Abstract
A series of phthalocyanine-C60 dyads 2a–d was synthesized. Key steps in their synthesis are preparation of the low symmetry phthalocyanine intermediate by the statistical condensation of two phthalonitriles, and the final esterification of the fullerene derivative bearing a free COOH group. Structural characterization of the molecules in solution was performed by NMR spectroscopy, UV–vis spectroscopy and cyclic voltammetry. Preliminary studies suggest formation of liquid crystalline (LC) mesophases for some of the prepared dyads. To the best of our knowledge, this is the first example of LC phthalocyanine-C60 dyads.

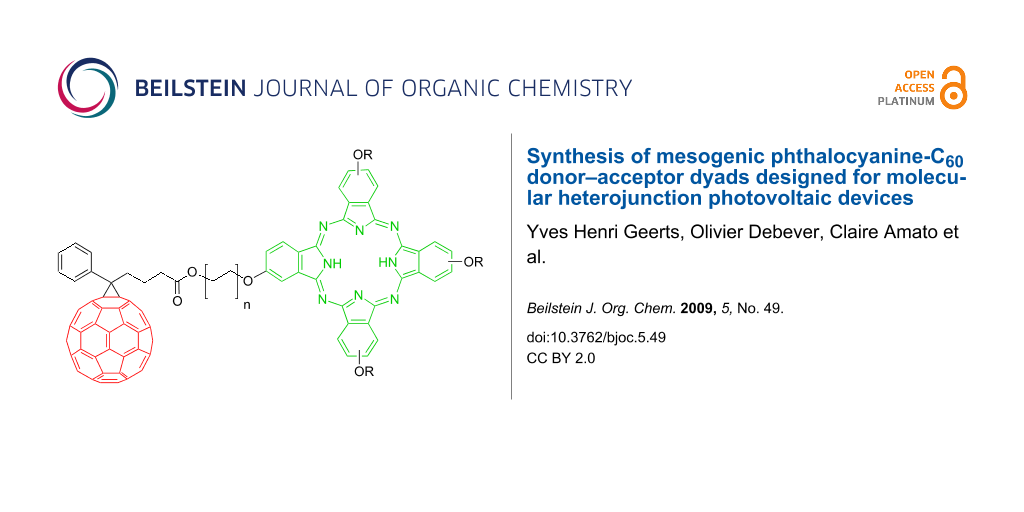
Graphical Abstract
Introduction
Among sustainable energy technologies, photovoltaic (PV) conversion of solar energy is considered as a promising solution. Although currently the market is dominated by inorganic PV devices, development of organic PV materials is driven by their compatibility with solution processing and hence the potentially low cost of large-scale production by printing technologies [1-3]. One of the most popular concepts in the design of organic PV devices is the “bulk heterojunction” architecture, featuring blends of the two immiscible materials: a donor and an acceptor of electrons [4-7]. After absorption of a photon, an initially formed exciton is dissociated at the donor/acceptor interface into a positive charge (hole) and a negative charge (electron), which are transported to the electrodes. Hence, a critical issue in bulk heterojunction PV devices is the control of morphology of materials, in order to provide both the efficient exciton generation and the rapid charge carrier transport. The logical step in the development of this architecture is “molecular heterojunction”, that is, creation of covalent linkages between donor and acceptor components [6]. The main idea is that in material well-organized on the molecular level, donor and acceptor moieties will create the separate pathways for the transport of holes and electrons, respectively. One can intuitively expect that for such high order of self-organization, liquid crystalline (LC) materials will be beneficial, because they combine order and fluidity, which allows the self-healing of structural defects [8].
One of the most widely used molecules in the bulk heterojunction PV devices is fullerene C60, firstly in the pristine form and later as easily soluble derivatives such as phenylcyclopropa[6,6]C60-butanoic acid methyl ester (PCBM, 1, Figure 1) [9-12]. It has been used in a combination with a plethora of conjugated polymers [13-18] as well as small molecules [19-21]. Furthermore, a number of C60-containing covalent ensembles were studied within the “molecular heterojunction” concept [22-29].
![[1860-5397-5-49-1]](/bjoc/content/figures/1860-5397-5-49-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Phthalocyanine-C60 dyads 2a–d described in this paper, C60-derivative 1 (PCBM) and previously reported mesogenic phthalocyanine 3.
Figure 1: Phthalocyanine-C60 dyads 2a–d described in this paper, C60-derivative 1 (PCBM) and previously repor...
Phthalocyanines (Pc) have found a number of industrial applications as dyes and pigments due to their bright blue or green colors combined with extraordinary thermal and photochemical stability [30]. Phthalocyanines bearing flexible peripheral substituents form columnar mesophases [8,31], which demonstrate very efficient charge transport along the columns [32,33]. A unique combination of properties makes phthalocyanines excellent candidates as active materials for photovoltaic devices.
Within the concept of a “molecular heterojunction”, a number of phthalocyanine-fullerene dyads and triads were synthesized [34-38] and long-lived photoinduced charge transfer was demonstrated in these type of materials [39]. Although examples of mesogenic fullerene derivatives have been previously reported [40-44], mesogenic Pc-C60 dyads are unknown, most probably due to the tedious purification of the unsymmetrically substituted phthalocyanines bearing long peripheral substituents. A possible elegant solution to this problem was demonstrated by Torres and co-workers, who obtained LC mesophases of several phthalocyanine-C60 dyads by blending them with a mesogenic phthalocyanine derivative [45]. In this paper we report the synthesis of the first example, to the best of our knowledge, of mesogenic phthalocyanine-fullerene dyads.
Results and Discussion
The structure of dyads 2a–d is depicted in Figure 1. We decided to combine in one molecule the phenylcyclopropa[6,6]C60 moiety of PCBM as an acceptor and a phthalocyanine bearing three swallow-tail alkoxy groups as a donor. The choice of the substituents on the phthalocyanine core was dictated by the previous finding that the pseudosymmetrical phthalocyanine 3 bearing four such substituents is very soluble in many organic solvents, forms columnar mesophases over a broad temperature range and, at the same time, possesses a reasonably low clearing point (ca. 180 °C) [46]. Finally, a critical issue in obtaining a mesogenic phthalocyanine-C60 dyad can be a difficulty in accommodating a bulky C60 molecule in the columnar LC mesophases [45]. To this end, we envisaged the variation of the linker between the fullerene and the phthalocyanine unit.
The synthesis of dyads 2a–d is summarized in Scheme 1 and Scheme 2. The key step, which also represents a bottle-neck in terms of yield (see below), is the preparation of the low-symmetry phthalocyanines 4a–d. To this end, we originally decided to explore the statistical condensation of phthalonitriles 5 and 6a–d (Scheme 1). Previously, synthesis of 4-alkoxyphthalonitriles such as 5 via aromatic nucleophilic substitution in 4-nitrophthalonitrile with the corresponding alcohols in the presence of LiOH in DMSO was reported [46]. However, the yields of phthalonitriles 5 were moderate (below 50%) and their separation from multiple side products was cumbersome. Hence, we preferred to prepare 5 (80% yield) by Williamson reaction between phenol 7 and bromide 8. The latter was synthesized from the corresponding commercially available alcohol by the well-known general method (CBr4/PPh3) [47]. A similar procedure was then used to synthesize phthalonitriles 6a–d (80–86% yield) starting from the corresponding commercially available ω-bromoalcohols.
![[1860-5397-5-49-i1]](/bjoc/content/inline/1860-5397-5-49-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of low-symmetry phthalocyanines 4a–d.
Scheme 1: Synthesis of low-symmetry phthalocyanines 4a–d.
