Abstract
Background
Tashiromine (1) is a naturally occurring indolizidine alkaloid. It has been the subject of thirteen successful total syntheses to date. Our own approach centres on the stereoselective construction of the indolizidine core by capture of an electrophilic acyliminium species by a pendant allylsilane. The key cyclisation precursor is constructed using olefin cross-metathesis chemistry, which has the potential to facilitate both racemic and asymmetric approaches, depending upon the choice of the allylsilane metathesis partner.
Results
The use of the allyltrimethylsilane cross-metathesis approach enables the rapid construction of the key cyclisation precursor 3 (3 steps from commercial materials), which undergoes acid-induced cyclisation to give the desired bicyclic indolizidine skeleton as a 96:4 mixture of diastereomers. Simple functional group interconversions allowed the completion of the total synthesis of racemic tashiromine in six steps (19% overall yield). Three chiral α-alkoxyallylsilanes (12, 14 and 15) were prepared in enantioenriched form and their cross-metathesis reactions studied as part of a putative asymmetric approach to tashiromine. In the event, α-hydroxysilane 12 underwent isomerisation under the reaction conditions to acylsilane 17, while silanes 14 and 15 were unreactive towards metathesis.
Conclusion
A concise, stereoselective total synthesis of racemic tashiromine has been developed. Attempts to translate this into an asymmetric synthesis have thus far been unsuccessful.

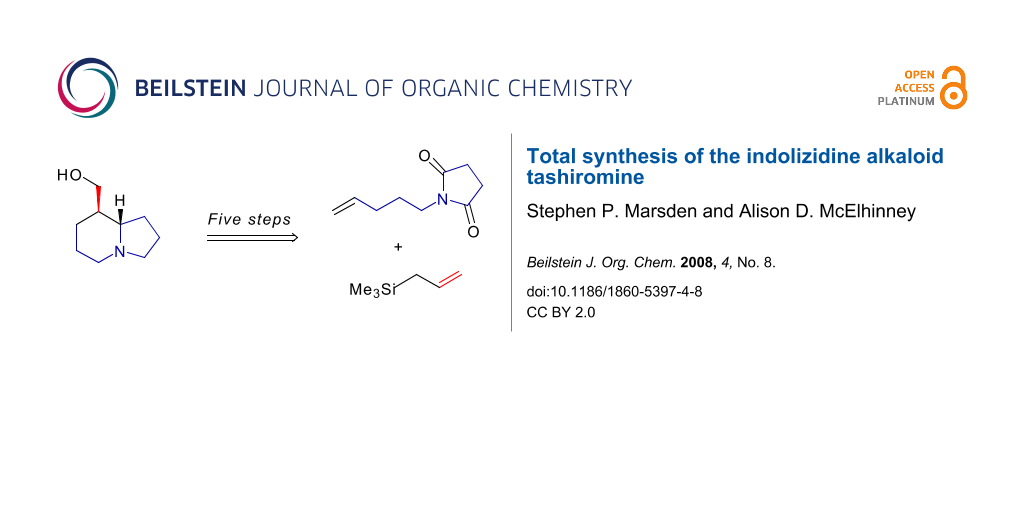
Graphical Abstract
Background
Tashiromine (1) is a naturally occurring indolizidine, isolated from an Asian deciduous shrub Maackia tashiroi [1]. As one of the structurally simpler indolizidine alkaloids [2], tashiromine has been a popular target for synthetic chemists, and to date has succumbed to total synthesis on thirteen occasions [3-15]. A wide variety of reactions have been employed to assemble the core indolizidine structure, including radical cyclisations [3]; nucleophilic addition to imines [5,14,15]; electrophilic alkylation of pyrroles [7,13]; alkylation of enamines [6], β-amino esters [8] and pyrrolidinyllithiums [12]; stereoselective reduction of enamines [4,9] and pyridinium salts [11]; and titanium-mediated reductive imide-olefin cyclisation [10]. Our own approach [14] utilises an intramolecular addition of an allylsilane to an N-acyliminium ion to deliver the [4.3.0]-azabicyclic (indolizidine) skeleton 2 (Scheme 1), wherein the pendant vinyl group acts as a handle to install the hydroxymethyl sidechain found in tashiromine. The synthesis of azabicyclic assemblies by intramolecular allylsilane/N-acyliminium cyclisations was first studied by Hiemstra and Speckamp [16], who prepared their functionalised allylsilane cyclisation precursors (such as 3) by alkylation of cyclic imides with reagent 4 (X = OMs). This, in turn, was prepared in four steps by alkylation of an acetylide anion with commercially available iodomethyltrimethylsilane, followed by partial reduction of the alkyne. Alternative synthetic approaches to 4 (X = OMs, I) involve olefination of aldehydes using the Seyferth-Fleming phosphorane [17] or nickel-catalysed 1,2-metallate rearrangement of lithiated dihydropyran [18]. Our approach was informed by the prior work by our own group [19-24] and others [25-38] on the use of olefin metathesis to generate functionalised allylsilanes. Specifically, cross-metathesis of N-pentenylsuccinimide 5 with allyltrimethylsilane (6) [39] followed by chemoselective partial reduction of the imide would give the cyclisation precursor 3 in short order. Further, the use of chiral allylsilanes as cross-metathesis partners would potentially facilitate an asymmetric approach to the total synthesis of 1. We report herein full details of the successful synthesis of racemic tashiromine 1 by this strategy [14], as well as our initial attempts towards an asymmetric variant.
![[1860-5397-4-8-i1]](/bjoc/content/inline/1860-5397-4-8-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Retrosynthesis for tashiromine.
Scheme 1: Retrosynthesis for tashiromine.
