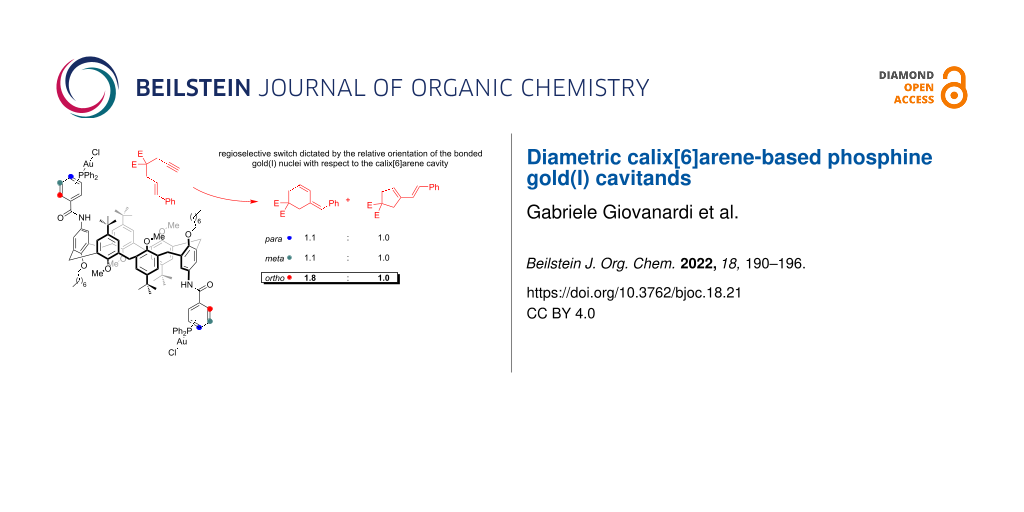
Abstract
We report the synthesis and characterization, in low polarity solvents, of a novel class of diametric phosphine gold(I) cavitands characterized by a 1,2,3-alternate geometry. Preliminary catalytic studies were performed on a model cycloisomerization of 1,6-enynes as a function of the relative orientation of the bonded gold(I) nuclei with respect to the macrocyclic cavity.

Graphical Abstract
Introduction
One of the latest challenges in supramolecular chemistry is the design and development of novel macrocyclic-based entities able to influence the catalytic activities of the metal center [1-3]. In this context, phosphines represent the most exploited class of ligands in homogeneous catalysis [4]. Noteworthy, reason of their wide applicability is the possibility of controlling the steric and electronic properties by proper functionalizations, hence tuning the catalytic properties of the bonded metal. This crucial aspect prompted their application in supramolecular chemistry as well. Thus, a recent evolution of their chemistry concerns the development of novel architectures in which P(III) compounds are incorporated in cavity-shaped macrocycles [5-8]. In this scenario, calix[4]- [9-13] and resorcin[4]arene [14-17] are the most exploited cavitands due to their inherent limited flexibility and already proved their ability to control the catalytic activity of late-transition metals and particularly gold(I) catalysts [18-25]. This occurs via strong steric interactions, often outside the macrocycle (Figure 1a) [11], that affect the first coordination sphere of the metal or by creating a spatial confinement around the metal that is thus directed towards the inner cavity (Figure 1b) [26,27]. Contrarily, calix[6]arene macrocycles are less exploited in catalysis [28]. The larger macrocycle size, its conformational adaptability, and the possibility to selectively functionalize the macrocycle offered several opportunities to design synthetic receptors and prototypes of nanodevices, instead [29]. In this context, we recently devised a new family of triphosphine calix[6]arene gold(I) complexes (Figure 1c) [30]. These cavitands are able to form (pseudo)rotaxane species, by threading viologen-based guests, with a conformational control operated by the sulfonamido hydrogen-bonding donor domain [31,32]. Furthermore, their catalytic activity was demonstrated in promoting gold(I)-catalyzed cycloisomerization of 1,6-enynes, with ample scope and high regioselectivity. However, preliminary studies suggested that the catalytic event occurs outside the macrocyclic cavity. In order to get more insights on the role of the cavity to dictate the position of the metal centers, we reasoned on the possibility to design a novel generation of diametric phosphine gold(I) cavitands exploiting a calix[6]arene scaffold characterized by a 1,2,3-alternate conformation. As working hypothesis, this geometry would segregate two catalytically active gold(I) nuclei to the opposite sides of the macrocycle, offering them the possibility to approach the cavity, thus exerting any control over the catalytic manifold (Figure 1d).
