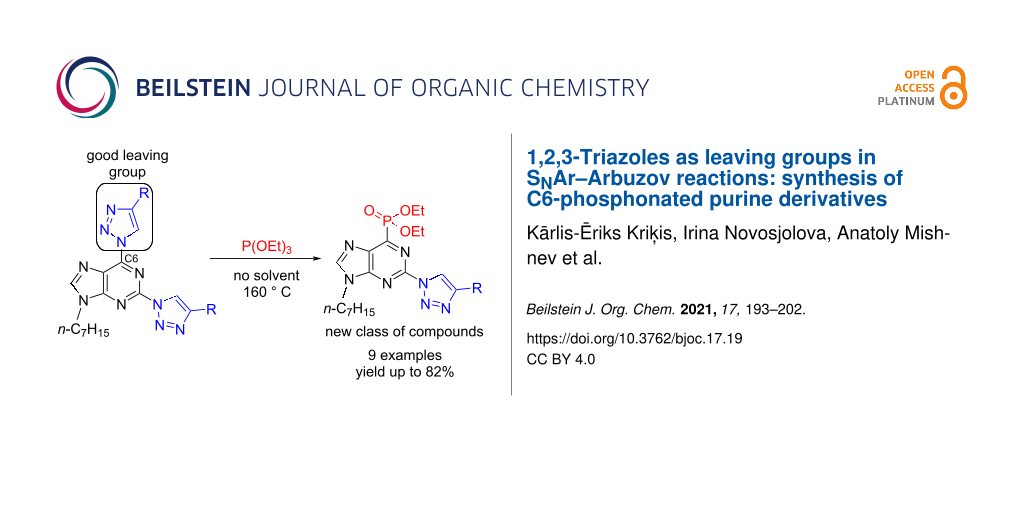
Abstract
A new method for C–N bond transformations into C–P bonds was developed using 1,2,3-triazoles as leaving groups in SNAr–Arbuzov reactions. A series of C6-phosphonated 2-triazolylpurine derivatives was synthesized for the first time, with the isolated yields reaching up to 82% in the C–P-bond-forming event. The SNAr–Arbuzov reaction of 2,6-bistriazolylpurines follows the general regioselectivity pattern of the C6-position being more reactive towards substitution, which was unambiguously proved by X-ray analysis of diethyl (9-heptyl-2-(4-phenyl-1H-1,2,3-triazol-1-yl)-9H-purin-6-yl)phosphonate.

Graphical Abstract
Introduction
Acyclic nucleoside phosphonates (ANPs) are an important compound class due to their biological activity profile [1-6]. Compounds bearing a phosphonate moiety in their N9 side chain are well known as antiviral agents, such as adefovir, tenofovir, and cidofovir [7]. Lately, it was found that ANPs possess inhibitory activity against hypoxanthine-guanine-xanthine phosphoribosyltransferase of the parasite Plasmodium falciparum, and several research groups are focused on the development of this topic [8-11].
On the contrary, only a few examples can be found in the literature where a phosphorus-containing substituent is directly attached to the purine ring [12,13]. In 2008, an SNAr–Arbuzov reaction was developed for 6-chloropurine derivatives under microwave irradiation (Scheme 1) [12]. In 2011, a single example of a C6-phosphonate, B (X = NH2; R1 = 2’-C-methylribose; R2 = Et), was synthesized among other compounds as a potential anti-hepatitis C virus agent and showed 19% inhibition at 10 μM in Huh7 cells (Scheme 1) [13]. Additionally, there are a few examples of C8-phosphonate synthesis. They can be obtained by 1) the reaction of a lithiated C8 position with diethyl chlorophosphate (C→D, Scheme 1) [14] and 2) an intermolecular [15] or intramolecular [16] photochemical reaction between 8-bromopurine derivatives and phosphite (E→F and G→H, respectively, Scheme 1). Further, the synthesis of C8-phosphonates of 7- and 9-deazapurines via C–H phosphonation has been reported [17].
![[1860-5397-17-19-i1]](/bjoc/content/inline/1860-5397-17-19-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Structural diversity and synthetic methods of purinylphosphonates. MWI = microwave irradiation; LG = leaving group.
Scheme 1: Structural diversity and synthetic methods of purinylphosphonates. MWI = microwave irradiation; LG ...
On the other hand, azolylpurines are an important compound class that combines two recognized structural motifs of drug design – purines and azoles. Derivatives of this class are known for their activity against Mycobacterium tuberculosis and also as agonists and antagonists of adenosine receptors [18].
In 2013, we developed an efficient approach for the synthesis of ribo- and arabino-2,6-bistriazolylpurine nucleosides and showed that the triazolyl ring in the C6 position of purine acts as a good leaving group in SNAr reactions with S- and N-nucleophiles [19-21]. It is worth to note that 2/6-amino-6/2-triazolylpurines possess high levels of fluorescence [19,22-24].
Herein, we describe an extension for SNAr reactions that makes use of the 1,2,3-triazole leaving group of 2,6-bistriazolylpurines. This led to a discovery of novel C–P bond formations from C–N bonds in SNAr–Arbuzov reactions (I→J, Scheme 1). The obtained series of compounds combines three structural motifs that are important in terms of medicinal chemistry in one molecule: purine, triazole, and phosphonate.
Results and Discussion
Synthetic approaches towards C6-phosphonated 2-triazolylpurines
Aiming to synthesize C6-phosphonated 2-triazolylpurines, we designed two synthetic routes (Scheme 2). Pathway A included: 1) a known SNAr–Arbuzov reaction between 2,6-dichloropurine derivative 1 and P(OEt)3 [12], 2) substitution of chlorine at the purine C2 position by azide, and 3) copper-catalyzed azide–alkyne 1,3-dipolar cycloaddition (CuAAC) with different alkynes. Pathway B included: 1) the two-step synthesis of 2,6-bistriazolylpurine derivatives 6 from 2,6-dichloropurine derivative 1 [22] and 2) the SNAr–Arbuzov reaction with phosphite.
![[1860-5397-17-19-i2]](/bjoc/content/inline/1860-5397-17-19-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthetic routes for the formation of purinylphosphonates 4.
Scheme 2: Synthetic routes for the formation of purinylphosphonates 4.