Results and Discussion
Metathesis precursor 5 was prepared by alkylation of the sodium salt of succinimide with 5-bromo-1-pentene in near quantitative yield (Scheme 2, see Supporting Information File 1 for full experimental data). The key cross-metathesis reaction of 5 was carried out using a fourfold excess of allyltrimethylsilane (6) and 5 mol% of Grubbs’ second generation catalyst in refluxing dichloromethane. The desired product 7 was formed in 73% yield as an inseparable 3:1 mixture of E- and Z-isomers. Partial reduction with sodium borohydride generated the cyclisation precursor 3 in 86% yield, again as a 3:1 mixture of olefin isomers. Exposure of this mixture to trifluoroacetic acid in dichloromethane at room temperature gave the bicyclic amide 2 in 85% yield as a 96:4 mixture of diastereomers. The identity of the major diastereomer was confirmed by comparison of the spectral data with those of Hiemstra [16]: specifically, the signal for the (ring-fusion) proton at C6 for the major diastereomer appeared as a doublet of triplets with δ = 3.19 ppm, whereas the corresponding signal for the minor diastereomer appeared at δ = 3.67 ppm. The stereochemical outcome of this reaction was rationalised on the basis of the model shown in Scheme 2, whereby nucleophilic addition of the allylsilane to the N-acyliminium ion occurs through a chair-like transition state with the nascent alkene equatorially disposed.
![[1860-5397-4-8-i2]](/bjoc/content/inline/1860-5397-4-8-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Stereoselective construction of the indolizidine core 2.
Scheme 2: Stereoselective construction of the indolizidine core 2.
All that remained to complete the synthesis of tashiromine 1 was to effect the oxidative cleavage of the C5 vinyl substituent, then carry out a global reduction of the resulting carbonyl function and the amide. In the event, attempts to form a C5 aldehyde using either ozonolytic or dihydroxylation/periodate alkene cleavage protocols were unsuccessful, with complex mixtures being obtained in both cases. We suspected that the problem lay in the potential for the desired aldehyde to undergo retro-Mannich fragmentation, and so elected to carry out a reductive work-up to the ozonolysis procedure (Scheme 3). The desired alcohol 8 was obtained in a crude form and immediately subjected to reduction with lithium aluminium hydride to give our target tashiromine 1 in 36% yield over two steps. Our stereochemical assignment for the cyclisation of 3 was further corroborated by the agreement of the spectral data for 1 with those previously published in the literature [3-5,9-12]. Additionally, the spectral data for the diastereomeric epi-tashiromine have been reported and differ significantly from those recorded for 1 [10].
![[1860-5397-4-8-i3]](/bjoc/content/inline/1860-5397-4-8-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Completion of the total synthesis of tashiromine 1.
Scheme 3: Completion of the total synthesis of tashiromine 1.
Having completed our target synthesis, our next goal was to investigate an asymmetric approach to tashiromine. Specifically, we envisaged that cyclisation precursors of type 9 ought to be readily available by cross-metathesis of 5 with an appropriate chiral allylsilane followed by chemoselective partial reduction by borohydride. Thereafter, exposure to acid would generate an N-acyliminium ion, which would cyclise through a chair-like transition state with the nascent alkenyl side-chain equatorially disposed, as in the racemic series (Figure 1). The absolute stereochemistry of the newly established asymmetric centres would be controlled by allylic strain arguments, assuming that the well-established precedent for anti-SE2’ attack of the iminium on the allylsilane was upheld here [40]. Thus, the predicted major stereoisomer 10 would have (5S,6S) stereochemistry and an E-configured side-chain, while cyclisation to the predicted minor (5R,6R) isomer 11 would be disfavoured by A1,3-interactions between the R1 group and vinylic proton (leading to the Z-configured side-chain). This would represent an immolative transfer of chirality approach to tashiromine, since the olefinic side-chains would be cleaved to install the hydroxymethyl side-chain required by the natural product.
![[1860-5397-4-8-1]](/bjoc/content/figures/1860-5397-4-8-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Rationale for stereoselective assembly of the indolizidine core using chiral allylsilanes.
Figure 1: Rationale for stereoselective assembly of the indolizidine core using chiral allylsilanes.
Our approach centred on the readily availability of chiral α-hydroxysilane 12 in enantioenriched format [41]. Protection of the hydroxyl group, either before or after cross-metathesis, would allow access to chiral allylsilanes 9 with R1 being an alkoxy or acyloxy group. Furthermore, this would generate products 10 and/or 11 with a readily oxidised enol-ether/ester side chain for progression to tashiromine. We were, of course, mindful that these functions could potentially act as nucleophiles themselves in the acidic medium of the electrophilic cyclisation, and the investigation of such chemoselectivity issues provided a further impetus for this study. Acylsilane 13 was therefore prepared from propargyl alcohol in four steps then subjected to asymmetric reduction with (−)-DIPCl according to Buynak et al. (Scheme 4) [41]. The desired hydroxysilane 12 was obtained in 53% yield and with 91% ee as determined by chiral HPLC analysis. Compound 12 was converted by standard methods to the acetate 14 and the tetrahydropyranyl ether 15. The latter compound was formed as a 1.3:1 mixture of diastereomers which were partially separated by column chromatography — all subsequent reactions were carried out on diastereomerically pure material for ease of analysis.
![[1860-5397-4-8-i4]](/bjoc/content/inline/1860-5397-4-8-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Asymmetric synthesis of chiral (alkoxy)allylsilanes.
Scheme 4: Asymmetric synthesis of chiral (alkoxy)allylsilanes.
With the requisite enantioenriched allylsilanes in hand, we next investigated their behaviour in olefin cross-metathesis reactions. Unfortunately, neither 14 nor 15 reacted with 5 under the standard cross-metathesis conditions used for trimethylsilane 6; the use of more forcing conditions (elevated temperature and higher catalyst loadings) did not effect the desired transformation, the only product observed being that of homodimerisation of 5 (Scheme 5).
![[1860-5397-4-8-i5]](/bjoc/content/inline/1860-5397-4-8-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Attempted cross-metathesis of (alkoxy)allylsilanes.