Several strategies for the synthesis of low-symmetry A3B phthalocyanines (where A and B refer to the chemically different isoindole units comprising the phthalocyanine core) were reported earlier, including expansion of subphthalocyanines or solid-phase synthesis on polymer supports [48]. However, operationally simplest and shortest route is the statistical condensation of the two different phthalonitriles. This method inevitably produces a mixture of the desired A3B phthalocyanines with the products of compositions A4, A2B2, AB3 and B4. In theory, 3 : 1 molar ratio of the two dinitriles is required to access A3B products. However, using larger excess of one of dinitriles (often 9 : 1 stoichiometry is used) is more practical. Although the amount of the A4 phthalocyanine in the reaction mixture grows, the formation of cross-condensation products other than A3B is essentially suppressed (see Figure SI2 in the Supporting Information for the graphical presentation of the product distribution). Hence, the isolation of the desired A3B is limited to the separation from the A4. Another factor that facilitates the separation by chromatography is rather different polarity of the substituents on A and B units. In our case, the oligomethylene linker with a terminal OH group is much more polar than the bulky swallow-tail alkyl group, and we did not expect difficulties in the separation of condensation products.
However, attempted condensation of 5 and 6a–d gave no desired phthalocyanine derivative. We reasoned that the unprotected OH group in one of the reaction components may affect the condensation, and converted alcohols 6a–d into acetals 9a–d (84–93% yield) by treatment with an excess of methoxyethoxymethyl chloride (MEMCl) in the presence of i-Pr2EtN as a base. The general choice of acetal as a protective group was dictated by its excellent stability in the presence of the strong bases, required for the cyclotetramerization of phthalonitriles. Among various popular acetal protective groups, the relatively polar MEM moiety was chosen to facilitate the chromatographic separation of the unsymmetrical phthalocyanines 10a–d from the major side product 3: the latter bears only relatively apolar, bulky swallow-tail alkoxy groups. In addition, the signal of the terminal methyl group of the MEM moiety serves as a convenient “marker” in rather complex 1H NMR spectra of phthalocyanines 10a–d (see below). Condensation of 5 and 9a–d in n-C5H11OH/n-C5H11OLi upon reflux afforded the desired A3B phthalocyanines 10a–d (13–24% after column chromatography) together with pseudo-symmetrical A4 phthalocyanine 3 (35–51%). The seemingly modest yields of 10a–d actually correspond to those reported earlier for other A3B-type phthalocyanines: yields higher than 20% are rare in such reactions [45,49-51]. The MEM protective group in 10a–d was then quantitatively removed to give 4a–d upon treatment with pyridinium tosylate (PPTS) in t-BuOH according to previously reported general method [52].
The structure of the phthalocyanines 4a–d was confirmed by 1H and 13C NMR, and HR-MALDI mass spectrometry. 1H NMR spectra of low-symmetry phthalocyanines 4a–d and 10a–d in CDCl3 closely resemble those of 3 and feature a series of three unstructured multiplets from the protons of 1,2,4-substituted benzene rings. Inner-core protons in 4a–d and 10a–d are observed as a broad and concentration-sensitive high-field signal. It should be noted that 3 (as well as any phthalocyanine derivative prepared from a 4-substituted phthalonitrile) represents a mixture of four regioisomers [48,53], while lower-symmetry 4a–d and 10a–d are actual mixtures of as many as eight regioisomers each. This greatly complicates their NMR spectra (see Supporting Information). Furthermore, each regioisomer is a mixture of different diastereoisomers due to the presence of the asymmetric carbon atom in every peripheral substituent. However, although 2-decyltetradecyl substituents in 3 and 10a–d are formally chiral, they should actually be treated as pseudo-achiral since the difference between the two of the substituents at the asymmetric carbon (the two linear alkyl groups of different length) is very small. As was shown previously, the complex diastereochemical composition does not influence the behavior of phthalocyanines bearing similar swallow-tail branched alkyl chains [46,54-57]. UV–vis absorption spectra of 4a–d (Figure 2) are characteristic for metal-free phthalocyanine derivatives: they feature two intense long-wave absorption bands (termed Q-bands).
In the final synthetic step, reaction between 4a–d and the acid 11 (prepared by acid-catalyzed hydrolysis of the commercially available methyl ester 1, PCBM) [10,58] afforded the corresponding dyads 2a–d (Scheme 2). Although in general esterification appears as a trivial synthetic transformation, esterification of 11 is greatly complicated by its poor solubility in virtually all organic solvents. After some experimenting, we found that the classical dicyclohexylcarbodiimide/N,N-dimethylaminopyridine (DCC/DMAP) protocol gives the best results, provided the acid 11 was first sonicated in o-dichlorobenzene for two hours prior to reaction, and then an alcohol 4a–d, DCC and DMAP were added to the reaction mixture. After purification by column chromatography, the dyads 2a–d were isolated in yields up to 45%. Again, the relatively modest yields are comparable with or superior to those previously reported for the esterification of poorly soluble acid 11 with various alcohols [22,50,59].
![[1860-5397-5-49-i2]](/bjoc/content/inline/1860-5397-5-49-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
The structure of dyads 2a–d was confirmed by NMR, MALDI MS and UV–vis absorption spectroscopy. 1H NMR spectra of the dyads 2a–d essentially correspond to the superposition of the spectra of the phthalocyanine 4a–d and PCBM (1), with the exception of the signal of CH2OC=O in dyads 2a–d (ca. 4.10 ppm), which is shifted downfield vs. the signal of CH2OH in phthalocyanines 4a–d (ca. 3.80 ppm). 13C NMR spectrum of the dyad 2c also essentially represents a superposition of the spectra of phthalocyanine 4c and PCBM (1). However, its detailed interpretation is greatly complicated because of a large number of signals, broadening of the signals due to the complex isomeric composition of phthalocyanine (see above), and overlaps of signals (see Supporting Information for the original NMR spectra). For this reason, only selected 13C chemical shifts are given in the Experimental Part. UV–vis absorption spectra of the dyads 2a–d also appear as superposition of the spectra of 4a–d and 1: next to the intense Q-band (maxima at 670 and 707 nm) and B-band (between ca. 300 and ca. 450 nm) of phthalocyanine moiety, very strong high energy absorption of the cyclopropanated C60 derivative with the maximum at ca. 260 nm is observed (Figure 2).
![[1860-5397-5-49-2]](/bjoc/content/figures/1860-5397-5-49-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: UV–vis absorption spectra of 2a (black), 4a (blue) and 1 (red) in CH2Cl2.
Figure 2: UV–vis absorption spectra of 2a (black), 4a (blue) and 1 (red) in CH2Cl2.
Cyclic voltammetry measurements of dyads 2a and 2d in CH2Cl2 produced essentially identical results. Wave potentials do not change upon elongation of the spacer between the donor and the acceptor (Table 1). Furthermore, the voltammograms effectively correspond to the superposition of those for PCBM (1) and the phthalocyanine 3, and do not display supplementary waves (Figure 3). The reduction part of the voltammograms of 2a,d displays five quasi-reversible reduction waves, three due to the C60 moiety and two due to the phthalocyanine fragment. In the oxidation part of the voltammograms of 2a,d, several poorly resolved waves identical to those of phthalocyanine 3 are observed at potentials up to +1.6 V vs. saturated calomel electrode (SCE). These observations, together with UV–vis spectra, suggest the absence of intramolecular charge transfer in the ground state for dyads 2a–d.
![[1860-5397-5-49-3]](/bjoc/content/figures/1860-5397-5-49-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Cyclic voltammograms of 1 (red), 2a (grey), 2d (blue) and 3 (black) in CH2Cl2 (c = 10−4 M), scan rate 100 mV s−1.
Figure 3: Cyclic voltammograms of 1 (red), 2a (grey), 2d (blue) and 3 (black) in CH2Cl2 (c = 10−4 M), scan ra...