![[1860-5397-18-21-1]](/bjoc/content/figures/1860-5397-18-21-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Selected examples of: a) calix[4]arene-; b) resorcin[4]arene-; c) calix[6]arene-gold(I) macrocyclic catalysts. d) Working hypothesis for novel calix[6]arene phosphine cavitands.
Figure 1: Selected examples of: a) calix[4]arene-; b) resorcin[4]arene-; c) calix[6]arene-gold(I) macrocyclic...
Results and Discussion
Synthesis and characterization
The synthesis of novel macrocyclic calix[6]arene ligands was first attempted starting from the known dioctyloxydinitro derivative DN (Scheme 1) [33]. Reduction of the nitro groups with hydrazine, in the presence of catalytic amounts of Pd/C (10 mol %) led to the corresponding diamino intermediate. This latter could be subsequently reacted with the desired phosphino benzoic acid derivative through a user-friendly amide coupling in the presence of EDC·HCl and catalytic amounts of DMAP in CH2Cl2. Under these conditions, the corresponding diphosphine intermediates A (para), B (meta), and C (ortho) were isolated in moderate yields. Finally, gold(I) catalysts could be obtained via conventional protocols using (Me2S)AuCl. Notably, the organometallic macrocycles A,B,C(AuCl)2 could be isolated via column chromatography separation.
![[1860-5397-18-21-i1]](/bjoc/content/inline/1860-5397-18-21-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: i) NH2NH2∙H2O, Pd/C in EtOH, 80 °C (quant.); ii) diphenylphosphinobenzoic acid, EDC∙HCl, DMAP (cat.), CH2Cl2, 0 °C to rt [A, 60%; B, 55%, C, 53%); iii) (Me2S)AuCl in CH2Cl2, 0 °C to rt (A(AuCl)2, 93%; B(AuCl)2, 74%; C(AuCl)2, 69%).
Scheme 1: i) NH2NH2∙H2O, Pd/C in EtOH, 80 °C (quant.); ii) diphenylphosphinobenzoic acid, EDC∙HCl, DMAP (cat....
Gold(I) catalysts were subsequently fully characterized by NMR analysis and high-resolution mass spectrometry. The conformation of the catalysts, in low polarity solvents, is dominated by the 1,2,3-alternate conformation assumed by the DN intermediate, as previously demonstrated in our recent contributions [33,34]. Hence, the most notable features of 1H NMR for A(AuCl)2 are represented by a pattern for the methylene bridging protons in a 1:1:1 integration ratio (Figure 2). These include: i) two doublets at 4.2 and 3.6 ppm with a geminal coupling of 2J = 14.2 Hz for the a/a' couple and ii) a singlet at 3.92 ppm for the b/b' couple, typical of an anti-orientation [35]. This situation suggests a single inversion point which confers to the macrocycle a high symmetrical geometry. Finally, a single broad peak for the four methoxy groups ($) appears at 2.96 ppm. In analogy with parental diureido and dithioureido calix[6]arenes, we were able to observe the presence of a second minor cone conformer, in a ≈4:1 ratio, highlighted by the presence of a second, single resonance for the methoxy groups ($*) at 3.11 ppm. An analogous situation was observed for B(AuCl)2 and C(AuCl)2 as well. However, here the singlets for the b/b’ couple overlap with the signals of the octyloxy chains (£) at 3.91 and 3.87 ppm, respectively.
![[1860-5397-18-21-2]](/bjoc/content/figures/1860-5397-18-21-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Stacked-plot, mid-field expanded region of the 1H NMR spectrum (400 MHz, 298 K) of A(AuCl)2, B(AuCl)2, and C(AuCl)2 in CDCl3. At the bottom are schematic representations of calix[6]arene macrocycles. The rectangle identifies the phenolic ring substituted with the octyloxy chains, while the circle identifies those with methoxy groups.
Figure 2: Stacked-plot, mid-field expanded region of the 1H NMR spectrum (400 MHz, 298 K) of A(AuCl)2, B(AuCl)...
