Abstract
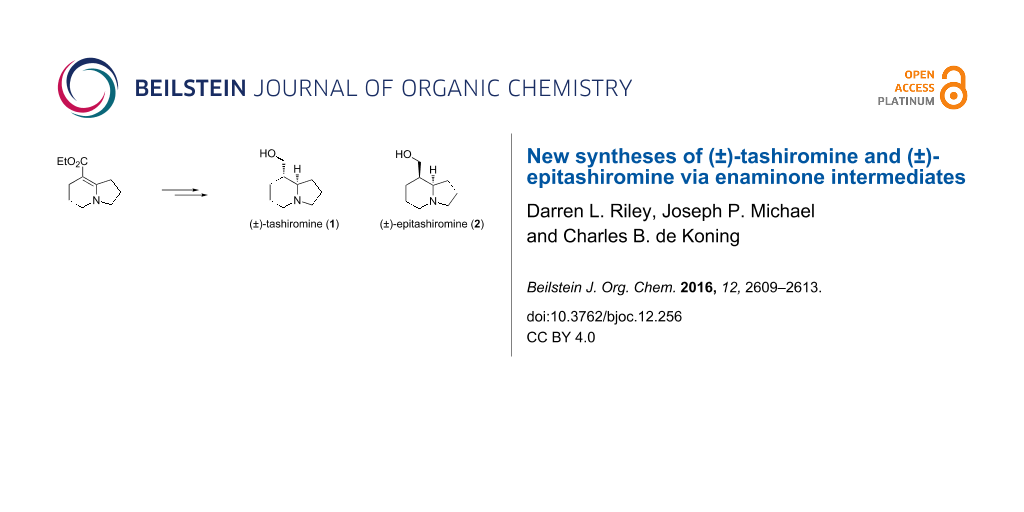
The syntheses of the naturally occurring indolizidine alkaloid (±)-tashiromine and its unnatural epimer (±)-epitashiromine are demonstrated through the use of enaminone chemistry. The impact of various electron-withdrawing substituents at the C-8 position of the indolizidine core on the preparation of the bicyclic system is described.

Graphical Abstract
Introduction
Tashiromine (1) is a monosubstituted indolizidine alkaloid originally isolated from the Asian deciduous shrub Maackia tashiroi [1] and subsequently from the genera Poecilanthe [2] and Crotalaria [3] (Figure 1). To date this natural product and its unnatural epimer 2 have been the targets of numerous enantioselective and racemic syntheses [4-9]. Typical approaches to accessing the alkaloid’s indolizidine skeleton have included a key cyclisation onto the nitrogen atom of either a piperidine or a pyrrolidine-based precursor, with few reports of alternative approaches.
![[1860-5397-12-256-1]](/bjoc/content/figures/1860-5397-12-256-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structures of (±)-tashiromine (1) and (±)-epitashiromine (2) showing the systematic numbering of the indolizidine skeleton. The epimer 2 is often referred to as 5-epitashiromine, in accordance with the alternative numbering of the bicyclic system as a 1-azabicyclo[4.3.0]nonane.
Figure 1: Structures of (±)-tashiromine (1) and (±)-epitashiromine (2) showing the systematic numbering of th...
The approach adopted by our research group to the synthesis of these and related bicyclic alkaloids takes advantage of enaminone chemistry [10-19]. Our interest in the enaminone manifold stems mainly from its ability to display both ambident nucleophilic and electrophilic reactivity, properties that have frequently been exploited in the synthesis of alkaloids and other nitrogen-containing heterocycles [20-23]. The enaminone and related units that form the basis of our own approach most commonly comprise a nitrogen atom in a pyrrolidine or piperidine ring conjugated at C-2 through an exocyclic vinyl fragment to an ester (vinylogous urethane) or another electron-withdrawing substituent (usually ketone, nitrile, nitro or sulfone). When suitable carbon chains bearing terminal electrophilic substituents are attached to the nitrogen, alkylating or acylating cyclisation onto the enamine carbon site leads to the formation of bicyclic systems with nitrogen located at the bridgehead. Our approach to the synthesis of indolizidines is thus unusual in that it entails the formation of the C-7/C-8 bond. In this article we describe further extensions to our route to substituted indolizidines, and an application to the synthesis of racemic tashiromine (1) and epitashiromine (2).