Scheme 5: Attempted cross-metathesis of (alkoxy)allylsilanes.
Finally, we examined the behaviour of alcohol 12 under cross-metathesis conditions. In the event, two isomerised products were isolated from this reaction (Scheme 6): the internal alkene 16 (formed in 99% yield as a ca. 3:1 mixture of E:Z isomers) and the acylsilane 17. The formation of isomerised alkenes accompanying (or instead of) metathesis processes using ruthenium-based catalysts is well documented [42-63], as is the formation of carbonyl compounds by isomerisation of the corresponding allylic alcohols [64-68]. At this stage we therefore reluctantly abandoned our investigations into the asymmetric synthesis of tashiromine.
![[1860-5397-4-8-i6]](/bjoc/content/inline/1860-5397-4-8-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Competing isomerisation processes in attempted cross-metathesis of (hydroxy)allylsilane 12.
Scheme 6: Competing isomerisation processes in attempted cross-metathesis of (hydroxy)allylsilane 12.
Conclusion
A concise, stereoselective total synthesis of racemic tashiromine has been developed (six steps from succinimide, 19% overall yield) in which the key steps are the preparation of a functionalised allylsilane by olefin cross-metathesis and the construction of the indolizidine core by intramolecular addition of the allylsilane to an N-acyliminium ion. Attempts to translate this into an asymmetric synthesis utilising cross-metathesis reactions of chiral α-alkoxysilanes have thus far been unsuccessful.
Experimental
Experimental protocols for the synthesis of tashiromine 1 and the preparation of silanes 12, 14, 15 and 17 available as Supporting Information File 1.
Supporting Information
| Supporting Information File 1: Supporting Information. Full experimental details and compound characterisation data for all new compounds described. | ||
| Format: PDF | Size: 105.4 KB | Download |
References
-
Ohmiya, S.; Kubo, H.; Otomasu, H.; Saito, K.; Murakoshi, I. Heterocycles 1990, 30, 537–542.
Return to citation in text: [1] -
Michael, J. P. Nat. Prod. Rep. 2007, 24, 191–222. doi:10.1039/b509525p
And references therein.
Return to citation in text: [1] -
Beckwith, A. L. J.; Westwood, S. W. Tetrahedron 1989, 45, 5269–5282. doi:10.1016/S0040-4020(01)81101-7
Return to citation in text: [1] [2] [3] -
Haddad, M.; Célérier, J. P.; Haviari, G.; Lhommet, G.; Dhimane, H.; Pommelet, J. C.; Chuche, J. Heterocycles 1990, 31, 1251–1260.
Return to citation in text: [1] [2] [3] -
Nagao, Y.; Dai, W.-M.; Ochiai, M.; Tsukagoshi, S.; Fujita, E. J. Org. Chem. 1990, 55, 1148–1156. doi:10.1021/jo00291a012
Return to citation in text: [1] [2] [3] -
Paulvannan, K.; Stille, J. R. J. Org. Chem. 1994, 59, 1613–1620. doi:10.1021/jo00086a009
Return to citation in text: [1] [2] -
Gage, J. L.; Branchaud, B. P. Tetrahedron Lett. 1997, 38, 7007–7010. doi:10.1016/S0040-4039(97)01638-9
Return to citation in text: [1] [2] -
Ha, D.-C.; Park, S.-H.; Choi, K.-S.; Yun, C.-S. Bull. Korean Chem. Soc. 1998, 19, 728–730.
Return to citation in text: [1] [2] -
David, O.; Blot, J.; Bellec, C.; Fargeau-Bellassoued, M.-C.; Haviari, G.; Célérier, J.-P.; Lhommet, G.; Gramain, J.-C.; Gardette, D. J. Org. Chem. 1999, 64, 3122–3131. doi:10.1021/jo982169p
Return to citation in text: [1] [2] [3] -
Kim, S.-H.; Kim, S.-I.; Lai, S.; Cha, J. K. J. Org. Chem. 1999, 64, 6771–6775. doi:10.1021/jo9907383
Return to citation in text: [1] [2] [3] [4] -
Bates, R. W.; Boonsombat, J. J. Chem. Soc., Perkin Trans. 1 2001, 654–656. doi:10.1039/b100407g
Return to citation in text: [1] [2] [3] -
Dieter, R. K.; Watson, R. Tetrahedron Lett. 2002, 43, 7725–7728. doi:10.1016/S0040-4039(02)01835-X
Return to citation in text: [1] [2] [3] -
Banwell, M. G.; Beck, D. A. S.; Smith, J. A. Org. Biomol. Chem. 2004, 2, 157–159. doi:10.1039/b312552a
Return to citation in text: [1] [2] -
McElhinney, A. D.; Marsden, S. P. Synlett 2005, 2528–2530. doi:10.1055/s-2005-917072
Return to citation in text: [1] [2] [3] [4] -
Bélanger, G.; Larouche-Gauthier, R.; Ménard, F.; Nantel, M.; Barabé, F. J. Org. Chem. 2006, 71, 704–712. doi:10.1021/jo052141v
Return to citation in text: [1] [2] -
Hiemstra, H.; Sno, M. H. A. M.; Vijn, R. J.; Speckamp, W. N. J. Org. Chem. 1985, 50, 4014–4020. doi:10.1021/jo00221a011
Return to citation in text: [1] [2] -
Paquette, L. A.; Mendez-Andino, J. L. J. Org. Chem. 1998, 63, 9061–9068. doi:10.1021/jo981683s
Return to citation in text: [1] -
Bergmeier, S. C.; Seth, P. P. J. Org. Chem. 1997, 62, 2671–2674. doi:10.1021/jo962307f
Return to citation in text: [1] -
Cassidy, J. H.; Marsden, S. P.; Stemp, G. Synlett 1997, 1411–1413. doi:10.1055/s-1997-1062
Return to citation in text: [1] -
Miles, S. M.; Marsden, S. P.; Leatherbarrow, R. J.; Coates, W. J. J. Org. Chem. 2004, 69, 6874–6882. doi:10.1021/jo048971a
Return to citation in text: [1] -
Miles, S. M.; Marsden, S. P.; Leatherbarrow, R. J.; Coates, W. J. Chem. Commun. 2004, 2292–2293. doi:10.1039/b409528f
Return to citation in text: [1] -
Akindele, T.; Marsden, S. P.; Cumming, J. G. Org. Lett. 2005, 7, 3685–3688. doi:10.1021/ol051292h
Return to citation in text: [1] -
Akindele, T.; Marsden, S. P.; Cumming, J. G. Tetrahedron Lett. 2005, 46, 7235–7238. doi:10.1016/j.tetlet.2005.08.046
Return to citation in text: [1] -
Cassidy, J. H.; Farthing, C. N.; Marsden, S. P.; Pedersen, A.; Slater, M.; Stemp, G. Org. Biomol. Chem. 2006, 4, 4118–4126. doi:10.1039/b612256f
Return to citation in text: [1] -
Teare, H.; Huguet, F.; Tredwell, M.; Thibaudeau, S.; Luthra, S.; Gouverneur, V. ARKIVOC 2007, No. x, 232–244.