The presence of these two major conformers, in slow exchange on the NMR timescale, was finally confirmed by variable temperature NMR analysis performed for A(AuCl)2 using tetrachloroethane-d2 as the solvent (Figure 3).
![[1860-5397-18-21-3]](/bjoc/content/figures/1860-5397-18-21-3.svg?scale=2.4&max-width=1024&background=FFFFFF)
Figure 3: Stacked plot 1H NMR (tetrachloroethane-d2) of A(AuCl)2 at variable temperature.
Figure 3: Stacked plot 1H NMR (tetrachloroethane-d2) of A(AuCl)2 at variable temperature.
Catalytic studies
To probe the role of the cavity and the influence of the position of the gold(I) nuclei implanted on the calix[6]arene scaffold, we carried out the synthesis of three monomeric gold catalyst analogues A’,B’,C’(AuCl). The synthesis of these compounds was performed using the previously optimized protocol, starting from a 4-(octyloxy)aniline intermediate (Scheme 2).
![[1860-5397-18-21-i2]](/bjoc/content/inline/1860-5397-18-21-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of the monomeric gold catalyst analogues A’,B’,C’(AuCl). Conditions: i) diphenylphosphinobenzoic acid, EDC∙HCl, DMAP (cat.), CH2Cl2, 0 °C to rt (A’, 71%; B’, 76%; C’, 69%); ii) (Me2S)AuCl in DCM, 0 °C to rt (A’(AuCl), 94%; B’(AuCl), 92%; C’(AuCl), 96%).
Scheme 2: Synthesis of the monomeric gold catalyst analogues A’,B’,C’(AuCl). Conditions: i) diphenylphosphino...
Subsequently, due to the general interest in controlling the reactivity of gold(I)-catalyzed transformations by means of supramolecular macrocycles, we choose a cycloisomerization of 1,6-enynes as a model reaction [36]. Substrate 1a was reacted in the presence of monomeric gold(I) catalyst A’(AuCl) (2 mol %), using AgSbF6 as the chloride scavenger [37]. After 4 h, NMR analysis of the crude reaction mixture revealed high conversion of the starting material with the formation of the 6-endo-dig rearranged diene 2a and the parental regioisomer 2b in a 1:1 ratio. Noteworthy, this latter is formed by an initial 5-exo-dig cyclization step (entry 1, Table 1) [38,39]. This result was compared with the one obtained using the macrocyclic analogue A(AuCl)2 (1 mol %). Hence, we did not observe a significant variation in the product distribution (entry 2, Table 1).
Table 1: Ligand effect in gold(I)-catalyzed cycloisomerization of 1a.
![[Graphic 1]](/bjoc/content/inline/1860-5397-18-21-i3.svg?max-width=637&scale=1.0)
|
|||
| Entrya | [Au] | conv. [%] | 2a/2b |
| 1 | A’(AuCl) (2 mol %) | 89 | 1.0:1.0 |
| 2 | A(AuCl)2 (1 mol %) | 88 | 1.1:1.0 |
| 3 | B’(AuCl) (2 mol %) | 86 | 1.2:1.0 |
| 4 | B(AuCl)2 (1 mol %) | 91 | 1.1:1.0 |
| 5 | C’(AuCl) (2 mol %) | 86 | 1.5:1.0 |
| 6 | C(AuCl)2 (1 mol %) | 89 | 1.8:1.0 |
aReaction conditions: 1a (0.2 mmol), AgSbF6 (2.0 mol %), CH2Cl2 (0.1 M), 4 h.
Analogously, the reactivity in the presence of meta-substituted catalysts B’(AuCl) and B(AuCl)2 was investigated. Also in this case, the reactivity and selectivity of the parental catalysts were comparable (entries 3 and 4, Table 1). Taken together, these outcomes suggest that the role of the macrocycle for catalysts A/B(AuCl)2 is not determining in changing the product distribution and that the catalytically active gold(I) nuclei are too far to be influenced by the cavity or by the macrocycle itself.