Results and Discussion
One of the most versatile methods for preparing the enaminones of interest is the Eschenmoser sulfide contraction of thiolactams with α-halocarbonyl compounds [24,25], which was originally described by Eschenmoser and co-workers in 1970–1971 [26,27]. We were interested to examine the impact of different electron-withdrawing substituents at what would become the 8-position of the indolizidine skeleton. To this end we chose to investigate the reaction of 3-(2-thioxo-1-pyrrolidinyl)propyl acetate (3), which we had previously used as a precursor to enaminones and indolizidines [28], with three different reaction partners, namely, α-bromoacetone, ethyl α-bromoacetate and α-bromoacetonitrile. The preparation of 3 in three steps from commercially available 3-amino-1-propanol and γ-butyrolactone (4) and its subsequent transformation into the substituted enamines is shown in Scheme 1.
![[1860-5397-12-256-i1]](/bjoc/content/inline/1860-5397-12-256-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Reagents and conditions: (i) NH2(CH2)3OH, 250 °C (sealed tube), 18 h, 81%; (ii) Ac2O, pyridine, 0 °C, 10 min, then, rt, 18 h, 87%; (iii) Na2CO3, P2S5, THF, 5 h, 90%; (iv) BrCH2R, CH3CN, rt, 24 h; (v) PPh3, NEt3, CH3CN, 5 h; (vi) K2CO3, MeOH, rt, 3 h.
Scheme 1: Reagents and conditions: (i) NH2(CH2)3OH, 250 °C (sealed tube), 18 h, 81%; (ii) Ac2O, pyridine, 0 °...
As described previously [28], 3-amino-1-propanol was condensed with γ-butyrolactone (4) in a sealed Carius tube at 250 °C, affording alcohol 5 in 81% yield. The subsequent protection of the primary alcohol as acetate 6 (87% yield), followed by thionation with phosphorus pentasulfide using the Brillon procedure [29] afforded thiolactam 3 in 90% yield. The succeeding Eschenmoser reaction with α-halocarbonyl compounds afforded enaminones 7a and 7b in good yields (90–95%), but the reaction with α-bromoacetonitrile gave enaminonitrile 7c in only 44% yield. The deacetylation of 7a–c with potassium carbonate in methanol afforded the corresponding alcohols 8a–c in good yields (82–89%; Table 1). The data for the vinylogous urethane 8b agreed with those for the same compound which we had previously prepared by alternative methods [13,14]. For comparison, Table 1 also includes our previously reported results [19] with an α-bromo Weinreb acetamide, 2-bromo-N-methoxy-N-methylacetamide.
Table 1: Yields for compounds shown in Scheme 1 and Scheme 2.
| R = COMe (yield, %) | R = CO2Et (yield, %) | R = CN (yield, %) | R = CO[N(OMe)Me] (yield, %) [19] |
|---|---|---|---|
| 7a (95) | 7b (90) | 7c (44) | 7d (85) |
| 8a (82) | 8b (85) | 8c (89) | 8d (83) |
| 9a (27)a | 9b (59) | 9c (72) | 9d (64) |
| 12a (0) | 12b (72; dr 85:15) | 12c (85; dr 92:8)b | 12d (25; dr 95:5)c |
aContaminated with triphenylphosphine traces. bBest result; reduction not reproducible. cBased on the N-hydroxypropyl enaminone 8d.
