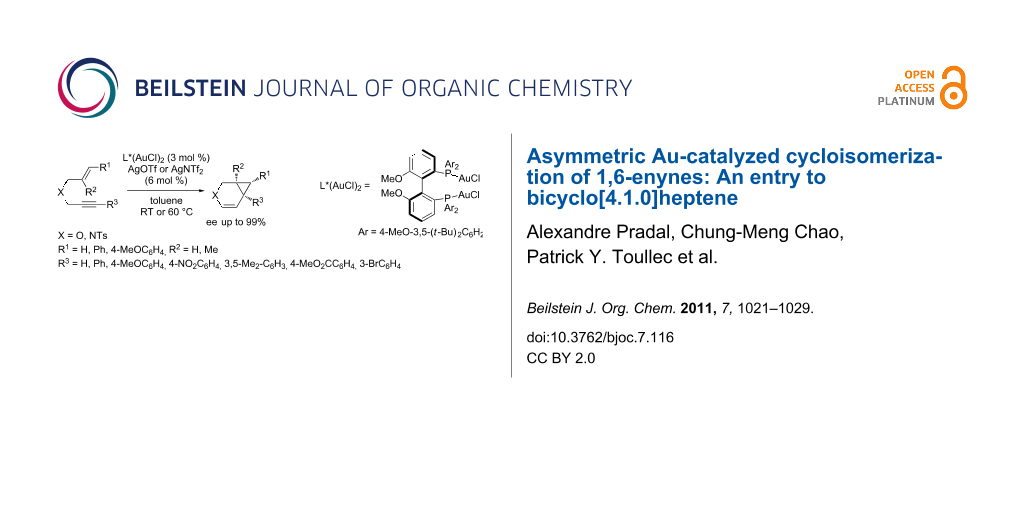
Abstract
A comprehensive study on the asymmetric gold-catalyzed cycloisomerization reaction of heteroatom tethered 1,6-enynes is described. The cycloisomerization reactions were conducted in the presence of the chiral cationic Au(I) catalyst consisting of (R)-4-MeO-3,5-(t-Bu)2-MeOBIPHEP-(AuCl)2 complex and silver salts (AgOTf or AgNTf2) in toluene under mild conditions to afford functionalized bicyclo[4.1.0]heptene derivatives. The reaction conditions were found to be highly substrate-dependent, the best results being obtained in the case of oxygen-tethered enynes. The formation of bicyclic derivatives, including cyclopropyl pentasubstituted ones, was reported in moderate to good yields and in enantiomeric excesses up to 99%.

Graphical Abstract
Introduction
Metal-catalyzed cycloisomerization reactions of 1,n-enynes have emerged as efficient processes that contribute to sustainable development and atom economy concepts [1-8]. In the last ten years, they have provided extremely efficient access to cyclic skeletons with a broad range of functional moieties. Among them, the synthesis of oxa- and azabicyclo[4.1.0]heptenes starting from heteroatom-linked 1,6-enynes has been recently a field of high interest considering the fundamental skeleton rearrangement research of 1,n-enynes [1-11] and the potential applications in biological active and natural products [12,13]. In 1995, Blum et al. described a novel PtCl4-catalyzed cycloisomerization reaction of allyl propynyl ethers leading to oxabicyclo[4.1.0]heptenes [14] (Scheme 1, reaction 1). The group of Murai observed a similar reactivity in the presence of PtCl2, although in a lower yield [15]. These seminal contributions were then followed by several comprehensive studies involving carbophilic complexes such as platinum or gold [16-22] that led to the formation of complex bicyclic and tricyclic compounds [23-40]. The first asymmetric version was described by Shibata’s group in 2005 in the presence of a chiral iridium catalyst [41] (Scheme 1, reaction 2). We and others recently pursued the improvement and development of this enantioselective process, by employing platinum [42-44], rhodium [45] or gold [46-48] complexes. Following our previous work with chiral gold catalysts [46], we report a comprehensive study on gold-catalyzed enantioselective synthesis of bicyclo[4.1.0]heptenes, focusing on the scope and limitations of such systems.
![[1860-5397-7-116-i1]](/bjoc/content/inline/1860-5397-7-116-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: First reports on the racemic and asymmetric synthesis of bicyclo[4.1.0]heptenes.
Scheme 1: First reports on the racemic and asymmetric synthesis of bicyclo[4.1.0]heptenes.
Results and Discussion
Optimization of the catalytic system
Based on our ongoing program on asymmetric gold catalysis [46,49-52], and on literature reports [53-55], we selected 4-MeO-3,5-(t-Bu)2-MeOBIPHEP-(AuCl)2 complex [56-58] as the best candidate for such a transformation. Initial experiments were performed using N-tosyl allyl substrate 1a and oxygen-linked propargylic 1,6-enyne 2a as model substrates (Table 1). The reaction of 1a was evaluated in various solvents and proceeded smoothly leading to the desired alkene 3a [59]. The reaction kinetics and stereoselectivity were found to be highly solvent-dependent, the enantiomeric excesses (ee) varying from 31% to 78% at room temperature (Table 1, entries 1–3). The reaction kinetic was very slow at room temperature in ether and toluene, but high ee’s were obtained. Increasing the temperature to 40 °C in toluene or ether had a positive effect both on the conversion and on the ee’s (Table 1, entries 4 and 5). The reaction was also conducted at 60 °C or 70 °C with good conversions and ee’s (Table 1, entries 6–8), the best results being obtained in toluene. At 80 °C in toluene, a decrease in the stereoselectivity was observed as the ee dropped to 91% (Table 1, entry 9). The reactivity of oxygen-tethered enynes such as 2a was different to that for 1a as a complete conversion was observed at room temperature in toluene, dichloromethane, ether and tetrahydrofuran (Table 1, entries 10–13). A better ee was obtained in toluene compared to other solvents. Cyclopropyl alkene 4a was isolated in 56% yield and 96% ee at 0 °C in toluene (Table 1, entry 14). Toluene was therefore chosen for further studies.
Table 1: Cycloisomerization reaction of nitrogen- and oxygen-linked 1,6-enynes 1a and 2a.
![[Graphic 1]](/bjoc/content/inline/1860-5397-7-116-i5.svg?max-width=637&scale=1.0)
|
|||||||
| Entry | Substrate | Solvent | T [°C] | t [h] | Conv. (Yield) [%]a | Product | ee [%]b |
|---|---|---|---|---|---|---|---|
| 1 | 1a | CH2Cl2 | RT | 36 | 78 | 3a | 31 (−) |
| 2 | 1a | Et2O | RT | 39 | 17 | 3a | 75 (−) |
| 3 | 1a | toluene | RT | 39 | 11 | 3a | 78 (−) |
| 4 | 1a | Et2O | 40 | 41 | 28 | 3a | 90 (−) |
| 5 | 1a | toluene | 40 | 96 | 100 (47) | 3a | 98 (−) |
| 6 | 1a | toluene | 60 | 96 | 100 (74) | 3a | 98 (−) |
| 7 | 1a | THF | 60 | 96 | 69 | 3a | 74 (−) |
| 8 | 1a | toluene | 70 | 96 | 100 (83) | 3a | 96 (−) |
| 9 | 1a | toluene | 80 | 48 | 66 | 3a | 91 (−) |
| 10 | 2a | toluene | RT | 30 | 100 (57) | 4a | 92 (−) |
| 11 | 2a | CH2Cl2 | RT | 25 | 100 (26) | 4a | 70 (−) |
| 12 | 2a | Et2O | RT | 25 | 100 (35) | 4a | 91 (−) |
| 13 | 2a | THF | RT | 25 | 100 (43) | 4a | 85 (−) |
| 14 | 2a | toluene | 0 | 120 | 100 (56) | 4a | 96 (−) |
aDetermined by 1H NMR, bdetermined by HPLC.