Preliminary investigation of the dyads 2a–d by polarized optical microscopy (POM) showed that the length of the spacer between the phthalocyanine and the fullerene moieties significantly affects the thermotropic properties of the material. Dyads 2a,b are liquid at room temperature, while dyads 2c,d display at room temperature fluidic birefringent textures confirming the occurrence of liquid crystalline (LC) mesophases. Upon heating, transitions to isotropic liquid were observed at 120 °C and 90 °C for 2c and 2d, respectively. For 2c this transition was only detectable by POM and not by differential scanning calorimetry (DSC), while for 2d DSC on heating revealed an endothermic peak at 90 °C [enthalpy 2.7 kJ/mol] in perfect agreement with POM observation. The small enthalpy value supports the transition from LC mesophase to isotropic liquid. However, assignment of the nature of LC mesophases for 1c,d from the available data was not obvious, since the textures observed by POM were not characteristic. The major conclusion is nonetheless that the length of the spacer in 2a–d between the C60 fragment and the mesogenic phthalocyanine moiety decisively affects the supramolecular structure of the material.
Conclusion
We report the first example, to the best of our knowledge, of mesogenic phthalocyanine-C60 dyads. The key step in their synthesis is the preparation of the low-symmetry phthalocyanines, bearing three mesogenic swallow-tail substituents and an OH-terminated polymethylene linker. These key intermediates were synthesized by experimentally simple statistical condensation of two phthalonitriles, giving comparatively high yields (up to 24%) for this type of reaction. Upon the favorable combination of the length of the linker with the length of peripheral substituents on the phthalocyanine, bulky C60 moiety can be accommodated in the LC mesophase. Detailed analysis of this supramolecular organization, as well as deeper insight in the structure-property relationships of dyads 2, will be the subject of a separate publication. Other objectives of our ongoing research include: studies of macroscopic alignment in films of dyads 2, studies of charge carrier mobility in aligned films, and fabrication and evaluation of photovoltaic devices.
Experimental
General details. All chemicals were purchased from Aldrich, Acros or TCI and used without further purification. Acid 11 was prepared according to the published procedure [10]. TLC was performed on precoated plates with silica gel 60 F254 (Merck), visualization with UV (λ = 254 nm). Column chromatography was performed on silica gel (0.040–0.063 mm, Macherey–Nagel). 1H NMR and 13C NMR spectra were recorded on Bruker Avance 300 or Varian VNMRS 400 spectrometers; chemical shifts are given in ppm relative to Me4Si (internal standard); coupling constants J are given in Hz. MALDI mass spectra were recorded on a Waters MALDI-QTOF Premier, using a 350 mW laser with dithranol (1,8-dihydroxy-10H-anthracen-9-one) as matrix for phthalocyanines and with DCB (trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene]malonitrile) as matrix for fullerene derivatives; EI and ESI mass spectra were recorded on a Waters AutoSpec 6. UV–Visible absorption spectra were recorded on a HP 8453 spectrophotometer in a quartz cell (optical path of 1 cm). For POM experiments, a NIKON Eclipse 80i microscope equipped with a digital camera DS Camera Head D5-5M was used; temperature was controlled by a Linkam Scientific Instruments GS350 hot stage. For DSC, a Perkin Elmer Diamond DSC calorimeter was used.
Electrochemistry. Cyclic voltammetry experiments were performed with a computer controlled Autolab potentiostat. Measurements were carried out at room temperature in a three-electrode single-compartment cell (10 mL), in CH2Cl2 solutions (concentration 10−4 M), containing Bu4NPF6 (0.1 M) as supporting electrolyte, at a scan rate of 100 mV s−1. A glassy carbon, polished by a slurry-suspension of alumina on micro-cloth and washed by Milli-Q water before each experiment, was used as a working electrode. A spiral platinum wire was employed as counter electrode and an Ag/AgCl/KCl(sat) used as reference electrode was connected to the cell solution via a salt bridge containing a saturated aqueous KCl solution. The Ag/AgCl electrode was checked against the ferrocene/ferrocinium (Fc/Fc+) couple (E°Fc/Fc+ = 0.425 V vs. Ag/AgCl) before and after each experiment. All potentials are reported versus saturated calomel electrode (SCE) (E°Fc/Fc+ = 0.405 V vs. SCE). Before each measurement, solutions were deaerated by 20 min nitrogen bubbling.
4-(2-Decyltetradecyloxy)phthalonitrile (5). To a solution of phenol 7 (1.80 g, 12.5 mmol) and bromide 8 (4.18 g, 10 mmol) in dry DMF (60 mL) was added dry K2CO3 (1.72 g, 12.5 mmol). The mixture was stirred at 90 °C under Ar for 7 h. After cooling to r.t., the mixture was poured into water (60 mL) and extracted with AcOEt (3 × 200 mL). The combined organic layers were washed with aqueous NaHCO3 (5%), dried with MgSO4 and concentrated in vacuum. Column chromatography (SiO2, CH2Cl2) afforded pure 5 as a light yellow solid (4.81 mg, 80%); Rf 0.7 (CH2Cl2). Analytical data identical to those previously reported [46].
4-{10-[(2-Methoxyethoxy)methoxy]decyloxy}phthalonitrile (9c). A solution of phthalonitrile 6c (570 mg, 1.47 mmol) and i-Pr2EtN (0.77 mL, 4.5 mmol) in CH2Cl2 (30 mL) was treated dropwise with MEMCl (0.33 mL, 2.94 mmol) and the resulting mixture was stirred overnight at r.t. The mixture was then treated with aqueous saturated NaHCO3 (35 mL); the organic layer was separated, the aqueous layer was extracted with CH2Cl2 (2 × 40 mL), combined organic layers were dried over MgSO4 and evaporated. Column chromatography of the residue (CH2Cl2/AcOEt 4 : 1, Rf 0.69) afforded 9c (479 mg, 84%) as a light yellow solid; mp 43 °C; Rf 0.64 (CH2Cl2/AcOEt 9 : 1); 1H NMR (300 MHz, CDCl3, 25 °C): δ = 7.68 (d, 3JH,H = 8.8 Hz, 1H), 7.24 (d, 4JH,H = 2.6 Hz, 1H), 7.14 (dd, 3JH,H = 8.8 Hz, 4JH,H = 2.6 Hz, 1H), 4.69 (s, 2H, OCH2O), 4.03 (t, 3JH,H = 6.5 Hz, 2H, CH2O), 3.68 (t, 3JH,H = 5.1 Hz, 2H, CH2O), 3.52–3.60 (m, 4H, OCH2CH2O), 3.38 (s, 3H, MeO), 1.81 (quint, 3JH,H = 7.2 Hz, 2H), 1.26–1.64 (m, 14H); 13C NMR (100 MHz, CDCl3, 25 °C): δ = 162.2, 135.1, 119.5, 119.3, 117.4, 115.7, 115.3, 107.0, 95.5, 71.8, 69.3, 67.9, 66.7, 59.0, 29.7, 29.4, 29.4, 29.4, 29.2, 28.7, 26.2, 25.8; HR-MS (EI): m/z calcd. for C22H32N2O4 ([M]+): 388.2362, found 388.2374.
