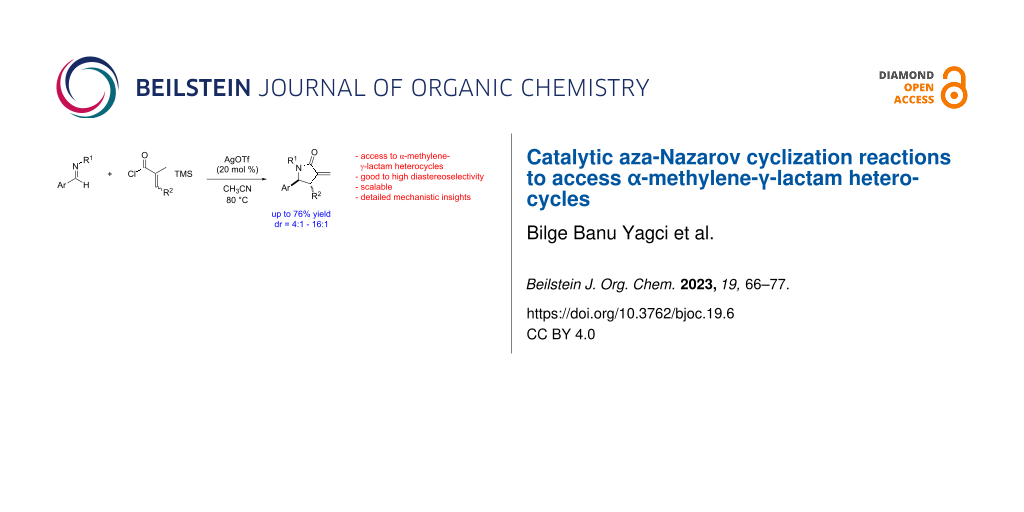
Abstract
We have developed a catalytic aza-Nazarov reaction of N-acyliminium salts generated in situ from the reaction of a variety of cyclic and acyclic imines with α,β-unsaturated acyl chlorides to afford substituted α-methylene-γ-lactam heterocycles. The reactions proceed effectively in the presence of catalytic (20 mol %) amounts of AgOTf as an anion exchange agent or hydrogen-bond donors such as squaramides and thioureas as anion-binding organocatalysts. The aza-Nazarov cyclization of 3,4-dihydroisoquinolines with α,β-unsaturated acyl chlorides gives tricyclic lactam products 7 in up to 79% yield with full diastereocontrol (dr = >99:1). The use of acyclic imines in a similar catalytic aza-Nazarov reaction with 20 mol % of AgOTf results in the formation of α-methylene-γ-lactam heterocycles 19 in up to 76% yield and with good to high diastereoselectivities (4.3:1 to 16:1). We have demonstrated the scalability of the reaction with a gram-scale example. The relative stereochemistry of the α-methylene-γ-lactam products 19 has been determined via the single-crystal X-ray analysis of lactam 19l. In order to shed light on the details of the reaction mechanism, we have performed carefully designed mechanistic studies which consist of experiments on the effect of β-silicon stabilization, the alkene geometry of the α,β-unsaturated acyl chloride reactants, and adventitious water on the success of the catalytic aza-Nazarov reaction.

Graphical Abstract
Introduction
The rapid construction of aliphatic heterocycles from acyclic building blocks via cyclization or cycloaddition reactions constitutes one of the main pillars of organic synthesis [1]. In this respect, the all-carbon Nazarov cyclization of divinyl ketones represents a direct method for the synthesis of five-membered carbocycles [2-9]. Variants of the Nazarov cyclization with substrates bearing one or more heteroatoms in the dienone core have the potential to provide efficient access to a variety of heterocyclic scaffolds. Among such variants, the aza-Nazarov reaction appears as a highly suitable cyclization reaction that presents opportunities for the construction of a diverse array of five-membered nitrogen-heterocycles [10].
Whereas an aza-Nazarov-type cyclization may be operative in some earlier examples [11-15], general interest in this area increased after the elegant studies of Würthwein and co-workers where they utilized the aza-Nazarov cyclization for the construction of multisubstituted pyrrole derivatives (Scheme 1a) [16-19]. Another seminal work in this field was the aza-Nazarov cyclization of N-acyliminium salts 1 reported by Klumpp and co-workers in 2007 (Scheme 1b) [20]. This reaction was promoted with the use of superstoichiometric amounts of TfOH (trifluoromethanesulfonic acid), and the N-acyliminium cation was proposed to be protonated with the super acidic TfOH to form a dicationic species, which was shown by computational studies to be crucial for the success of this transformation. In a later study, the same research group showed that benzamides 2 bearing an acetal group could be used as substrates in an aza-Nazarov cyclization with the intermediacy of in situ-generated N-acyliminium ions (Scheme 1b) [21]. The first catalytic aza-Nazarov reaction was reported by Tius and co-workers in 2010, which involved the kinetic resolution of azirine derivatives via an enantioselective organocatalytic aza-Nazarov cyclization affording six-membered heterocycles after a ring expansion of the cyclization products [22]. Rasapalli and co-workers recently developed an efficient aza-Nazarov cyclization of quinazolinonyl enones promoted by TfOH or MsOH (methanesulfonic acid) for the syntheses of C-aryl luotonins and vasicinones (Scheme 1c) [23,24]. Moreover, an aza-Nazarov cyclization was utilized for the construction of a variety of heterocyclic frameworks such as aminopyrroles [25,26], N-hydroxyoxindoles [27], indolizines [28], pyrido[1,2-a]indoles [29,30], indoles [31,32], and pyrrole-fused heterocyclic tricycles [33]. The involvement of Nazarov and in particular aza-Nazarov reactions in the cyclization of alkynes that go through metal carbene intermediates has recently been reviewed by Gao and co-workers [34].
![[1860-5397-19-6-i1]](/bjoc/content/inline/1860-5397-19-6-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Examples of aza-Nazarov reactions.
Scheme 1: Examples of aza-Nazarov reactions.
In 2019, we reported a highly effective aza-Nazarov cyclization for the synthesis of tricyclic α-methylene-γ-lactams 7 as single diastereomers and in good to high yields through the use of a catalytic amount of AgOTf (silver trifluoromethanesulfonate) as an anion-exchange agent (Scheme 1d) [35]. In this transformation, treatment of 3,4-dihydroisoquinolines 5 with acyl chlorides 6 initially gives N-acyliminium salts 8, which are activated upon treatment with AgOTf resulting in an anion metathesis of Cl− with OTf−. This activation is proposed to facilitate the desired aza-Nazarov reaction to afford the cyclized intermediate 9, which is stabilized by the trimethylsilyl (TMS) group via the β-silicon stabilization effect [36-38] as supported by computational studies [35]. The desilylation of intermediate 9 with the chloride of another molecule of 8 would result in the formation of final aza-Nazarov product 7 along with TMSCl (Scheme 1d). Five-membered nitrogen-heterocycles [39-45] and specifically α-methylene-γ-lactams as a subset of this compound class, are popular targets in heterocyclic chemistry and drug discovery [46-56]. Against this background, we herein disclose a full account of our studies on the catalytic aza-Nazarov reaction starting from cyclic and acyclic imines and TMS-substituted α,β-unsaturated acyl chlorides to yield α-methylene-γ-lactam heterocycles with high diastereoselectivities. We also report the results of our detailed mechanistic studies along with the necessary control experiments in order to shed light on the mechanism and specific features of this transformation.