Synthesis of 1,6-enynes
We prepared various oxygen-tethered 1,6-enynes according to classic methodologies employing a Williamson alkylation reaction and/or a Sonogashira cross-coupling [60,61] (Scheme 2 and Scheme 3). The known enyne 5 [62,63] was engaged in Pd-catalyzed coupling in the presence of diversely functionalized aryl iodides (Scheme 2). The corresponding substituted alkynes 2b–e [46] were isolated in 71–85% yield. An analogous 1,6-enyne 6 [64] was also reacted with 3-bromoiodobenzene under the same reaction conditions and led to the formation of substrate 2f in 58% isolated yield. We also envisaged preparing two trisubstituted alkenes 2g and 2h by an alkylation/Sonogashira sequence starting from commercially available substrates 7 and 8.
![[1860-5397-7-116-i2]](/bjoc/content/inline/1860-5397-7-116-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of oxygen-tethered 1,6-enynes.
Scheme 2: Synthesis of oxygen-tethered 1,6-enynes.
![[1860-5397-7-116-i3]](/bjoc/content/inline/1860-5397-7-116-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
We also selected some nitrogen-tethered 1,6-enynes 1b–e from the literature [23,41-44] and synthesized them to evaluate the efficiency of the gold chiral catalytic system (Scheme 3).
Scope and limitations of the catalytic system
The prepared heteroatom-linked 1,6-enynes were then engaged in the cycloisomerization process in the presence of Au(I) cationic catalyst generated by mixing (R)-4-MeO-3,5-(t-Bu)2-MeOBIPHEP-(AuCl)2 complex and silver salts (Table 2). Anticipating the moderate reactivity of nitrogen-tethered enynes 1, the reactions were conducted at 60 °C in toluene (Table 2, entries 1–5). The substitution of the aromatic ring on the alkyne moiety led to a substantial decrease of both isolated yields and ee’s, as the presence of several by-products was detected, presumably due to degradation or polymerization [15]. A good ee was achieved in the case of enyne 1c, by using AgNTf2 [65] instead of AgOTf (Table 2, entry 2 compared to entry 1). The substitution of the allylic side chain seemed to slow down the degradation process, as the cyclic alkene 3e was isolated in 61% yield (Table 2, entry 4). In the case of non-substituted enyne 1b (Table 2, entry 5), the bicyclic alkenyl derivative 3b was isolated in low yield and ee: The synthesis of 3b was accompanied by the formation of known 1,3- and 1,4-dienes (5% and 10% isolated yield respectively) resulting from 5-exo- and 6-endo cycloisomerization reactions [20,66]. Thus, the gold catalytic system cannot compete with the results obtained for the cyclizations of nitrogen-tethered enynes in the presence of iridium, platinum or rhodium catalysts [41-45]. The cycloisomerization process was found to be highly stereoselective in the case of oxygen-tethered enynes (Table 2, entries 6–12). In all cases, the ee’s were greater than 90% and in one case as high as 98%. The stability of the resulting bicyclic alkenes 4 was generally only moderate, which led to low isolated yields. In the case of 1,6-enynes 2c and 2d, the low yields (25% and 32% respectively) could be improved by switching from AgOTf salt to AgNTf2, presumably due to the experimentally observed lower hygroscopicity of bistriflimide complex (Table 2, entry 7 compared to 8 and 9 compared to 10). The functionalized derivatives 4c and 4d were obtained in 64% and 63% yields respectively and in excellent ee’s (Table 2, entries 8 and 10). The compatibility with another functional group on the aromatic ring such as bromine (Table 2, entry 11), and with a different allylic side chain (Table 2, entry 12) was also evaluated: The corresponding bicyclic adducts 4e and 4f were isolated in modest to good yield and 95% ee.
Table 2: Cycloisomerization reaction of nitrogen- and oxygen-linked 1,6-enynes.
![[Graphic 2]](/bjoc/content/inline/1860-5397-7-116-i6.svg?max-width=637&scale=1.0)
|
|||||||
| Entry | Enyne | t [h] | Yield [%]a | Product | ee [%]b | ||
|---|---|---|---|---|---|---|---|
| 1c |
![[Graphic 3]](/bjoc/content/inline/1860-5397-7-116-i7.svg?max-width=637&scale=1.0)
|
1c | 17 | 8 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-7-116-i8.svg?max-width=637&scale=1.0)
|
3c | 77 |
| 2c,d |
![[Graphic 5]](/bjoc/content/inline/1860-5397-7-116-i9.svg?max-width=637&scale=1.0)
|
1c | 17 | 8 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-7-116-i10.svg?max-width=637&scale=1.0)
|
3c | 89 |
| 3c |
![[Graphic 7]](/bjoc/content/inline/1860-5397-7-116-i11.svg?max-width=637&scale=1.0)
|
1d | 24 | 7 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-7-116-i12.svg?max-width=637&scale=1.0)
|
3d | 35 |
| 4c |
![[Graphic 9]](/bjoc/content/inline/1860-5397-7-116-i13.svg?max-width=637&scale=1.0)
|
1e | 16 | 61 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-7-116-i14.svg?max-width=637&scale=1.0)
|
3e | 13 |
| 5c |
![[Graphic 11]](/bjoc/content/inline/1860-5397-7-116-i15.svg?max-width=637&scale=1.0)
|
1b | 16 | 23 |
![[Graphic 12]](/bjoc/content/inline/1860-5397-7-116-i16.svg?max-width=637&scale=1.0)
|
3b | 22 |
| 6 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-7-116-i17.svg?max-width=637&scale=1.0)
|
2b | 3.5 | 54 |
![[Graphic 14]](/bjoc/content/inline/1860-5397-7-116-i18.svg?max-width=637&scale=1.0)
|
4b | 93 (+) |
| 7 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-7-116-i19.svg?max-width=637&scale=1.0)
|
2c | 15 | 25 |
![[Graphic 16]](/bjoc/content/inline/1860-5397-7-116-i20.svg?max-width=637&scale=1.0)
|
4c | 94 (−) |
| 8d |
![[Graphic 17]](/bjoc/content/inline/1860-5397-7-116-i21.svg?max-width=637&scale=1.0)
|
2c | 15 | 64 |
![[Graphic 18]](/bjoc/content/inline/1860-5397-7-116-i22.svg?max-width=637&scale=1.0)
|
4c | 94 (−) |
| 9 |
![[Graphic 19]](/bjoc/content/inline/1860-5397-7-116-i23.svg?max-width=637&scale=1.0)
|
2d | 15 | 32 |
![[Graphic 20]](/bjoc/content/inline/1860-5397-7-116-i24.svg?max-width=637&scale=1.0)
|
4d | 96 (−) |
| 10d |
![[Graphic 21]](/bjoc/content/inline/1860-5397-7-116-i25.svg?max-width=637&scale=1.0)
|
2d | 15 | 63 |
![[Graphic 22]](/bjoc/content/inline/1860-5397-7-116-i26.svg?max-width=637&scale=1.0)
|
4d | 98 (−) |
| 11 |
![[Graphic 23]](/bjoc/content/inline/1860-5397-7-116-i27.svg?max-width=637&scale=1.0)
|
2e | 30 | 59 |
![[Graphic 24]](/bjoc/content/inline/1860-5397-7-116-i28.svg?max-width=637&scale=1.0)
|
4e | 95 (−) |
| 12 |
![[Graphic 25]](/bjoc/content/inline/1860-5397-7-116-i29.svg?max-width=637&scale=1.0)
|
2f | 1 | 37 |
![[Graphic 26]](/bjoc/content/inline/1860-5397-7-116-i30.svg?max-width=637&scale=1.0)
|
4f | 95 (−) |
aIsolated yield, bdetermined by HPLC, c60 °C, dAgNTf2.
Considering the observed highly stereoselective reactions of oxygen-tethered 1,6-enynes, we decided to study the challenging asymmetric synthesis of pentasubstituted cyclopropyl derivatives [67-70] (Scheme 4). The bicyclic derivative 4h was obtained in moderate yield and 73% ee. Conducting the reaction at 0 °C and using AgNTf2 as a chloride scavenger led to the formation of the alkenyl functionalized derivative 4g in 36% isolated yield and excellent 99% ee.
