; Kairisalo, P.; Thaler, H.; Koivisto, M.; Tuomi, S. J. Heterocycl. Chem. 1980, 17, 797–798. doi:10.1002/jhet.5570170436
Return to citation in text: [1] -
Petty, A.; Idippily, N.; Bobba, V.; Geldenhuys, W. J.; Zhong, B.; Su, B.; Wang, B. Eur. J. Med. Chem. 2018, 143, 1261–1276. doi:10.1016/j.ejmech.2017.10.026
Return to citation in text: [1] -
Chan, T. R.; Hilgraf, R.; Sharpless, K. B.; Fokin, V. V. Org. Lett. 2004, 6, 2853–2855. doi:10.1021/ol0493094
Return to citation in text: [1]
| 1. | Karan, R.; Agarwal, P.; Sinha, M.; Mahato, N. ChemEngineering 2021, 5, 73. doi:10.3390/chemengineering5040073 |
| 2. | Gomaa, H. A. M. Chem. Biol. Drug Des. 2022, 100, 639–655. doi:10.1111/cbdd.14129 |
| 9. | Tamatam, R.; Kim, S.-H.; Shin, D. Front. Chem. (Lausanne, Switz.) 2023, 11, 1140562. doi:10.3389/fchem.2023.1140562 |
| 10. | Shui, H.; Zhong, Y.; Ouyang, L.; Luo, N.; Luo, R. Synthesis 2022, 54, 2876–2884. doi:10.1055/a-1755-4700 |
| 24. | Nguyen, V. D.; Nguyen, V. T.; Haug, G. C.; Dang, H. T.; Arman, H. D.; Ermler, W. C.; Larionov, O. V. ACS Catal. 2019, 9, 4015–4024. doi:10.1021/acscatal.9b00464 |
| 8. | Khan, I.; Ibrar, A.; Ahmed, W.; Saeed, A. Eur. J. Med. Chem. 2015, 90, 124–169. doi:10.1016/j.ejmech.2014.10.084 |
| 25. | Feng, J.; Zhang, Z.; Wallace, M. B.; Stafford, J. A.; Kaldor, S. W.; Kassel, D. B.; Navre, M.; Shi, L.; Skene, R. J.; Asakawa, T.; Takeuchi, K.; Xu, R.; Webb, D. R.; Gwaltney, S. L., II. J. Med. Chem. 2008, 51, 4357. doi:10.1021/jm8006799 |
| 5. | Li, B.; Wang, Z.; Su, S.-J.; Guo, F.; Cao, Y.; Zhang, Y. Adv. Opt. Mater. 2019, 7, 1801496. doi:10.1002/adom.201801496 |
| 6. | Li, P.; Xiang, Y.; Gong, S.; Lee, W.-K.; Huang, Y.-H.; Wang, C.-Y.; Yang, C.; Wu, C.-C. J. Mater. Chem. C 2021, 9, 12633–12641. doi:10.1039/d1tc02633j |
| 7. | Li, B.; Li, Z.; Guo, F.; Song, J.; Jiang, X.; Wang, Y.; Gao, S.; Wang, J.; Pang, X.; Zhao, L.; Zhang, Y. ACS Appl. Mater. Interfaces 2020, 12, 14233–14243. doi:10.1021/acsami.9b20162 |
| 21. | El Guesmi, N.; Berionni, G.; Asghar, B. H. Monatsh. Chem. 2013, 144, 1537–1545. doi:10.1007/s00706-013-1030-7 |
| 22. | Marenich, A. V.; Cramer, C. J.; Truhlar, D. G. J. Phys. Chem. B 2009, 113, 6378–6396. doi:10.1021/jp810292n |
| 3. | Minarini, A.; Bolognesi, M. L.; Tumiatti, V.; Melchiorre, C. Expert Opin. Drug Discovery 2006, 1, 395–407. doi:10.1517/17460441.1.5.395 |
| 4. | Leonardi, A.; Motta, G.; Boi, C.; Testa, R. Quinazolinyl-amino derivatives having α-antagonist activity. WO Patent WO95/25726, Sept 28, 1995. |
| 2. | Gomaa, H. A. M. Chem. Biol. Drug Des. 2022, 100, 639–655. doi:10.1111/cbdd.14129 |
| 3. | Minarini, A.; Bolognesi, M. L.; Tumiatti, V.; Melchiorre, C. Expert Opin. Drug Discovery 2006, 1, 395–407. doi:10.1517/17460441.1.5.395 |
| 8. | Khan, I.; Ibrar, A.; Ahmed, W.; Saeed, A. Eur. J. Med. Chem. 2015, 90, 124–169. doi:10.1016/j.ejmech.2014.10.084 |
| 23. | Mizukawa, Y.; Ikegami-Kawai, M.; Horiuchi, M.; Kaiser, M.; Kojima, M.; Sakanoue, S.; Miyagi, S.; Nanga Chick, C.; Togashi, H.; Tsubuki, M.; Ihara, M.; Usuki, T.; Itoh, I. Bioorg. Med. Chem. 2021, 33, 116018. doi:10.1016/j.bmc.2021.116018 |