2-{10-[(2-Methoxyethoxy)methoxy]decyloxy}-9(10),16(17),23(24)-tri(2-decyltetradecyloxy)-29H,31H-phthalocyanine (10c). Li (125 mg, 17.7 mmol) was dissolved under Ar in n-pentanol (10 mL) and the mixture was heated to reflux until Li was completely dissolved. After cooling to r.t., a solution of phthalonitriles 5 (3.41 g, 7.1 mmol) and 9c (305 mg, 0.79 mmol) in n-pentanol (15 mL) was added dropwise and the mixture was heated to reflux overnight. After cooling to r.t., AcOH was added dropwise until pH 5–6 was reached. The green solid was filtered and extensively washed with MeOH. Column chromatography of the residue afforded first 3 (eluted with CH2Cl2, 1.76 g, 50%) and then 10c (eluted with CH2Cl2/AcOEt 1 : 1, 347 mg, 24%). Green solid; Rf 0.75 (CH2Cl2/AcOEt 1:1). 1H NMR (300 MHz, CDCl3, 25 °C): δ = 8.50–8.90 (m, 4H), 8.10–8.50 (m, 4H), 7.30–7.60 (m, 4H), 4.71 (s, 2H, OCH2O), 4.25–4.50 (m, 8H, ArOCH2), 3.65–3.80 (m, 4H, CH2OCH2OCH2), 3.58–3.63 (m, 2H, OCH2CH2O), 3.36 (s, 3H, MeO), 2.10–2.23 (m, 5H, ArOCH2CH2 + ArOCH2CH), 1.15–1.90 (m, 134H), 0.85 (t, 3JH,H = 6.8 Hz, 18H), −2.84 (s, 2H, NH); 13C NMR (100 MHz, CDCl3, 25 °C): δ = 160.2 (br), 147.0 (br), 136.6 (br), 127.8 (br), 122.2 (br), 117.0 (br), 104.0 (br), 94.6 (OCH2O), 70.9 (br, CH2O), 70.7 (br, CH2O), 67.4 (br, CH2O), 66.9 (CH2O), 65.7 (CH2O), 58.0 (MeO), 37.5 (br), 30.5–31.0 (several CH2), 28.3–29.4 (several CH2), 26.3 (CH2), 25.2–25.5 (several CH2), 21.5–21.8 (several CH2), 13.1 (Me); HR-MS (MALDI): m/z calcd. for C118H190N8O7 ([M]+): 1831.4757, found 1831.4748.
2-(10-Hydroxy-decyloxy)-9(10),16(17),23(24)-tri(2-decyltetradecyloxy)-29H,31H-phthalocyanine (4c). A stirred solution of the phthalocyanine 10c (275 mg, 0.15 mmol) and PPTS (188 mg, 0.75 mmol) in t-BuOH (20 mL) was heated to reflux overnight. After cooling to r.t., the reaction mixture was concentrated in vacuum. The resulting solid was suspended in MeOH (50 mL), then filtered, extensively washed with MeOH and dried to give 4c (261 mg, 100%). Green solid; 1H NMR (300 MHz, CDCl3, 25 °C): δ = 8.90–9.20 (m, 4H), 8.45–8.80 (m, 4H), 7.50–7.75 (m, 4H), 4.35–4.55 (m, 8H, ArOCH2), 3.72 (t, 3JH,H = 6.0 Hz, 2H, CH2OH), 2.03–2.20 (m, 5H, ArOCH2CH2 + ArOCH2CH), 1.10–1.95 (m, 134H), 0.80–0.93 (m, 18H), −3.07 (s, 2H, NH); 13C NMR (100 MHz, CDCl3, 25 °C): δ = 161.0 (br), 147.6 (br), 137.4 (br), 128.6 (br), 123.0 (br), 118.2 (br), 104.8 (br), 71.6 (br, CH2O), 68.4 (br, CH2O), 63.1 (CH2O), 38.5 (br), 32.9 (br), 31.6–32.2 (several CH2), 29.4–30.5 (several CH2), 27.3 (CH2), 26.3–26.6 (several CH2), 25.9, 22.6–22.9 (several CH2), 14.2 (Me); HR-MS (MALDI): m/z calcd. for C114H182N8O5 ([M]+): 1743.4233, found 1743.4240.
Phthalocyanine-C60 dyad 2c. A solution of the phthalocyanine 4c (64 mg, 0.037 mmol) and the acid 11 (50 mg, 0.056 mmol) in o-dichlorobenzene (7 mL) was sonicated for 2 h. After cooling to 0 °C, DCC (31 mg, 0.15 mmol) and DMAP (18 mg, 0.15 mmol) were added, the mixture was allowed to reach r.t. and was stirred overnight. The resulting mixture was concentrated and the residue was purified by column chromatography (CH2Cl2/hexane 3 : 2) to give 2c (46 mg, 47%). Green solid; Rf 0.67 (CH2Cl2/hexane 3 : 2); 1H NMR (300 MHz, CDCl3, 25 °C): δ = 8.25–8.80 (m, 4H), 7.90–8.20 (m, 4H), 7.72 (d, 3JH,H = 7.4 Hz, 2H), 7.30–7.60 (m, 7H), 4.20–4.50 (m, 8H, ArOCH2), 4.05–4.15 (m, 2H, COOCH2), 2.67 (t, 3JH,H = 7.5 Hz, 2H, CH2COO), 2.39 (t, 3JH,H = 7.0 Hz, 2H, PhCCH2), 1.98–2.25 (m, 7H, ArOCH2CH2 + ArOCH2CH + CH2CH2COO), 1.10–1.90 (m, 134H), 0.80–0.95 (m, 18H), −3.36 (s, 2H, NH); 13C NMR (100 MHz, CDCl3, 25 °C), selected signals: δ = 173.1 (C=O), 160.3 (br, arom. C–O), 79.7 (fullerene C(sp3)), 71.8 (br, CH2O), 68.4 (br, CH2O), 64.7 (CH2OC=O), 51.7 (PhC), 33.5 and 34.0 (CH2C=O and PhCCH2); HR-MS (MALDI): m/z calcd. for C185H192N8O6 ([M]+): 2621.4965 (M+), found 2621.5530.
Supporting Information
Experimental procedures and analytical data for derivatives 2a,b,d, 4a,b,d, 6a–d, 9a,b,d, 10a,b,d.
| Supporting Information File 1: Experimental details | ||
| Format: DOC | Size: 2.9 MB | Download |
References
-
Spanggaard, H.; Krebs, F. C. Sol. Energy Mater. Sol. Cells 2004, 83, 125–146. doi:10.1016/j.solmat.2004.02.021
Return to citation in text: [1] -
Pivrikas, A.; Sariciftci, N. S.; Juška, G.; Österbacka, R. Prog. Photovoltaics 2007, 15, 677–696. doi:10.1002/pip.791
Return to citation in text: [1] -
Hoth, C. N.; Schilinsky, P.; Choulis, S. A.; Brabec, C. J. Nano Lett. 2008, 8, 2806–2813. doi:10.1021/nl801365k
Return to citation in text: [1] -
Hiramoto, M.; Fujiwara, H.; Yokoyama, M. Appl. Phys. Lett. 1991, 58, 1062–1064. doi:10.1063/1.104423
Return to citation in text: [1] -
Halls, J. J. M.; Walsh, C. A.; Greenham, N. C.; Marseglia, E. A.; Friend, R. H.; Moratti, S. C.; Holmes, A. B. Nature 1995, 376, 498–500. doi:10.1038/376498a0
Return to citation in text: [1] -
Yu, G.; Gao, J.; Hummelen, J. C.; Wudl, F.; Heeger, A. J. Science 1995, 270, 1789–1791. doi:10.1126/science.270.5243.1789
Return to citation in text: [1] [2] -
Janssen, R. A. J.; Hummelen, J. C.; Sariciftci, N. S. MRS Bull. 2005, 30, 33–36.