Results and Discussion
In the initial phase of our work, we used AgOTf as an anion-exchange agent in order to promote the desired aza-Nazarov cyclization [35]. The reaction between 3,4-dihydoisoquinoline (5a) and acyl chloride 6a proceeds well at 23 °C in CH2Cl2 with the use of a stoichiometric amount (1.3 equiv) of AgOTf providing the aza-Nazarov product 7a in 57% yield and as single diastereomer, whereas no cyclization was observed in the absence of AgOTf (Table 1, entries 1 and 2). When the same reaction was tested at 80 °C in CH3CN, a substoichiometric amount of AgOTf (20 mol %) was found to be sufficient to efficiently promote the reaction to give product 7a in 79% yield (Table 1, entry 3). When AgOTf was not used under these conditions, the reaction was observed to proceed only with 13% yield (Table 1, entry 4). The scope of the reaction with 3,4-dihydroisoquinoline substrates as cyclic imines was investigated under these conditions [35]. In the current work, we first opted to examine the activities of other Lewis acids in the aza-Nazarov cyclization of imine 5a with acyl chloride 6b. The reason of using acyl chloride 6b with an isobutyl side chain is its low volatility in contrast to the highly volatile compound 6a. The aza-Nazarov product 7b was isolated in 61% yield with 20 mol % of AgOTf at 80 °C (Table 1, entry 5). The use of TMSOTf as a Si-based Lewis acid catalyst with 20 mol % loading afforded the cyclization product 7b in 47% yield (Table 1, entry 6), whereas the activity of Zn(OTf)2 was observed to be lower providing the desired product in only 36% yield (entry 7). Surprisingly, NaBF4 appeared to be effective for the aza-Nazarov reaction, and the α-methylene-γ-lactam product 7b was isolated in 54% yield (Table 1, entry 8). The exchange of chloride with the non-coordinating BF4− anion, driven by the precipitation of NaCl, is proposed to be responsible for this positive result.
Table 1: Screening of Lewis acids and hydrogen-bond donors (HBD) for the aza-Nazarov cyclization.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-19-6-i10.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Lewis acid or HBD (equiv) | Solvent | T (°C) | Product | Yield (%)b |
| 1 | AgOTf (1.3) | CH2Cl2 | 23 | 7a | 57 |
| 2 | – | CH2Cl2 | 23 | 7a | <1 |
| 3 | AgOTf (0.2) | CH3CN | 80 | 7a | 79 |
| 4 | – | CH3CN | 80 | 7a | 13 |
| 5c | AgOTf (0.2) | CH3CN | 80 | 7b | 61 |
| 6 | TMSOTf (0.2) | CH3CN | 80 | 7b | 47 |
| 7 | Zn(OTf)2 (0.2) | CH3CN | 80 | 7b | 36 |
| 8 | NaBF4 (0.2) | CH3CN | 80 | 7b | 54 |
| 9 | 10 (0.2) | CH2Cl2 | 23 | 7b | <1 |
| 10 | 10 (1.2) | CH2Cl2 | 23 | 7b | 15 |
| 11 | 10 (0.2) | CH3CN | 80 | 7b | 40 |
| 12 | 11 (1.2) | CH2Cl2 | 23 | 7b | 22 |
| 13 | 11 (0.2) | CH3CN | 80 | 7b | 48 |
| 14 | 12 (1.2) | CH2Cl2 | 23 | 7b | 17 |
| 15 | 12 (0.2) | CH3CN | 80 | 7b | 59 |
| 16 | 13 (1.2) | CH2Cl2 | 23 | 7b | 25 |
a1.3 equiv of acyl chloride 6 and 1.0 equiv of imine 5a were reacted for 22 h. bYields refer to isolated product yields after purification by column chromatography. cReaction scale: 1.0 mmol.
![[Graphic 2]](/bjoc/content/inline/1860-5397-19-6-i11.svg?max-width=637&scale=1.18182)
Next, we turned our attention to the use of hydrogen-bond donors as anion-binding catalysts [57,58]. To this end, we first tested the achiral thiourea derivative 10, which exhibited very low activity at 23 °C when used in catalytic and stoichiometric amounts (Table 1, entries 9 and 10) [59,60]. The yield of the aza-Nazarov product 7b increased to 40% when 20 mol % of thiourea 10 were used at 80 °C in CH3CN (Table 1, entry 11). With this promising result in hand, we next examined the use of squaramides, which were shown to be highly effective hydrogen-bonding catalysts in a broad range of transformations [61-64]. When achiral squaramide derivatives 11 [65] and 12 [66] were tested in stoichiometric amounts at 23 °C in CH2Cl2, the aza-Nazarov product 7b was formed in low yields (22 and 17%, Table 1, entries 12 and 14, respectively). However, the yields of the isolated products increased to 48 and 59% with the use of catalytic amounts of 11 and 12, respectively, at 80 °C (Table 1, entries 13 and 15). Finally, we investigated the activity of the achiral thiophosphoric triamide 13 as a triple hydrogen-bond donor [67]. With the use of 1.2 equiv of 13 at 23 °C, product 7b was isolated in 25% yield (Table 1, entry 16). Overall, while AgOTf still appeared to be the best reagent to be used in catalytic amount to promote the aza-Nazarov reactions investigated in this study, the results discussed above showcase the potential of strong hydrogen-bond donors as effective anion binding catalysts for this transformation. Finally, it should be noted that all aza-Nazarov products that are presented in Table 1, were obtained as single diastereomers.
In order to assess the scalability of the aza-Nazarov cyclization, we investigated the synthesis of the previously unreported lactam 7c via the reaction between imine 5b and acyl chloride 6b on a gram scale (Scheme 2). Acyl chloride 6b was prepared in four steps starting from the commercially available triethyl phosphonoacetate (14, Scheme 2a). The deprotonation of 14 with NaH followed by alkylation using (iodomethyl)trimethylsilane afforded phosphonate 15 in 60% yield (7.0 g). Its subsequent Horner–Wadsworth–Emmons reaction with isovaleraldehyde resulted in the formation of ester 16 in 90% yield (4.8 g, dr (diastereomeric ratio) = 2.6:1). Hydrolysis of the α,β-unsaturated ester 16 under basic conditions followed by acidification gave carboxylic acid 17 in 60% yield. Finally, the α,β-unsaturated acyl chloride 6b was obtained via the treatment of acid 17 with oxalyl chloride at 60 °C in 95% yield (2.6 g) and with a dr (E:Z) of 3:1 (Scheme 2a). When imine 5b was reacted with 6b on an 8.0 mmol scale with the use of AgOTf (20 mol %) in acetonitrile at 80 °C, the aza-Nazarov product 7c was isolated in 61% yield (1.42 g) as a single diastereomer. This result clearly demonstrates the preparative value of the catalytic aza-Nazarov cyclization developed in this work.
![[1860-5397-19-6-i2]](/bjoc/content/inline/1860-5397-19-6-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Aza-Nazarov cyclization on gram scale.
Scheme 2: Aza-Nazarov cyclization on gram scale.