| 15. | Chen, X.; Luo, X.; Wang, K.; Liang, F.; Wang, P. Synlett 2021, 32, 733–737. doi:10.1055/a-1294-0158 |
| 16. | Xie, D.; Liu, Y.; Liu, X.; Yang, Q.; Peng, Y. Eur. J. Org. Chem. 2024, 27, e202300993. doi:10.1002/ejoc.202300993 |
| 19. | Zaķis, J. M.; Ozols, K.; Novosjolova, I.; Vilšķe̅rsts, R.; Mishnev, A.; Turks, M. J. Org. Chem. 2020, 85, 4753–4771. doi:10.1021/acs.joc.9b03518 |
| 13. | Tamatam, R.; Shin, D. Molecules 2023, 28, 3227. doi:10.3390/molecules28073227 |
| 14. | Faisal, M.; Saeed, A. Front. Chem. (Lausanne, Switz.) 2021, 8, 594717. doi:10.3389/fchem.2020.594717 |
| 20. | Bottini, A.; De, S. K.; Wu, B.; Tang, C.; Varani, G.; Pellecchia, M. Chem. Biol. Drug Des. 2015, 86, 663–673. doi:10.1111/cbdd.12534 |
| 12. | Gheidari, D.; Mehrdad, M.; Maleki, S. Sustainable Chem. Pharm. 2022, 27, 100696. doi:10.1016/j.scp.2022.100696 |
| 11. | Connolly, D. J.; Cusack, D.; O'Sullivan, T. P.; Guiry, P. J. Tetrahedron 2005, 61, 10153–10202. doi:10.1016/j.tet.2005.07.010 |
| 17. | Jeminejs, A.; Goliškina, S. M.; Novosjolova, I.; Stepanovs, D.; Bizdēna, Ē.; Turks, M. Synthesis 2021, 53, 1543–1556. doi:10.1055/s-0040-1706568 |
| 18. | Jeminejs, A.; Novosjolova, I.; Bizdēna, Ē.; Turks, M. Org. Biomol. Chem. 2021, 19, 7706–7723. doi:10.1039/d1ob01315g |
| 25. | Feng, J.; Zhang, Z.; Wallace, M. B.; Stafford, J. A.; Kaldor, S. W.; Kassel, D. B.; Navre, M.; Shi, L.; Skene, R. J.; Asakawa, T.; Takeuchi, K.; Xu, R.; Webb, D. R.; Gwaltney, S. L., II. J. Med. Chem. 2008, 51, 4357. doi:10.1021/jm8006799 |
| 19. | Zaķis, J. M.; Ozols, K.; Novosjolova, I.; Vilšķe̅rsts, R.; Mishnev, A.; Turks, M. J. Org. Chem. 2020, 85, 4753–4771. doi:10.1021/acs.joc.9b03518 |
| 26. | Horn, A.; Kazmaier, U. Eur. J. Org. Chem. 2018, 2531–2536. doi:10.1002/ejoc.201701645 |
| 31. | Honkanen, E.; Pippuri, A.; Kairisalo, P.; Thaler, H.; Koivisto, M.; Tuomi, S. J. Heterocycl. Chem. 1980, 17, 797–798. doi:10.1002/jhet.5570170436 |
| 32. | Petty, A.; Idippily, N.; Bobba, V.; Geldenhuys, W. J.; Zhong, B.; Su, B.; Wang, B. Eur. J. Med. Chem. 2018, 143, 1261–1276. doi:10.1016/j.ejmech.2017.10.026 |
| 33. | Chan, T. R.; Hilgraf, R.; Sharpless, K. B.; Fokin, V. V. Org. Lett. 2004, 6, 2853–2855. doi:10.1021/ol0493094 |
| 28. | Līpiņš, D. D.; Jeminejs, A.; Novosjolova, I.; Turks, M. The use of the sulfonyl group dance in quinazolines for the synthesis of 2-amino substituted 6,7-dimetoxyquinazolines. Latvian Patent LVP2023/000121, Nov 27, 2023. |
| 29. | Joshi, S. V.; Soni, M. N.; Fulwala, K. M.; Jalani, H. B.; Prajapati, K. K. Process for preparing anhydrous terazosin hydrochloride. Indian Patent 2004MU01090, April 27, 2007. |
| 30. | Schwartz, E.; Mendelovici, M.; Gershon, N. Process for the preparation of terazosin hydrochloride dihydrate. U.S. Patent US6248888B1, June 19, 2001. |
| 26. | Horn, A.; Kazmaier, U. Eur. J. Org. Chem. 2018, 2531–2536. doi:10.1002/ejoc.201701645 |
| 27. | Sebris, A.; Turks, M. Chem. Heterocycl. Compd. 2019, 55, 1041–1043. doi:10.1007/s10593-019-02574-7 |
© 2024 Līpiņš et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.