![[1860-5397-7-116-i4]](/bjoc/content/inline/1860-5397-7-116-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of pentasubstituted bicyclic cyclopropanes.
Scheme 4: Synthesis of pentasubstituted bicyclic cyclopropanes.
Conclusion
In conclusion, we have contributed to the development of an asymmetric gold-catalyzed cycloisomerization reaction allowing the formation of oxa- and aza-bicyclo[4.1.0]heptene derivatives. The combination of chiral Au(I) complex (R)-4-MeO-3,5-(t-Bu)2-MeOBIPHEP-(AuCl)2 associated to silver salts promotes the enantioselective rearrangement of oxygen and nitrogen-tethered 1,6-enynes in toluene at room temperature or 60 °C. The cycloisomerization reactions were found to be highly substrate-dependent as low yield and ee’s were generally obtained in the case of nitrogen-tethered enynes. The enantiomerically enriched functionalized oxabicyclo[4.1.0]heptenes were isolated in moderate yields but with excellent ee values ranging from 73% to 99%. This methodology was successfully applied to the synthesis of pentasubstituted cyclopropyl heterobicycles.
Experimental
All reactions were carried out under an argon atmosphere. 1H NMR and 13C NMR were recorded on a Bruker AV 300 instrument. All signals were expressed as ppm (δ) and internally referenced to residual proton solvent signals. Coupling constants (J) are reported in Hz and refer to apparent peak multiplicities. Enantiomeric excesses were determined by high pressure liquid chromatography analyses (HPLC) on Waters instruments (Waters 486 detector, 717 autosampler equipped with Daicel Chiralcel OD-H, OJ and Chiralpak IA, AD, λ = 215 nm). Optical rotation measurements were conducted on a Perkin–Elmer 241 polarimeter at 589 nm. Enynes 5 [71], 2a [72], 2b–e [46], 1a [73], 1b [74], 1c,d [75], 1e [73], and 6 [64] were prepared according to published procedures. 1H, 13C NMR and mass spectrometry data for compounds 3a,b [23], 3c,d [75], 3e [76] and 4a–e [46] were described elsewhere.
(E)-1-Bromo-3-(3-(3-(4-methoxyphenyl)allyloxy)prop-1-ynyl)benzene (2f): CuI (46 mg, 0.1 equiv) and PdCl2(PPh3)2 (86 mg, 0.05 equiv) were placed in a Schlenk tube under argon. Distilled diisopropylamine (3 mL) was added and the reaction mixture was stirred at RT for 5 min. 1-Bromo-3-iodobenzene (0.4 mL, 1.3 equiv) was added and the reaction mixture was stirred for 5 min. Enyne 6, dissolved in 2 mL of distilled diisopropylamine was added and the reaction mixture stirred for 3 h at RT. After hydrolysis with sat. aq. NH4Cl solution, the aqueous phase was extracted with EtOAc. The organic layer was successively washed with sat. aq. NH4Cl solution and brine. The organic layer was then dried with MgSO4, filtered and the solvents were evaporated under reduced pressure. The crude product was purified by silica gel chromatography (cyclohexane/ethyl acetate 90:10) to give 2f as a colorless oil (509 mg, 58%). TLC (cyclohexane/ethyl acetate 70:30) Rf 0.77; 1H NMR (300 MHz, CDCl3) δ 3.71 (s, 3H), 4.17 (dd, J = 6.3, 1.2 Hz, 2H), 4.30 (s, 2H), 6.08 (dt, J = 15.9, 6.3 Hz, 1H), 6.52 (d, J = 15.9 Hz, 1H), 6.76 (d, J = 8.8 Hz, 2H), 7.07 (t, J = 7.9 Hz, 1H), 7.16–7.33 (m, 3H), 7.36 (dt, J = 8.0, 1.1 Hz, 1H), 7.5 (t, J = 1.6 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 55.6, 57.9, 71.0, 85.1, 87.1, 114.4 (2C), 122.4, 123.2, 125.1, 128.1 (2C), 129.7, 130.1, 130.7, 132.0, 133.6, 134.9, 159.8.
(E)-1-Methoxy-4-(3-(2-methyl-3-phenylallyloxy)prop-1-ynyl)benzene (2g): Following the same procedure as for the synthesis of 2f, in the presence of CuI (103 mg, 0.1 equiv) and PdCl2(PPh3)2 (190 mg, 0.05 equiv), 1-methoxy-4-iodobenzene (1.52 g, 1.2 equiv) in distilled diisopropylamine (10 mL), (E)-(2-methyl-3-(prop-2-ynyloxy)prop-1-enyl)benzene [77] (1 g, 1 equiv) was transformed to 2g (896 mg) in 57% yield. TLC (cyclohexane/ethyl acetate 90:10) Rf 0.71; 1H NMR (300 MHz, CDCl3) δ 2.20 (d, J = 1.2 Hz, 3H), 4.07 (s, 3H), 4.44 (d, J = 0.9 Hz, 2H), 4.66 (s, 2H), 6.83 (s, 1H), 7.11 (m, 2H), 7.68–7.48 (m, 7H); 13C NMR (75 MHz, CDCl3) δ 15.7, 55.3, 57.8, 76.0, 83.8, 86.2, 113.9, 114.8, 126.5, 127.8, 128.1 (2C), 128.9 (2C), 133.3 (2C), 134.5, 137.4, 159.7.
(E)-1-Bromo-3-(3-(2-methyl-3-phenylallyloxy)prop-1-ynyl)benzene (2h): Following the same procedure as for the synthesis of 2f, in the presence of CuI (62 mg, 0.1 equiv) and PdCl2(PPh3)2 (115 mg, 0.05 equiv), 1-bromo-3-iodobenzene (0.53 mL, 1.3 equiv) in distilled diisopropylamine (6.8 mL), (E)-(2-methyl-3-(prop-2-ynyloxy)prop-1-enyl)benzene [77] (611 mg, 1 equiv) was transformed to 2h (851 mg) in 76% yield. TLC (cyclohexane/ethyl acetate 90:10) Rf 0.63; 1H NMR (300 MHz, CDCl3) δ 2.08 (d, J = 1.3 Hz, 3H), 4.31 (d, J = 0.8 Hz, 2H), 4.53 (s, 2H), 6.71 (d, J = 1.0 Hz, 1H), 7.30 (t, J = 7.8 Hz, 1H), 7.36–7.56 (m, 6H), 7.58 (dt, J = 8.0, 0.8 Hz, 1H), 7.75 (t, J = 1.6 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 16.0, 58.0 (2C), 85.1, 87.1, 122.5, 125.1, 127.0, 128.3, 128.5 (2C), 129.3 (2C), 130.1, 130.7, 132.0, 134.7, 134.9, 137.7.
General procedure for Au(I)-catalyzed cycloisomerization reactions: A mixture of L-(AuCl)2 (L = (R)-4-MeO-3,5-(t-Bu)2MeOBIPHEP) (3 mol %) and AgOTf (or AgNTf2) (6 mol %) in distilled toluene (0.5 M) was stirred under an argon atmosphere at room temperature for 30 min. Enyne (1 equiv) was then added and the mixture stirred until completion of the reaction. The mixture was then filtered through a short pad of silica to eliminate the catalyst (EtOAc) and the solvents were concentrated under reduced pressure. The crude product was purified by silica gel flash chromatography (petroleum ether/ethyl acetate 98:2 to 80:20 v/v) if necessary.