Finally, the catalytic reaction was attempted using C’(AuCl). We thus observed a selectivity towards product 2a (1.5:1) which might be caused by the different orientation of the phosphine ligand implanted on the aromatic ring (entry 5, Table 1). Interestingly, this effect was substantially improved with the use of the calix[6]arene-based complex C(AuCl)2 (entry 6, Table 1). Overall, the ortho-substituted macrocycle C(AuCl)2 displayed an enhanced selectivity, with respect to the parental macrocycles A,B(AuCl)2, that arise from the proximity of the two gold(I) nuclei to the calix[6]arene scaffold. Although just preliminary, these results indicate that the conformational properties of this class of macrocycles can influence the selectivity in gold(I)-catalyzed cycloisomerization of 1,6-enynes.
Conclusion
We reported the synthesis of a novel family of diametric diphosphine gold(I) complexes whose geometry in low-polarity solvents is controlled by the 1,2,3-alternate conformation of the calix[6]arene precursor. These catalysts are able to tune the selectivity of catalytic cycloisomerization of 1,6-enynes as a function of the relative orientation of the bonded gold(I) nuclei with respect to the macrocycle. Further studies are currently under progress to outline the role of the macrocycle and to verify if a possible re-orientation of the gold(I) nuclei towards the center of the aromatic cavity can play any role in dictating the selectivity of the catalytic transformation.
Experimental
General procedure for catalysis: In a 10 mL two-necked round-bottomed flask containing A(AuCl)2 (1.0 mol %, 4.4 mg) or A’(AuCl) (2.0 mol %, 3.0 mg), dry CH2Cl2 (2.0 mL) was added under nitrogen atmosphere. Subsequently, a tip of spatula (micro spatula, Heyman type 16 cm) of AgSbF6 (≈2.0 mol %, ≈2 mg) was added along with 20 mg of 4 Å molecular sieves. The flask was covered with an aluminum foil and the mixture stirred for 5 minutes. Subsequently, 1a (0.2 mmol, 63.0 mg) was added and the reaction mixture was stirred for 4 hours. After completion, the mixture was diluted with 20 mL of CH2Cl2, filtered through a pad of celite, and transferred in a 100 mL flask where it was concentrated under reduced atmosphere. Conversions and selectivities were determined by 1H NMR analysis (data confirmed by performing the reaction twice). 1H NMR (2a) (400 MHz, CDCl3) δ 7.48–7.19 (m, 5H), 6.63 (dtd, J = 10.1, 2.1, 1.0 Hz, 1H), 6.37 (s, 1H), 5.90 (dtd, J = 10.1, 4.0, 1.6 Hz, 1H), 4.30–4.21 (m, 4H), 2.98 (d, J = 1.6 Hz, 2H), 2.78 (ddd, J = 4.0, 2.1, 0.9 Hz, 2H), 1.32–1.24 (m, 6H). 1H NMR (2b) (400 MHz, CDCl3) δ 7.48–7.20 (m, 5H), 6.93 (d, J = 16.2 Hz, 1H), 6.48 (d, J = 16.2 Hz, 1H), 5.73 (ddd, J = 2.7, 1.9, 0.9 Hz, 1H), 4.23–4.14 (m, 4H), 3.28 (dd, J = 1.9, 0.9 Hz, 2H), 3.18 (dd, J = 1.9, 0.9 Hz, 2H), 1.32–1.21 (m, 6H).