The SNAr–Arbuzov reaction between 2,6-dichloropurine derivative 1 and triethylphosphite gave product 2a in 82% yield (Scheme 3) [12]. Next, attempts to substitute the chlorine atom at the purine C2 position were made using either NaN3 or TBAN3. Azidation experiments were tried in solvents such as EtOH, MeOH, and MeCN in temperature diapasons up to 100 °C, but no conversion of the staring material 2a (R1 = Et) was observed. The change of the solvent to DMF or DMSO resulted in the cleavage of one ethyl ester group [25], but still the SNAr reaction at C2 was not effective. LC–MS analysis of the crude reaction mixtures revealed the presence of the products 7a and 8a (Scheme 3). When the latter mixture was submitted to CuAAC with phenylacetylene (CuI/Et3N/AcOH/EtOH (or DCM), CuSO4∙5H2O/sodium ascorbate/EtOH (or DMF)), no triazole formation at the purine C2 position was observed.
![[1860-5397-17-19-i3]](/bjoc/content/inline/1860-5397-17-19-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of phosphonates 2, 7, and 8.
Scheme 3: Synthesis of phosphonates 2, 7, and 8.
We briefly tried to optimize the Cl→N3 SNAr process at the purine C2 position, and that way, the isopropyl phosphonate 2b was also obtained. It is known that both chloride and azide can cleave phosphonate esters [25-28], but the chloride source would not interfere with the SNAr process at C2. Hence, we compared the reaction outcome and rates when DMSO-d6 solutions of the starting materials 2a and 2b were treated either with NaN3 or NaCl in parallel experiments. The reaction mixtures were directly analyzed by 1H and 31P NMR spectroscopy using 1,2,3-trimethoxybenzene as an internal standard (Tables S1 and S2 as well as Figures S1 and S2 in Supporting Information File 1). The reaction between the diethyl phosphonate 2a and NaN3 gave a mixture of products 3a, 7a, and 8a already after 15 min. A significant amount of the azido monoester 8a (39%) was formed in only 48 h (Scheme 4, Figure 1, and Table S1 in Supporting Information File 1). The cleavage of the ester groups in the presence of NaCl was slower than in the presence of NaN3 (Figure 1 and Table S2 in Supporting Information File 1). Further, the cleavage of the sterically bulky isopropyl ester from phosphonate 2b showed a similar pattern: 5% conversation to monoester 7b was observed with NaCl after 48 h (Scheme 4, Figure 1, and Table S2 in Supporting Information File 1), but the reaction with NaN3 resulted in a mixture of products, which contained 45% of 2-azido monoester 8b (Scheme 4).
![[1860-5397-17-19-i4]](/bjoc/content/inline/1860-5397-17-19-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of phosphonic acid monoesters 3 and 7–9 as well as phosphonic acid 10.
Scheme 4: Synthesis of phosphonic acid monoesters 3 and 7–9 as well as phosphonic acid 10.
![[1860-5397-17-19-1]](/bjoc/content/figures/1860-5397-17-19-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Screenings of the rate for the ester group cleavage (conversion determined by NMR spectroscopy) in the reactions between dialkyl (2-chloro-9-heptyl-9H-purin-6-yl)phosphonates 2a and 2b, respectively, with NaN3 (a, c) and NaCl (b, d). Reaction conditions: DMSO-d6, 90 °C.
Figure 1: Screenings of the rate for the ester group cleavage (conversion determined by NMR spectroscopy) in ...
Based on the previous observations, we forced the SNAr reaction of the Cl atom at the C2 position of purine with an excess of NaN3, and after chromatographic isolation. We obtained the pure azido-substituted phosphonate monoesters 9a and 9b in 28 and 23% yield, respectively (Scheme 4). The products 9a and 9b were further submitted to CuAAC reactions, but the desired triazole derivatives were not obtained. Furthermore, the hydrolysis of the dialkyl ester groups were performed with TMSI [29,30], and phosphonic acid 10 was obtained. The latter was inert to the SNAr reaction with NaN3 at C2 (Scheme 4).
SNAr–Arbuzov reaction between 2,6-bistriazolylpurines and P(OEt)3
Next, we switched to pathway B (Scheme 2) and prepared 2,6-diazidopurine derivative 5 from 2,6-dichloropurine (11) via a Mitsunobu alkylation and SNAr reaction with NaN3 (Scheme 5) [22]. 2,6-Bistriazolylpurine derivatives 6a–i were obtained in CuAAC reactions with various alkynes in 35–76% yield (Table 1). We found that a combination of CuI with an amine buffer system [31-37] suites substrate 5 better than the previously used CuSO4∙5H2O and sodium ascorbate catalytic system [22]. Most probably, this is due to the solubility issues of the starting material 5 in aqueous solutions, as used in the Cu(II) and ascorbate protocol. In some cases, the use of Et3N lowered the yield of 2,6-bistriazolylpurines 6c and 6f–i due to the competing Glaser coupling [38,39] and the reduction of 2,6-diazide 5 by the Cu(I) species [40,41]. The bistriazolyl derivatives 6a–i were easily crystalized from MeOH, EtOH, or a hexane/EtOH mixture or purified by column chromatography.
![[1860-5397-17-19-i5]](/bjoc/content/inline/1860-5397-17-19-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Synthesis of 2,6-bistriazolylpurine derivatives 6a–i.
Scheme 5: Synthesis of 2,6-bistriazolylpurine derivatives 6a–i.
Table 1: Synthesis of 2,6-bistriazolylpurines 6a–i (5→6a–i) according to Scheme 5.
| entry | R | additive | t, h | yield of 6, % |
| 1 |
![[Graphic 1]](/bjoc/content/inline/1860-5397-17-19-i7.svg?max-width=637&scale=1.0)
|
DIPEA | 2 | 6a, 76 |
| 2 |
![[Graphic 2]](/bjoc/content/inline/1860-5397-17-19-i8.svg?max-width=637&scale=1.0)
|
DIPEA | 9 | 6b, 73 |
| 3 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-17-19-i9.svg?max-width=637&scale=1.0)
|
Et3N | 3.5 | 6c, 57 |
| 4 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-17-19-i10.svg?max-width=637&scale=1.0)
|
Et3N | 3 | 6d, 70 |
| 5 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-17-19-i11.svg?max-width=637&scale=1.0)
|
Et3N | 12 | 6e, 70 |
| 6 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-17-19-i12.svg?max-width=637&scale=1.0)
|
Et3N | 1.5 | 6f, 46 |
| 7 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-17-19-i13.svg?max-width=637&scale=1.0)
|
Et3N | 15 | 6g, 35 |
| 8 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-17-19-i14.svg?max-width=637&scale=1.0)
|
Et3N | 3 | 6h, 58 |
| 9 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-17-19-i15.svg?max-width=637&scale=1.0)
|
Et3N | 9 | 6i, 35 |
The obtained 2,6-bistriazolylpurine derivatives 6a–i were explored as substrates for the SNAr–Arbuzov reaction with P(OEt)3. In attempts to perform the SNAr–Arbuzov reaction in common laboratory solvents, such as toluene, MeCN, and DCM, and in the presence of 1–20 equiv of P(OEt)3, the formation of the desired phosphonates 4 was not observed (Scheme 6). We started an optimization of the reaction conditions using substrate 6d, and reactions in neat phosphite at various temperatures were tried (Table 2). The conversion of starting material 6d was monitored by HPLC, and after completion, product 4d was precipitated from the reaction mixture by hexane. For entries 1 and 3 in Table 2, an extra purification step by silica gel column chromatography was required. For compound 4d, the optimal reaction conditions were 2 hours in neat P(OEt)3 at 160 °C.