A key step in the planned reaction sequence was the alkylating cyclisation of the liberated alcohols 8a–c to produce the indolizidine nucleus (Scheme 2). The cyclisation was achieved by initially treating these deprotected enaminones with imidazole and triphenylphosphine at ambient temperature in acetonitrile followed by iodine under refluxing conditions [30]. The products from vinylogous urethane 8b and vinylogous cyanamide 8c were easily purified under standard chromatographic conditions, affording the corresponding bicyclic systems 9b and 9c in 59% and 72% yields, respectively (Scheme 2 and Table 1). We had previously found that the bicyclic vinylogous urea 9d (64%) required a more careful flash chromatographic separation [19]. Unfortunately, the bicyclic vinylogous amide 9a could not be separated adequately from the triphenylphosphine residues under standard chromatographic and recrystallisation conditions. In an attempt to circumvent the issues associated with the purification of the cyclic enamines, we investigated the conversion of alcohols 8c and 8d into the corresponding tosylates followed by treatment with sodium iodide as an alternative for the cyclisation step. The tosylations afforded 10c and 10d in 19% and 71% yields, respectively, with small amounts of the corresponding chlorides 11c and 11d present. However, we were unable to facilitate the desired cyclisation of these compounds to access the indolizidine skeleton, even after in situ conversion of the intermediates into the corresponding iodides. We reverted to using the original procedure, but adjusted the purification protocol by first separating both the products and triphenylphosphine oxide residues from the baseline impurities by column chromatography, then removing most of the remaining triphenylphosphine residues by simple recrystallisation from hexane.
![[1860-5397-12-256-i2]](/bjoc/content/inline/1860-5397-12-256-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Reagents and conditions: (i) Imidazole, PPh3, I2, CH3CN–PhCH3, reflux, 1 h; (ii) p-TsCl, NEt3, DMAP, CH2Cl2, rt, 18 h, (10c) 19%, (10d) 71%; (iii) H2 (1 atm), Adams catalyst, AcOH, rt, 24 h.
Scheme 2: Reagents and conditions: (i) Imidazole, PPh3, I2, CH3CN–PhCH3, reflux, 1 h; (ii) p-TsCl, NEt3, DMAP...
The cyclised enaminones 9b–d underwent catalytic hydrogenation in the presence of Adams catalyst (PtO2·xH2O) under mildly acidic conditions. The reduction can proceed either by direct cis-hydrogenation of the C=C bond, or by hydrogenation of the bicyclic iminium system formed by C-protonation of the enaminone. In the latter case, the preferred formation of the product with a cis relationship of the hydrogen atoms between C-8 and C-8a in the free base has been argued on stereoelectronic grounds [31], although the formation of some trans-product cannot be precluded. In the event, the products 12b–d were obtained as racemic mixtures of diastereomers in which, as expected, the major diastereomer resulted from cis addition of hydrogen across the double bond. This was confirmed in the case of the reduced ester 12b, which was obtained in a good yield of 72% and a diastereomeric ratio of 85:15. The NMR spectroscopic data for the two diastereomers (8R*,8aR*)-12b' and (8R*,8aS*)-12b", pure samples of which could be obtained for characterisation by careful chromatography, agreed with those reported for the same isomers by Kiss et al. [9].
The reduction of the alkene unit in the bicyclic vinylogous cyanamide 9c was not reproducible. In the best case we isolated the nitrile 12c in 85% yield. Despite repeating the reaction several times and altering reaction times and hydrogen pressure in an attempt to optimise the reaction conditions, in most cases the nitrile group also appeared to be fully or partially reduced. In the case of the reduction of 9a, which still had significant triphenylphosphine residues present, we were unable to isolate any product. Interestingly, we were also never able to isolate the reduced product 12d when the bicyclic vinylogous urea 9d contained anything more than trace amounts of triphenylphosphine residues; our best yield for the product was only 25% [19].
Reduction of the diastereomeric mixture of esters 12b to the corresponding alcohols was achieved with a slurry of lithium aluminium hydride in diethyl ether. The reduction afforded a mixture of (±)-tashiromine (1) and its epimer (±)-epitashiromine (2) in an overall yield of 87% and in a 13:87 ratio, which matches the diastereoisomeric ratio in the precursor (Scheme 3). Pure samples for characterisation could be obtained by flash column chromatography on silica gel using a 95:4.75:0.25 mixture of methanol/dichloromethane/ammonium hydroxide. The spectroscopic data compared well with previously published results [7]. In particular, 13C chemical shifts for both tashiromine and epitashiromine were within ±1.0 ppm of those reported by Dieter et al. [32] and Kim et al. [33].