Following the success of the newly developed aza-Nazarov reaction with 3,4-dihydroisoquinoline derivatives as cyclic imines [35], we next opted to investigate its effectiveness with acyclic imine substrates. The aza-Nazarov reactions of such imines were found to proceed successfully, albeit with slightly lower diastereoselectivities (ca. 4:1 to 16:1 dr) compared to their cyclic counterparts (Scheme 3). The reaction of the aldimine formed via the condensation of benzaldehyde and n-propylamine with acyl chloride 6b gave the aza-Nazarov product 19a in 46% yield and with a dr of 10:1 when 20 mol % AgOTf were used as anion exchange reagent in CH3CN at 80 °C. Aldimines with electron-donating substituents were found to be suitable substrates for this transformation affording the α-methylene-γ-lactam products 19b–d in good yields (54–69%). We were pleased to see that the CF3-substituted aza-Nazarov product 19e was isolated as single diastereomer in 76% yield along with only 8% of the minor diastereomer. Surprisingly, aza-Nazarov products 19f with a 4-Cl substituent on the phenyl ring as well as 19g and 19h bearing a strongly electron-withdrawing NO2 substituent at the 4- and 3-positions were obtained in lower yields (35, 39 and 20%, respectively) under the optimized conditions. We next turned our attention to the use of heteroaromatic side chains on the imine component. Gratifyingly, the reaction was found to tolerate the presence of furan and thiophene rings where 2-furyl- and thienyl-substituted aza-Nazarov products 19i and 19j were isolated in 59 and 53% yields, and with a dr of 11:1 and 6:1, respectively. Inspired by the success of the thiophene-containing imine reactant, we also wanted to check the reactivity of a thiophene-fused dihydropyridine as a cyclic imine. The tricyclic α-methylene-γ-lactam product 19k was obtained in 40% yield and as single diastereomer. It should be noted that due to the low solubility of the imine substrate in CH3CN, this reaction was carried out in CH2Cl2 at 23 °C and with the use of 1.3 equivalents of AgOTf. Finally, we sought to check the reactivity of other substituents on the amine component of acyclic imine substrates. With the use of benzyl-substituted aldimine, the aza-Nazarov product 19l was obtained in 49% yield and with 10:1 dr. Similarly, the aza-Nazarov cyclization of the allyl-substituted imine substrate afforded α-methylene-γ-lactam product 19m in 56% yield.
![[1860-5397-19-6-i3]](/bjoc/content/inline/1860-5397-19-6-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Scope of the aza-Nazarov cyclization with acyclic imines. aThe syntheses of aza-Nazarov products 19b, 19c, and 19f were described previously [35]. bThe reaction was carried out at 23 °C in CH2Cl2 using 1.3 equiv of AgOTf.
Scheme 3: Scope of the aza-Nazarov cyclization with acyclic imines. aThe syntheses of aza-Nazarov products 19b...
In our previous study, the relative stereochemistry of a tricyclic aza-Nazarov product obtained from a 3,4-dihydroisoquinoline derivative was secured by single-crystal X-ray analysis [35]. In the present work, the α-methylene-γ-lactam products 19 were in most cases isolated as oils after column chromatography. Fortunately, lactam product 19l turned out to be a solid, and high-quality crystals could be obtained via slow vapor diffusion of pentane into its CH2Cl2 solution. Single-crystal X-ray analysis revealed that the relative stereochemistry of lactam 19l with the isobutyl and phenyl groups in a trans arrangement (Figure 1, CCDC 2116978). This finding confirms that both cyclic and acyclic imines undergo the aza-Nazarov cyclization developed in this work via the same stereochemical arrangement.
![[1860-5397-19-6-1]](/bjoc/content/figures/1860-5397-19-6-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: X-ray crystal structure of compound 19l.
Figure 1: X-ray crystal structure of compound 19l.
Another intriguing aspect of the aza-Nazarov reactions performed in the current work with acyclic imines is the formation of the cis isomer as the minor diastereomer in contrast to the reactions of 3,4-dihydroisoquinoline derivatives where the aza-Nazarov products are obtained as single diastereomers. This reaction outcome was attributed to a potential cis–trans isomerization of the C–N double bond upon iminium formation (Scheme 4). In this respect, the mixing of imine 18 with the α,β-unsaturated acyl chloride 6 is expected to form the iminium ion 20a with Z-configuration. A chloride-mediated iminium E–Z isomerization may take place through the intermediacy of α-chloroamide 21. The aza-Nazarov reaction of the more stable E-iminium ion 20b is expected to proceed faster due to steric considerations giving the major diastereomer 19 whereas the less stable Z-iminium ion 20a would provide the minor diastereomer 22. In the case of aza-Nazarov reactions of 3,4-dihydroisoquinoline derivatives, an E–Z isomerization of the iminium intermediates is not possible due to their cyclic nature, which leads to the formation of the aza-Nazarov products as single diastereomers.
![[1860-5397-19-6-i4]](/bjoc/content/inline/1860-5397-19-6-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Proposed mechanism for the formation of diastereomers 19 and 22.
Scheme 4: Proposed mechanism for the formation of diastereomers 19 and 22.
The aza-Nazarov reactions that we examined so far involved the use of α,β-unsaturated acyl chlorides having a -CH2TMS group at the α-position. Next, we decided to test the reactivity of an α,β-unsaturated acyl chloride possessing a TMS group at the β-position, which would still be suitable to take advantage of the β-silicon effect. For this purpose, we prepared the known acyl chloride 23 in four steps starting from propargyl alcohol (Scheme 5) [68]. Trimethylsilylation of propargyl alcohol (24, 89% yield) followed by reduction of the alkyne using LiAlH4 afforded the allylic alcohol 26 as a single (E) diastereomer. Oxidation of 26 with Jones reagent afforded the carboxylic acid 27 (84% over two steps) and its subsequent treatment with oxalyl chloride gave the desired β-TMS-substituted acyl chloride 23 in 91% yield.
![[1860-5397-19-6-i5]](/bjoc/content/inline/1860-5397-19-6-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Preparation of acyl chloride 23.
Scheme 5: Preparation of acyl chloride 23.
Based on our previous observations, the reaction of imine 5a with acyl chloride 23 was expected to give N-acyliminium ion 28, the aza-Nazarov cyclization of which would provide the final product 30 through the intermediacy of 29 (Scheme 6). According to our hypothesis, the carbocation of intermediate 29 would be stabilized by the β-silicon effect similar to intermediate 9 (vide supra). Unfortunately, screening a variety of conditions including the use of stoichiometric and substoichiometric amounts of Lewis acids such as AgOTf and BF3·OEt2 with and without heating (23 and 80 °C) did not afford any targeted aza-Nazarov product 30 (for details see Table S1 in Supporting Information File 1). The reactivity differences between acyl chlorides 23 and 6 in the examined aza-Nazarov cyclizations can be understood when the electron densities on the two olefin moieties are considered. Indeed, while both of the proposed intermediates 29 and 9 (Scheme 1) can benefit from the β-silicon stabilization effect, the olefinic group of intermediate 8 is expected to be more nucleophilic than that of intermediate 28 given the fact that allylsilanes possess a significantly higher nucleophilicity than vinylsilanes [69]. Therefore, the electron richness of the nucleophilic alkene appears to be an important parameter in addition to the β-silicon effect in this aza-Nazarov cyclization.
![[1860-5397-19-6-i6]](/bjoc/content/inline/1860-5397-19-6-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Aza-Nazarov reaction tested using β-TMS-substituted acyl chloride 23.
Scheme 6: Aza-Nazarov reaction tested using β-TMS-substituted acyl chloride 23.
During the course of our studies, we observed occasionally the formation of aldehyde-containing side products, the amount of which increased when the aza-Nazarov cyclizations did not proceed efficiently. We proposed that, if the aza-Nazarov cyclization of an in situ-formed iminium intermediate is not efficient under certain reaction conditions, then its hydrolysis with adventitious water, which might be present in the reaction medium, would lead to the formation of an aldehyde side product. Unfortunately, our attempts to isolate such a side product in pure form from a crude reaction mixture failed. However, when a mixture of imine 5a and methacryloyl chloride (31) was stirred in a biphasic mixture of CH2Cl2 and aqueous NaHCO3 solution, we were able to isolate and fully characterize aldehyde 32 which would form via the hydrolysis of iminium ion 33 (Scheme 7). Pleasingly, the same approach worked successfully with the acyl chloride 6b where aldehyde product 34 was isolated in diastereomerically pure form (Scheme 7). The 1H NMR spectral data of 34 matched those observed in the spectra of the crude reaction mixtures. These findings underscore the importance of maintaining strictly anhydrous reaction conditions during the aza-Nazarov cyclization for achieving high reaction yields.