Return to citation in text: [1] -
Sergeyev, S.; Pisula, W.; Geerts, Y. H. Chem. Soc. Rev. 2007, 36, 1902–1929. doi:10.1039/b417320c
Return to citation in text: [1] [2] -
Wienk, M. M.; Kroon, J. M.; Verhees, W. J. H.; Knol, J.; Hummelen, J. C.; van Hal, P. A.; Janssen, R. A. J. Angew. Chem., Int. Ed. 2003, 42, 3371–3375. doi:10.1002/anie.200351647
Return to citation in text: [1] -
Hummelen, J. C.; Knight, B. W.; LePeq, F.; Wudl, F.; Yao, J.; Wilkins, C. L. J. Org. Chem. 1995, 60, 532–538. doi:10.1021/jo00108a012
Return to citation in text: [1] [2] [3] -
Janssen, R. A. J.; Hummelen, J. C.; Wudl, F. J. Am. Chem. Soc. 1995, 117, 544–545. doi:10.1021/ja00106a068
Return to citation in text: [1] -
Yang, C.; Cho, S.; Heeger, A. J.; Wudl, F. Angew. Chem., Int. Ed. 2009, 48, 1592–1595. doi:10.1002/anie.200805228
Return to citation in text: [1] -
Thompson, B. C.; Fréchet, J. M. J. Angew. Chem., Int. Ed. 2008, 47, 58–77. doi:10.1002/anie.200702506
Return to citation in text: [1] -
Wienk, M. M.; Turbiez, M.; Gilot, J.; Janssen, R. A. J. Adv. Mater. 2008, 20, 2556–2560. doi:10.1002/adma.200800456
Return to citation in text: [1] -
Yi, H.; Johnson, R. G.; Iraqi, A.; Mohamad, D.; Royce, R.; Lidzey, D. G. Macromol. Rapid Commun. 2008, 29, 1804–1809. doi:10.1002/marc.200800440
Return to citation in text: [1] -
He, Y.; Wu, W.; Zhao, G.; Liu, Y.; Li, Y. Macromolecules 2008, 41, 9760–9766. doi:10.1021/ma801923c
Return to citation in text: [1] -
Zhang, F.; Bijleveld, J.; Perzon, E.; Tvingstedt, K.; Barrau, S.; Inganäs, O.; Andersson, M. R. J. Mater. Chem. 2008, 18, 5468–5474. doi:10.1039/b811957k
Return to citation in text: [1] -
Zhou, E.; Nakamura, M.; Nishizawa, T.; Zhang, Y.; Wei, Q.; Tajima, K.; Yang, C.; Hashimoto, K. Macromolecules 2008, 41, 8302–8305. doi:10.1021/ma802052w
Return to citation in text: [1] -
Silvestri, F.; Irwin, M. D.; Beverina, L.; Facchetti, A.; Pagani, G. A.; Marks, T. J. J. Am. Chem. Soc. 2008, 130, 17640–17641. doi:10.1021/ja8067879
Return to citation in text: [1] -
Kronenberg, N. M.; Deppisch, M.; Würthner, F.; Lademann, H. W. A.; Deing, K.; Meerholz, K. Chem. Commun. 2008, 6489–6491. doi:10.1039/b813341g
Return to citation in text: [1] -
Tamayo, A. B.; Walker, B.; Nguyen, T.-Q. J. Phys. Chem. C 2008, 112, 11545–11551. doi:10.1021/jp8031572
Return to citation in text: [1] -
Schuster, D. I.; MacMahon, S.; Guldi, D. M.; Echegoyen, L.; Braslavsky, S. E. Tetrahedron 2006, 62, 1928–1936. doi:10.1016/j.tet.2005.07.127
Return to citation in text: [1] [2] -
Narutaki, M.; Takimiya, K.; Otsubo, T.; Harima, Y.; Zhang, H.; Araki, Y.; Ito, O. J. Org. Chem. 2006, 71, 1761–1768. doi:10.1021/jo051821v
Return to citation in text: [1] -
Nakamura, T.; Araki, Y.; Ito, O.; Murata, Y.; Komatsu, K. J. Photochem. Photobiol., A 2006, 178, 242–250.
Return to citation in text: [1] -
Fujitsuka, M.; Makinoshima, T.; Takamizawa, A.; Araki, Y.; Ito, O.; Obara, Y.; Aso, Y.; Otsubo, T. Bull. Chem. Soc. Jpn. 2006, 79, 1860–1868. doi:10.1246/bcsj.79.1860
Return to citation in text: [1] -
Schuster, D. I.; Li, K.; Guldi, D. M.; Palkar, A.; Echegoyen, L.; Stanisky, C.; Cross, R. J.; Niemi, M.; Tkachenko, N. V.; Lemmetyinen, H. J. Am. Chem. Soc. 2007, 129, 15973–15982. doi:10.1021/ja074684n
Return to citation in text: [1] -
Guldi, D. M.; Spänig, F.; Kreher, D.; Perepichka, I. F.; van der Pol, C.; Bryce, M. R.; Ohkubo, K.; Fukuzumi, S. Chem.–Eur. J. 2008, 14, 250–258. doi:10.1002/chem.200700837
Return to citation in text: [1] -
Kawauchi, H.; Suzuki, S.; Kozaki, M.; Okada, K.; Islam, D.-M. S.; Araki, Y.; Ito, O.; Yamanaka, K.-i. J. Phys. Chem. A 2008, 112, 5878–5884. doi:10.1021/jp800716e
Return to citation in text: [1] -
Guldi, D. M.; Illescas, B. M.; Atienza, C. M.; Wielopolski, M.; Martín, N. Chem. Soc. Rev. 2009, 38, 1587–1597. doi:10.1039/b900402p
Return to citation in text: [1] -
Gregory, P. J. Porphyrins Phthalocyanines 2000, 4, 432–437. doi:10.1002/(SICI)1099-1409(200006/07)4:4<432::AID-JPP254>3.0.CO;2-N
Return to citation in text: [1] -
Laschat, S.; Baro, A.; Steinke, N.; Giesselmann, F.; Hägele, C.; Scalia, G.; Judele, R.; Kapatsina, E.; Sauer, S.; Schreivogel, A.; Tosoni, M. Angew. Chem., Int. Ed. 2007, 46, 4832–4887. doi:10.1002/anie.200604203
Return to citation in text: [1] -
Warman, J. M.; de Haas, M. P.; Dicker, G.; Grozema, F. C.; Piris, J.; Debije, M. G. Chem. Mater. 2004, 16, 4600–4609. doi:10.1021/cm049577w
Return to citation in text: [1] -
Iino, H.; Hanna, J.; Bushby, R. J.; Movaghar, B.; Whitaker, B. J.; Cook, M. J. Appl. Phys. Lett. 2005, 87, 132102. doi:10.1063/1.2056608
Return to citation in text: [1] -
Kahnt, A.; Guldi, D. M.; de la Escosura, A.; Martínez-Díaz, M. V.; Torres, T. J. Mater. Chem. 2008, 18, 77–82. doi:10.1039/b712751k
Return to citation in text: [1] -
Guldi, D. M.; Zilbermann, I.; Gouloumis, A.; Vázquez, P.; Torres, T. J. Phys. Chem. B 2004, 108, 18485–18494. doi:10.1021/jp047105z
Return to citation in text: [1] -
Guldi, D. M.; Gouloumis, A.; Vazquez, P.; Torres, T. Chem. Commun. 2002, 2056–2057. doi:10.1039/b205620h
Return to citation in text: [1] -