Return to citation in text: [1] -
Thibaudeau, S.; Gouverneur, V. Org. Lett. 2003, 5, 4891–4893. doi:10.1021/ol035991a
Return to citation in text: [1] -
Álvarez-Corral, M.; López-Sánchez, C.; Jiménez-González, L.; Rosales, A.; Muñoz-Dorado, M.; Rodríguez-García, I. Beilstein J. Org. Chem. 2007, 3, No. 5. doi:10.1186/1860-5397-3-5
Return to citation in text: [1] -
Jiménez-González, L.; García-Muñoz, S.; Álvarez-Corral, M.; Muñoz-Dorado, M.; Rodríguez-García, I. Chem.–Eur. J. 2007, 13, 557–568. doi:10.1002/chem.200601017
Return to citation in text: [1] -
Jiménez-González, L.; García-Muñoz, S.; Álvarez-Corral, M.; Muñoz-Dorado, M.; Rodríguez-García, I. Chem.–Eur. J. 2006, 12, 8762–8769. doi:10.1002/chem.200600332
Return to citation in text: [1] -
García-Muñoz, S.; Jiménez-González, L.; Álvarez-Corral, M.; Muñoz-Dorado, M.; Rodríguez-García, I. Synlett 2005, 3011–3013. doi:10.1055/s-2005-921913
Return to citation in text: [1] -
Jiménez-González, L.; Álvarez-Corral, M.; Muñoz-Dorado, M.; Rodríguez-García, I. Chem. Commun. 2005, 2689–2691. doi:10.1039/b500919g
Return to citation in text: [1] -
Vedrenne, E.; Dupont, H.; Oualef, S.; Elkaïm, L.; Grimaud, L. Synlett 2005, 670–672. doi:10.1055/s-2005-862375
Return to citation in text: [1] -
He, A.; Yan, B.; Thanavaro, A.; Spilling, C. D.; Rath, N. P. J. Org. Chem. 2004, 69, 8643–8651. doi:10.1021/jo0490090
Return to citation in text: [1] -
BouzBouz, S.; De Lemos, E.; Cossy, J. Adv. Synth. Catal. 2002, 344, 627–630. doi:10.1002/1615-4169(200208)344:6/7<627::AID-ADSC627>3.0.CO;2-C
Return to citation in text: [1] -
Meyer, C.; Cossy, J. Tetrahedron Lett. 1997, 38, 7861–7864. doi:10.1016/S0040-4039(97)10147-2
Return to citation in text: [1] -
Taylor, R. E.; Engelhardt, F. C.; Schmitt, M. J.; Yuan, H. J. Am. Chem. Soc. 2001, 123, 2964–2969. doi:10.1021/ja0037163
Return to citation in text: [1] -
Taylor, R. E.; Engelhardt, F. C.; Yuan, H. Org. Lett. 1999, 1, 1257–1260. doi:10.1021/ol990221d
Return to citation in text: [1] -
Chang, S.; Grubbs, R. H. Tetrahedron Lett. 1997, 38, 4757–4760. doi:10.1016/S0040-4039(97)01031-9
Return to citation in text: [1] -
Crowe, W. E.; Goldberg, D. R.; Zhang, Z. J. Tetrahedron Lett. 1996, 37, 2117–2120. doi:10.1016/0040-4039(96)00230-4
Return to citation in text: [1] -
Fleming, I.; Barbero, A.; Walter, D. Chem. Rev. 1997, 97, 2063–2192. doi:10.1021/cr941074u
Return to citation in text: [1] -
Buynak, J. D.; Strickland, J. B.; Lamb, G. W.; Khasnis, D.; Modi, S.; Williams, D.; Zhang, H. J. Org. Chem. 1991, 56, 7076–7083. doi:10.1021/jo00025a024
Return to citation in text: [1] [2] -
Miller, S. J.; Blackwell, H. E.; Grubbs, R. H. J. Am. Chem. Soc. 1996, 118, 9606–9614. doi:10.1021/ja961626l
Return to citation in text: [1] -
Hoye, T. R.; Promo, M. A. Tetrahedron Lett. 1999, 40, 1429–1432. doi:10.1016/S0040-4039(98)02697-5
Return to citation in text: [1] -
Maynard, H. D.; Grubbs, R. H. Tetrahedron Lett. 1999, 40, 4137–4140. doi:10.1016/S0040-4039(99)00726-1
Return to citation in text: [1] -
Edwards, S. D.; Lewis, T.; Taylor, R. J. K. Tetrahedron Lett. 1999, 40, 4267–4270. doi:10.1016/S0040-4039(99)00703-0
Return to citation in text: [1] -
Fürstner, A.; Thiel, O. R.; Ackermann, L.; Schanz, H. J.; Nolan, S. P. J. Org. Chem. 2000, 65, 2204–2207. doi:10.1021/jo9918504
Return to citation in text: [1] -
Cadot, C.; Dalko, P. I.; Cossy, J. Tetrahedron Lett. 2002, 43, 1839–1841. doi:10.1016/S0040-4039(02)00141-7
Return to citation in text: [1] -
Arisawa, M.; Terada, Y.; Nakagawa, M.; Nishida, A. Angew. Chem., Int. Ed. 2002, 41, 4732–4734. doi:10.1002/anie.200290031
Return to citation in text: [1] -