![[1860-5397-19-6-i7]](/bjoc/content/inline/1860-5397-19-6-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Hydrolysis of N-acyliminium intermediates.
Scheme 7: Hydrolysis of N-acyliminium intermediates.
Regarding the reaction between imine 5a and acyl chloride 6 to yield final product 7 two pathways can be envisaged as shown in Scheme 8a. According to the originally proposed pathway I, the initial formation of N-acyliminium ion 8 followed by an aza-Nazarov cyclization would lead to product 7. Alternatively, an aza-Hosomi–Sakurai-type reaction may be considered as the initial step between imine 5a and the allylsilane moiety of 6 to give intermediate 35, even though the nucleophilicity of the allylsilane is expected to be low due to the presence of the electron-withdrawing acyl chloride functionality (pathway II, Scheme 8a). The subsequent lactam formation in intermediate 35 would give the final product 7. While general reactivity considerations and the aforementioned formation of the hydrolysis side product 34 support pathway I, we sought to design an experiment to rule out pathway II. To this end, we investigated the potential reaction between imine 5a and α,β-unsaturated ester 36 in the presence of a variety of Lewis acids (Scheme 8b). The reason of using ester 36 with an n-propyl group at the β-position is that it does not have the volatility issues of the ethyl-substituted ester, and that it is sterically less hindered than the isobutyl-substituted ester. When a broad range of Lewis and Brønsted acids such as AgOTf, Zn(OTf)2, ZnBr2, CuCl2, BF3·OEt2, and Tf2NH were tested at different temperatures (Table S2 in Supporting Information File 1) in the reaction between imine 5a and ester 36, the reactants were observed to remain intact, and cyclization product 7 or any other side products, which may be indicative of an aza-Hosomi–Sakurai reaction, were not observed (Scheme 8b). These results strongly support pathway I for the formation of 7 starting from 5a and acyl chloride 6.
![[1860-5397-19-6-i8]](/bjoc/content/inline/1860-5397-19-6-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: (a) Two possible pathways for the formation of 7 and (b) investigation of the reaction between imine 5a and ester 36.
Scheme 8: (a) Two possible pathways for the formation of 7 and (b) investigation of the reaction between imin...
In the present work, the α,β-unsaturated acyl chlorides were generally used as mixtures of diastereomers (typically dr = 3:1 or 4:1) due to the difficulties in the isolation of pure (E)- and (Z)-isomers. The aza-Nazarov cyclization of such diastereomeric mixtures always resulted in the formation of products as single diastereomers with cyclic imines (e.g., imine 5a) [35]. We initially attributed this observed diastereoselectivity to the reaction taking place only with the major (E)-isomer of the acyl chloride based on steric considerations. However, in order to show lack of reactivity of the minor (Z)-isomer, we sought to isolate the two diastereomers in pure form. After extensive efforts, a mixture of benzotrifluoride (PhCF3) and hexanes was identified as a suitable mobile phase for the column chromatographic separation of the diastereomeric esters 16a and 16b. With the successful isolation of these esters in pure form, both were converted to the corresponding acyl chlorides 6ba and 6bb via an initial basic hydrolysis followed by treatment with oxalyl chloride (Scheme 9a). As expected, the aza-Nazarov product 7b was obtained as single diastereomer in 52% yield when the major acyl chloride 6ba with (E)-configuration was subjected to the reaction conditions (Scheme 9b). Surprisingly, the minor (Z)-isomer 6bb also gave the same diastereomerically pure aza-Nazarov product 7b, albeit in a lower yield (24%). The fact that the other diastereomer of the aza-Nazarov product 7b, which would be formed via the reaction of acyl chloride 6bb with (Z)-olefin configuration, was not observed in this transformation points to a potential E–Z isomerization during the reaction. This type of olefin E–Z isomerization is likely to take place after the N-acyliminium formation, possibly via a Michael/retro-Michael addition sequence between the chloride (Cl−) anion and the electron-deficient olefin of the N-acyliminium intermediate at 80 °C (Scheme 9c). In conclusion, these results show that while both isomers of the acyl chloride (6ba and 6bb) are capable of undergoing the aza-Nazarov cyclization with imine 5a affording the same cyclization product, the major (E)-isomer is understandably more reactive due to the favorable sterics of the C–C-bond-forming cyclization step.
![[1860-5397-19-6-i9]](/bjoc/content/inline/1860-5397-19-6-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: (a) Preparation of acyl chlorides 6ba and 6bb in diastereomerically pure forms, (b) aza-Nazarov cyclization of (E)-6ba and (Z)-6bb under the optimized reaction conditions and (c) tentative mechanism for the olefin E–Z isomerization.
Scheme 9: (a) Preparation of acyl chlorides 6ba and 6bb in diastereomerically pure forms, (b) aza-Nazarov cyc...
Conclusion
In summary, we developed an effective aza-Nazarov reaction of 3,4-dihydroisoquinolines and acyclic imines with α,β-unsaturated acyl chlorides to yield α-methylene-γ-lactam heterocyclic products. Whereas a variety of metal-based Lewis acids and organic hydrogen-bond donors were shown to be capable of catalyzing the reaction via anion binding, AgOTf stands out as the optimal Lewis acid for this transformation. The aza-Nazarov cyclization of 3,4-dihydroisoquinolines with TMS-substituted α,β-unsaturated acyl chlorides proceeds efficiently in the presence of AgOTf (20 mol %) in CH3CN at 80 °C to afford tricyclic lactam products in up to 79% yield and as single diastereomers. Moreover, acyclic imines were shown to be competent substrates for this reaction where substituted α-methylene-γ-lactam heteocycles were obtained in up to 76% yield and with dr values of 4.3:1 to 16:1. Detailed mechanistic and control experiments provided valuable insights on the reaction mechanism including the importance of the β-silicon effect and the alkene geometry of the α,β-unsaturated acyl chloride reactants on reactivity, different potential modes of cyclization, and the effect of adventitious water on the aza-Nazarov cyclization.
Acknowledgements
The authors acknowledge the Scientific and Technological Research Application and Research Center, Sinop University, Turkey, for the use of the Bruker D8 Quest diffractometer. The data presented in this article are partly taken from the M.Sc. theses of Bilge Banu Yagci (Bilkent University, 2021) and Selin Ezgi Donmez (Bilkent University, 2019).
References
-
Ma, S., Ed. Handbook of Cyclization Reactions; Wiley-VCH: Weinheim, Germany, 2009; Vol. 1–2.
Return to citation in text: [1] -
Nazarov, I. N.; Zaretskaya, I. I. Izv. Akad. Nauk SSSR, Ser. Khim. 1942, 200–209.