![[1860-5397-12-256-i3]](/bjoc/content/inline/1860-5397-12-256-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Reagents and conditions: (i) LiAlH4, Et2O, 3 h, 87%.
Scheme 3: Reagents and conditions: (i) LiAlH4, Et2O, 3 h, 87%.
Conclusion
A concise total synthesis of (±)-tashiromine (1) and (±)-epitashiromine (2) using enaminone chemistry is reported. The NMR spectroscopic data correlated well with those in previously reported syntheses. The synthetic approach indicated that there was reasonable tolerance of functionality at the 8-position, with only the nitrile experiencing reproducibility issues during the cyclisation step. Key to the success of the synthesis however is the ability to remove efficiently the triphenylphosphine residues after the cyclisation step as the catalytic hydrogenation appeared to be adversely effected by the presence of these residues.
Supporting Information
| Supporting Information File 1: Experimental procedures and copies of NMR spectra. | ||
| Format: PDF | Size: 1.9 MB | Download |
References
-
Ohmiya, S.; Kubo, H.; Otomasu, H.; Saito, K.; Murakoshi, I. Heterocycles 1990, 30, 537–542. doi:10.3987/COM-89-S47
Return to citation in text: [1] -
Greinwald, R.; Bachmann, P.; Lewis, G.; Witte, L.; Czygan, F.-C. Biochem. Syst. Ecol. 1995, 23, 547–553. doi:10.1016/0305-1978(95)00050-5
Return to citation in text: [1] -
Asres, K.; Sporer, F.; Wink, M. Biochem. Syst. Ecol. 2004, 32, 915–930. doi:10.1016/j.bse.2004.03.004
Return to citation in text: [1] -
Michael, J. P. Simple Indolizidine and Quinolizidine Alkaloids. In The Alkaloids: Chemistry and Biology; Knölker, H.-J., Ed.; Academic Press: Cambridge, MA, 2016; Vol. 75, pp 1–498. doi:10.1016/bs.alkal.2014.12.001
for a comprehensive survey of both formal and total enantioselective and racemic syntheses of tashiromine and epitashiromine up to 2013. For more recent syntheses, see references [5-9].
Return to citation in text: [1] -
Koley, D.; Srinivas, K.; Krishna, Y.; Gupta, A. RSC Adv. 2014, 4, 3934–3937. doi:10.1039/C3RA44762F
Return to citation in text: [1] [2] -
Davies, S. G.; Fletcher, A. M.; Foster, E. M.; Houlsby, I. T. T.; Roberts, P. M.; Schofield, T. M.; Thomson, J. E. Org. Biomol. Chem. 2014, 12, 9223–9235. doi:10.1039/C4OB01737D
Return to citation in text: [1] [2] -
Koley, D.; Krishna, Y.; Srinivas, K.; Khan, A. A.; Kant, R. Angew. Chem., Int. Ed. 2014, 53, 13196–13200. doi:10.1002/anie.201407185
Return to citation in text: [1] [2] [3] -
Gavhane, K. B.; Bhorkade, S. B.; Kulkarni, M. G. Tetrahedron: Asymmetry 2015, 26, 746–750. doi:10.1016/j.tetasy.2015.05.011
Return to citation in text: [1] [2] -
Kiss, L.; Forró, E.; Fülöp, F. Beilstein J. Org. Chem. 2015, 11, 596–603. doi:10.3762/bjoc.11.66
Return to citation in text: [1] [2] [3] -
Michael, J. P.; de Koning, C. B.; Gravestock, D.; Hosken, G. D.; Howard, A. S.; Jungmann, C. M.; Krause, R. W. M.; Parsons, A. S.; Pelly, S. C.; Stanbury, T. V. Pure Appl. Chem. 1999, 71, 979–988. doi:10.1351/pac199971060979
and references cited therein. For more recent examples of our approach to the synthesis of alkaloids via enaminones, see references [11-19].