Sutton, A. E.; Seigal, B. A.; Finnegan, D. F.; Snapper, M. L. J. Am. Chem. Soc. 2002, 124, 13390–13391. doi:10.1021/ja028044q
Return to citation in text: [1] -
Wipf, P.; Rector, S. R.; Takahashi, H. J. Am. Chem. Soc. 2002, 124, 14848–14849. doi:10.1021/ja028603t
Return to citation in text: [1] -
Schmidt, B. Eur. J. Org. Chem. 2003, 816–819. doi:10.1002/ejoc.200390124
Return to citation in text: [1] -
Sworen, J. C.; Pawlow, J. H.; Case, W.; Lever, J.; Wagener, K. B. J. Mol. Catal. A 2003, 194, 69–78. doi:10.1016/S1381-1169(02)00524-1
Return to citation in text: [1] -
Lehman, S. E., Jr.; Schwendeman, J. E.; O'Donnell, P. M.; Wagener, K. B. Inorg. Chim. Acta 2003, 345, 190–198. doi:10.1016/S0020-1693(02)01307-5
Return to citation in text: [1] -
Peczuh, M. W.; Snyder, N. L. Tetrahedron Lett. 2003, 44, 4057–4061. doi:10.1016/S0040-4039(03)00849-9
Return to citation in text: [1] -
Alcaide, B.; Almendros, P.; Alonso, J. M. Chem.–Eur. J. 2003, 9, 5793–5799. doi:10.1002/chem.200305236
Return to citation in text: [1] -
Kotha, S.; Mandal, K. Tetrahedron Lett. 2004, 45, 1391–1394. doi:10.1016/j.tetlet.2003.12.075
Return to citation in text: [1] -
Schmidt, B. J. Org. Chem. 2004, 69, 7672–7687. doi:10.1021/jo048937w
Return to citation in text: [1] -
Hong, S. H.; Sanders, D. P.; Lee, C. W.; Grubbs, R. H. J. Am. Chem. Soc. 2005, 127, 17160–17161. doi:10.1021/ja052939w
Return to citation in text: [1] -
Bennasar, M. L.; Roca, T.; Monerris, M.; García-Díaz, D. J. Org. Chem. 2006, 71, 7028–7034. doi:10.1021/jo061180j
Return to citation in text: [1] -
Hanessian, S.; Giroux, S.; Larsson, A. Org. Lett. 2006, 8, 5481–5484. doi:10.1021/ol062167o
Return to citation in text: [1] -
Courchay, F. C.; Sworen, J. C.; Ghiviriga, I.; Abboud, K. A.; Wagener, K. B. Organometallics 2006, 25, 6074–6086. doi:10.1021/om0509894
Return to citation in text: [1] -
Raju, R.; Allen, L. J.; Le, T.; Taylor, C. D.; Howell, A. R. Org. Lett. 2007, 9, 1699–1701. doi:10.1021/ol070574+
Return to citation in text: [1] -
Moïse, J.; Arseniyadis, S.; Cossy, J. Org. Lett. 2007, 9, 1695–1698. doi:10.1021/ol0703940
Return to citation in text: [1] -
Gurjar, M. K.; Yakambram, P. Tetrahedron Lett. 2001, 42, 3633–3636. doi:10.1016/S0040-4039(01)00449-X
Return to citation in text: [1] -
Greenwood, E. S.; Parsons, P. J.; Young, M. J. Synth. Commun. 2003, 33, 223–228. doi:10.1081/SCC-120015704
Return to citation in text: [1] -
Werner, H.; Grünwald, C.; Stüer, W.; Wolf, J. Organometallics 2003, 22, 1558–1560. doi:10.1021/om0210006
Return to citation in text: [1] -
Edlin, C. D.; Faulkner, J.; Fengas, D.; Knight, C. K.; Parker, J.; Preece, I.; Quayle, P.; Richards, S. N. Synlett 2005, 572–576. doi:10.1055/s-2005-862384
Return to citation in text: [1] -
Finnegan, D.; Seigal, B. A.; Snapper, M. L. Org. Lett. 2006, 8, 2603–2606. doi:10.1021/ol060918g
Return to citation in text: [1]
| 64. | Gurjar, M. K.; Yakambram, P. Tetrahedron Lett. 2001, 42, 3633–3636. doi:10.1016/S0040-4039(01)00449-X |
| 65. | Greenwood, E. S.; Parsons, P. J.; Young, M. J. Synth. Commun. 2003, 33, 223–228. doi:10.1081/SCC-120015704 |
| 66. | Werner, H.; Grünwald, C.; Stüer, W.; Wolf, J. Organometallics 2003, 22, 1558–1560. doi:10.1021/om0210006 |
| 67. | Edlin, C. D.; Faulkner, J.; Fengas, D.; Knight, C. K.; Parker, J.; Preece, I.; Quayle, P.; Richards, S. N. Synlett 2005, 572–576. doi:10.1055/s-2005-862384 |
| 68. | Finnegan, D.; Seigal, B. A.; Snapper, M. L. Org. Lett. 2006, 8, 2603–2606. doi:10.1021/ol060918g |
| 1. | Ohmiya, S.; Kubo, H.; Otomasu, H.; Saito, K.; Murakoshi, I. Heterocycles 1990, 30, 537–542. |