Return to citation in text: [1] -
Habermas, K. L.; Denmark, S. E.; Jones, T. K. Org. React. 1994, 45, 1–158. doi:10.1002/0471264180.or045.01
Return to citation in text: [1] -
Grant, T. N.; Rieder, C. J.; West, F. G. Chem. Commun. 2009, 5676–5688. doi:10.1039/b908515g
Return to citation in text: [1] -
Vaidya, T.; Eisenberg, R.; Frontier, A. J. ChemCatChem 2011, 3, 1531–1548. doi:10.1002/cctc.201100137
Return to citation in text: [1] -
Shimada, N.; Stewart, C.; Tius, M. A. Tetrahedron 2011, 67, 5851–5870. doi:10.1016/j.tet.2011.05.062
Return to citation in text: [1] -
West, F. G.; Scadeng, O.; Wu, Y.-K.; Fradette, R. J.; Joy, S. The Nazarov Cyclization. In Comprehensive Organic Synthesis II; Knochel, P., Ed.; Elsevier: Amsterdam, Netherlands, 2014; Vol. 5, pp 827–866. doi:10.1016/b978-0-08-097742-3.00519-x
Return to citation in text: [1] -
Vinogradov, M. G.; Turova, O. V.; Zlotin, S. G. Org. Biomol. Chem. 2017, 15, 8245–8269. doi:10.1039/c7ob01981e
Return to citation in text: [1] -
Yadykov, A. V.; Shirinian, V. Z. Adv. Synth. Catal. 2020, 362, 702–723. doi:10.1002/adsc.201901001
Return to citation in text: [1] -
Di Grandi, M. J. Org. Biomol. Chem. 2014, 12, 5331–5345. doi:10.1039/c4ob00804a
Return to citation in text: [1] -
Hurt, C. R.; Filipescu, N. J. Am. Chem. Soc. 1972, 94, 3649–3651. doi:10.1021/ja00765a077
Return to citation in text: [1] -
Matoba, K.; Itoh, K.; Kondo, K.; Yamazaki, T.; Nagata, M. Chem. Pharm. Bull. 1981, 29, 2442–2450. doi:10.1248/cpb.29.2442
Return to citation in text: [1] -
Matoba, K.; Miyata, Y.; Yamazaki, T. Chem. Pharm. Bull. 1983, 31, 476–481. doi:10.1248/cpb.31.476
Return to citation in text: [1] -
Atfah, A.; Y., M.; Abu-Shuheil; Hill, J. Tetrahedron 1990, 46, 6483–6500. doi:10.1016/s0040-4020(01)96016-8
Return to citation in text: [1] -
Ciufolini, M. A.; Roschangar, F. Tetrahedron 1997, 53, 11049–11060. doi:10.1016/s0040-4020(97)00365-7
Return to citation in text: [1] -
Dieker, J.; Fröhlich, R.; Würthwein, E.-U. Eur. J. Org. Chem. 2006, 5339–5356. doi:10.1002/ejoc.200600602
Return to citation in text: [1] -
Ghavtadze, N.; Fröhlich, R.; Würthwein, E.-U. Eur. J. Org. Chem. 2008, 3656–3667. doi:10.1002/ejoc.200800384
Return to citation in text: [1] -
Narayan, R.; Fröhlich, R.; Würthwein, E.-U. J. Org. Chem. 2012, 77, 1868–1879. doi:10.1021/jo202477h
Return to citation in text: [1] -
Narayan, R.; Daniliuc, C.-G.; Würthwein, E.-U. Eur. J. Org. Chem. 2012, 6021–6032. doi:10.1002/ejoc.201200913
Return to citation in text: [1] -
Klumpp, D. A.; Zhang, Y.; O'Connor, M. J.; Esteves, P. M.; de Almeida, L. S. Org. Lett. 2007, 9, 3085–3088. doi:10.1021/ol0711570
Return to citation in text: [1] -
Sai, K. K. S.; O’Connor, M. J.; Klumpp, D. A. Tetrahedron Lett. 2011, 52, 2195–2198. doi:10.1016/j.tetlet.2010.11.164
Return to citation in text: [1] -
Shimada, N.; Ashburn, B. O.; Basak, A. K.; Bow, W. F.; Vicic, D. A.; Tius, M. A. Chem. Commun. 2010, 46, 3774–3775. doi:10.1039/b927564a
Return to citation in text: [1] -
Rasapalli, S.; Sammeta, V. R.; Murphy, Z. F.; Huang, Y.; Boerth, J. A.; Golen, J. A.; Savinov, S. N. Org. Lett. 2019, 21, 9824–9828. doi:10.1021/acs.orglett.9b03586
Return to citation in text: [1] -
Rasapalli, S.; Sammeta, V. R.; Murphy, Z. F.; Golen, J. A.; Agama, K.; Pommier, Y.; Savinov, S. N. Bioorg. Med. Chem. Lett. 2021, 41, 127998. doi:10.1016/j.bmcl.2021.127998
Return to citation in text: [1] -
You, X.; Xie, X.; Sun, R.; Chen, H.; Li, S.; Liu, Y. Org. Chem. Front. 2014, 1, 940–946. doi:10.1039/c4qo00159a
Return to citation in text: [1] -
Shu, C.; Wang, Y.-H.; Shen, C.-H.; Ruan, P.-P.; Lu, X.; Ye, L.-W. Org. Lett. 2016, 18, 3254–3257. doi:10.1021/acs.orglett.6b01503
Return to citation in text: [1] -
Ji, W.; Liu, Y. A.; Liao, X. Angew. Chem., Int. Ed. 2016, 55, 13286–13289. doi:10.1002/anie.201607177
Return to citation in text: [1] -
Shi, Y.; Gevorgyan, V. Chem. Commun. 2015, 51, 17166–17169. doi:10.1039/c5cc07598j
Return to citation in text: [1] -
Naredla, R. R.; Zheng, C.; Nilsson Lill, S. O.; Klumpp, D. A. J. Am. Chem. Soc. 2011, 133, 13169–13175. doi:10.1021/ja2046364
Return to citation in text: [1] -
Karthikeyan, I.; Arunprasath, D.; Sekar, G. Chem. Commun. 2015, 51, 1701–1704. doi:10.1039/c4cc08783f
Return to citation in text: [1] -
Li, S.; Wang, Y.; Wu, Z.; Shi, W.; Lei, Y.; Davies, P. W.; Shu, W. Org. Lett. 2021, 23, 7209–7214. doi:10.1021/acs.orglett.1c02519
Return to citation in text: [1] -
Mori-Quiroz, L. M.; Comadoll, C. G.; Super, J. E.; Clift, M. D. Org. Lett. 2021, 23, 7008–7013. doi:10.1021/acs.orglett.1c00867
Return to citation in text: [1] -
Liu, Z.; Liu, X.; Yang, S.; Miao, X.; Li, D.; Wang, D. J. Org. Chem. 2022, 87, 10319–10332. doi:10.1021/acs.joc.2c01379
Return to citation in text: [1] -
Ru, G.-X.; Zhang, T.-T.; Zhang, M.; Jiang, X.-L.; Wan, Z.-K.; Zhu, X.-H.; Shen, W.-B.; Gao, G.-Q. Org. Biomol. Chem. 2021, 19, 5274–5283. doi:10.1039/d1ob00744k
Return to citation in text: [1] -
Donmez, S. E.; Soydaş, E.; Aydın, G.; Şahin, O.; Bozkaya, U.; Türkmen, Y. E. Org. Lett. 2019, 21, 554–558. doi:10.1021/acs.orglett.8b03886