Supporting Information
| Supporting Information File 1: Experimental procedures, characterization data of compounds and copies of NMR spectra. | ||
| Format: PDF | Size: 2.6 MB | Download |
References
-
Kaya, Z.; Bentouhami, E.; Pelzer, K.; Armspach, D. Coord. Chem. Rev. 2021, 445, 214066. doi:10.1016/j.ccr.2021.214066
Return to citation in text: [1] -
Olivo, G.; Capocasa, G.; Del Giudice, D.; Lanzalunga, O.; Di Stefano, S. Chem. Soc. Rev. 2021, 50, 7681–7724. doi:10.1039/d1cs00175b
Return to citation in text: [1] -
Gramage-Doria, R.; Armspach, D.; Matt, D. Coord. Chem. Rev. 2013, 257, 776–816. doi:10.1016/j.ccr.2012.10.006
Return to citation in text: [1] -
Kamer, P. C. J.; van Leeuwen, P. W. N. M., Eds. Phosphorus(III) Ligands in Homogeneous Catalysis: Design and Synthesis; John Wiley & Sons: Chichester, UK, 2012. doi:10.1002/9781118299715
Return to citation in text: [1] -
Orton, G. R. F.; Pilgrim, B. S.; Champness, N. R. Chem. Soc. Rev. 2021, 50, 4411–4431. doi:10.1039/d0cs01556c
Return to citation in text: [1] -
Shet, H.; Parmar, U.; Bhilare, S.; Kapdi, A. R. Org. Chem. Front. 2021, 8, 1599–1656. doi:10.1039/d0qo01194k
Return to citation in text: [1] -
Matt, D.; Harrowfield, J. ChemCatChem 2021, 13, 153–168. doi:10.1002/cctc.202001242
Return to citation in text: [1] -
Sémeril, D.; Matt, D. Coord. Chem. Rev. 2014, 279, 58–95. doi:10.1016/j.ccr.2014.06.019
Return to citation in text: [1] -
Schöttle, C.; Guan, E.; Okrut, A.; Grosso-Giordano, N. A.; Palermo, A.; Solovyov, A.; Gates, B. C.; Katz, A. J. Am. Chem. Soc. 2019, 141, 4010–4015. doi:10.1021/jacs.8b13013
Return to citation in text: [1] -
Karpus, A.; Yesypenko, O.; Boiko, V.; Poli, R.; Daran, J.-C.; Voitenko, Z.; Kalchenko, V.; Manoury, E. Eur. J. Org. Chem. 2016, 3386–3394. doi:10.1002/ejoc.201600208
Return to citation in text: [1] -
Elaieb, F.; Hedhli, A.; Sémeril, D.; Matt, D. Eur. J. Org. Chem. 2016, 1867–1873. doi:10.1002/ejoc.201600055
Return to citation in text: [1] [2] -
Monnereau, L.; Sémeril, D.; Matt, D.; Toupet, L. Adv. Synth. Catal. 2009, 351, 1629–1636. doi:10.1002/adsc.200900074
Return to citation in text: [1] -
Sémeril, D.; Matt, D.; Toupet, L. Chem. – Eur. J. 2008, 14, 7144–7155. doi:10.1002/chem.200800747
Return to citation in text: [1] -
Chavagnan, T.; Sémeril, D.; Matt, D.; Toupet, L. Eur. J. Org. Chem. 2017, 313–323. doi:10.1002/ejoc.201601278
Return to citation in text: [1] -
Chavagnan, T.; Sémeril, D.; Matt, D.; Harrowfield, J.; Toupet, L. Chem. – Eur. J. 2015, 21, 6678–6681. doi:10.1002/chem.201500177
Return to citation in text: [1] -
El Moll, H.; Sémeril, D.; Matt, D.; Toupet, L. Adv. Synth. Catal. 2010, 352, 901–908. doi:10.1002/adsc.200900767
Return to citation in text: [1] -
El Moll, H.; Sémeril, D.; Matt, D.; Youinou, M.-T.; Toupet, L. Org. Biomol. Chem. 2009, 7, 495–501. doi:10.1039/b813373e
Return to citation in text: [1] -
Martín‐Torres, I.; Ogalla, G.; Yang, J.-M.; Rinaldi, A.; Echavarren, A. M. Angew. Chem., Int. Ed. 2021, 60, 9339–9344. doi:10.1002/anie.202017035
Return to citation in text: [1] -