Return to citation in text: [1] -
Michael, J. P.; Gravestock, D. J. Chem. Soc., Perkin Trans. 1 2000, 1919–1928. doi:10.1039/b001853h
Return to citation in text: [1] [2] -
Michael, J. P.; de Koning, C. B.; Petersen, R. L.; Stanbury, T. V. Tetrahedron Lett. 2001, 42, 7513–7516. doi:10.1016/S0040-4039(01)01597-0
Return to citation in text: [1] [2] -
Michael, J. P.; de Koning, C. B.; San Fat, C.; Nattrass, G. L. ARKIVOC 2002, No. ix, 62–77.
Return to citation in text: [1] [2] [3] -
Michael, J. P.; de Koning, C. B.; Malefetse, T. J.; Yillah, I. Org. Biomol. Chem. 2004, 2, 3510–3517. doi:10.1039/B413379J
Return to citation in text: [1] [2] [3] -
Michael, J. P.; de Koning, C. B.; van der Westhuyzen, C. W. Org. Biomol. Chem. 2005, 3, 836–847. doi:10.1039/b418062c
Return to citation in text: [1] [2] -
Michael, J. P.; de Koning, C. B.; Pienaar, D. P. Synlett 2006, 383–386. doi:10.1055/s-2006-926233
Return to citation in text: [1] [2] -
Michael, J. P.; de Koning, C. B.; Mudzunga, T. T.; Petersen, R. L. Synlett 2006, 3284–3288. doi:10.1055/s-2006-951532
Return to citation in text: [1] [2] -
Michael, J. P.; Accone, C.; de Koning, C. B.; van der Westhuyzen, C. W. Beilstein J. Org. Chem. 2008, 4, No. 5. doi:10.1186/1860-5397-4-5
Return to citation in text: [1] [2] -
de Koning, C. B.; Michael, J. P.; Riley, D. L. Heterocycles 2009, 79, 935–953. doi:10.3987/COM-08-S(D)68
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Elassar, A.-Z. A.; El-Khair, A. A. Tetrahedron 2003, 59, 8463–8480. doi:10.1016/S0040-4020(03)01201-8
Return to citation in text: [1] -
Cheng, Y.; Huang, Z.-T.; Wang, M.-X. Curr. Org. Chem. 2004, 8, 325–351. doi:10.2174/1385272043485936
Return to citation in text: [1] -
Negri, G.; Kascheres, C.; Kascheres, A. J. J. Heterocycl. Chem. 2004, 41, 461–491. doi:10.1002/jhet.5570410402
Return to citation in text: [1] -
Wood, J. L.; Elliott, M. C.; Wordingham, S. V. Trends Heterocycl. Chem. 2006, 10, 73–95.
Return to citation in text: [1] -
Hussaini, S. R.; Chamala, R. R.; Wang, Z. Tetrahedron 2015, 71, 6017–6086. doi:10.1016/j.tet.2015.06.026
Return to citation in text: [1] -
Shiosaki, K. The Eschenmoser Coupling Reaction. In Comprehensive Organic Synthesis; Trost, B. M., Ed.; Pergamon Press: Oxford, 1991; Vol. 2, pp 865–892. doi:10.1016/B978-0-08-052349-1.00051-2
Return to citation in text: [1] -
Eschenmoser, A. Q. Rev., Chem. Soc. 1970, 24, 366–415. doi:10.1039/QR9702400366
Return to citation in text: [1] -
Roth, M.; Dubs, P.; Götschi, E.; Eschenmoser, A. Helv. Chim. Acta 1971, 54, 710–734. doi:10.1002/hlca.19710540229
Return to citation in text: [1] -
Michael, J. P.; Parsons, A. S. S. Afr. J. Chem. 1993, 46, 65–69.