Gouloumis, A.; Liu, S.-G.; Sastre, A.; Vazquez, P.; Echegoyen, L.; Torres, T. Chem.–Eur. J. 2000, 6, 3600–3607. doi:10.1002/1521-3765(20001002)6:19<3600::AID-CHEM3600>3.3.CO;2-S
Return to citation in text: [1] -
Isosomppi, M.; Tkachenko, N. V.; Efimov, A.; Vahasalo, H.; Jukola, J.; Vainiotalo, P.; Lemmetyinen, H. Chem. Phys. Lett. 2006, 430, 36–40. doi:10.1016/j.cplett.2006.08.107
Return to citation in text: [1] -
Loi, M. A.; Denk, P.; Hoppe, H.; Neugebauer, H.; Winder, C.; Meissner, D.; Brabec, C.; Sariciftci, N. S.; Gouloumis, A.; Vazquez, P.; Torres, T. J. Mater. Chem. 2003, 13, 700–704. doi:10.1039/b212621d
Return to citation in text: [1] -
Donnio, B.; Buathong, S.; Bury, I.; Guillon, D. Chem. Soc. Rev. 2007, 36, 1495–1513. doi:10.1039/b605531c
Return to citation in text: [1] -
Nakanishi, T.; Shen, Y.; Wang, J.; Yagai, S.; Funahashi, M.; Kato, T.; Fernandes, P.; Möhwald, H.; Kurth, D. G. J. Am. Chem. Soc. 2008, 130, 9236–9237. doi:10.1021/ja803593j
Return to citation in text: [1] -
Mamlouk, H.; Heinrich, B.; Bourgogne, C.; Donnio, B.; Guillon, D.; Felder-Flesch, D. J. Mater. Chem. 2007, 17, 2199–2205. doi:10.1039/b700460e
Return to citation in text: [1] -
Lenoble, J.; Campidelli, S.; Maringa, N.; Donnio, B.; Guillon, D.; Yevlampieva, N.; Deschenaux, R. J. Am. Chem. Soc. 2007, 129, 9941–9952. doi:10.1021/ja071012o
Return to citation in text: [1] -
Maringa, N.; Lenoble, J.; Donnio, B.; Guillon, D.; Deschenaux, R. J. Mater. Chem. 2008, 18, 1524–1534. doi:10.1039/b717105f
Return to citation in text: [1] -
de la Escosura, A.; Martinez-Diaz, M. V.; Barbera, J.; Torres, T. J. Org. Chem. 2008, 73, 1475–1480. doi:10.1021/jo7022763
Return to citation in text: [1] [2] [3] -
Tant, J.; Geerts, Y. H.; Lehmann, M.; de Cupere, V.; Zucchi, G.; Laursen, B. W.; Bjørnholm, T.; Lemaur, V.; Marcq, V.; Burquel, A.; Hennebicq, E.; Gardebien, F.; Viville, P.; Beljonne, D.; Lazzaroni, R.; Cornil, J. J. Phys. Chem. B 2005, 109, 20315–20323. doi:10.1021/jp054778o
Return to citation in text: [1] [2] [3] [4] -
Appel, R. Angew. Chem., Int. Ed. Engl. 1975, 14, 801–811. doi:10.1002/anie.197508011
Return to citation in text: [1] -
de la Torre, G.; Claessens, C. G.; Torres, T. Eur. J. Org. Chem. 2000, 2821–2830. doi:10.1002/1099-0690(200008)2000:16<2821::AID-EJOC2821>3.0.CO;2-2
Return to citation in text: [1] [2] -
Huang, X.; Liu, Y.; Wang, S.; Zhou, S.; Zhu, D. Chem.–Eur. J. 2002, 8, 4179–4184. doi:10.1002/1521-3765(20020916)8:18<4179::AID-CHEM4179>3.0.CO;2-L
Return to citation in text: [1] -
Sastre, A.; Gouloumis, A.; Vázquez, P.; Torres, T.; Doan, V.; Schwartz, B. J.; Wudl, F.; Echegoyen, L.; Rivera, J. Org. Lett. 1999, 1, 1807–1810. doi:10.1021/ol991063t
Return to citation in text: [1] [2] -
Humberstone, P.; Clarkson, G. J.; McKeown, N. B.; Tracher, K. E. J. Mater. Chem. 1996, 6, 315–322. doi:10.1039/jm9960600315
Return to citation in text: [1] -
Banwell, M. G.; Edwards, A. J.; Harfoot, G. J.; Jolliffe, K. A. Tetrahedron 2004, 60, 535–547. doi:10.1016/j.tet.2003.10.122
Return to citation in text: [1] -
Rager, C.; Schmid, G.; Hanack, M. Chem.–Eur. J. 1999, 5, 280–288. doi:10.1002/(SICI)1521-3765(19990104)5:1<280::AID-CHEM280>3.0.CO;2-0
Return to citation in text: [1] -
Sergeyev, S.; Pouzet, E.; Debever, O.; Levin, J.; Gierschner, J.; Cornil, J.; Gomez-Aspe, R.; Geerts, Y. H. J. Mater. Chem. 2007, 17, 1777–1784. doi:10.1039/b617856a
Return to citation in text: [1] -
Sergeyev, S.; Debever, O.; Pouzet, E.; Geerts, Y. H. J. Mater. Chem. 2007, 17, 3002–3007. doi:10.1039/b704344a
Return to citation in text: [1] -
Tylleman, B.; Gómez-Aspe, R.; Gbabode, G.; Geerts, Y. H.; Sergeyev, S. Tetrahedron 2008, 64, 4155–4161. doi:10.1016/j.tet.2008.03.004
Return to citation in text: [1] -
Tylleman, B.; Gbabode, G.; Amato, C.; Buess-Herman, C.; Lemaur, V.; Cornil, J.; Gómez-Aspe, R.; Geerts, Y. H.; Sergeyev, S. Chem. Mater. 2009, 21, 2789–2797. doi:10.1021/cm900383c
Return to citation in text: [1] -
Drees, M.; Hoppe, H.; Winder, C.; Neugebauer, H.; Sariciftci, N. S.; Schwinger, W.; Schäffler, F.; Topf, C.; Scharber, M. C.; Zhud, Z.; Gaudianad, R. J. Mater. Chem. 2005, 15, 5158–5163. doi:10.1039/b505361g
Return to citation in text: [1] -
Giacalone, F.; Segura, J. L.; Martin, N. J. Org. Chem. 2002, 67, 3529–3532. doi:10.1021/jo020088u
Return to citation in text: [1]
| 46. | Tant, J.; Geerts, Y. H.; Lehmann, M.; de Cupere, V.; Zucchi, G.; Laursen, B. W.; Bjørnholm, T.; Lemaur, V.; Marcq, V.; Burquel, A.; Hennebicq, E.; Gardebien, F.; Viville, P.; Beljonne, D.; Lazzaroni, R.; Cornil, J. J. Phys. Chem. B 2005, 109, 20315–20323. doi:10.1021/jp054778o |
| 1. | Spanggaard, H.; Krebs, F. C. Sol. Energy Mater. Sol. Cells 2004, 83, 125–146. doi:10.1016/j.solmat.2004.02.021 |
| 2. | Pivrikas, A.; Sariciftci, N. S.; Juška, G.; Österbacka, R. Prog. Photovoltaics 2007, 15, 677–696. doi:10.1002/pip.791 |
| 3. | Hoth, C. N.; Schilinsky, P.; Choulis, S. A.; Brabec, C. J. Nano Lett. 2008, 8, 2806–2813. doi:10.1021/nl801365k |
| 9. | Wienk, M. M.; Kroon, J. M.; Verhees, W. J. H.; Knol, J.; Hummelen, J. C.; van Hal, P. A.; Janssen, R. A. J. Angew. Chem., Int. Ed. 2003, 42, 3371–3375. doi:10.1002/anie.200351647 |