6-(3-Bromophenyl)-7-(4-methoxyphenyl)-3-oxabicyclo[4.1.0]hept-4-ene (4f): TLC (cyclohexane/ethyl acetate 80:20) Rf 0.70; 1H NMR (300 MHz, CDCl3) δ 2.29 (d, J = 5.4 Hz, 1H), 2.65 (d, J = 6.0 Hz, 1H), 3.61 (s, 3H), 3.96 (dd, J = 10.6, 1.9 Hz, 1H), 4.30 (d, J = 10.6 Hz, 1H), 5.21 (d, J = 6.0 Hz, 1H), 6.18 (d, J = 6.0 Hz, 1H), 6.52–6.65 (m, 4H), 6.86–6.92 (m, 2H), 7.12–7.19 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 29.9, 30.3, 37.3, 55.5, 61.6, 111.0, 113.7 (2C), 122.5, 128.7, 129.0 (2C), 129.4, 129.9, 130.1, 133.0, 141.2, 142.8, 158.2; HPLC (Chiralpack AD, hexane/propan-2-ol (97:3), flow rate 1.0 mL/min, λ = 215 nm): retention times 7 and 7.5 min, ee 95%; [α]D23 −18.6 (c 1, CHCl3).
6-(4-Methoxyphenyl)-1-methyl-7-phenyl-3-oxabicyclo[4.1.0]hept-4-ene (4g): TLC (cyclohexane/ethyl acetate 90:10) Rf 0.66; 1H NMR (300 MHz, CDCl3) δ 1.18 (s, 3H), 2.84 (s, 1H), 3.73 (d, J = 10.4 Hz, 1H), 3.80 (s, 3H), 4.15 (d, J = 10.4 Hz, 1H), 5.19 (d, J = 5.8, 1 Hz, 1H), 6.21 (d, J = 5.8 Hz,1H), 6.80–6.86 (m, 4H), 7.01–7.04 (m, 2H), 7.12–7.15 (m, 3H); 13C NMR (75 MHz, CDCl3) δ 12.9, 31.6, 35.2, 38.2, 55.2, 67.8, 113.7, 114.5, 125.4, 127.5 (2C), 130.0 (2C), 130.7, 132.1 (2C), 137.5, 140.0, 158.1; HPLC (Chiralcel OJ, hexane/propan-2-ol (99/1), flow rate 1.0 mL/min, λ = 215 nm): retention times 20.3 and 27.1 min, ee 99%; [α]D23 +26.1 (c 1, CHCl3).
6-(3-Bromophenyl)-1-methyl-7-phenyl-3-oxabicyclo[4.1.0]hept-4-ene (4h): TLC (cyclohexane/ethyl acetate 90:10) Rf 0.71; 1H NMR (300 MHz, CDCl3) δ 1.10 (s, 3H), 2.80 (s, 1H), 3.64 (d, J = 10.5 Hz, 1H), 4.07 (d, J = 10.5 Hz, 1H), 5.09 (d, J = 5.8 Hz, 1H), 6.16 (d, J = 5.8 Hz, 1H), 6.75 (d, J = 2.1 Hz, 1H), 6.78 (d, J = 4.0 Hz, 1H), 6.92 (dt, J = 7.7, 1.4 Hz, 1H), 7.00–7.20 (m, 5H), 7.29 (dt, J = 7.8, 1.2 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 14.4, 33.2, 36.5, 39.7, 68.9, 114.6, 123.5, 127.2, 129.0 (2C), 131.1 (2C), 131.2, 131.3 (2C), 135.3, 138.2, 142.0, 142.5; HPLC (Chiralpak IA, hexane/propan-2-ol (99.9:0.1), flow rate 0.5 mL/min, λ = 215 nm): retention times 11.8 and 12.6 min, ee 73%; [α]D23 +11.7 (c 1, CHCl3).
Supporting Information
| Supporting Information File 1: Spectral data. | ||
| Format: PDF | Size: 1.2 MB | Download |
Acknowledgements
This work was supported by the Centre National de la Recherche Scientifique and the Ministère de l’Education et de la Recherche. Chung-Meng Chao is grateful to the Ministère de l’Education et de la Recherche for a grant (2006–2009). Alexandre Pradal is grateful to National Research Agency (ANR-09-JCJC-0078) for a grant (2009–2012). The authors thank Dr. M. Scalone (Hoffmann–La Roche) for the generous donation of 4-MeO-3,5-(t-Bu)2-MeOBIPHEP ligand. Johnson-Matthey is acknowledged for a generous donation of HAuCl4.
References
-
Belmont, P.; Parker, E. Eur. J. Org. Chem. 2009, 6075. doi:10.1002/ejoc.200900790
Return to citation in text: [1] [2] -
Jiménez-Núñez, E.; Echavarren, A. M. Chem. Rev. 2008, 108, 3326. doi:10.1021/cr0684319
Return to citation in text: [1] [2] -
Michelet, V.; Toullec, P. Y.; Genêt, J.-P. Angew. Chem., Int. Ed. 2008, 47, 4268. doi:10.1002/anie.200701589
Return to citation in text: [1] [2] -
Zhang, L.; Sun, J.; Kozmin, S. A. Adv. Synth. Catal. 2006, 348, 2271. doi:10.1002/adsc.200600368
Return to citation in text: [1] [2] -
Nevado, C.; Echavarren, A. M. Synthesis 2005, 167. doi:10.1055/s-2005-861781
Return to citation in text: [1] [2] -
Fairlamb, I. J. S. Angew. Chem., Int. Ed. 2004, 43, 1048. doi:10.1002/anie.200301699
Return to citation in text: [1] [2] -
Aubert, C.; Buisine, O.; Malacria, M. Chem. Rev. 2002, 102, 813. doi:10.1021/cr980054f
Return to citation in text: [1] [2] -
Trost, B. M.; Krische, M. J. Synlett 1998, 1. doi:10.1055/s-1998-1557
Return to citation in text: [1] [2] -
Soriano, E.; Marco-Contelles, J. Acc. Chem. Res. 2009, 42, 1026. doi:10.1021/ar800200m
Return to citation in text: [1] -
Soriano, E.; Ballesteros, P.; Marco-Contelles, J. J. Org. Chem. 2004, 69, 8018. doi:10.1021/jo048828h
Return to citation in text: [1] -
He, R.-X.; Li, M.; Li, X.-Y. THEOCHEM 2005, 717, 21. doi:10.1016/j.theochem.2004.10.067
Return to citation in text: [1] -
Mandal, A. K.; Borude, D. P.; Armugasamy, R.; Soni, N. R.; Jawalker, D. G.; Mahajan, S. W.; Ratman, K. R.; Goghare, A. D. Tetrahedron 1986, 42, 5715. doi:10.1016/S0040-4020(01)88177-1
And references cited therein.
Return to citation in text: [1] -
Tessier, J. R. In Recent Advances in the Chemistry of Insect Control; James, N. F., Ed.; Royal Soc. Chem.: London, 1985; p 26.