Return to citation in text: [1] [2] -
Brillon, D. Synth. Commun. 1990, 20, 3085–3095. doi:10.1080/00397919008051529
Return to citation in text: [1] -
Garegg, P. J.; Samuelsson, B. J. Chem. Soc., Perkin Trans. 1 1980, 2866–2869. doi:10.1039/p19800002866
Return to citation in text: [1] -
Stevens, R. V. Acc. Chem. Res. 1984, 17, 289–296. doi:10.1021/ar00104a005
Return to citation in text: [1] -
Dieter, R. K.; Chen, N.; Watson, R. T. Tetrahedron 2005, 61, 3221–3230. doi:10.1016/j.tet.2005.01.094
Return to citation in text: [1] -
Kim, S.-H.; Kim, S.-I.; Lai, S.; Cha, J. K. J. Org. Chem. 1999, 64, 6771–6775. doi:10.1021/jo9907383
Return to citation in text: [1]
| 19. | de Koning, C. B.; Michael, J. P.; Riley, D. L. Heterocycles 2009, 79, 935–953. doi:10.3987/COM-08-S(D)68 |
| 9. | Kiss, L.; Forró, E.; Fülöp, F. Beilstein J. Org. Chem. 2015, 11, 596–603. doi:10.3762/bjoc.11.66 |
| 1. | Ohmiya, S.; Kubo, H.; Otomasu, H.; Saito, K.; Murakoshi, I. Heterocycles 1990, 30, 537–542. doi:10.3987/COM-89-S47 |
| 10. |
Michael, J. P.; de Koning, C. B.; Gravestock, D.; Hosken, G. D.; Howard, A. S.; Jungmann, C. M.; Krause, R. W. M.; Parsons, A. S.; Pelly, S. C.; Stanbury, T. V. Pure Appl. Chem. 1999, 71, 979–988. doi:10.1351/pac199971060979
and references cited therein. For more recent examples of our approach to the synthesis of alkaloids via enaminones, see references [11-19]. |
| 11. | Michael, J. P.; Gravestock, D. J. Chem. Soc., Perkin Trans. 1 2000, 1919–1928. doi:10.1039/b001853h |
| 12. | Michael, J. P.; de Koning, C. B.; Petersen, R. L.; Stanbury, T. V. Tetrahedron Lett. 2001, 42, 7513–7516. doi:10.1016/S0040-4039(01)01597-0 |
| 13. | Michael, J. P.; de Koning, C. B.; San Fat, C.; Nattrass, G. L. ARKIVOC 2002, No. ix, 62–77. |
| 14. | Michael, J. P.; de Koning, C. B.; Malefetse, T. J.; Yillah, I. Org. Biomol. Chem. 2004, 2, 3510–3517. doi:10.1039/B413379J |
| 15. | Michael, J. P.; de Koning, C. B.; van der Westhuyzen, C. W. Org. Biomol. Chem. 2005, 3, 836–847. doi:10.1039/b418062c |
| 16. | Michael, J. P.; de Koning, C. B.; Pienaar, D. P. Synlett 2006, 383–386. doi:10.1055/s-2006-926233 |
| 17. | Michael, J. P.; de Koning, C. B.; Mudzunga, T. T.; Petersen, R. L. Synlett 2006, 3284–3288. doi:10.1055/s-2006-951532 |
| 18. | Michael, J. P.; Accone, C.; de Koning, C. B.; van der Westhuyzen, C. W. Beilstein J. Org. Chem. 2008, 4, No. 5. doi:10.1186/1860-5397-4-5 |
| 19. | de Koning, C. B.; Michael, J. P.; Riley, D. L. Heterocycles 2009, 79, 935–953. doi:10.3987/COM-08-S(D)68 |
| 30. | Garegg, P. J.; Samuelsson, B. J. Chem. Soc., Perkin Trans. 1 1980, 2866–2869. doi:10.1039/p19800002866 |