| 10. | Hummelen, J. C.; Knight, B. W.; LePeq, F.; Wudl, F.; Yao, J.; Wilkins, C. L. J. Org. Chem. 1995, 60, 532–538. doi:10.1021/jo00108a012 |
| 11. | Janssen, R. A. J.; Hummelen, J. C.; Wudl, F. J. Am. Chem. Soc. 1995, 117, 544–545. doi:10.1021/ja00106a068 |
| 12. | Yang, C.; Cho, S.; Heeger, A. J.; Wudl, F. Angew. Chem., Int. Ed. 2009, 48, 1592–1595. doi:10.1002/anie.200805228 |
| 45. | de la Escosura, A.; Martinez-Diaz, M. V.; Barbera, J.; Torres, T. J. Org. Chem. 2008, 73, 1475–1480. doi:10.1021/jo7022763 |
| 8. | Sergeyev, S.; Pisula, W.; Geerts, Y. H. Chem. Soc. Rev. 2007, 36, 1902–1929. doi:10.1039/b417320c |
| 46. | Tant, J.; Geerts, Y. H.; Lehmann, M.; de Cupere, V.; Zucchi, G.; Laursen, B. W.; Bjørnholm, T.; Lemaur, V.; Marcq, V.; Burquel, A.; Hennebicq, E.; Gardebien, F.; Viville, P.; Beljonne, D.; Lazzaroni, R.; Cornil, J. J. Phys. Chem. B 2005, 109, 20315–20323. doi:10.1021/jp054778o |
| 6. | Yu, G.; Gao, J.; Hummelen, J. C.; Wudl, F.; Heeger, A. J. Science 1995, 270, 1789–1791. doi:10.1126/science.270.5243.1789 |
| 39. | Loi, M. A.; Denk, P.; Hoppe, H.; Neugebauer, H.; Winder, C.; Meissner, D.; Brabec, C.; Sariciftci, N. S.; Gouloumis, A.; Vazquez, P.; Torres, T. J. Mater. Chem. 2003, 13, 700–704. doi:10.1039/b212621d |
| 4. | Hiramoto, M.; Fujiwara, H.; Yokoyama, M. Appl. Phys. Lett. 1991, 58, 1062–1064. doi:10.1063/1.104423 |
| 5. | Halls, J. J. M.; Walsh, C. A.; Greenham, N. C.; Marseglia, E. A.; Friend, R. H.; Moratti, S. C.; Holmes, A. B. Nature 1995, 376, 498–500. doi:10.1038/376498a0 |
| 6. | Yu, G.; Gao, J.; Hummelen, J. C.; Wudl, F.; Heeger, A. J. Science 1995, 270, 1789–1791. doi:10.1126/science.270.5243.1789 |
| 7. | Janssen, R. A. J.; Hummelen, J. C.; Sariciftci, N. S. MRS Bull. 2005, 30, 33–36. |
| 40. | Donnio, B.; Buathong, S.; Bury, I.; Guillon, D. Chem. Soc. Rev. 2007, 36, 1495–1513. doi:10.1039/b605531c |
| 41. | Nakanishi, T.; Shen, Y.; Wang, J.; Yagai, S.; Funahashi, M.; Kato, T.; Fernandes, P.; Möhwald, H.; Kurth, D. G. J. Am. Chem. Soc. 2008, 130, 9236–9237. doi:10.1021/ja803593j |
| 42. | Mamlouk, H.; Heinrich, B.; Bourgogne, C.; Donnio, B.; Guillon, D.; Felder-Flesch, D. J. Mater. Chem. 2007, 17, 2199–2205. doi:10.1039/b700460e |
| 43. | Lenoble, J.; Campidelli, S.; Maringa, N.; Donnio, B.; Guillon, D.; Yevlampieva, N.; Deschenaux, R. J. Am. Chem. Soc. 2007, 129, 9941–9952. doi:10.1021/ja071012o |
| 44. | Maringa, N.; Lenoble, J.; Donnio, B.; Guillon, D.; Deschenaux, R. J. Mater. Chem. 2008, 18, 1524–1534. doi:10.1039/b717105f |
| 30. | Gregory, P. J. Porphyrins Phthalocyanines 2000, 4, 432–437. doi:10.1002/(SICI)1099-1409(200006/07)4:4<432::AID-JPP254>3.0.CO;2-N |
| 32. | Warman, J. M.; de Haas, M. P.; Dicker, G.; Grozema, F. C.; Piris, J.; Debije, M. G. Chem. Mater. 2004, 16, 4600–4609. doi:10.1021/cm049577w |
| 33. | Iino, H.; Hanna, J.; Bushby, R. J.; Movaghar, B.; Whitaker, B. J.; Cook, M. J. Appl. Phys. Lett. 2005, 87, 132102. doi:10.1063/1.2056608 |
| 22. | Schuster, D. I.; MacMahon, S.; Guldi, D. M.; Echegoyen, L.; Braslavsky, S. E. Tetrahedron 2006, 62, 1928–1936. doi:10.1016/j.tet.2005.07.127 |
| 23. | Narutaki, M.; Takimiya, K.; Otsubo, T.; Harima, Y.; Zhang, H.; Araki, Y.; Ito, O. J. Org. Chem. 2006, 71, 1761–1768. doi:10.1021/jo051821v |
| 24. | Nakamura, T.; Araki, Y.; Ito, O.; Murata, Y.; Komatsu, K. J. Photochem. Photobiol., A 2006, 178, 242–250. |
| 25. | Fujitsuka, M.; Makinoshima, T.; Takamizawa, A.; Araki, Y.; Ito, O.; Obara, Y.; Aso, Y.; Otsubo, T. Bull. Chem. Soc. Jpn. 2006, 79, 1860–1868. doi:10.1246/bcsj.79.1860 |
| 26. | Schuster, D. I.; Li, K.; Guldi, D. M.; Palkar, A.; Echegoyen, L.; Stanisky, C.; Cross, R. J.; Niemi, M.; Tkachenko, N. V.; Lemmetyinen, H. J. Am. Chem. Soc. 2007, 129, 15973–15982. doi:10.1021/ja074684n |
| 27. | Guldi, D. M.; Spänig, F.; Kreher, D.; Perepichka, I. F.; van der Pol, C.; Bryce, M. R.; Ohkubo, K.; Fukuzumi, S. Chem.–Eur. J. 2008, 14, 250–258. doi:10.1002/chem.200700837 |
| 28. | Kawauchi, H.; Suzuki, S.; Kozaki, M.; Okada, K.; Islam, D.-M. S.; Araki, Y.; Ito, O.; Yamanaka, K.-i. J. Phys. Chem. A 2008, 112, 5878–5884. doi:10.1021/jp800716e |
| 29. | Guldi, D. M.; Illescas, B. M.; Atienza, C. M.; Wielopolski, M.; Martín, N. Chem. Soc. Rev. 2009, 38, 1587–1597. doi:10.1039/b900402p |
| 34. | Kahnt, A.; Guldi, D. M.; de la Escosura, A.; Martínez-Díaz, M. V.; Torres, T. J. Mater. Chem. 2008, 18, 77–82. doi:10.1039/b712751k |
| 35. | Guldi, D. M.; Zilbermann, I.; Gouloumis, A.; Vázquez, P.; Torres, T. J. Phys. Chem. B 2004, 108, 18485–18494. doi:10.1021/jp047105z |
| 36. | Guldi, D. M.; Gouloumis, A.; Vazquez, P.; Torres, T. Chem. Commun. 2002, 2056–2057. doi:10.1039/b205620h |
| 37. | Gouloumis, A.; Liu, S.-G.; Sastre, A.; Vazquez, P.; Echegoyen, L.; Torres, T. Chem.–Eur. J. 2000, 6, 3600–3607. doi:10.1002/1521-3765(20001002)6:19<3600::AID-CHEM3600>3.3.CO;2-S |
| 38. | Isosomppi, M.; Tkachenko, N. V.; Efimov, A.; Vahasalo, H.; Jukola, J.; Vainiotalo, P.; Lemmetyinen, H. Chem. Phys. Lett. 2006, 430, 36–40. doi:10.1016/j.cplett.2006.08.107 |
| 19. | Silvestri, F.; Irwin, M. D.; Beverina, L.; Facchetti, A.; Pagani, G. A.; Marks, T. J. J. Am. Chem. Soc. 2008, 130, 17640–17641. doi:10.1021/ja8067879 |