Return to citation in text: [1] -
Blum, J.; Beer-Kraft, H.; Badrieh, Y. J. Org. Chem. 1995, 60, 5567. doi:10.1021/jo00122a043
Return to citation in text: [1] -
Chatani, N.; Furukawa, N.; Sakurai, H.; Murai, S. Organometallics 1996, 15, 901. doi:10.1021/om950832j
Return to citation in text: [1] [2] -
Lloyd-Jones, G. C. Org. Biomol. Chem. 2003, 1, 215. doi:10.1039/b209175p
Return to citation in text: [1] -
Bruneau, C. Angew. Chem., Int. Ed. 2005, 44, 2328. doi:10.1002/anie.200462568
Return to citation in text: [1] -
Chianese, A. R.; Lee, S. J.; Gagné, M. R. Angew. Chem., Int. Ed. 2007, 46, 4042. doi:10.1002/anie.200603954
Return to citation in text: [1] -
Fürstner, A.; Davies, P. W. Angew. Chem., Int. Ed. 2007, 46, 3410. doi:10.1002/anie.200604335
Return to citation in text: [1] -
Lee, S. I.; Chatani, N. Chem. Commun. 2009, 371. doi:10.1039/b812466c
Return to citation in text: [1] [2] -
Fürstner, A. Chem. Soc. Rev. 2009, 38, 3208. doi:10.1039/b816696j
Return to citation in text: [1] -
Toullec, P. Y.; Michelet, V. Top. Curr. Chem. 2011, 1. doi:10.1007/128_2010_116
Return to citation in text: [1] -
Fürstner, A.; Szillat, H.; Stelzer, F. J. Am. Chem. Soc. 2000, 122, 6785. doi:10.1021/ja001034+
Return to citation in text: [1] [2] [3] -
Fürstner, A.; Stelzer, F.; Szillat, H. J. Am. Chem. Soc. 2001, 123, 11863. doi:10.1021/ja0109343
Return to citation in text: [1] -
Méndez, M.; Muñoz, M. P.; Nevado, C.; Cárdenas, D. J.; Echavarren, A. M. J. Am. Chem. Soc. 2001, 123, 10511. doi:10.1021/ja0112184
Return to citation in text: [1] -
Nevado, C.; Ferrer, C.; Echavarren, A. M. Org. Lett. 2004, 6, 3191. doi:10.1021/ol0486573
Return to citation in text: [1] -
Lee, S. I.; Kim, S. M.; Choi, M. R.; Kim, S. Y.; Chung, Y. K. J. Org. Chem. 2006, 71, 9366. doi:10.1021/jo061254r
Return to citation in text: [1] -
Ferrer, C.; Raducan, M.; Nevado, C.; Claverie, C. K.; Echavarren, A. M. Tetrahedron 2007, 63, 6306. doi:10.1016/j.tet.2007.02.122
Return to citation in text: [1] -
Ye, L.; Chen, Q.; Zhang, J.; Michelet, V. J. Org. Chem. 2009, 74, 9550. doi:10.1021/jo902083r
Return to citation in text: [1] -
Harrak, Y.; Simonneau, A.; Malacria, M.; Gandon, V.; Fensterbank, L. Chem. Commun. 2010, 46, 865. doi:10.1039/b919240a
Return to citation in text: [1] -
Mamane, V.; Gress, T.; Krause, H.; Fürstner, A. J. Am. Chem. Soc. 2004, 126, 8654. doi:10.1021/ja048094q
Return to citation in text: [1] -
Harrak, Y.; Blaszylowski, C.; Bernard, M.; Cariou, K.; Mainetti, E.; Mouriès, V.; Dhimane, A.-L.; Fensterbank, L.; Malacria, M. J. Am. Chem. Soc. 2004, 126, 8656. doi:10.1021/ja0474695
Return to citation in text: [1] -
Blaszykowski, C.; Harrak, Y.; Gonçalves, M.-H.; Cloarec, J.-M.; Dhimane, A.-L.; Fensterbank, L.; Malacria, M. Org. Lett. 2004, 6, 3771. doi:10.1021/ol048463n
Return to citation in text: [1] -
Luzung, M. R.; Markham, J. P.; Toste, F. D. J. Am. Chem. Soc. 2004, 126, 10858. doi:10.1021/ja046248w
Return to citation in text: [1] -
Gagosz, F. Org. Lett. 2005, 7, 4129. doi:10.1021/ol051397k
Return to citation in text: [1] -
Sun, J.; Conley, M. P.; Zhang, L.; Kozmin, S. A. J. Am. Chem. Soc. 2006, 128, 9705. doi:10.1021/ja063384n
Return to citation in text: [1] -
Couty, S.; Meyer, C.; Cossy, J. Angew. Chem., Int. Ed. 2006, 45, 6726. doi:10.1002/anie.200602270
Return to citation in text: [1] -
Blaszylowski, C.; Harrak, Y.; Brancour, C.; Nakama, K.; Dhimane, A.-L.; Fensterbank, L.; Malacria, M. Synthesis 2007, 13, 2037. doi:10.1055/s-2007-983736
Return to citation in text: [1] -
Couty, S.; Meyer, C.; Cossy, J. Tetrahedron 2009, 65, 1809. doi:10.1016/j.tet.2008.10.108
Return to citation in text: [1] -
Horino, Y.; Yamamoto, T.; Ueda, K.; Kuroda, S.; Toste, F. D. J. Am. Chem. Soc. 2009, 131, 2809. doi:10.1021/ja808780r
Return to citation in text: [1] -
Shibata, T.; Kobayashi, Y.; Maekawa, S.; Toshida, N.; Takagi, K. Tetrahedron 2005, 61, 9018. doi:10.1016/j.tet.2005.07.039
Return to citation in text: [1] [2] [3] -
Brissy, D.; Skander, M.; Jullien, H.; Retailleau, P.; Marinetti, A. Org. Lett. 2009, 11, 2137. doi:10.1021/ol900724z
Return to citation in text: [1] [2] [3] -
Brissy, D.; Skander, M.; Retailleau, P.; Marinetti, A. Organometallics 2007, 26, 5782. doi:10.1021/om700839y
Return to citation in text: [1] [2] [3] -
Brissy, D.; Skander, M.; Retailleau, P.; Frison, G.; Marinetti, A. Organometallics 2009, 28, 140. doi:10.1021/om800743r
Return to citation in text: [1] [2] [3] -
Nishimura, T.; Kawamoto, T.; Nagaosa, M.; Kumamoto, H.; Hayashi, T. Angew. Chem., Int. Ed. 2010, 49, 1638. doi:10.1002/anie.200906792
Return to citation in text: [1] [2] -
Chao, C.-M.; Beltrami, D.; Toullec, P. Y.; Michelet, V. Chem. Commun. 2009, 6988. doi:10.1039/b913554e
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Deschamps, N. M.; Elitzin, V. I.; Liu, B.; Mitchell, M. B.; Sharp, M. J.; Tabet, E. A. J. Org. Chem. 2011, 76, 712. doi:10.1021/jo102098y
Return to citation in text: [1] -
Teller, H.; Fürstner, A. Chem.–Eur. J. 2011, 17, 7764. doi:10.1002/chem.201101346
Return to citation in text: [1] -
Chao, C.-M.; Vitale, M. R.; Toullec, P. Y.; Genêt, J.-P.; Michelet, V. Chem.–Eur. J. 2009, 15, 1319. doi:10.1002/chem.200802341
Return to citation in text: [1] -
Chao, C.-M.; Genin, E.; Toullec, P. Y.; Genêt, J.-P.; Michelet, V. J. Organomet. Chem. 2009, 694, 538. doi:10.1016/j.jorganchem.2008.08.008
Return to citation in text: [1] -
Andreiadis, E. S.; Vitale, M. R.; Mézailles, N.; Le Goff, X.; Le Floch, P.; Toullec, P. Y.; Michelet, V. Dalton Trans. 2010, 39, 10608. doi:10.1039/c0dt00399a
Return to citation in text: [1] -
Pradal, A.; Chao, C.-M.; Vitale, M.; Toullec, P. Y.; Michelet, V. Tetrahedron 2011, 67, 4371. doi:10.1016/j.tet.2011.03.071
Return to citation in text: [1] -
Widenhoefer, R. A. Chem.–Eur. J. 2008, 14, 5382. doi:10.1002/chem.200800219
Return to citation in text: [1] -
Sengupta, S.; Shi, X. ChemCatChem 2010, 2, 609. doi:10.1002/cctc.201000070
Return to citation in text: [1] -
Pradal, A.; Toullec, P. Y.; Michelet, V. Synthesis 2011, 10, 1501. doi:10.1055/s-0030-1258465
Return to citation in text: [1] -
Liu, C.; Widenhoefer, R. A. Org. Lett. 2007, 9, 1935. doi:10.1021/ol070483c
Return to citation in text: [1] -
Muñoz, M. P.; Adrio, J.; Carretero, J. C.; Echavarren, A. M. Organometallics 2005, 24, 1293. doi:10.1021/om0491645
Return to citation in text: [1] -
Schmid, R.; Broger, E. A.; Cereghetti, M.; Crameri, Y.; Foricher, J.; Lalonde, M.; Müller, R. K.; Scalone, M.; Schoettel, G.; Zutter, U. Pure Appl. Chem. 1996, 68, 131. doi:10.1351/pac199668010131
For the synthesis and applications in hydrogenation reactions of (R)-4-MeO-3,5-(t-Bu)2-MeOBIPHEP (and references cited therein).