| 5. | Nagao, Y.; Dai, W.-M.; Ochiai, M.; Tsukagoshi, S.; Fujita, E. J. Org. Chem. 1990, 55, 1148–1156. doi:10.1021/jo00291a012 |
| 14. | McElhinney, A. D.; Marsden, S. P. Synlett 2005, 2528–2530. doi:10.1055/s-2005-917072 |
| 15. | Bélanger, G.; Larouche-Gauthier, R.; Ménard, F.; Nantel, M.; Barabé, F. J. Org. Chem. 2006, 71, 704–712. doi:10.1021/jo052141v |
| 17. | Paquette, L. A.; Mendez-Andino, J. L. J. Org. Chem. 1998, 63, 9061–9068. doi:10.1021/jo981683s |
| 3. | Beckwith, A. L. J.; Westwood, S. W. Tetrahedron 1989, 45, 5269–5282. doi:10.1016/S0040-4020(01)81101-7 |
| 18. | Bergmeier, S. C.; Seth, P. P. J. Org. Chem. 1997, 62, 2671–2674. doi:10.1021/jo962307f |
| 3. | Beckwith, A. L. J.; Westwood, S. W. Tetrahedron 1989, 45, 5269–5282. doi:10.1016/S0040-4020(01)81101-7 |
| 4. | Haddad, M.; Célérier, J. P.; Haviari, G.; Lhommet, G.; Dhimane, H.; Pommelet, J. C.; Chuche, J. Heterocycles 1990, 31, 1251–1260. |
| 5. | Nagao, Y.; Dai, W.-M.; Ochiai, M.; Tsukagoshi, S.; Fujita, E. J. Org. Chem. 1990, 55, 1148–1156. doi:10.1021/jo00291a012 |
| 6. | Paulvannan, K.; Stille, J. R. J. Org. Chem. 1994, 59, 1613–1620. doi:10.1021/jo00086a009 |
| 7. | Gage, J. L.; Branchaud, B. P. Tetrahedron Lett. 1997, 38, 7007–7010. doi:10.1016/S0040-4039(97)01638-9 |
| 8. | Ha, D.-C.; Park, S.-H.; Choi, K.-S.; Yun, C.-S. Bull. Korean Chem. Soc. 1998, 19, 728–730. |
| 9. | David, O.; Blot, J.; Bellec, C.; Fargeau-Bellassoued, M.-C.; Haviari, G.; Célérier, J.-P.; Lhommet, G.; Gramain, J.-C.; Gardette, D. J. Org. Chem. 1999, 64, 3122–3131. doi:10.1021/jo982169p |
| 10. | Kim, S.-H.; Kim, S.-I.; Lai, S.; Cha, J. K. J. Org. Chem. 1999, 64, 6771–6775. doi:10.1021/jo9907383 |
| 11. | Bates, R. W.; Boonsombat, J. J. Chem. Soc., Perkin Trans. 1 2001, 654–656. doi:10.1039/b100407g |
| 12. | Dieter, R. K.; Watson, R. Tetrahedron Lett. 2002, 43, 7725–7728. doi:10.1016/S0040-4039(02)01835-X |
| 13. | Banwell, M. G.; Beck, D. A. S.; Smith, J. A. Org. Biomol. Chem. 2004, 2, 157–159. doi:10.1039/b312552a |
| 14. | McElhinney, A. D.; Marsden, S. P. Synlett 2005, 2528–2530. doi:10.1055/s-2005-917072 |
| 15. | Bélanger, G.; Larouche-Gauthier, R.; Ménard, F.; Nantel, M.; Barabé, F. J. Org. Chem. 2006, 71, 704–712. doi:10.1021/jo052141v |
| 14. | McElhinney, A. D.; Marsden, S. P. Synlett 2005, 2528–2530. doi:10.1055/s-2005-917072 |
| 2. |
Michael, J. P. Nat. Prod. Rep. 2007, 24, 191–222. doi:10.1039/b509525p
And references therein. |
| 16. | Hiemstra, H.; Sno, M. H. A. M.; Vijn, R. J.; Speckamp, W. N. J. Org. Chem. 1985, 50, 4014–4020. doi:10.1021/jo00221a011 |
| 12. | Dieter, R. K.; Watson, R. Tetrahedron Lett. 2002, 43, 7725–7728. doi:10.1016/S0040-4039(02)01835-X |
| 11. | Bates, R. W.; Boonsombat, J. J. Chem. Soc., Perkin Trans. 1 2001, 654–656. doi:10.1039/b100407g |
| 8. | Ha, D.-C.; Park, S.-H.; Choi, K.-S.; Yun, C.-S. Bull. Korean Chem. Soc. 1998, 19, 728–730. |
| 10. | Kim, S.-H.; Kim, S.-I.; Lai, S.; Cha, J. K. J. Org. Chem. 1999, 64, 6771–6775. doi:10.1021/jo9907383 |
| 6. | Paulvannan, K.; Stille, J. R. J. Org. Chem. 1994, 59, 1613–1620. doi:10.1021/jo00086a009 |
| 7. | Gage, J. L.; Branchaud, B. P. Tetrahedron Lett. 1997, 38, 7007–7010. doi:10.1016/S0040-4039(97)01638-9 |
| 13. | Banwell, M. G.; Beck, D. A. S.; Smith, J. A. Org. Biomol. Chem. 2004, 2, 157–159. doi:10.1039/b312552a |
| 4. | Haddad, M.; Célérier, J. P.; Haviari, G.; Lhommet, G.; Dhimane, H.; Pommelet, J. C.; Chuche, J. Heterocycles 1990, 31, 1251–1260. |
| 9. | David, O.; Blot, J.; Bellec, C.; Fargeau-Bellassoued, M.-C.; Haviari, G.; Célérier, J.-P.; Lhommet, G.; Gramain, J.-C.; Gardette, D. J. Org. Chem. 1999, 64, 3122–3131. doi:10.1021/jo982169p |