| 20. | Kronenberg, N. M.; Deppisch, M.; Würthner, F.; Lademann, H. W. A.; Deing, K.; Meerholz, K. Chem. Commun. 2008, 6489–6491. doi:10.1039/b813341g |
| 21. | Tamayo, A. B.; Walker, B.; Nguyen, T.-Q. J. Phys. Chem. C 2008, 112, 11545–11551. doi:10.1021/jp8031572 |
| 13. | Thompson, B. C.; Fréchet, J. M. J. Angew. Chem., Int. Ed. 2008, 47, 58–77. doi:10.1002/anie.200702506 |
| 14. | Wienk, M. M.; Turbiez, M.; Gilot, J.; Janssen, R. A. J. Adv. Mater. 2008, 20, 2556–2560. doi:10.1002/adma.200800456 |
| 15. | Yi, H.; Johnson, R. G.; Iraqi, A.; Mohamad, D.; Royce, R.; Lidzey, D. G. Macromol. Rapid Commun. 2008, 29, 1804–1809. doi:10.1002/marc.200800440 |
| 16. | He, Y.; Wu, W.; Zhao, G.; Liu, Y.; Li, Y. Macromolecules 2008, 41, 9760–9766. doi:10.1021/ma801923c |
| 17. | Zhang, F.; Bijleveld, J.; Perzon, E.; Tvingstedt, K.; Barrau, S.; Inganäs, O.; Andersson, M. R. J. Mater. Chem. 2008, 18, 5468–5474. doi:10.1039/b811957k |
| 18. | Zhou, E.; Nakamura, M.; Nishizawa, T.; Zhang, Y.; Wei, Q.; Tajima, K.; Yang, C.; Hashimoto, K. Macromolecules 2008, 41, 8302–8305. doi:10.1021/ma802052w |
| 8. | Sergeyev, S.; Pisula, W.; Geerts, Y. H. Chem. Soc. Rev. 2007, 36, 1902–1929. doi:10.1039/b417320c |
| 31. | Laschat, S.; Baro, A.; Steinke, N.; Giesselmann, F.; Hägele, C.; Scalia, G.; Judele, R.; Kapatsina, E.; Sauer, S.; Schreivogel, A.; Tosoni, M. Angew. Chem., Int. Ed. 2007, 46, 4832–4887. doi:10.1002/anie.200604203 |
| 47. | Appel, R. Angew. Chem., Int. Ed. Engl. 1975, 14, 801–811. doi:10.1002/anie.197508011 |
| 45. | de la Escosura, A.; Martinez-Diaz, M. V.; Barbera, J.; Torres, T. J. Org. Chem. 2008, 73, 1475–1480. doi:10.1021/jo7022763 |
| 46. | Tant, J.; Geerts, Y. H.; Lehmann, M.; de Cupere, V.; Zucchi, G.; Laursen, B. W.; Bjørnholm, T.; Lemaur, V.; Marcq, V.; Burquel, A.; Hennebicq, E.; Gardebien, F.; Viville, P.; Beljonne, D.; Lazzaroni, R.; Cornil, J. J. Phys. Chem. B 2005, 109, 20315–20323. doi:10.1021/jp054778o |
| 22. | Schuster, D. I.; MacMahon, S.; Guldi, D. M.; Echegoyen, L.; Braslavsky, S. E. Tetrahedron 2006, 62, 1928–1936. doi:10.1016/j.tet.2005.07.127 |
| 50. | Sastre, A.; Gouloumis, A.; Vázquez, P.; Torres, T.; Doan, V.; Schwartz, B. J.; Wudl, F.; Echegoyen, L.; Rivera, J. Org. Lett. 1999, 1, 1807–1810. doi:10.1021/ol991063t |
| 59. | Giacalone, F.; Segura, J. L.; Martin, N. J. Org. Chem. 2002, 67, 3529–3532. doi:10.1021/jo020088u |
| 10. | Hummelen, J. C.; Knight, B. W.; LePeq, F.; Wudl, F.; Yao, J.; Wilkins, C. L. J. Org. Chem. 1995, 60, 532–538. doi:10.1021/jo00108a012 |
| 46. | Tant, J.; Geerts, Y. H.; Lehmann, M.; de Cupere, V.; Zucchi, G.; Laursen, B. W.; Bjørnholm, T.; Lemaur, V.; Marcq, V.; Burquel, A.; Hennebicq, E.; Gardebien, F.; Viville, P.; Beljonne, D.; Lazzaroni, R.; Cornil, J. J. Phys. Chem. B 2005, 109, 20315–20323. doi:10.1021/jp054778o |
| 54. | Sergeyev, S.; Pouzet, E.; Debever, O.; Levin, J.; Gierschner, J.; Cornil, J.; Gomez-Aspe, R.; Geerts, Y. H. J. Mater. Chem. 2007, 17, 1777–1784. doi:10.1039/b617856a |
| 55. | Sergeyev, S.; Debever, O.; Pouzet, E.; Geerts, Y. H. J. Mater. Chem. 2007, 17, 3002–3007. doi:10.1039/b704344a |
| 56. | Tylleman, B.; Gómez-Aspe, R.; Gbabode, G.; Geerts, Y. H.; Sergeyev, S. Tetrahedron 2008, 64, 4155–4161. doi:10.1016/j.tet.2008.03.004 |
| 57. | Tylleman, B.; Gbabode, G.; Amato, C.; Buess-Herman, C.; Lemaur, V.; Cornil, J.; Gómez-Aspe, R.; Geerts, Y. H.; Sergeyev, S. Chem. Mater. 2009, 21, 2789–2797. doi:10.1021/cm900383c |
| 10. | Hummelen, J. C.; Knight, B. W.; LePeq, F.; Wudl, F.; Yao, J.; Wilkins, C. L. J. Org. Chem. 1995, 60, 532–538. doi:10.1021/jo00108a012 |
| 58. | Drees, M.; Hoppe, H.; Winder, C.; Neugebauer, H.; Sariciftci, N. S.; Schwinger, W.; Schäffler, F.; Topf, C.; Scharber, M. C.; Zhud, Z.; Gaudianad, R. J. Mater. Chem. 2005, 15, 5158–5163. doi:10.1039/b505361g |
| 52. | Banwell, M. G.; Edwards, A. J.; Harfoot, G. J.; Jolliffe, K. A. Tetrahedron 2004, 60, 535–547. doi:10.1016/j.tet.2003.10.122 |
| 48. | de la Torre, G.; Claessens, C. G.; Torres, T. Eur. J. Org. Chem. 2000, 2821–2830. doi:10.1002/1099-0690(200008)2000:16<2821::AID-EJOC2821>3.0.CO;2-2 |
| 53. | Rager, C.; Schmid, G.; Hanack, M. Chem.–Eur. J. 1999, 5, 280–288. doi:10.1002/(SICI)1521-3765(19990104)5:1<280::AID-CHEM280>3.0.CO;2-0 |
| 48. | de la Torre, G.; Claessens, C. G.; Torres, T. Eur. J. Org. Chem. 2000, 2821–2830. doi:10.1002/1099-0690(200008)2000:16<2821::AID-EJOC2821>3.0.CO;2-2 |
| 45. | de la Escosura, A.; Martinez-Diaz, M. V.; Barbera, J.; Torres, T. J. Org. Chem. 2008, 73, 1475–1480. doi:10.1021/jo7022763 |
| 49. | Huang, X.; Liu, Y.; Wang, S.; Zhou, S.; Zhu, D. Chem.–Eur. J. 2002, 8, 4179–4184. doi:10.1002/1521-3765(20020916)8:18<4179::AID-CHEM4179>3.0.CO;2-L |
| 50. | Sastre, A.; Gouloumis, A.; Vázquez, P.; Torres, T.; Doan, V.; Schwartz, B. J.; Wudl, F.; Echegoyen, L.; Rivera, J. Org. Lett. 1999, 1, 1807–1810. doi:10.1021/ol991063t |
| 51. | Humberstone, P.; Clarkson, G. J.; McKeown, N. B.; Tracher, K. E. J. Mater. Chem. 1996, 6, 315–322. doi:10.1039/jm9960600315 |
© 2009 Geerts et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)