Return to citation in text: [1] -
The absolute configuration was proposed in analogy to the data concerning 4a and to literature [45].
Return to citation in text: [1] -
Chinchilla, R.; Nájera, C. Chem. Rev. 2007, 107, 874. doi:10.1021/cr050992x
Return to citation in text: [1] -
Doucet, H.; Hierso, J.-C. Angew. Chem., Int. Ed. 2007, 46, 834. doi:10.1002/anie.200602761
Return to citation in text: [1] -
Nevado, C.; Charruault, L.; Michelet, V.; Nieto-Oberhuber, C.; Muñoz, M. P.; Méndez, M.; Rager, M.-N.; Genêt, J.-P.; Echavarren, A. M. Eur. J. Org. Chem. 2003, 706. doi:10.1002/ejoc.200390110
Return to citation in text: [1] -
Galland, J.-C.; Savignac, M.; Genêt, J.-P. Tetrahedron Lett. 1997, 38, 8695. doi:10.1016/S0040-4039(97)10337-9
Return to citation in text: [1] -
Michelet, V.; Charruault, L.; Gladiali, S.; Genêt, J.-P. Pure Appl. Chem. 2006, 78, 397. doi:10.1351/pac200678020397
Return to citation in text: [1] [2] -
Mézailles, N.; Ricard, L.; Gagosz, F. Org. Lett. 2005, 7, 4133. doi:10.1021/ol0515917
Return to citation in text: [1] -
Nakai, H.; Chatani, N. Chem. Lett. 2007, 36, 1494. doi:10.1246/cl.2007.1494
Return to citation in text: [1] -
Charette, A. B.; Beauchemin, A. Org. React. 2001, 58, 1.
Return to citation in text: [1] -
Lebel, H.; Marcoux, J.-F.; Molinaro, C.; Charette, A. B. Chem. Rev. 2003, 103, 977. doi:10.1021/cr010007e
Return to citation in text: [1] -
Pelissier, H. Tetrahedron 2008, 64, 7041. doi:10.1016/j.tet.2008.04.079
Return to citation in text: [1] -
Doyle, M. P.; Forbes, D. C. Chem. Rev. 1998, 98, 911. doi:10.1021/cr940066a
Return to citation in text: [1] -
Trost, B. M.; Edstrom, E. D.; Carter-Petillo, M. B. J. Org. Chem. 1989, 54, 4489. doi:10.1021/jo00280a006
Return to citation in text: [1] -
Kudoh, T.; Mori, T.; Shirahama, M.; Yamada, M.; Ishikawa, T.; Saito, S.; Kobayashi, H. J. Am. Chem. Soc. 2007, 129, 4939. doi:10.1021/ja066485u
Return to citation in text: [1] -
Morimoto, T.; Fuji, K.; Tsutsumi, K.; Kakiuchi, K. J. Am. Chem. Soc. 2002, 124, 3806. doi:10.1021/ja0126881
Return to citation in text: [1] [2] -
Oppolzer, W.; Pimm, A.; Stammen, B.; Hume, W. E. Helv. Chim. Acta 1997, 80, 623. doi:10.1002/hlca.19970800302
Return to citation in text: [1] -
Tang, Y.; Deng, L.; Zhang, Y.; Dong, G.; Chen, J.; Yang, Z. Org. Lett. 2005, 7, 1657. doi:10.1021/ol050410y
Return to citation in text: [1] [2] -
Shibata, T.; Arai, Y.; Tahara, Y.-K. Org. Lett. 2005, 7, 4955. doi:10.1021/ol051876j
Return to citation in text: [1] -
Mikami, K.; Hatano, M. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 5767. doi:10.1073/pnas.0307217101
Return to citation in text: [1] [2]
| 72. | Kudoh, T.; Mori, T.; Shirahama, M.; Yamada, M.; Ishikawa, T.; Saito, S.; Kobayashi, H. J. Am. Chem. Soc. 2007, 129, 4939. doi:10.1021/ja066485u |
| 46. | Chao, C.-M.; Beltrami, D.; Toullec, P. Y.; Michelet, V. Chem. Commun. 2009, 6988. doi:10.1039/b913554e |
| 73. | Morimoto, T.; Fuji, K.; Tsutsumi, K.; Kakiuchi, K. J. Am. Chem. Soc. 2002, 124, 3806. doi:10.1021/ja0126881 |
| 1. | Belmont, P.; Parker, E. Eur. J. Org. Chem. 2009, 6075. doi:10.1002/ejoc.200900790 |
| 2. | Jiménez-Núñez, E.; Echavarren, A. M. Chem. Rev. 2008, 108, 3326. doi:10.1021/cr0684319 |
| 3. | Michelet, V.; Toullec, P. Y.; Genêt, J.-P. Angew. Chem., Int. Ed. 2008, 47, 4268. doi:10.1002/anie.200701589 |
| 4. | Zhang, L.; Sun, J.; Kozmin, S. A. Adv. Synth. Catal. 2006, 348, 2271. doi:10.1002/adsc.200600368 |
| 5. | Nevado, C.; Echavarren, A. M. Synthesis 2005, 167. doi:10.1055/s-2005-861781 |
| 6. | Fairlamb, I. J. S. Angew. Chem., Int. Ed. 2004, 43, 1048. doi:10.1002/anie.200301699 |
| 7. | Aubert, C.; Buisine, O.; Malacria, M. Chem. Rev. 2002, 102, 813. doi:10.1021/cr980054f |
| 8. | Trost, B. M.; Krische, M. J. Synlett 1998, 1. doi:10.1055/s-1998-1557 |
| 15. | Chatani, N.; Furukawa, N.; Sakurai, H.; Murai, S. Organometallics 1996, 15, 901. doi:10.1021/om950832j |
| 56. | Liu, C.; Widenhoefer, R. A. Org. Lett. 2007, 9, 1935. doi:10.1021/ol070483c |
| 57. | Muñoz, M. P.; Adrio, J.; Carretero, J. C.; Echavarren, A. M. Organometallics 2005, 24, 1293. doi:10.1021/om0491645 |
| 58. |
Schmid, R.; Broger, E. A.; Cereghetti, M.; Crameri, Y.; Foricher, J.; Lalonde, M.; Müller, R. K.; Scalone, M.; Schoettel, G.; Zutter, U. Pure Appl. Chem. 1996, 68, 131. doi:10.1351/pac199668010131
For the synthesis and applications in hydrogenation reactions of (R)-4-MeO-3,5-(t-Bu)2-MeOBIPHEP (and references cited therein). |
| 76. | Shibata, T.; Arai, Y.; Tahara, Y.-K. Org. Lett. 2005, 7, 4955. doi:10.1021/ol051876j |
| 14. | Blum, J.; Beer-Kraft, H.; Badrieh, Y. J. Org. Chem. 1995, 60, 5567. doi:10.1021/jo00122a043 |
| 59. | The absolute configuration was proposed in analogy to the data concerning 4a and to literature [45]. |
| 46. | Chao, C.-M.; Beltrami, D.; Toullec, P. Y.; Michelet, V. Chem. Commun. 2009, 6988. doi:10.1039/b913554e |
| 12. |
Mandal, A. K.; Borude, D. P.; Armugasamy, R.; Soni, N. R.; Jawalker, D. G.; Mahajan, S. W.; Ratman, K. R.; Goghare, A. D. Tetrahedron 1986, 42, 5715. doi:10.1016/S0040-4020(01)88177-1
And references cited therein. |