![[1860-5397-17-19-i6]](/bjoc/content/inline/1860-5397-17-19-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: SNAr–Arbuzov reaction between the bistriazolylpurines 6a–i and P(OEt)3.
Scheme 6: SNAr–Arbuzov reaction between the bistriazolylpurines 6a–i and P(OEt)3.
Table 2: Optimization of the SNAr–Arbuzov reaction conditions for 6d→4d according to Scheme 6.
| entry | T, °C | t, h | conversion of 6d, %a | yield, %b |
| 1 | 140 | 1 | 9 | 30 |
| 2 | 19 | |||
| 3 | 49 | |||
| 4 | 75 | |||
| 5 | 85 | |||
| 6 | 91 | |||
| 2 | 150 | 1 | 63 | 50 |
| 2 | 81 | |||
| 3 | 91 | |||
| 3 | 160 | 1 | 85 | 67 |
| 2 | 96 | |||
| 4 | 170 | 1 | 92 | 50 |
| 2 | 96 | |||
aThe conversion was determined by HPLC analysis (column: XBridge C18, 4.6 × 150 mm, particle size 3.5 μm, flow rate 1 mL/min. Gradient: 30–95% B 5 min, 95% B 5 min, 95–30% B 2 min. Eluent A: 0.1% TFA in water with 5 vol % MeCN; eluent B: MeCN). bIsolated yield after purification.
With the experimental conditions in hand, the SNAr–Arbuzov reaction between 2,6-bistriazolylpurines 6a–i and P(OEt)3 provided a library of novel purine phosphonates 4a–i in 27–82% yield (Table 3). The products 4a, 4d, 4e, and 4i were easily precipitated from hexane left at −20 °C within 10 hours and were then filtered and washed with cold hexane. The product purity, if necessary, was further improved by column chromatography. Some phosphonates, for example, 4b, 4c, and 4f, were reductant to precipitate from hexane and were purified solely by silica gel column chromatography. At the preparative level, the excess of P(OEt)3 was evaporated under vacuum (5 mbar) over 4–5 hours at 50 °C before further purification.
Table 3: SNAr–Arbuzov reactions between 2,6-bistriazolylpurines 6a–i and P(OEt)3 according to Scheme 6.
| entry | R | t, h | yield of 4, % |
| 1 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-17-19-i16.svg?max-width=637&scale=1.0)
|
3 | 4a, 72 |
| 2 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-17-19-i17.svg?max-width=637&scale=1.0)
|
20 | 4b, 44 |
| 3 |
![[Graphic 12]](/bjoc/content/inline/1860-5397-17-19-i18.svg?max-width=637&scale=1.0)
|
14 | 4c, 30 |
| 4 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-17-19-i19.svg?max-width=637&scale=1.0)
|
6 | 4d, 76 |
| 5 |
![[Graphic 14]](/bjoc/content/inline/1860-5397-17-19-i20.svg?max-width=637&scale=1.0)
|
2 | 4e, 82 |
| 6 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-17-19-i21.svg?max-width=637&scale=1.0)
|
23 | 4f, 40 |
| 7 |
![[Graphic 16]](/bjoc/content/inline/1860-5397-17-19-i22.svg?max-width=637&scale=1.0)
|
9 | 4g, 80 |
| 8 |
![[Graphic 17]](/bjoc/content/inline/1860-5397-17-19-i23.svg?max-width=637&scale=1.0)
|
8 | 4h, 27 |
| 9 |
![[Graphic 18]](/bjoc/content/inline/1860-5397-17-19-i24.svg?max-width=637&scale=1.0)
|
14 | 4i, 70 |
The regioselectivity of the newly developed SNAr–Arbuzov reaction was unambiguously established by X-ray analysis of the product 4d, which was crystalized from a mixture of hexane and DCM using the slow-evaporation technique (Figure 2). This follows the previously reported regioselective C6-substitution of 2,6-bistriazolylpurines in SNAr transformations.
![[1860-5397-17-19-2]](/bjoc/content/figures/1860-5397-17-19-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Single-crystal X-ray analysis of diethyl (9-heptyl-2-(4-phenyl-1H-1,2,3-triazol-1-yl)-9H-purin-6-yl)phosphonate (4d). CCDC-2044976.
Figure 2: Single-crystal X-ray analysis of diethyl (9-heptyl-2-(4-phenyl-1H-1,2,3-triazol-1-yl)-9H-purin-6-yl...
Conclusion
We have developed a novel SNAr–Arbuzov transformation that makes use of 1,2,3-triazole as a leaving group. This has permitted to obtain a novel series of C6-phosphonated 2-triazolylpurine derivatives. It was also demonstrated that there is no alternative SNAr protocol towards the designed products. The synthetic intermediates, (2-chloro-9H-purin-6-yl)phosphonates, of the alternative pathway are sluggish in substitution reactions with NaN3, and the burdensomely obtained (2-azido-9H-purin-6-yl)phosphonates fail to undergo CuAAC reactions. The developed SNAr–Arbuzov reaction between 2,6-bistriazolylpurine derivatives and trialkyl phosphites is C6-regioselective, as proved by single-crystal X-ray analysis. This is similar to the previously observed substitution pattern in SNAr reactions of 2,6-bistriazolylpurine derivatives with simple N- and S-nucleophiles.
Experimental
General information
Commercially available reagents were used as received. The reactions and the purity of the synthesized compounds were monitored by HPLC and TLC analysis using silica gel 60 F254 aluminum plates (Merck). Visualization was accomplished by UV light. Column chromatography was performed on silica gel (60 Å, 40−63 μm, ROCC). The yield of the products refers to chromatographically and spectroscopically homogeneous materials.
Melting points were recorded with a Fisher Digital Melting Point Analyzer Model 355 apparatus. The infrared spectra were recorded in hexachlorobutadiene (4000−2000 cm−1) and paraffin oil (2000−450 cm−1) with an FTIR Perkin-Elmer Spectrum 100 spectrometer.