Rusali, L. E.; Schramm, M. P. Tetrahedron Lett. 2020, 61, 152333. doi:10.1016/j.tetlet.2020.152333
Return to citation in text: [1] -
Inoue, M.; Kamiguchi, S.; Ugawa, K.; Hkiri, S.; Bouffard, J.; Sémeril, D.; Iwasawa, T. Eur. J. Org. Chem. 2019, 6261–6268. doi:10.1002/ejoc.201901058
Return to citation in text: [1] -
Ho, T. D.; Schramm, M. P. Eur. J. Org. Chem. 2019, 5678–5684. doi:10.1002/ejoc.201900829
Return to citation in text: [1] -
Inoue, M.; Ugawa, K.; Maruyama, T.; Iwasawa, T. Eur. J. Org. Chem. 2018, 5304–5311. doi:10.1002/ejoc.201800948
Return to citation in text: [1] -
Endo, N.; Inoue, M.; Iwasawa, T. Eur. J. Org. Chem. 2018, 1136–1140. doi:10.1002/ejoc.201701613
Return to citation in text: [1] -
Schramm, M. P.; Kanaura, M.; Ito, K.; Ide, M.; Iwasawa, T. Eur. J. Org. Chem. 2016, 813–820. doi:10.1002/ejoc.201501426
Return to citation in text: [1] -
Endo, N.; Kanaura, M.; Schramm, M. P.; Iwasawa, T. Eur. J. Org. Chem. 2016, 2514–2521. doi:10.1002/ejoc.201600362
Return to citation in text: [1] -
Mitschke, B.; Turberg, M.; List, B. Chem 2020, 6, 2515–2532. doi:10.1016/j.chempr.2020.09.007
Return to citation in text: [1] -
Mouarrawis, V.; Plessius, R.; van der Vlugt, J. I.; Reek, J. N. H. Front. Chem. (Lausanne, Switz.) 2018, 6, 623. doi:10.3389/fchem.2018.00623
Return to citation in text: [1] -
Homden, D. M.; Redshaw, C. Chem. Rev. 2008, 108, 5086–5130. doi:10.1021/cr8002196
Return to citation in text: [1] -
Cera, G.; Arduini, A.; Secchi, A.; Credi, A.; Silvi, S. Chem. Rec. 2021, 21, 1161–1181. doi:10.1002/tcr.202100012
Return to citation in text: [1] -
Cera, G.; Giovanardi, G.; Secchi, A.; Arduini, A. Chem. – Eur. J. 2021, 27, 10261–10266. doi:10.1002/chem.202101323
Return to citation in text: [1] -
Cera, G.; Cester Bonati, F.; Bazzoni, M.; Secchi, A.; Arduini, A. Org. Biomol. Chem. 2021, 19, 1546–1554. doi:10.1039/d0ob02393k
Return to citation in text: [1] -
Cera, G.; Bazzoni, M.; Arduini, A.; Secchi, A. Org. Lett. 2020, 22, 3702–3705. doi:10.1021/acs.orglett.0c01191
Return to citation in text: [1] -
Bazzoni, M.; Zanichelli, V.; Casimiro, L.; Massera, C.; Credi, A.; Secchi, A.; Silvi, S.; Arduini, A. Eur. J. Org. Chem. 2019, 3513–3524. doi:10.1002/ejoc.201900211
Return to citation in text: [1] [2] -
Cera, G.; Bazzoni, M.; Andreoni, L.; Cester Bonati, F.; Massera, C.; Silvi, S.; Credi, A.; Secchi, A.; Arduini, A. Eur. J. Org. Chem. 2021, 5788–5798. doi:10.1002/ejoc.202101080
Return to citation in text: [1] -
van Duynhoven, J. P. M.; Janssen, R. G.; Verboom, W.; Franken, S. M.; Casnati, A.; Pochini, A.; Ungaro, R.; de Mendoza, J.; Nieto, P. M. J. Am. Chem. Soc. 1994, 116, 5814–5822. doi:10.1021/ja00092a036
Return to citation in text: [1] -
Nieto-Oberhuber, C.; Paz Muñoz, M.; López, S.; Jiménez-Núñez, E.; Nevado, C.; Herrero-Gómez, E.; Raducan, M.; Echavarren, A. M. Chem. – Eur. J. 2006, 12, 1677–1693. doi:10.1002/chem.200501088
Return to citation in text: [1] -
A model reaction performed with AgSbF6 (2 mol %) as the catalyst, in the absence of any gold(I) complex, did not lead to any conversion of starting material 1a.