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] -
Wierschke, S. G.; Chandrasekhar, J.; Jorgensen, W. L. J. Am. Chem. Soc. 1985, 107, 1496–1500. doi:10.1021/ja00292a008
Return to citation in text: [1] -
Lambert, J. B.; Zhao, Y.; Emblidge, R. W.; Salvador, L. A.; Liu, X.; So, J.-H.; Chelius, E. C. Acc. Chem. Res. 1999, 32, 183–190. doi:10.1021/ar970296m
Return to citation in text: [1] -
Denmark, S. E.; Jones, T. K. J. Am. Chem. Soc. 1982, 104, 2642–2645. doi:10.1021/ja00373a055
Return to citation in text: [1] -
Unsworth, W. P.; Coulthard, G.; Kitsiou, C.; Taylor, R. J. K. J. Org. Chem. 2014, 79, 1368–1376. doi:10.1021/jo402768r
Return to citation in text: [1] -
Unsworth, W. P.; Taylor, R. J. K. Synlett 2016, 27, 2051–2064. doi:10.1055/s-0035-1562095
Return to citation in text: [1] -
Cusumano, A. Q.; Boudreau, M. W.; Pierce, J. G. J. Org. Chem. 2017, 82, 13714–13721. doi:10.1021/acs.joc.7b02572
Return to citation in text: [1] -
Massaro, N. P.; Pierce, J. G. Org. Lett. 2020, 22, 5079–5084. doi:10.1021/acs.orglett.0c01650
Return to citation in text: [1] -
Wei, J.; Shaw, J. T. Org. Lett. 2007, 9, 4077–4080. doi:10.1021/ol701911u
Return to citation in text: [1] -
Laws, S. W.; Howard, S. Y.; Mato, R.; Meng, S.; Fettinger, J. C.; Shaw, J. T. Org. Lett. 2019, 21, 5073–5077. doi:10.1021/acs.orglett.9b01664
Return to citation in text: [1] -
Klychnikov, M. K.; Pohl, R.; Císařová, I.; Jahn, U. Beilstein J. Org. Chem. 2021, 17, 688–704. doi:10.3762/bjoc.17.58
Return to citation in text: [1] -
Yamaguchi, R.; Mochizuki, K.; Kozima, S.; Takaya, H. Chem. Lett. 1994, 23, 1809–1812. doi:10.1246/cl.1994.1809
Return to citation in text: [1] -
Singh, V.; Kanojiya, S.; Batra, S. Tetrahedron 2006, 62, 10100–10110. doi:10.1016/j.tet.2006.08.045
Return to citation in text: [1] -
Krawczyk, H.; Albrecht, Ł.; Wojciechowski, J.; Wolf, W. M.; Krajewska, U.; Różalski, M. Tetrahedron 2008, 64, 6307–6314. doi:10.1016/j.tet.2008.04.090
Return to citation in text: [1] -
Umemura, S.; McLaughlin, M.; Micalizio, G. C. Org. Lett. 2009, 11, 5402–5405. doi:10.1021/ol9022134
Return to citation in text: [1] -
Shen, A.; Liu, M.; Jia, Z.-S.; Xu, M.-H.; Lin, G.-Q. Org. Lett. 2010, 12, 5154–5157. doi:10.1021/ol102148b
Return to citation in text: [1] -
Companyó, X.; Geant, P.-Y.; Mazzanti, A.; Moyano, A.; Rios, R. Tetrahedron 2014, 70, 75–82. doi:10.1016/j.tet.2013.11.028
Return to citation in text: [1] -
Sengoku, T.; Hayashi, D.; Takahashi, M.; Yoda, H. Eur. J. Org. Chem. 2018, 1813–1820. doi:10.1002/ejoc.201800084
Return to citation in text: [1] -
Sengoku, T.; Shirai, A.; Takano, A.; Inuzuka, T.; Sakamoto, M.; Takahashi, M.; Yoda, H. J. Org. Chem. 2019, 84, 12532–12541. doi:10.1021/acs.joc.9b02038
Return to citation in text: [1] -
Sengoku, T.; Makino, K.; Iijima, A.; Inuzuka, T.; Yoda, H. Beilstein J. Org. Chem. 2020, 16, 2769–2775. doi:10.3762/bjoc.16.227
Return to citation in text: [1] -
Jackson, P. A.; Schares, H. A. M.; Jones, K. F. M.; Widen, J. C.; Dempe, D. P.; Grillet, F.; Cuellar, M. E.; Walters, M. A.; Harki, D. A.; Brummond, K. M. J. Med. Chem. 2020, 63, 14951–14978. doi:10.1021/acs.jmedchem.0c01464
Return to citation in text: [1] -
Hernández-Guadarrama, A.; Cuevas, F.; Montoya-Balbás, I. J.; Román-Bravo, P.; Linzaga-Elizalde, I. Tetrahedron Lett. 2022, 107, 154105. doi:10.1016/j.tetlet.2022.154105
Return to citation in text: [1] -
Taylor, M. S.; Jacobsen, E. N. J. Am. Chem. Soc. 2004, 126, 10558–10559. doi:10.1021/ja046259p
Return to citation in text: [1] -
Brak, K.; Jacobsen, E. N. Angew. Chem., Int. Ed. 2013, 52, 534–561. doi:10.1002/anie.201205449
Return to citation in text: [1] -
Schreiner, P. R.; Wittkopp, A. Org. Lett. 2002, 4, 217–220. doi:10.1021/ol017117s
Return to citation in text: [1] -
Wittkopp, A.; Schreiner, P. R. Chem. – Eur. J. 2003, 9, 407–414. doi:10.1002/chem.200390042
Return to citation in text: [1] -
Malerich, J. P.; Hagihara, K.; Rawal, V. H. J. Am. Chem. Soc. 2008, 130, 14416–14417. doi:10.1021/ja805693p
Return to citation in text: [1] -
Zhu, Y.; Malerich, J. P.; Rawal, V. H. Angew. Chem., Int. Ed. 2010, 49, 153–156. doi:10.1002/anie.200904779
Return to citation in text: [1] -
Yang, K. S.; Nibbs, A. E.; Türkmen, Y. E.; Rawal, V. H. J. Am. Chem. Soc. 2013, 135, 16050–16053. doi:10.1021/ja409012q
Return to citation in text: [1] -
Storer, R. I.; Aciro, C.; Jones, L. H. Chem. Soc. Rev. 2011, 40, 2330–2346. doi:10.1039/c0cs00200c
Return to citation in text: [1] -
Rostami, A.; Colin, A.; Li, X. Y.; Chudzinski, M. G.; Lough, A. J.; Taylor, M. S. J. Org. Chem. 2010, 75, 3983–3992. doi:10.1021/jo100104g
Return to citation in text: [1] -
Busschaert, N.; Kirby, I. L.; Young, S.; Coles, S. J.; Horton, P. N.; Light, M. E.; Gale, P. A. Angew. Chem., Int. Ed. 2012, 51, 4426–4430. doi:10.1002/anie.201200729
Return to citation in text: [1] -
Rodriguez, A. A.; Yoo, H.; Ziller, J. W.; Shea, K. J. Tetrahedron Lett. 2009, 50, 6830–6833. doi:10.1016/j.tetlet.2009.09.129
Return to citation in text: [1] -
Wilson, S. R.; Di Grandi, M. J. J. Org. Chem. 1991, 56, 4766–4772. doi:10.1021/jo00015a036