1H, 13C, and 31P NMR spectra were recorded with Bruker Avance 300 or Bruker Avance 500 spectrometers in CDCl3, DMSO-d6, and MeOD-d4. Chemical shifts (δ) are reported in ppm and coupling constants (J) in Hz. The proton (CDCl3 δ = 7.26 ppm, DMSO-d6 δ = 2.50 ppm, MeOD-d4 δ = 3.31 ppm, AcOD-d4 δ = 11.65 ppm) and carbon signals (CDCl3 δ = 77.16 ppm, DMSO-d6 δ = 39.52 ppm, MeOD-d4 δ = 49.00 ppm, AcOD-d4 δ = 178.99 ppm) for residual nondeuterated solvents were used as an internal reference for 1H and 13C NMR spectra, respectively. 1H NMR were recorded at 500 and 300 MHz and 13C NMR spectra at 125.7 and 75.5 MHz. 31P NMR spectra were recorded at 121 and 202 MHz with H3PO4 (85%) as an external standard (H3PO4 δP = 0.00 ppm). The multiplicity is assigned as follows: s – singlet, d (for 1H NMR) and D (for 13C NMR) – doublet, t – triplet, q – quartet, m – multiplet. Nontrivial peak assignments were confirmed by 1H,1H–COSY, 1H,1H-HMBC, and/or 1H,13C-HSQC 2D NMR experiments for representative products of each compound class.
Crystallographic diffraction data were collected with a NoniusKappa CCD diffractometer (Mo Kα, λ = 0.71073 Å) equipped with a low-temperature Oxford Cryosystems Cryostream Plus device.
HPLC analysis was performed using an Agilent Technologies 1200 Series system equipped with an XBridge C18 column, 4.6 × 150 mm, particle size 3.5 μm, with a flow rate of 1 mL/min, using eluent A–0.1% TFA/H2O with 5 vol % MeCN and eluent B–MeCN as the mobile phase. The wavelength of detection was 260 nm. Gradient: 30–95% B 5 min, 95% B 5 min, 95–30% B 2 min. LC–MS spectra were recorded with a Waters Acquity UPLC system equipped with an Acquilty UPLC BEH C18 1.7 μm, 2.1 × 50 mm column, using 0.1% TFA/H2O and MeCN as the mobile phase. HRMS analyses were performed on an Agilent 1290 Infinity series UPLC system equipped with an Extend C18 RRHD 2.1 × 50 mm, 1.8 μm column, connected to an Agilent 6230 TOF LC–MS mass spectrometer.
General procedures and product characterization
The synthesis and characterization of the starting materials 1 and 5 and of 2,6-bistriazolylpurine derivative 6d have been reported earlier [22].
General procedure for the SNAr–Arbuzov reaction: synthesis of 9-alkyl-2-chloro-9H-purine C6-phosphonates 2
Diethyl (2-chloro-9-heptyl-9H-purin-6-yl)phosphonate (2a): 2,6-Dichloro-9-heptyl-9H-purine (1, 1.03 g, 3.59 mmol, 1.0 equiv) was dissolved in P(OEt)3 (12 mL) and stirred for 3 h at 160 °C (HPLC control). Then, the solution was cooled to room temperature, hexane (40 mL) was added, and the mixture was left in the freezer (−20 °C) for 10 h. The precipitated colorless crystals of 2a were filtered and washed with cold hexane (4 × 5 mL). Colorless crystals (1.15 g, 82%). mp 57–59 °C; IR ν̃max (cm−1): 2924, 2858, 1334, 1243, 1021, 981; 1H NMR (CDCl3, 300 MHz) δ 8.18 (s, 1H, H–C(8)), 4.41 (quintet, 3J = 7.1 Hz, 4H, 2×H2C–O–P), 4.25 (t, 3J = 7.2 Hz, 2H, –CH2(1’)–), 2.02–1.77 (m, 2H, –CH2(2’)–), 1.40 (t, 3J = 7.1 Hz, 6H, 2×(–CH3)), 1.35–1.10 (m, 8H, 4×(–CH2–)), 0.85 (t, 3J = 6.6 Hz, 3H, –CH3(7’)); 13C NMR (CDCl3, 75.5 MHz) δ 154.3 (D, 3JC–P = 11.7 Hz), 154.2 (D, 3JC–P = 7.7 Hz), 152.5 (D, 1JC–P = 203.6 Hz), 147.5, 134.3 (D, 2JC–P = 21.4 Hz), 64.2 (D, 2JC–P = 6.1 Hz), 44.2, 31.5, 29.8, 28.6, 26.5, 22.5, 16.4 (D, 3JC–P = 6.2 Hz), 14.4; 31P NMR (CDCl3, 121 MHz) δ 5.3; HRMS-ESI (m/z): [M + H]+ calcd for C16H27ClN4O3P, 389.1504; found, 389.1508.
General procedure for the synthesis of 9-alkyl-2,6-bistriazolyl-9H-purine derivatives 6
Dimethyl 1,1'-(9-heptyl-9H-purine-2,6-diyl)bis(1H-1,2,3-triazole-4-carboxylate) (6a): CuI (0.06 g, 0.30 mmol, 0.12 equiv) was added to a stirred solution of 2,6-diazido-9-heptyl-9H-purine (5, 0.76 g, 2.53 mmol, 1.0 equiv) in DCM (35 mL), followed by the addition of triethylamine (0.39 mL, 2.78 mmol, 1.1 equiv), methyl propiolate (0.68 mL, 7.59 mmol, 3.0 equiv), and acetic acid (0.16 mL, 2.78 mmol, 1.1 equiv). The reaction mixture was stirred for 2 h at room temperature. Then, the mixture was washed with brine (1 × 7 mL) and an aqueous solution of NaHS (2 × 5 mL). The inorganic phase was back-extracted with DCM (2 × 3 mL). The organic phase was collected, dried over anhydrous Na2SO4, filtered through Celite®, and evaporated under reduced pressure. Silica gel column chromatography (DCM/MeCN, gradient: 20→33%) provided the product 6a (0.91 g, 76%) as a brown amorphous solid. Rf 0.20 (DCM/MeCN 4:1); HPLC: tR 5.72 min; IR ν̃max (cm−1): 2953, 2930, 1728, 1434, 1223, 1025, 774; 1H NMR (500 MHz, CDCl3) δ 9.63, 9.25 (2s, 2H, 2×H–C(triazole)), 8.40 (s, 1H, H–C(8)), 4.47 (t, 3J = 7.0 Hz, 2H, H2–C(1’)), 4.08 (s, 6H, 2×OMe), 2.10–1.93 (m, 2H, H2–C(2’)), 1.49–1.15 (m, 8H, 4×(–CH2–)), 0.85 (t, 3J = 6.9 Hz, 3H, H3–C(7’)); 13C NMR (125.7 MHz, CDCl3) δ 160.8, 160.5, 156.2, 148.5, 148.0, 144.8, 140.6, 140.5, 128.4, 127.5, 122.9, 52.74, 52.66, 45.1, 31.6, 29.9, 28.7, 26.7, 22.6, 14.1; HRMS-ESI (m/z): [M + H]+ calcd for C20H25N10O4, 469.2055; found, 469.2022.