| 4. |
Michael, J. P. Simple Indolizidine and Quinolizidine Alkaloids. In The Alkaloids: Chemistry and Biology; Knölker, H.-J., Ed.; Academic Press: Cambridge, MA, 2016; Vol. 75, pp 1–498. doi:10.1016/bs.alkal.2014.12.001
for a comprehensive survey of both formal and total enantioselective and racemic syntheses of tashiromine and epitashiromine up to 2013. For more recent syntheses, see references [5-9]. |
| 5. | Koley, D.; Srinivas, K.; Krishna, Y.; Gupta, A. RSC Adv. 2014, 4, 3934–3937. doi:10.1039/C3RA44762F |
| 6. | Davies, S. G.; Fletcher, A. M.; Foster, E. M.; Houlsby, I. T. T.; Roberts, P. M.; Schofield, T. M.; Thomson, J. E. Org. Biomol. Chem. 2014, 12, 9223–9235. doi:10.1039/C4OB01737D |
| 7. | Koley, D.; Krishna, Y.; Srinivas, K.; Khan, A. A.; Kant, R. Angew. Chem., Int. Ed. 2014, 53, 13196–13200. doi:10.1002/anie.201407185 |
| 8. | Gavhane, K. B.; Bhorkade, S. B.; Kulkarni, M. G. Tetrahedron: Asymmetry 2015, 26, 746–750. doi:10.1016/j.tetasy.2015.05.011 |
| 9. | Kiss, L.; Forró, E.; Fülöp, F. Beilstein J. Org. Chem. 2015, 11, 596–603. doi:10.3762/bjoc.11.66 |
| 19. | de Koning, C. B.; Michael, J. P.; Riley, D. L. Heterocycles 2009, 79, 935–953. doi:10.3987/COM-08-S(D)68 |
| 3. | Asres, K.; Sporer, F.; Wink, M. Biochem. Syst. Ecol. 2004, 32, 915–930. doi:10.1016/j.bse.2004.03.004 |
| 19. | de Koning, C. B.; Michael, J. P.; Riley, D. L. Heterocycles 2009, 79, 935–953. doi:10.3987/COM-08-S(D)68 |
| 11. | Michael, J. P.; Gravestock, D. J. Chem. Soc., Perkin Trans. 1 2000, 1919–1928. doi:10.1039/b001853h |
| 12. | Michael, J. P.; de Koning, C. B.; Petersen, R. L.; Stanbury, T. V. Tetrahedron Lett. 2001, 42, 7513–7516. doi:10.1016/S0040-4039(01)01597-0 |
| 13. | Michael, J. P.; de Koning, C. B.; San Fat, C.; Nattrass, G. L. ARKIVOC 2002, No. ix, 62–77. |
| 14. | Michael, J. P.; de Koning, C. B.; Malefetse, T. J.; Yillah, I. Org. Biomol. Chem. 2004, 2, 3510–3517. doi:10.1039/B413379J |
| 15. | Michael, J. P.; de Koning, C. B.; van der Westhuyzen, C. W. Org. Biomol. Chem. 2005, 3, 836–847. doi:10.1039/b418062c |
| 16. | Michael, J. P.; de Koning, C. B.; Pienaar, D. P. Synlett 2006, 383–386. doi:10.1055/s-2006-926233 |
| 17. | Michael, J. P.; de Koning, C. B.; Mudzunga, T. T.; Petersen, R. L. Synlett 2006, 3284–3288. doi:10.1055/s-2006-951532 |
| 18. | Michael, J. P.; Accone, C.; de Koning, C. B.; van der Westhuyzen, C. W. Beilstein J. Org. Chem. 2008, 4, No. 5. doi:10.1186/1860-5397-4-5 |
| 19. | de Koning, C. B.; Michael, J. P.; Riley, D. L. Heterocycles 2009, 79, 935–953. doi:10.3987/COM-08-S(D)68 |