Return to citation in text: [1] -
Laub, H. A.; Mayr, H. Chem. – Eur. J. 2014, 20, 1103–1110. doi:10.1002/chem.201303215
Return to citation in text: [1]
| 66. | Busschaert, N.; Kirby, I. L.; Young, S.; Coles, S. J.; Horton, P. N.; Light, M. E.; Gale, P. A. Angew. Chem., Int. Ed. 2012, 51, 4426–4430. doi:10.1002/anie.201200729 |
| 67. | Rodriguez, A. A.; Yoo, H.; Ziller, J. W.; Shea, K. J. Tetrahedron Lett. 2009, 50, 6830–6833. doi:10.1016/j.tetlet.2009.09.129 |
| 35. | Donmez, S. E.; Soydaş, E.; Aydın, G.; Şahin, O.; Bozkaya, U.; Türkmen, Y. E. Org. Lett. 2019, 21, 554–558. doi:10.1021/acs.orglett.8b03886 |
| 1. | Ma, S., Ed. Handbook of Cyclization Reactions; Wiley-VCH: Weinheim, Germany, 2009; Vol. 1–2. |
| 16. | Dieker, J.; Fröhlich, R.; Würthwein, E.-U. Eur. J. Org. Chem. 2006, 5339–5356. doi:10.1002/ejoc.200600602 |
| 17. | Ghavtadze, N.; Fröhlich, R.; Würthwein, E.-U. Eur. J. Org. Chem. 2008, 3656–3667. doi:10.1002/ejoc.200800384 |
| 18. | Narayan, R.; Fröhlich, R.; Würthwein, E.-U. J. Org. Chem. 2012, 77, 1868–1879. doi:10.1021/jo202477h |
| 19. | Narayan, R.; Daniliuc, C.-G.; Würthwein, E.-U. Eur. J. Org. Chem. 2012, 6021–6032. doi:10.1002/ejoc.201200913 |
| 33. | Liu, Z.; Liu, X.; Yang, S.; Miao, X.; Li, D.; Wang, D. J. Org. Chem. 2022, 87, 10319–10332. doi:10.1021/acs.joc.2c01379 |
| 11. | Hurt, C. R.; Filipescu, N. J. Am. Chem. Soc. 1972, 94, 3649–3651. doi:10.1021/ja00765a077 |
| 12. | Matoba, K.; Itoh, K.; Kondo, K.; Yamazaki, T.; Nagata, M. Chem. Pharm. Bull. 1981, 29, 2442–2450. doi:10.1248/cpb.29.2442 |
| 13. | Matoba, K.; Miyata, Y.; Yamazaki, T. Chem. Pharm. Bull. 1983, 31, 476–481. doi:10.1248/cpb.31.476 |
| 14. | Atfah, A.; Y., M.; Abu-Shuheil; Hill, J. Tetrahedron 1990, 46, 6483–6500. doi:10.1016/s0040-4020(01)96016-8 |
| 15. | Ciufolini, M. A.; Roschangar, F. Tetrahedron 1997, 53, 11049–11060. doi:10.1016/s0040-4020(97)00365-7 |
| 34. | Ru, G.-X.; Zhang, T.-T.; Zhang, M.; Jiang, X.-L.; Wan, Z.-K.; Zhu, X.-H.; Shen, W.-B.; Gao, G.-Q. Org. Biomol. Chem. 2021, 19, 5274–5283. doi:10.1039/d1ob00744k |
| 10. | Di Grandi, M. J. Org. Biomol. Chem. 2014, 12, 5331–5345. doi:10.1039/c4ob00804a |
| 29. | Naredla, R. R.; Zheng, C.; Nilsson Lill, S. O.; Klumpp, D. A. J. Am. Chem. Soc. 2011, 133, 13169–13175. doi:10.1021/ja2046364 |
| 30. | Karthikeyan, I.; Arunprasath, D.; Sekar, G. Chem. Commun. 2015, 51, 1701–1704. doi:10.1039/c4cc08783f |
| 35. | Donmez, S. E.; Soydaş, E.; Aydın, G.; Şahin, O.; Bozkaya, U.; Türkmen, Y. E. Org. Lett. 2019, 21, 554–558. doi:10.1021/acs.orglett.8b03886 |
| 2. | Nazarov, I. N.; Zaretskaya, I. I. Izv. Akad. Nauk SSSR, Ser. Khim. 1942, 200–209. |
| 3. | Habermas, K. L.; Denmark, S. E.; Jones, T. K. Org. React. 1994, 45, 1–158. doi:10.1002/0471264180.or045.01 |
| 4. | Grant, T. N.; Rieder, C. J.; West, F. G. Chem. Commun. 2009, 5676–5688. doi:10.1039/b908515g |
| 5. | Vaidya, T.; Eisenberg, R.; Frontier, A. J. ChemCatChem 2011, 3, 1531–1548. doi:10.1002/cctc.201100137 |
| 6. | Shimada, N.; Stewart, C.; Tius, M. A. Tetrahedron 2011, 67, 5851–5870. doi:10.1016/j.tet.2011.05.062 |
| 7. | West, F. G.; Scadeng, O.; Wu, Y.-K.; Fradette, R. J.; Joy, S. The Nazarov Cyclization. In Comprehensive Organic Synthesis II; Knochel, P., Ed.; Elsevier: Amsterdam, Netherlands, 2014; Vol. 5, pp 827–866. doi:10.1016/b978-0-08-097742-3.00519-x |
| 8. | Vinogradov, M. G.; Turova, O. V.; Zlotin, S. G. Org. Biomol. Chem. 2017, 15, 8245–8269. doi:10.1039/c7ob01981e |
| 9. | Yadykov, A. V.; Shirinian, V. Z. Adv. Synth. Catal. 2020, 362, 702–723. doi:10.1002/adsc.201901001 |
| 31. | Li, S.; Wang, Y.; Wu, Z.; Shi, W.; Lei, Y.; Davies, P. W.; Shu, W. Org. Lett. 2021, 23, 7209–7214. doi:10.1021/acs.orglett.1c02519 |
| 32. | Mori-Quiroz, L. M.; Comadoll, C. G.; Super, J. E.; Clift, M. D. Org. Lett. 2021, 23, 7008–7013. doi:10.1021/acs.orglett.1c00867 |
| 23. | Rasapalli, S.; Sammeta, V. R.; Murphy, Z. F.; Huang, Y.; Boerth, J. A.; Golen, J. A.; Savinov, S. N. Org. Lett. 2019, 21, 9824–9828. doi:10.1021/acs.orglett.9b03586 |
| 24. | Rasapalli, S.; Sammeta, V. R.; Murphy, Z. F.; Golen, J. A.; Agama, K.; Pommier, Y.; Savinov, S. N. Bioorg. Med. Chem. Lett. 2021, 41, 127998. doi:10.1016/j.bmcl.2021.127998 |
| 27. | Ji, W.; Liu, Y. A.; Liao, X. Angew. Chem., Int. Ed. 2016, 55, 13286–13289. doi:10.1002/anie.201607177 |
| 68. | Wilson, S. R.; Di Grandi, M. J. J. Org. Chem. 1991, 56, 4766–4772. doi:10.1021/jo00015a036 |
| 22. | Shimada, N.; Ashburn, B. O.; Basak, A. K.; Bow, W. F.; Vicic, D. A.; Tius, M. A. Chem. Commun. 2010, 46, 3774–3775. doi:10.1039/b927564a |
| 28. | Shi, Y.; Gevorgyan, V. Chem. Commun. 2015, 51, 17166–17169. doi:10.1039/c5cc07598j |
| 69. | Laub, H. A.; Mayr, H. Chem. – Eur. J. 2014, 20, 1103–1110. doi:10.1002/chem.201303215 |
| 21. | Sai, K. K. S.; O’Connor, M. J.; Klumpp, D. A. Tetrahedron Lett. 2011, 52, 2195–2198. doi:10.1016/j.tetlet.2010.11.164 |
| 35. | Donmez, S. E.; Soydaş, E.; Aydın, G.; Şahin, O.; Bozkaya, U.; Türkmen, Y. E. Org. Lett. 2019, 21, 554–558. doi:10.1021/acs.orglett.8b03886 |