General procedure for the SNAr–Arbuzov reaction: synthesis of 9-alkyl-2-triazolyl-9H-purine C6-phosphonates 4
Methyl 1-(6-(diethoxyphosphoryl)-9-heptyl-9H-purin-2-yl)-1H-1,2,3-triazole-4-carboxylate (4a): Dimethyl 1,1'-(9-heptyl-9H-purine-2,6-diyl)bis(1H-1,2,3-triazole-4-carboxylate) (6a, 0.20 g, 0.43 mmol, 1.0 equiv) was dissolved in P(OEt)3 (2 mL) and stirred for 3 hours at 160 °C. Then, the solution was cooled to room temperature, hexane (10 mL) was added, and the mixture was left in the freezer (−20 °C) for 10 h. The brown solids were filtered, washed with cold hexane (4 × 5 mL), then dissolved from the filter with DCM (10 mL) and purified by silica gel column chromatography (DCM/MeOH, gradient: 3→5%). Orange powder (0.148 g, 72%). Rf 0.2 (DCM/MeOH 25:1); IR ν̃max (cm−1): 2980, 2930, 1250, 1143, 1102, 990; 1H NMR (500 MHz, CDCl3) δ 9.19 (s, 1H, H–C(triazole)), 8.32 (s, 1H, H–C(8)), 4.53–4.42 (m, 4H, 2×H2C–O–P), 4.40 (t, 3J = 7.3 Hz, 2H, H2–C(1’)), 4.00 (s, 3H, H3C–O–CO), 2.07–1.84 (m, 2H, H2–C(2’)), 1.45 (t, 3J = 7.1 Hz, 6H, 2×(–CH3)), 1.37–1.15 (m, 8H, 4×(–CH2–)), 0.85 (t, 3J = 6.9 Hz, 3H, H3–C(7’)); 13C NMR (125.7 MHz, CDCl3) δ 160.9, 154.0 (D, 3JC–P = 11.1 Hz), 152.4 (D, 1JC–P = 220.6 Hz), 148.6, 148.3 (D, 3JC–P = 23.4 Hz), 140.3, 135.3 (D, 2JC–P = 20.8 Hz), 127.4, 64.5 (D, 2JC–P = 6.2 Hz), 52.6, 44.6, 31.6, 29.9, 28.7, 26.7, 22.7, 16.6 (D, 3JC–P = 5.9 Hz), 14.1; 31P NMR (202 MHz, CDCl3) δ 5.3; HRMS-ESI (m/z): [M + H]+ calcd for C20H30N7O5P, 480.2119; found, 480.2121.
References
-
Gazivoda, T.; Plevnik, M.; Plavec, J.; Kraljević, S.; Kralj, M.; Pavelić, K.; Balzarini, J.; De Clercq, E.; Mintas, M.; Raić-Malić, S. Bioorg. Med. Chem. 2005, 13, 131–139. doi:10.1016/j.bmc.2004.09.052
Return to citation in text: [1] -
Holý, A. Antiviral Res. 2006, 71, 248–253. doi:10.1016/j.antiviral.2006.06.002
Return to citation in text: [1] -
De Clercq, E. Biochem. Pharmacol. 2011, 82, 99–109. doi:10.1016/j.bcp.2011.03.027
Return to citation in text: [1] -
Macchi, B.; Romeo, G.; Chiacchio, U.; Frezza, C.; Giofrè, S. V.; Marino-Merlo, F.; Mastino, A. Phosphonated Nucleoside Analogues as Antiviral Agents. In Therapy of Viral Infections; Diederich, W. E.; Steuber, H., Eds.; Topics in Medicinal Chemistry, Vol. 15; Springer: Berlin, Heidelberg, Germany, 2013; pp 53–91. doi:10.1007/7355_2013_28
Return to citation in text: [1] -
Pradere, U.; Garnier-Amblard, E. C.; Coats, S. J.; Amblard, F.; Schinazi, R. F. Chem. Rev. 2014, 114, 9154–9218. doi:10.1021/cr5002035
Return to citation in text: [1] -
Hocková, D.; Rosenbergová, Š.; Ménová, P.; Páv, O.; Pohl, R.; Novák, P.; Rosenberg, I. Org. Biomol. Chem. 2015, 13, 4449–4458. doi:10.1039/c4ob02265c
Return to citation in text: [1] -
Andrei, G.; Topalis, D.; De Schutter, T.; Snoeck, R. Antiviral Res. 2015, 114, 21–46. doi:10.1016/j.antiviral.2014.10.012
Return to citation in text: [1] -
Klejch, T.; Pohl, R.; Janeba, Z.; Sun, M.; Keough, D. T.; Guddat, L. W.; Hocková, D. Tetrahedron 2018, 74, 5886–5897. doi:10.1016/j.tet.2018.08.014
Return to citation in text: [1] -
Klejch, T.; Keough, D. T.; Chavchich, M.; Travis, J.; Skácel, J.; Pohl, R.; Janeba, Z.; Edstein, M. D.; Avery, V. M.; Guddat, L. W.; Hocková, D. Eur. J. Med. Chem. 2019, 183, 111667. doi:10.1016/j.ejmech.2019.111667
Return to citation in text: [1] -
Keough, D. T.; Hocková, D.; Janeba, Z.; Wang, T.-H.; Naesens, L.; Edstein, M. D.; Chavchich, M.; Guddat, L. W. J. Med. Chem. 2015, 58, 827–846. doi:10.1021/jm501416t
Return to citation in text: [1] -
Cheviet, T.; Wein, S.; Bourchenin, G.; Lagacherie, M.; Périgaud, C.; Cerdan, R.; Peyrottes, S. J. Med. Chem. 2020, 63, 8069–8087. doi:10.1021/acs.jmedchem.0c00131
Return to citation in text: [1] -
Qu, G.-R.; Xia, R.; Yang, X.-N.; Li, J.-G.; Wang, D.-C.; Guo, H.-M. J. Org. Chem. 2008, 73, 2416–2419. doi:10.1021/jo702680p
Return to citation in text: [1] [2] [3] [4] -
Zhang, H.-w.; Zhou, L.; Coats, S. J.; McBrayer, T. R.; Tharnish, P. M.; Bondada, L.; Detorio, M.; Amichai, S. A.; Johns, M. D.; Whitaker, T.; Schinazi, R. F. Bioorg. Med. Chem. Lett. 2011, 21, 6788–6792. doi:10.1016/j.bmcl.2011.09.034