| 2. | Greinwald, R.; Bachmann, P.; Lewis, G.; Witte, L.; Czygan, F.-C. Biochem. Syst. Ecol. 1995, 23, 547–553. doi:10.1016/0305-1978(95)00050-5 |
| 19. | de Koning, C. B.; Michael, J. P.; Riley, D. L. Heterocycles 2009, 79, 935–953. doi:10.3987/COM-08-S(D)68 |
| 29. | Brillon, D. Synth. Commun. 1990, 20, 3085–3095. doi:10.1080/00397919008051529 |
| 33. | Kim, S.-H.; Kim, S.-I.; Lai, S.; Cha, J. K. J. Org. Chem. 1999, 64, 6771–6775. doi:10.1021/jo9907383 |
| 26. | Eschenmoser, A. Q. Rev., Chem. Soc. 1970, 24, 366–415. doi:10.1039/QR9702400366 |
| 27. | Roth, M.; Dubs, P.; Götschi, E.; Eschenmoser, A. Helv. Chim. Acta 1971, 54, 710–734. doi:10.1002/hlca.19710540229 |
| 13. | Michael, J. P.; de Koning, C. B.; San Fat, C.; Nattrass, G. L. ARKIVOC 2002, No. ix, 62–77. |
| 14. | Michael, J. P.; de Koning, C. B.; Malefetse, T. J.; Yillah, I. Org. Biomol. Chem. 2004, 2, 3510–3517. doi:10.1039/B413379J |
| 5. | Koley, D.; Srinivas, K.; Krishna, Y.; Gupta, A. RSC Adv. 2014, 4, 3934–3937. doi:10.1039/C3RA44762F |
| 6. | Davies, S. G.; Fletcher, A. M.; Foster, E. M.; Houlsby, I. T. T.; Roberts, P. M.; Schofield, T. M.; Thomson, J. E. Org. Biomol. Chem. 2014, 12, 9223–9235. doi:10.1039/C4OB01737D |
| 7. | Koley, D.; Krishna, Y.; Srinivas, K.; Khan, A. A.; Kant, R. Angew. Chem., Int. Ed. 2014, 53, 13196–13200. doi:10.1002/anie.201407185 |
| 8. | Gavhane, K. B.; Bhorkade, S. B.; Kulkarni, M. G. Tetrahedron: Asymmetry 2015, 26, 746–750. doi:10.1016/j.tetasy.2015.05.011 |
| 9. | Kiss, L.; Forró, E.; Fülöp, F. Beilstein J. Org. Chem. 2015, 11, 596–603. doi:10.3762/bjoc.11.66 |
| 24. | Hussaini, S. R.; Chamala, R. R.; Wang, Z. Tetrahedron 2015, 71, 6017–6086. doi:10.1016/j.tet.2015.06.026 |
| 25. | Shiosaki, K. The Eschenmoser Coupling Reaction. In Comprehensive Organic Synthesis; Trost, B. M., Ed.; Pergamon Press: Oxford, 1991; Vol. 2, pp 865–892. doi:10.1016/B978-0-08-052349-1.00051-2 |
| 7. | Koley, D.; Krishna, Y.; Srinivas, K.; Khan, A. A.; Kant, R. Angew. Chem., Int. Ed. 2014, 53, 13196–13200. doi:10.1002/anie.201407185 |
| 20. | Elassar, A.-Z. A.; El-Khair, A. A. Tetrahedron 2003, 59, 8463–8480. doi:10.1016/S0040-4020(03)01201-8 |
| 21. | Cheng, Y.; Huang, Z.-T.; Wang, M.-X. Curr. Org. Chem. 2004, 8, 325–351. doi:10.2174/1385272043485936 |
| 22. | Negri, G.; Kascheres, C.; Kascheres, A. J. J. Heterocycl. Chem. 2004, 41, 461–491. doi:10.1002/jhet.5570410402 |
| 23. | Wood, J. L.; Elliott, M. C.; Wordingham, S. V. Trends Heterocycl. Chem. 2006, 10, 73–95. |
| 32. | Dieter, R. K.; Chen, N.; Watson, R. T. Tetrahedron 2005, 61, 3221–3230. doi:10.1016/j.tet.2005.01.094 |
© 2016 Riley et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)