| 13. | Tessier, J. R. In Recent Advances in the Chemistry of Insect Control; James, N. F., Ed.; Royal Soc. Chem.: London, 1985; p 26. |
| 46. | Chao, C.-M.; Beltrami, D.; Toullec, P. Y.; Michelet, V. Chem. Commun. 2009, 6988. doi:10.1039/b913554e |
| 49. | Chao, C.-M.; Vitale, M. R.; Toullec, P. Y.; Genêt, J.-P.; Michelet, V. Chem.–Eur. J. 2009, 15, 1319. doi:10.1002/chem.200802341 |
| 50. | Chao, C.-M.; Genin, E.; Toullec, P. Y.; Genêt, J.-P.; Michelet, V. J. Organomet. Chem. 2009, 694, 538. doi:10.1016/j.jorganchem.2008.08.008 |
| 51. | Andreiadis, E. S.; Vitale, M. R.; Mézailles, N.; Le Goff, X.; Le Floch, P.; Toullec, P. Y.; Michelet, V. Dalton Trans. 2010, 39, 10608. doi:10.1039/c0dt00399a |
| 52. | Pradal, A.; Chao, C.-M.; Vitale, M.; Toullec, P. Y.; Michelet, V. Tetrahedron 2011, 67, 4371. doi:10.1016/j.tet.2011.03.071 |
| 23. | Fürstner, A.; Szillat, H.; Stelzer, F. J. Am. Chem. Soc. 2000, 122, 6785. doi:10.1021/ja001034+ |
| 1. | Belmont, P.; Parker, E. Eur. J. Org. Chem. 2009, 6075. doi:10.1002/ejoc.200900790 |
| 2. | Jiménez-Núñez, E.; Echavarren, A. M. Chem. Rev. 2008, 108, 3326. doi:10.1021/cr0684319 |
| 3. | Michelet, V.; Toullec, P. Y.; Genêt, J.-P. Angew. Chem., Int. Ed. 2008, 47, 4268. doi:10.1002/anie.200701589 |
| 4. | Zhang, L.; Sun, J.; Kozmin, S. A. Adv. Synth. Catal. 2006, 348, 2271. doi:10.1002/adsc.200600368 |
| 5. | Nevado, C.; Echavarren, A. M. Synthesis 2005, 167. doi:10.1055/s-2005-861781 |
| 6. | Fairlamb, I. J. S. Angew. Chem., Int. Ed. 2004, 43, 1048. doi:10.1002/anie.200301699 |
| 7. | Aubert, C.; Buisine, O.; Malacria, M. Chem. Rev. 2002, 102, 813. doi:10.1021/cr980054f |
| 8. | Trost, B. M.; Krische, M. J. Synlett 1998, 1. doi:10.1055/s-1998-1557 |
| 9. | Soriano, E.; Marco-Contelles, J. Acc. Chem. Res. 2009, 42, 1026. doi:10.1021/ar800200m |
| 10. | Soriano, E.; Ballesteros, P.; Marco-Contelles, J. J. Org. Chem. 2004, 69, 8018. doi:10.1021/jo048828h |
| 11. | He, R.-X.; Li, M.; Li, X.-Y. THEOCHEM 2005, 717, 21. doi:10.1016/j.theochem.2004.10.067 |
| 53. | Widenhoefer, R. A. Chem.–Eur. J. 2008, 14, 5382. doi:10.1002/chem.200800219 |
| 54. | Sengupta, S.; Shi, X. ChemCatChem 2010, 2, 609. doi:10.1002/cctc.201000070 |
| 55. | Pradal, A.; Toullec, P. Y.; Michelet, V. Synthesis 2011, 10, 1501. doi:10.1055/s-0030-1258465 |
| 75. | Tang, Y.; Deng, L.; Zhang, Y.; Dong, G.; Chen, J.; Yang, Z. Org. Lett. 2005, 7, 1657. doi:10.1021/ol050410y |
| 42. | Brissy, D.; Skander, M.; Jullien, H.; Retailleau, P.; Marinetti, A. Org. Lett. 2009, 11, 2137. doi:10.1021/ol900724z |
| 43. | Brissy, D.; Skander, M.; Retailleau, P.; Marinetti, A. Organometallics 2007, 26, 5782. doi:10.1021/om700839y |
| 44. | Brissy, D.; Skander, M.; Retailleau, P.; Frison, G.; Marinetti, A. Organometallics 2009, 28, 140. doi:10.1021/om800743r |
| 46. | Chao, C.-M.; Beltrami, D.; Toullec, P. Y.; Michelet, V. Chem. Commun. 2009, 6988. doi:10.1039/b913554e |
| 47. | Deschamps, N. M.; Elitzin, V. I.; Liu, B.; Mitchell, M. B.; Sharp, M. J.; Tabet, E. A. J. Org. Chem. 2011, 76, 712. doi:10.1021/jo102098y |
| 48. | Teller, H.; Fürstner, A. Chem.–Eur. J. 2011, 17, 7764. doi:10.1002/chem.201101346 |
| 73. | Morimoto, T.; Fuji, K.; Tsutsumi, K.; Kakiuchi, K. J. Am. Chem. Soc. 2002, 124, 3806. doi:10.1021/ja0126881 |
| 41. | Shibata, T.; Kobayashi, Y.; Maekawa, S.; Toshida, N.; Takagi, K. Tetrahedron 2005, 61, 9018. doi:10.1016/j.tet.2005.07.039 |
| 46. | Chao, C.-M.; Beltrami, D.; Toullec, P. Y.; Michelet, V. Chem. Commun. 2009, 6988. doi:10.1039/b913554e |
| 64. | Michelet, V.; Charruault, L.; Gladiali, S.; Genêt, J.-P. Pure Appl. Chem. 2006, 78, 397. doi:10.1351/pac200678020397 |
| 23. | Fürstner, A.; Szillat, H.; Stelzer, F. J. Am. Chem. Soc. 2000, 122, 6785. doi:10.1021/ja001034+ |
| 24. | Fürstner, A.; Stelzer, F.; Szillat, H. J. Am. Chem. Soc. 2001, 123, 11863. doi:10.1021/ja0109343 |
| 25. | Méndez, M.; Muñoz, M. P.; Nevado, C.; Cárdenas, D. J.; Echavarren, A. M. J. Am. Chem. Soc. 2001, 123, 10511. doi:10.1021/ja0112184 |
| 26. | Nevado, C.; Ferrer, C.; Echavarren, A. M. Org. Lett. 2004, 6, 3191. doi:10.1021/ol0486573 |
| 27. | Lee, S. I.; Kim, S. M.; Choi, M. R.; Kim, S. Y.; Chung, Y. K. J. Org. Chem. 2006, 71, 9366. doi:10.1021/jo061254r |
| 28. | Ferrer, C.; Raducan, M.; Nevado, C.; Claverie, C. K.; Echavarren, A. M. Tetrahedron 2007, 63, 6306. doi:10.1016/j.tet.2007.02.122 |
| 29. | Ye, L.; Chen, Q.; Zhang, J.; Michelet, V. J. Org. Chem. 2009, 74, 9550. doi:10.1021/jo902083r |
| 30. | Harrak, Y.; Simonneau, A.; Malacria, M.; Gandon, V.; Fensterbank, L. Chem. Commun. 2010, 46, 865. doi:10.1039/b919240a |
| 31. | Mamane, V.; Gress, T.; Krause, H.; Fürstner, A. J. Am. Chem. Soc. 2004, 126, 8654. doi:10.1021/ja048094q |
| 32. | Harrak, Y.; Blaszylowski, C.; Bernard, M.; Cariou, K.; Mainetti, E.; Mouriès, V.; Dhimane, A.-L.; Fensterbank, L.; Malacria, M. J. Am. Chem. Soc. 2004, 126, 8656. doi:10.1021/ja0474695 |
| 33. | Blaszykowski, C.; Harrak, Y.; Gonçalves, M.-H.; Cloarec, J.-M.; Dhimane, A.-L.; Fensterbank, L.; Malacria, M. Org. Lett. 2004, 6, 3771. doi:10.1021/ol048463n |