Return to citation in text: [1] [2] -
Honjo, M.; Maruyama, T.; Horikawa, M.; Balzarini, J.; De Clercq, E. Chem. Pharm. Bull. 1987, 35, 3227–3234. doi:10.1248/cpb.35.3227
Return to citation in text: [1] -
Maruyama, T.; Honjo, M. Nucleosides Nucleotides 1988, 7, 203–211. doi:10.1080/07328318808070204
Return to citation in text: [1] -
Maruyama, T.; Adachi, Y.; Honjo, M. J. Org. Chem. 1988, 53, 4552–4555. doi:10.1021/jo00254a025
Return to citation in text: [1] -
Sabat, N.; Poštová Slavětínská, L.; Klepetářová, B.; Hocek, M. J. Org. Chem. 2016, 81, 9507–9514. doi:10.1021/acs.joc.6b01970
Return to citation in text: [1] -
Novosjolova, I.; Bizdēna, Ē.; Turks, M. Eur. J. Org. Chem. 2015, 3629–3649. doi:10.1002/ejoc.201403527
Return to citation in text: [1] -
Kovaļovs, A.; Novosjolova, I.; Bizdēna, Ē.; Bižāne, I.; Skardziute, L.; Kazlauskas, K.; Jursenas, S.; Turks, M. Tetrahedron Lett. 2013, 54, 850–853. doi:10.1016/j.tetlet.2012.11.095
Return to citation in text: [1] [2] -
Novosjolova, I.; Bizdēna, Ē.; Turks, M. Tetrahedron Lett. 2013, 54, 6557–6561. doi:10.1016/j.tetlet.2013.09.095
Return to citation in text: [1] -
Novosjolova, I.; Bizdēna, Ē.; Turks, M. Phosphorus, Sulfur Silicon Relat. Elem. 2015, 190, 1236–1241. doi:10.1080/10426507.2014.989435
Return to citation in text: [1] -
Šišuļins, A.; Bucevičius, J.; Tseng, Y.-T.; Novosjolova, I.; Traskovskis, K.; Bizdēna, Ē.; Chang, H.-T.; Tumkevičius, S.; Turks, M. Beilstein J. Org. Chem. 2019, 15, 474–489. doi:10.3762/bjoc.15.41
Return to citation in text: [1] [2] [3] [4] [5] -
Zayas, J.; Annoual, M.; Das, J. K.; Felty, Q.; Gonzalez, W. G.; Miksovska, J.; Sharifai, N.; Chiba, A.; Wnuk, S. F. Bioconjugate Chem. 2015, 26, 1519–1532. doi:10.1021/acs.bioconjchem.5b00300
Return to citation in text: [1] -
Dyrager, C.; Börjesson, K.; Dinér, P.; Elf, A.; Albinsson, B.; Wilhelmsson, L. M.; Grøtli, M. Eur. J. Org. Chem. 2009, 1515–1521. doi:10.1002/ejoc.200900018
Return to citation in text: [1] -
Holý, A. Synthesis 1998, 381–385. doi:10.1055/s-1998-2047
Return to citation in text: [1] [2] -
Rabinowitz, R. J. Am. Chem. Soc. 1960, 82, 4564–4567. doi:10.1021/ja01502a030
Return to citation in text: [1] -
Krawczyk, H. Synth. Commun. 1997, 27, 3151–3161. doi:10.1080/00397919708004173
Return to citation in text: [1] -
Abramov, V. S.; Sergeyeva, Y. V.; Chelpanova, I. V. J. Gen. Chem. USSR 1944, 14, 1030–1033.
Return to citation in text: [1] -
Olah, G. A.; Narang, S. C. Tetrahedron 1982, 38, 2225–2277. doi:10.1016/0040-4020(82)87002-6
Return to citation in text: [1] -
Zygmunt, J.; Kafarski, P.; Mastalerz, P. Synthesis 1978, 609–612. doi:10.1055/s-1978-24832
Return to citation in text: [1] -
Shao, C.; Wang, X.; Zhang, Q.; Luo, S.; Zhao, J.; Hu, Y. J. Org. Chem. 2011, 76, 6832–6836. doi:10.1021/jo200869a
Return to citation in text: [1] -
Hein, J. E.; Fokin, V. V. Chem. Soc. Rev. 2010, 39, 1302–1315. doi:10.1039/b904091a
Return to citation in text: [1] -
Liang, L.; Astruc, D. Coord. Chem. Rev. 2011, 255, 2933–2945. doi:10.1016/j.ccr.2011.06.028
Return to citation in text: [1] -
Singh, M. S.; Chowdhury, S.; Koley, S. Tetrahedron 2016, 72, 5257–5283. doi:10.1016/j.tet.2016.07.044
Return to citation in text: [1] -
Smith, N. W.; Polenz, B. P.; Johnson, S. B.; Dzyuba, S. V. Tetrahedron Lett. 2010, 51, 550–553. doi:10.1016/j.tetlet.2009.11.089
Return to citation in text: [1] -
Hein, C. D.; Liu, X.-M.; Wang, D. Pharm. Res. 2008, 25, 2216–2230. doi:10.1007/s11095-008-9616-1
Return to citation in text: [1] -
Bock, V. D.; Hiemstra, H.; van Maarseveen, J. H. Eur. J. Org. Chem. 2006, 51–68. doi:10.1002/ejoc.200500483
Return to citation in text: [1] -
Siemsen, P.; Livingston, R. C.; Diederich, F. Angew. Chem., Int. Ed. 2000, 39, 2632–2657. doi:10.1002/1521-3773(20000804)39:15<2632::aid-anie2632>3.0.co;2-f
Return to citation in text: [1] -
Sindhu, K. S.; Anilkumar, G. RSC Adv. 2014, 4, 27867–27887. doi:10.1039/c4ra02416h
Return to citation in text: [1] -
Zelenay, B.; Besora, M.; Monasterio, Z.; Ventura-Espinosa, D.; White, A. J. P.; Maseras, F.; Díez-González, S. Catal. Sci. Technol. 2018, 8, 5763–5773. doi:10.1039/c8cy00515j