Return to citation in text: [1] -
Fürstner, A.; Morency, L. Angew. Chem., Int. Ed. 2008, 47, 5030–5033. doi:10.1002/anie.200800934
Return to citation in text: [1] -
Jiménez-Núñez, E.; Echavarren, A. M. Chem. Rev. 2008, 108, 3326–3350. doi:10.1021/cr0684319
Return to citation in text: [1]
| 37. | A model reaction performed with AgSbF6 (2 mol %) as the catalyst, in the absence of any gold(I) complex, did not lead to any conversion of starting material 1a. |
| 38. | Fürstner, A.; Morency, L. Angew. Chem., Int. Ed. 2008, 47, 5030–5033. doi:10.1002/anie.200800934 |
| 39. | Jiménez-Núñez, E.; Echavarren, A. M. Chem. Rev. 2008, 108, 3326–3350. doi:10.1021/cr0684319 |
| 1. | Kaya, Z.; Bentouhami, E.; Pelzer, K.; Armspach, D. Coord. Chem. Rev. 2021, 445, 214066. doi:10.1016/j.ccr.2021.214066 |
| 2. | Olivo, G.; Capocasa, G.; Del Giudice, D.; Lanzalunga, O.; Di Stefano, S. Chem. Soc. Rev. 2021, 50, 7681–7724. doi:10.1039/d1cs00175b |
| 3. | Gramage-Doria, R.; Armspach, D.; Matt, D. Coord. Chem. Rev. 2013, 257, 776–816. doi:10.1016/j.ccr.2012.10.006 |
| 14. | Chavagnan, T.; Sémeril, D.; Matt, D.; Toupet, L. Eur. J. Org. Chem. 2017, 313–323. doi:10.1002/ejoc.201601278 |
| 15. | Chavagnan, T.; Sémeril, D.; Matt, D.; Harrowfield, J.; Toupet, L. Chem. – Eur. J. 2015, 21, 6678–6681. doi:10.1002/chem.201500177 |
| 16. | El Moll, H.; Sémeril, D.; Matt, D.; Toupet, L. Adv. Synth. Catal. 2010, 352, 901–908. doi:10.1002/adsc.200900767 |
| 17. | El Moll, H.; Sémeril, D.; Matt, D.; Youinou, M.-T.; Toupet, L. Org. Biomol. Chem. 2009, 7, 495–501. doi:10.1039/b813373e |
| 35. | van Duynhoven, J. P. M.; Janssen, R. G.; Verboom, W.; Franken, S. M.; Casnati, A.; Pochini, A.; Ungaro, R.; de Mendoza, J.; Nieto, P. M. J. Am. Chem. Soc. 1994, 116, 5814–5822. doi:10.1021/ja00092a036 |
| 9. | Schöttle, C.; Guan, E.; Okrut, A.; Grosso-Giordano, N. A.; Palermo, A.; Solovyov, A.; Gates, B. C.; Katz, A. J. Am. Chem. Soc. 2019, 141, 4010–4015. doi:10.1021/jacs.8b13013 |
| 10. | Karpus, A.; Yesypenko, O.; Boiko, V.; Poli, R.; Daran, J.-C.; Voitenko, Z.; Kalchenko, V.; Manoury, E. Eur. J. Org. Chem. 2016, 3386–3394. doi:10.1002/ejoc.201600208 |
| 11. | Elaieb, F.; Hedhli, A.; Sémeril, D.; Matt, D. Eur. J. Org. Chem. 2016, 1867–1873. doi:10.1002/ejoc.201600055 |
| 12. | Monnereau, L.; Sémeril, D.; Matt, D.; Toupet, L. Adv. Synth. Catal. 2009, 351, 1629–1636. doi:10.1002/adsc.200900074 |
| 13. | Sémeril, D.; Matt, D.; Toupet, L. Chem. – Eur. J. 2008, 14, 7144–7155. doi:10.1002/chem.200800747 |
| 36. | Nieto-Oberhuber, C.; Paz Muñoz, M.; López, S.; Jiménez-Núñez, E.; Nevado, C.; Herrero-Gómez, E.; Raducan, M.; Echavarren, A. M. Chem. – Eur. J. 2006, 12, 1677–1693. doi:10.1002/chem.200501088 |
| 5. | Orton, G. R. F.; Pilgrim, B. S.; Champness, N. R. Chem. Soc. Rev. 2021, 50, 4411–4431. doi:10.1039/d0cs01556c |
| 6. | Shet, H.; Parmar, U.; Bhilare, S.; Kapdi, A. R. Org. Chem. Front. 2021, 8, 1599–1656. doi:10.1039/d0qo01194k |