| 34. | Luzung, M. R.; Markham, J. P.; Toste, F. D. J. Am. Chem. Soc. 2004, 126, 10858. doi:10.1021/ja046248w |
| 35. | Gagosz, F. Org. Lett. 2005, 7, 4129. doi:10.1021/ol051397k |
| 36. | Sun, J.; Conley, M. P.; Zhang, L.; Kozmin, S. A. J. Am. Chem. Soc. 2006, 128, 9705. doi:10.1021/ja063384n |
| 37. | Couty, S.; Meyer, C.; Cossy, J. Angew. Chem., Int. Ed. 2006, 45, 6726. doi:10.1002/anie.200602270 |
| 38. | Blaszylowski, C.; Harrak, Y.; Brancour, C.; Nakama, K.; Dhimane, A.-L.; Fensterbank, L.; Malacria, M. Synthesis 2007, 13, 2037. doi:10.1055/s-2007-983736 |
| 39. | Couty, S.; Meyer, C.; Cossy, J. Tetrahedron 2009, 65, 1809. doi:10.1016/j.tet.2008.10.108 |
| 40. | Horino, Y.; Yamamoto, T.; Ueda, K.; Kuroda, S.; Toste, F. D. J. Am. Chem. Soc. 2009, 131, 2809. doi:10.1021/ja808780r |
| 74. | Oppolzer, W.; Pimm, A.; Stammen, B.; Hume, W. E. Helv. Chim. Acta 1997, 80, 623. doi:10.1002/hlca.19970800302 |
| 16. | Lloyd-Jones, G. C. Org. Biomol. Chem. 2003, 1, 215. doi:10.1039/b209175p |
| 17. | Bruneau, C. Angew. Chem., Int. Ed. 2005, 44, 2328. doi:10.1002/anie.200462568 |
| 18. | Chianese, A. R.; Lee, S. J.; Gagné, M. R. Angew. Chem., Int. Ed. 2007, 46, 4042. doi:10.1002/anie.200603954 |
| 19. | Fürstner, A.; Davies, P. W. Angew. Chem., Int. Ed. 2007, 46, 3410. doi:10.1002/anie.200604335 |
| 20. | Lee, S. I.; Chatani, N. Chem. Commun. 2009, 371. doi:10.1039/b812466c |
| 21. | Fürstner, A. Chem. Soc. Rev. 2009, 38, 3208. doi:10.1039/b816696j |
| 22. | Toullec, P. Y.; Michelet, V. Top. Curr. Chem. 2011, 1. doi:10.1007/128_2010_116 |
| 45. | Nishimura, T.; Kawamoto, T.; Nagaosa, M.; Kumamoto, H.; Hayashi, T. Angew. Chem., Int. Ed. 2010, 49, 1638. doi:10.1002/anie.200906792 |
| 75. | Tang, Y.; Deng, L.; Zhang, Y.; Dong, G.; Chen, J.; Yang, Z. Org. Lett. 2005, 7, 1657. doi:10.1021/ol050410y |
| 46. | Chao, C.-M.; Beltrami, D.; Toullec, P. Y.; Michelet, V. Chem. Commun. 2009, 6988. doi:10.1039/b913554e |
| 60. | Chinchilla, R.; Nájera, C. Chem. Rev. 2007, 107, 874. doi:10.1021/cr050992x |
| 61. | Doucet, H.; Hierso, J.-C. Angew. Chem., Int. Ed. 2007, 46, 834. doi:10.1002/anie.200602761 |
| 77. | Mikami, K.; Hatano, M. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 5767. doi:10.1073/pnas.0307217101 |
| 62. | Nevado, C.; Charruault, L.; Michelet, V.; Nieto-Oberhuber, C.; Muñoz, M. P.; Méndez, M.; Rager, M.-N.; Genêt, J.-P.; Echavarren, A. M. Eur. J. Org. Chem. 2003, 706. doi:10.1002/ejoc.200390110 |
| 63. | Galland, J.-C.; Savignac, M.; Genêt, J.-P. Tetrahedron Lett. 1997, 38, 8695. doi:10.1016/S0040-4039(97)10337-9 |
| 77. | Mikami, K.; Hatano, M. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 5767. doi:10.1073/pnas.0307217101 |
| 45. | Nishimura, T.; Kawamoto, T.; Nagaosa, M.; Kumamoto, H.; Hayashi, T. Angew. Chem., Int. Ed. 2010, 49, 1638. doi:10.1002/anie.200906792 |
| 67. | Charette, A. B.; Beauchemin, A. Org. React. 2001, 58, 1. |
| 68. | Lebel, H.; Marcoux, J.-F.; Molinaro, C.; Charette, A. B. Chem. Rev. 2003, 103, 977. doi:10.1021/cr010007e |
| 69. | Pelissier, H. Tetrahedron 2008, 64, 7041. doi:10.1016/j.tet.2008.04.079 |
| 70. | Doyle, M. P.; Forbes, D. C. Chem. Rev. 1998, 98, 911. doi:10.1021/cr940066a |
| 71. | Trost, B. M.; Edstrom, E. D.; Carter-Petillo, M. B. J. Org. Chem. 1989, 54, 4489. doi:10.1021/jo00280a006 |
| 20. | Lee, S. I.; Chatani, N. Chem. Commun. 2009, 371. doi:10.1039/b812466c |
| 66. | Nakai, H.; Chatani, N. Chem. Lett. 2007, 36, 1494. doi:10.1246/cl.2007.1494 |
| 41. | Shibata, T.; Kobayashi, Y.; Maekawa, S.; Toshida, N.; Takagi, K. Tetrahedron 2005, 61, 9018. doi:10.1016/j.tet.2005.07.039 |
| 42. | Brissy, D.; Skander, M.; Jullien, H.; Retailleau, P.; Marinetti, A. Org. Lett. 2009, 11, 2137. doi:10.1021/ol900724z |
| 43. | Brissy, D.; Skander, M.; Retailleau, P.; Marinetti, A. Organometallics 2007, 26, 5782. doi:10.1021/om700839y |
| 44. | Brissy, D.; Skander, M.; Retailleau, P.; Frison, G.; Marinetti, A. Organometallics 2009, 28, 140. doi:10.1021/om800743r |
| 45. | Nishimura, T.; Kawamoto, T.; Nagaosa, M.; Kumamoto, H.; Hayashi, T. Angew. Chem., Int. Ed. 2010, 49, 1638. doi:10.1002/anie.200906792 |
| 15. | Chatani, N.; Furukawa, N.; Sakurai, H.; Murai, S. Organometallics 1996, 15, 901. doi:10.1021/om950832j |
| 65. | Mézailles, N.; Ricard, L.; Gagosz, F. Org. Lett. 2005, 7, 4133. doi:10.1021/ol0515917 |
| 64. | Michelet, V.; Charruault, L.; Gladiali, S.; Genêt, J.-P. Pure Appl. Chem. 2006, 78, 397. doi:10.1351/pac200678020397 |
| 23. | Fürstner, A.; Szillat, H.; Stelzer, F. J. Am. Chem. Soc. 2000, 122, 6785. doi:10.1021/ja001034+ |
| 41. | Shibata, T.; Kobayashi, Y.; Maekawa, S.; Toshida, N.; Takagi, K. Tetrahedron 2005, 61, 9018. doi:10.1016/j.tet.2005.07.039 |
| 42. | Brissy, D.; Skander, M.; Jullien, H.; Retailleau, P.; Marinetti, A. Org. Lett. 2009, 11, 2137. doi:10.1021/ol900724z |
| 43. | Brissy, D.; Skander, M.; Retailleau, P.; Marinetti, A. Organometallics 2007, 26, 5782. doi:10.1021/om700839y |
| 44. | Brissy, D.; Skander, M.; Retailleau, P.; Frison, G.; Marinetti, A. Organometallics 2009, 28, 140. doi:10.1021/om800743r |
© 2011 Pradal et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)