Return to citation in text: [1] -
Ozols, K.; Cīrule, D.; Novosjolova, I.; Stepanovs, D.; Liepinsh, E.; Bizdēna, Ē.; Turks, M. Tetrahedron Lett. 2016, 57, 1174–1178. doi:10.1016/j.tetlet.2016.02.003
Return to citation in text: [1]
| 1. | Gazivoda, T.; Plevnik, M.; Plavec, J.; Kraljević, S.; Kralj, M.; Pavelić, K.; Balzarini, J.; De Clercq, E.; Mintas, M.; Raić-Malić, S. Bioorg. Med. Chem. 2005, 13, 131–139. doi:10.1016/j.bmc.2004.09.052 |
| 2. | Holý, A. Antiviral Res. 2006, 71, 248–253. doi:10.1016/j.antiviral.2006.06.002 |
| 3. | De Clercq, E. Biochem. Pharmacol. 2011, 82, 99–109. doi:10.1016/j.bcp.2011.03.027 |
| 4. | Macchi, B.; Romeo, G.; Chiacchio, U.; Frezza, C.; Giofrè, S. V.; Marino-Merlo, F.; Mastino, A. Phosphonated Nucleoside Analogues as Antiviral Agents. In Therapy of Viral Infections; Diederich, W. E.; Steuber, H., Eds.; Topics in Medicinal Chemistry, Vol. 15; Springer: Berlin, Heidelberg, Germany, 2013; pp 53–91. doi:10.1007/7355_2013_28 |
| 5. | Pradere, U.; Garnier-Amblard, E. C.; Coats, S. J.; Amblard, F.; Schinazi, R. F. Chem. Rev. 2014, 114, 9154–9218. doi:10.1021/cr5002035 |
| 6. | Hocková, D.; Rosenbergová, Š.; Ménová, P.; Páv, O.; Pohl, R.; Novák, P.; Rosenberg, I. Org. Biomol. Chem. 2015, 13, 4449–4458. doi:10.1039/c4ob02265c |
| 12. | Qu, G.-R.; Xia, R.; Yang, X.-N.; Li, J.-G.; Wang, D.-C.; Guo, H.-M. J. Org. Chem. 2008, 73, 2416–2419. doi:10.1021/jo702680p |
| 22. | Šišuļins, A.; Bucevičius, J.; Tseng, Y.-T.; Novosjolova, I.; Traskovskis, K.; Bizdēna, Ē.; Chang, H.-T.; Tumkevičius, S.; Turks, M. Beilstein J. Org. Chem. 2019, 15, 474–489. doi:10.3762/bjoc.15.41 |
| 12. | Qu, G.-R.; Xia, R.; Yang, X.-N.; Li, J.-G.; Wang, D.-C.; Guo, H.-M. J. Org. Chem. 2008, 73, 2416–2419. doi:10.1021/jo702680p |
| 13. | Zhang, H.-w.; Zhou, L.; Coats, S. J.; McBrayer, T. R.; Tharnish, P. M.; Bondada, L.; Detorio, M.; Amichai, S. A.; Johns, M. D.; Whitaker, T.; Schinazi, R. F. Bioorg. Med. Chem. Lett. 2011, 21, 6788–6792. doi:10.1016/j.bmcl.2011.09.034 |
| 12. | Qu, G.-R.; Xia, R.; Yang, X.-N.; Li, J.-G.; Wang, D.-C.; Guo, H.-M. J. Org. Chem. 2008, 73, 2416–2419. doi:10.1021/jo702680p |
| 8. | Klejch, T.; Pohl, R.; Janeba, Z.; Sun, M.; Keough, D. T.; Guddat, L. W.; Hocková, D. Tetrahedron 2018, 74, 5886–5897. doi:10.1016/j.tet.2018.08.014 |
| 9. | Klejch, T.; Keough, D. T.; Chavchich, M.; Travis, J.; Skácel, J.; Pohl, R.; Janeba, Z.; Edstein, M. D.; Avery, V. M.; Guddat, L. W.; Hocková, D. Eur. J. Med. Chem. 2019, 183, 111667. doi:10.1016/j.ejmech.2019.111667 |
| 10. | Keough, D. T.; Hocková, D.; Janeba, Z.; Wang, T.-H.; Naesens, L.; Edstein, M. D.; Chavchich, M.; Guddat, L. W. J. Med. Chem. 2015, 58, 827–846. doi:10.1021/jm501416t |
| 11. | Cheviet, T.; Wein, S.; Bourchenin, G.; Lagacherie, M.; Périgaud, C.; Cerdan, R.; Peyrottes, S. J. Med. Chem. 2020, 63, 8069–8087. doi:10.1021/acs.jmedchem.0c00131 |
| 19. | Kovaļovs, A.; Novosjolova, I.; Bizdēna, Ē.; Bižāne, I.; Skardziute, L.; Kazlauskas, K.; Jursenas, S.; Turks, M. Tetrahedron Lett. 2013, 54, 850–853. doi:10.1016/j.tetlet.2012.11.095 |
| 22. | Šišuļins, A.; Bucevičius, J.; Tseng, Y.-T.; Novosjolova, I.; Traskovskis, K.; Bizdēna, Ē.; Chang, H.-T.; Tumkevičius, S.; Turks, M. Beilstein J. Org. Chem. 2019, 15, 474–489. doi:10.3762/bjoc.15.41 |
| 23. | Zayas, J.; Annoual, M.; Das, J. K.; Felty, Q.; Gonzalez, W. G.; Miksovska, J.; Sharifai, N.; Chiba, A.; Wnuk, S. F. Bioconjugate Chem. 2015, 26, 1519–1532. doi:10.1021/acs.bioconjchem.5b00300 |
| 24. | Dyrager, C.; Börjesson, K.; Dinér, P.; Elf, A.; Albinsson, B.; Wilhelmsson, L. M.; Grøtli, M. Eur. J. Org. Chem. 2009, 1515–1521. doi:10.1002/ejoc.200900018 |
| 7. | Andrei, G.; Topalis, D.; De Schutter, T.; Snoeck, R. Antiviral Res. 2015, 114, 21–46. doi:10.1016/j.antiviral.2014.10.012 |
| 12. | Qu, G.-R.; Xia, R.; Yang, X.-N.; Li, J.-G.; Wang, D.-C.; Guo, H.-M. J. Org. Chem. 2008, 73, 2416–2419. doi:10.1021/jo702680p |
| 16. | Maruyama, T.; Adachi, Y.; Honjo, M. J. Org. Chem. 1988, 53, 4552–4555. doi:10.1021/jo00254a025 |