| 39. | Crowe, W. E.; Goldberg, D. R.; Zhang, Z. J. Tetrahedron Lett. 1996, 37, 2117–2120. doi:10.1016/0040-4039(96)00230-4 |
| 19. | Cassidy, J. H.; Marsden, S. P.; Stemp, G. Synlett 1997, 1411–1413. doi:10.1055/s-1997-1062 |
| 20. | Miles, S. M.; Marsden, S. P.; Leatherbarrow, R. J.; Coates, W. J. J. Org. Chem. 2004, 69, 6874–6882. doi:10.1021/jo048971a |
| 21. | Miles, S. M.; Marsden, S. P.; Leatherbarrow, R. J.; Coates, W. J. Chem. Commun. 2004, 2292–2293. doi:10.1039/b409528f |
| 22. | Akindele, T.; Marsden, S. P.; Cumming, J. G. Org. Lett. 2005, 7, 3685–3688. doi:10.1021/ol051292h |
| 23. | Akindele, T.; Marsden, S. P.; Cumming, J. G. Tetrahedron Lett. 2005, 46, 7235–7238. doi:10.1016/j.tetlet.2005.08.046 |
| 24. | Cassidy, J. H.; Farthing, C. N.; Marsden, S. P.; Pedersen, A.; Slater, M.; Stemp, G. Org. Biomol. Chem. 2006, 4, 4118–4126. doi:10.1039/b612256f |
| 25. | Teare, H.; Huguet, F.; Tredwell, M.; Thibaudeau, S.; Luthra, S.; Gouverneur, V. ARKIVOC 2007, No. x, 232–244. |
| 26. | Thibaudeau, S.; Gouverneur, V. Org. Lett. 2003, 5, 4891–4893. doi:10.1021/ol035991a |
| 27. | Álvarez-Corral, M.; López-Sánchez, C.; Jiménez-González, L.; Rosales, A.; Muñoz-Dorado, M.; Rodríguez-García, I. Beilstein J. Org. Chem. 2007, 3, No. 5. doi:10.1186/1860-5397-3-5 |
| 28. | Jiménez-González, L.; García-Muñoz, S.; Álvarez-Corral, M.; Muñoz-Dorado, M.; Rodríguez-García, I. Chem.–Eur. J. 2007, 13, 557–568. doi:10.1002/chem.200601017 |
| 29. | Jiménez-González, L.; García-Muñoz, S.; Álvarez-Corral, M.; Muñoz-Dorado, M.; Rodríguez-García, I. Chem.–Eur. J. 2006, 12, 8762–8769. doi:10.1002/chem.200600332 |
| 30. | García-Muñoz, S.; Jiménez-González, L.; Álvarez-Corral, M.; Muñoz-Dorado, M.; Rodríguez-García, I. Synlett 2005, 3011–3013. doi:10.1055/s-2005-921913 |
| 31. | Jiménez-González, L.; Álvarez-Corral, M.; Muñoz-Dorado, M.; Rodríguez-García, I. Chem. Commun. 2005, 2689–2691. doi:10.1039/b500919g |
| 32. | Vedrenne, E.; Dupont, H.; Oualef, S.; Elkaïm, L.; Grimaud, L. Synlett 2005, 670–672. doi:10.1055/s-2005-862375 |
| 33. | He, A.; Yan, B.; Thanavaro, A.; Spilling, C. D.; Rath, N. P. J. Org. Chem. 2004, 69, 8643–8651. doi:10.1021/jo0490090 |
| 34. | BouzBouz, S.; De Lemos, E.; Cossy, J. Adv. Synth. Catal. 2002, 344, 627–630. doi:10.1002/1615-4169(200208)344:6/7<627::AID-ADSC627>3.0.CO;2-C |
| 35. | Meyer, C.; Cossy, J. Tetrahedron Lett. 1997, 38, 7861–7864. doi:10.1016/S0040-4039(97)10147-2 |
| 36. | Taylor, R. E.; Engelhardt, F. C.; Schmitt, M. J.; Yuan, H. J. Am. Chem. Soc. 2001, 123, 2964–2969. doi:10.1021/ja0037163 |
| 37. | Taylor, R. E.; Engelhardt, F. C.; Yuan, H. Org. Lett. 1999, 1, 1257–1260. doi:10.1021/ol990221d |
| 38. | Chang, S.; Grubbs, R. H. Tetrahedron Lett. 1997, 38, 4757–4760. doi:10.1016/S0040-4039(97)01031-9 |
| 41. | Buynak, J. D.; Strickland, J. B.; Lamb, G. W.; Khasnis, D.; Modi, S.; Williams, D.; Zhang, H. J. Org. Chem. 1991, 56, 7076–7083. doi:10.1021/jo00025a024 |
| 42. | Miller, S. J.; Blackwell, H. E.; Grubbs, R. H. J. Am. Chem. Soc. 1996, 118, 9606–9614. doi:10.1021/ja961626l |
| 43. | Hoye, T. R.; Promo, M. A. Tetrahedron Lett. 1999, 40, 1429–1432. doi:10.1016/S0040-4039(98)02697-5 |
| 44. | Maynard, H. D.; Grubbs, R. H. Tetrahedron Lett. 1999, 40, 4137–4140. doi:10.1016/S0040-4039(99)00726-1 |
| 45. | Edwards, S. D.; Lewis, T.; Taylor, R. J. K. Tetrahedron Lett. 1999, 40, 4267–4270. doi:10.1016/S0040-4039(99)00703-0 |
| 46. | Fürstner, A.; Thiel, O. R.; Ackermann, L.; Schanz, H. J.; Nolan, S. P. J. Org. Chem. 2000, 65, 2204–2207. doi:10.1021/jo9918504 |