| 20. | Klumpp, D. A.; Zhang, Y.; O'Connor, M. J.; Esteves, P. M.; de Almeida, L. S. Org. Lett. 2007, 9, 3085–3088. doi:10.1021/ol0711570 |
| 25. | You, X.; Xie, X.; Sun, R.; Chen, H.; Li, S.; Liu, Y. Org. Chem. Front. 2014, 1, 940–946. doi:10.1039/c4qo00159a |
| 26. | Shu, C.; Wang, Y.-H.; Shen, C.-H.; Ruan, P.-P.; Lu, X.; Ye, L.-W. Org. Lett. 2016, 18, 3254–3257. doi:10.1021/acs.orglett.6b01503 |
| 35. | Donmez, S. E.; Soydaş, E.; Aydın, G.; Şahin, O.; Bozkaya, U.; Türkmen, Y. E. Org. Lett. 2019, 21, 554–558. doi:10.1021/acs.orglett.8b03886 |
| 35. | Donmez, S. E.; Soydaş, E.; Aydın, G.; Şahin, O.; Bozkaya, U.; Türkmen, Y. E. Org. Lett. 2019, 21, 554–558. doi:10.1021/acs.orglett.8b03886 |
| 35. | Donmez, S. E.; Soydaş, E.; Aydın, G.; Şahin, O.; Bozkaya, U.; Türkmen, Y. E. Org. Lett. 2019, 21, 554–558. doi:10.1021/acs.orglett.8b03886 |
| 36. | Wierschke, S. G.; Chandrasekhar, J.; Jorgensen, W. L. J. Am. Chem. Soc. 1985, 107, 1496–1500. doi:10.1021/ja00292a008 |
| 37. | Lambert, J. B.; Zhao, Y.; Emblidge, R. W.; Salvador, L. A.; Liu, X.; So, J.-H.; Chelius, E. C. Acc. Chem. Res. 1999, 32, 183–190. doi:10.1021/ar970296m |
| 38. | Denmark, S. E.; Jones, T. K. J. Am. Chem. Soc. 1982, 104, 2642–2645. doi:10.1021/ja00373a055 |
| 61. | Malerich, J. P.; Hagihara, K.; Rawal, V. H. J. Am. Chem. Soc. 2008, 130, 14416–14417. doi:10.1021/ja805693p |
| 62. | Zhu, Y.; Malerich, J. P.; Rawal, V. H. Angew. Chem., Int. Ed. 2010, 49, 153–156. doi:10.1002/anie.200904779 |
| 63. | Yang, K. S.; Nibbs, A. E.; Türkmen, Y. E.; Rawal, V. H. J. Am. Chem. Soc. 2013, 135, 16050–16053. doi:10.1021/ja409012q |
| 64. | Storer, R. I.; Aciro, C.; Jones, L. H. Chem. Soc. Rev. 2011, 40, 2330–2346. doi:10.1039/c0cs00200c |
| 65. | Rostami, A.; Colin, A.; Li, X. Y.; Chudzinski, M. G.; Lough, A. J.; Taylor, M. S. J. Org. Chem. 2010, 75, 3983–3992. doi:10.1021/jo100104g |
| 57. | Taylor, M. S.; Jacobsen, E. N. J. Am. Chem. Soc. 2004, 126, 10558–10559. doi:10.1021/ja046259p |
| 58. | Brak, K.; Jacobsen, E. N. Angew. Chem., Int. Ed. 2013, 52, 534–561. doi:10.1002/anie.201205449 |
| 59. | Schreiner, P. R.; Wittkopp, A. Org. Lett. 2002, 4, 217–220. doi:10.1021/ol017117s |
| 60. | Wittkopp, A.; Schreiner, P. R. Chem. – Eur. J. 2003, 9, 407–414. doi:10.1002/chem.200390042 |
| 35. | Donmez, S. E.; Soydaş, E.; Aydın, G.; Şahin, O.; Bozkaya, U.; Türkmen, Y. E. Org. Lett. 2019, 21, 554–558. doi:10.1021/acs.orglett.8b03886 |
| 35. | Donmez, S. E.; Soydaş, E.; Aydın, G.; Şahin, O.; Bozkaya, U.; Türkmen, Y. E. Org. Lett. 2019, 21, 554–558. doi:10.1021/acs.orglett.8b03886 |
| 39. | Unsworth, W. P.; Coulthard, G.; Kitsiou, C.; Taylor, R. J. K. J. Org. Chem. 2014, 79, 1368–1376. doi:10.1021/jo402768r |
| 40. | Unsworth, W. P.; Taylor, R. J. K. Synlett 2016, 27, 2051–2064. doi:10.1055/s-0035-1562095 |
| 41. | Cusumano, A. Q.; Boudreau, M. W.; Pierce, J. G. J. Org. Chem. 2017, 82, 13714–13721. doi:10.1021/acs.joc.7b02572 |
| 42. | Massaro, N. P.; Pierce, J. G. Org. Lett. 2020, 22, 5079–5084. doi:10.1021/acs.orglett.0c01650 |
| 43. | Wei, J.; Shaw, J. T. Org. Lett. 2007, 9, 4077–4080. doi:10.1021/ol701911u |
| 44. | Laws, S. W.; Howard, S. Y.; Mato, R.; Meng, S.; Fettinger, J. C.; Shaw, J. T. Org. Lett. 2019, 21, 5073–5077. doi:10.1021/acs.orglett.9b01664 |
| 45. | Klychnikov, M. K.; Pohl, R.; Císařová, I.; Jahn, U. Beilstein J. Org. Chem. 2021, 17, 688–704. doi:10.3762/bjoc.17.58 |
| 46. | Yamaguchi, R.; Mochizuki, K.; Kozima, S.; Takaya, H. Chem. Lett. 1994, 23, 1809–1812. doi:10.1246/cl.1994.1809 |
| 47. | Singh, V.; Kanojiya, S.; Batra, S. Tetrahedron 2006, 62, 10100–10110. doi:10.1016/j.tet.2006.08.045 |
| 48. | Krawczyk, H.; Albrecht, Ł.; Wojciechowski, J.; Wolf, W. M.; Krajewska, U.; Różalski, M. Tetrahedron 2008, 64, 6307–6314. doi:10.1016/j.tet.2008.04.090 |
| 49. | Umemura, S.; McLaughlin, M.; Micalizio, G. C. Org. Lett. 2009, 11, 5402–5405. doi:10.1021/ol9022134 |
| 50. | Shen, A.; Liu, M.; Jia, Z.-S.; Xu, M.-H.; Lin, G.-Q. Org. Lett. 2010, 12, 5154–5157. doi:10.1021/ol102148b |
| 51. | Companyó, X.; Geant, P.-Y.; Mazzanti, A.; Moyano, A.; Rios, R. Tetrahedron 2014, 70, 75–82. doi:10.1016/j.tet.2013.11.028 |
| 52. | Sengoku, T.; Hayashi, D.; Takahashi, M.; Yoda, H. Eur. J. Org. Chem. 2018, 1813–1820. doi:10.1002/ejoc.201800084 |
| 53. | Sengoku, T.; Shirai, A.; Takano, A.; Inuzuka, T.; Sakamoto, M.; Takahashi, M.; Yoda, H. J. Org. Chem. 2019, 84, 12532–12541. doi:10.1021/acs.joc.9b02038 |
| 54. | Sengoku, T.; Makino, K.; Iijima, A.; Inuzuka, T.; Yoda, H. Beilstein J. Org. Chem. 2020, 16, 2769–2775. doi:10.3762/bjoc.16.227 |
| 55. | Jackson, P. A.; Schares, H. A. M.; Jones, K. F. M.; Widen, J. C.; Dempe, D. P.; Grillet, F.; Cuellar, M. E.; Walters, M. A.; Harki, D. A.; Brummond, K. M. J. Med. Chem. 2020, 63, 14951–14978. doi:10.1021/acs.jmedchem.0c01464 |
| 56. | Hernández-Guadarrama, A.; Cuevas, F.; Montoya-Balbás, I. J.; Román-Bravo, P.; Linzaga-Elizalde, I. Tetrahedron Lett. 2022, 107, 154105. doi:10.1016/j.tetlet.2022.154105 |
© 2023 Yagci et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.