| 18. | Novosjolova, I.; Bizdēna, Ē.; Turks, M. Eur. J. Org. Chem. 2015, 3629–3649. doi:10.1002/ejoc.201403527 |
| 15. | Maruyama, T.; Honjo, M. Nucleosides Nucleotides 1988, 7, 203–211. doi:10.1080/07328318808070204 |
| 19. | Kovaļovs, A.; Novosjolova, I.; Bizdēna, Ē.; Bižāne, I.; Skardziute, L.; Kazlauskas, K.; Jursenas, S.; Turks, M. Tetrahedron Lett. 2013, 54, 850–853. doi:10.1016/j.tetlet.2012.11.095 |
| 20. | Novosjolova, I.; Bizdēna, Ē.; Turks, M. Tetrahedron Lett. 2013, 54, 6557–6561. doi:10.1016/j.tetlet.2013.09.095 |
| 21. | Novosjolova, I.; Bizdēna, Ē.; Turks, M. Phosphorus, Sulfur Silicon Relat. Elem. 2015, 190, 1236–1241. doi:10.1080/10426507.2014.989435 |
| 14. | Honjo, M.; Maruyama, T.; Horikawa, M.; Balzarini, J.; De Clercq, E. Chem. Pharm. Bull. 1987, 35, 3227–3234. doi:10.1248/cpb.35.3227 |
| 13. | Zhang, H.-w.; Zhou, L.; Coats, S. J.; McBrayer, T. R.; Tharnish, P. M.; Bondada, L.; Detorio, M.; Amichai, S. A.; Johns, M. D.; Whitaker, T.; Schinazi, R. F. Bioorg. Med. Chem. Lett. 2011, 21, 6788–6792. doi:10.1016/j.bmcl.2011.09.034 |
| 17. | Sabat, N.; Poštová Slavětínská, L.; Klepetářová, B.; Hocek, M. J. Org. Chem. 2016, 81, 9507–9514. doi:10.1021/acs.joc.6b01970 |
| 29. | Olah, G. A.; Narang, S. C. Tetrahedron 1982, 38, 2225–2277. doi:10.1016/0040-4020(82)87002-6 |
| 30. | Zygmunt, J.; Kafarski, P.; Mastalerz, P. Synthesis 1978, 609–612. doi:10.1055/s-1978-24832 |
| 25. | Holý, A. Synthesis 1998, 381–385. doi:10.1055/s-1998-2047 |
| 26. | Rabinowitz, R. J. Am. Chem. Soc. 1960, 82, 4564–4567. doi:10.1021/ja01502a030 |
| 27. | Krawczyk, H. Synth. Commun. 1997, 27, 3151–3161. doi:10.1080/00397919708004173 |
| 28. | Abramov, V. S.; Sergeyeva, Y. V.; Chelpanova, I. V. J. Gen. Chem. USSR 1944, 14, 1030–1033. |
| 40. | Zelenay, B.; Besora, M.; Monasterio, Z.; Ventura-Espinosa, D.; White, A. J. P.; Maseras, F.; Díez-González, S. Catal. Sci. Technol. 2018, 8, 5763–5773. doi:10.1039/c8cy00515j |
| 41. | Ozols, K.; Cīrule, D.; Novosjolova, I.; Stepanovs, D.; Liepinsh, E.; Bizdēna, Ē.; Turks, M. Tetrahedron Lett. 2016, 57, 1174–1178. doi:10.1016/j.tetlet.2016.02.003 |
| 22. | Šišuļins, A.; Bucevičius, J.; Tseng, Y.-T.; Novosjolova, I.; Traskovskis, K.; Bizdēna, Ē.; Chang, H.-T.; Tumkevičius, S.; Turks, M. Beilstein J. Org. Chem. 2019, 15, 474–489. doi:10.3762/bjoc.15.41 |
| 22. | Šišuļins, A.; Bucevičius, J.; Tseng, Y.-T.; Novosjolova, I.; Traskovskis, K.; Bizdēna, Ē.; Chang, H.-T.; Tumkevičius, S.; Turks, M. Beilstein J. Org. Chem. 2019, 15, 474–489. doi:10.3762/bjoc.15.41 |
| 38. | Siemsen, P.; Livingston, R. C.; Diederich, F. Angew. Chem., Int. Ed. 2000, 39, 2632–2657. doi:10.1002/1521-3773(20000804)39:15<2632::aid-anie2632>3.0.co;2-f |
| 39. | Sindhu, K. S.; Anilkumar, G. RSC Adv. 2014, 4, 27867–27887. doi:10.1039/c4ra02416h |
| 22. | Šišuļins, A.; Bucevičius, J.; Tseng, Y.-T.; Novosjolova, I.; Traskovskis, K.; Bizdēna, Ē.; Chang, H.-T.; Tumkevičius, S.; Turks, M. Beilstein J. Org. Chem. 2019, 15, 474–489. doi:10.3762/bjoc.15.41 |
| 31. | Shao, C.; Wang, X.; Zhang, Q.; Luo, S.; Zhao, J.; Hu, Y. J. Org. Chem. 2011, 76, 6832–6836. doi:10.1021/jo200869a |
| 32. | Hein, J. E.; Fokin, V. V. Chem. Soc. Rev. 2010, 39, 1302–1315. doi:10.1039/b904091a |
| 33. | Liang, L.; Astruc, D. Coord. Chem. Rev. 2011, 255, 2933–2945. doi:10.1016/j.ccr.2011.06.028 |
| 34. | Singh, M. S.; Chowdhury, S.; Koley, S. Tetrahedron 2016, 72, 5257–5283. doi:10.1016/j.tet.2016.07.044 |
| 35. | Smith, N. W.; Polenz, B. P.; Johnson, S. B.; Dzyuba, S. V. Tetrahedron Lett. 2010, 51, 550–553. doi:10.1016/j.tetlet.2009.11.089 |
| 36. | Hein, C. D.; Liu, X.-M.; Wang, D. Pharm. Res. 2008, 25, 2216–2230. doi:10.1007/s11095-008-9616-1 |
| 37. | Bock, V. D.; Hiemstra, H.; van Maarseveen, J. H. Eur. J. Org. Chem. 2006, 51–68. doi:10.1002/ejoc.200500483 |
© 2021 Kriķis et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the author(s) and source are credited and that individual graphics may be subject to special legal provisions.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc/terms)