| 47. | Cadot, C.; Dalko, P. I.; Cossy, J. Tetrahedron Lett. 2002, 43, 1839–1841. doi:10.1016/S0040-4039(02)00141-7 |
| 48. | Arisawa, M.; Terada, Y.; Nakagawa, M.; Nishida, A. Angew. Chem., Int. Ed. 2002, 41, 4732–4734. doi:10.1002/anie.200290031 |
| 49. | Sutton, A. E.; Seigal, B. A.; Finnegan, D. F.; Snapper, M. L. J. Am. Chem. Soc. 2002, 124, 13390–13391. doi:10.1021/ja028044q |
| 50. | Wipf, P.; Rector, S. R.; Takahashi, H. J. Am. Chem. Soc. 2002, 124, 14848–14849. doi:10.1021/ja028603t |
| 51. | Schmidt, B. Eur. J. Org. Chem. 2003, 816–819. doi:10.1002/ejoc.200390124 |
| 52. | Sworen, J. C.; Pawlow, J. H.; Case, W.; Lever, J.; Wagener, K. B. J. Mol. Catal. A 2003, 194, 69–78. doi:10.1016/S1381-1169(02)00524-1 |
| 53. | Lehman, S. E., Jr.; Schwendeman, J. E.; O'Donnell, P. M.; Wagener, K. B. Inorg. Chim. Acta 2003, 345, 190–198. doi:10.1016/S0020-1693(02)01307-5 |
| 54. | Peczuh, M. W.; Snyder, N. L. Tetrahedron Lett. 2003, 44, 4057–4061. doi:10.1016/S0040-4039(03)00849-9 |
| 55. | Alcaide, B.; Almendros, P.; Alonso, J. M. Chem.–Eur. J. 2003, 9, 5793–5799. doi:10.1002/chem.200305236 |
| 56. | Kotha, S.; Mandal, K. Tetrahedron Lett. 2004, 45, 1391–1394. doi:10.1016/j.tetlet.2003.12.075 |
| 57. | Schmidt, B. J. Org. Chem. 2004, 69, 7672–7687. doi:10.1021/jo048937w |
| 58. | Hong, S. H.; Sanders, D. P.; Lee, C. W.; Grubbs, R. H. J. Am. Chem. Soc. 2005, 127, 17160–17161. doi:10.1021/ja052939w |
| 59. | Bennasar, M. L.; Roca, T.; Monerris, M.; García-Díaz, D. J. Org. Chem. 2006, 71, 7028–7034. doi:10.1021/jo061180j |
| 60. | Hanessian, S.; Giroux, S.; Larsson, A. Org. Lett. 2006, 8, 5481–5484. doi:10.1021/ol062167o |
| 61. | Courchay, F. C.; Sworen, J. C.; Ghiviriga, I.; Abboud, K. A.; Wagener, K. B. Organometallics 2006, 25, 6074–6086. doi:10.1021/om0509894 |
| 62. | Raju, R.; Allen, L. J.; Le, T.; Taylor, C. D.; Howell, A. R. Org. Lett. 2007, 9, 1699–1701. doi:10.1021/ol070574+ |
| 63. | Moïse, J.; Arseniyadis, S.; Cossy, J. Org. Lett. 2007, 9, 1695–1698. doi:10.1021/ol0703940 |
| 40. | Fleming, I.; Barbero, A.; Walter, D. Chem. Rev. 1997, 97, 2063–2192. doi:10.1021/cr941074u |
| 41. | Buynak, J. D.; Strickland, J. B.; Lamb, G. W.; Khasnis, D.; Modi, S.; Williams, D.; Zhang, H. J. Org. Chem. 1991, 56, 7076–7083. doi:10.1021/jo00025a024 |
| 3. | Beckwith, A. L. J.; Westwood, S. W. Tetrahedron 1989, 45, 5269–5282. doi:10.1016/S0040-4020(01)81101-7 |
| 4. | Haddad, M.; Célérier, J. P.; Haviari, G.; Lhommet, G.; Dhimane, H.; Pommelet, J. C.; Chuche, J. Heterocycles 1990, 31, 1251–1260. |
| 5. | Nagao, Y.; Dai, W.-M.; Ochiai, M.; Tsukagoshi, S.; Fujita, E. J. Org. Chem. 1990, 55, 1148–1156. doi:10.1021/jo00291a012 |
| 9. | David, O.; Blot, J.; Bellec, C.; Fargeau-Bellassoued, M.-C.; Haviari, G.; Célérier, J.-P.; Lhommet, G.; Gramain, J.-C.; Gardette, D. J. Org. Chem. 1999, 64, 3122–3131. doi:10.1021/jo982169p |
| 10. | Kim, S.-H.; Kim, S.-I.; Lai, S.; Cha, J. K. J. Org. Chem. 1999, 64, 6771–6775. doi:10.1021/jo9907383 |
| 11. | Bates, R. W.; Boonsombat, J. J. Chem. Soc., Perkin Trans. 1 2001, 654–656. doi:10.1039/b100407g |
| 12. | Dieter, R. K.; Watson, R. Tetrahedron Lett. 2002, 43, 7725–7728. doi:10.1016/S0040-4039(02)01835-X |
| 10. | Kim, S.-H.; Kim, S.-I.; Lai, S.; Cha, J. K. J. Org. Chem. 1999, 64, 6771–6775. doi:10.1021/jo9907383 |
| 14. | McElhinney, A. D.; Marsden, S. P. Synlett 2005, 2528–2530. doi:10.1055/s-2005-917072 |
| 16. | Hiemstra, H.; Sno, M. H. A. M.; Vijn, R. J.; Speckamp, W. N. J. Org. Chem. 1985, 50, 4014–4020. doi:10.1021/jo00221a011 |
© 2008 Marsden and McElhinney; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)