| 7. | Matt, D.; Harrowfield, J. ChemCatChem 2021, 13, 153–168. doi:10.1002/cctc.202001242 |
| 8. | Sémeril, D.; Matt, D. Coord. Chem. Rev. 2014, 279, 58–95. doi:10.1016/j.ccr.2014.06.019 |
| 33. | Bazzoni, M.; Zanichelli, V.; Casimiro, L.; Massera, C.; Credi, A.; Secchi, A.; Silvi, S.; Arduini, A. Eur. J. Org. Chem. 2019, 3513–3524. doi:10.1002/ejoc.201900211 |
| 4. | Kamer, P. C. J.; van Leeuwen, P. W. N. M., Eds. Phosphorus(III) Ligands in Homogeneous Catalysis: Design and Synthesis; John Wiley & Sons: Chichester, UK, 2012. doi:10.1002/9781118299715 |
| 33. | Bazzoni, M.; Zanichelli, V.; Casimiro, L.; Massera, C.; Credi, A.; Secchi, A.; Silvi, S.; Arduini, A. Eur. J. Org. Chem. 2019, 3513–3524. doi:10.1002/ejoc.201900211 |
| 34. | Cera, G.; Bazzoni, M.; Andreoni, L.; Cester Bonati, F.; Massera, C.; Silvi, S.; Credi, A.; Secchi, A.; Arduini, A. Eur. J. Org. Chem. 2021, 5788–5798. doi:10.1002/ejoc.202101080 |
| 28. | Homden, D. M.; Redshaw, C. Chem. Rev. 2008, 108, 5086–5130. doi:10.1021/cr8002196 |
| 30. | Cera, G.; Giovanardi, G.; Secchi, A.; Arduini, A. Chem. – Eur. J. 2021, 27, 10261–10266. doi:10.1002/chem.202101323 |
| 26. | Mitschke, B.; Turberg, M.; List, B. Chem 2020, 6, 2515–2532. doi:10.1016/j.chempr.2020.09.007 |
| 27. | Mouarrawis, V.; Plessius, R.; van der Vlugt, J. I.; Reek, J. N. H. Front. Chem. (Lausanne, Switz.) 2018, 6, 623. doi:10.3389/fchem.2018.00623 |
| 31. | Cera, G.; Cester Bonati, F.; Bazzoni, M.; Secchi, A.; Arduini, A. Org. Biomol. Chem. 2021, 19, 1546–1554. doi:10.1039/d0ob02393k |
| 32. | Cera, G.; Bazzoni, M.; Arduini, A.; Secchi, A. Org. Lett. 2020, 22, 3702–3705. doi:10.1021/acs.orglett.0c01191 |
| 11. | Elaieb, F.; Hedhli, A.; Sémeril, D.; Matt, D. Eur. J. Org. Chem. 2016, 1867–1873. doi:10.1002/ejoc.201600055 |
| 18. | Martín‐Torres, I.; Ogalla, G.; Yang, J.-M.; Rinaldi, A.; Echavarren, A. M. Angew. Chem., Int. Ed. 2021, 60, 9339–9344. doi:10.1002/anie.202017035 |
| 19. | Rusali, L. E.; Schramm, M. P. Tetrahedron Lett. 2020, 61, 152333. doi:10.1016/j.tetlet.2020.152333 |
| 20. | Inoue, M.; Kamiguchi, S.; Ugawa, K.; Hkiri, S.; Bouffard, J.; Sémeril, D.; Iwasawa, T. Eur. J. Org. Chem. 2019, 6261–6268. doi:10.1002/ejoc.201901058 |
| 21. | Ho, T. D.; Schramm, M. P. Eur. J. Org. Chem. 2019, 5678–5684. doi:10.1002/ejoc.201900829 |
| 22. | Inoue, M.; Ugawa, K.; Maruyama, T.; Iwasawa, T. Eur. J. Org. Chem. 2018, 5304–5311. doi:10.1002/ejoc.201800948 |
| 23. | Endo, N.; Inoue, M.; Iwasawa, T. Eur. J. Org. Chem. 2018, 1136–1140. doi:10.1002/ejoc.201701613 |
| 24. | Schramm, M. P.; Kanaura, M.; Ito, K.; Ide, M.; Iwasawa, T. Eur. J. Org. Chem. 2016, 813–820. doi:10.1002/ejoc.201501426 |
| 25. | Endo, N.; Kanaura, M.; Schramm, M. P.; Iwasawa, T. Eur. J. Org. Chem. 2016, 2514–2521. doi:10.1002/ejoc.201600362 |
| 29. | Cera, G.; Arduini, A.; Secchi, A.; Credi, A.; Silvi, S. Chem. Rec. 2021, 21, 1161–1181. doi:10.1002/tcr.202100012 |
© 2022 Giovanardi et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.