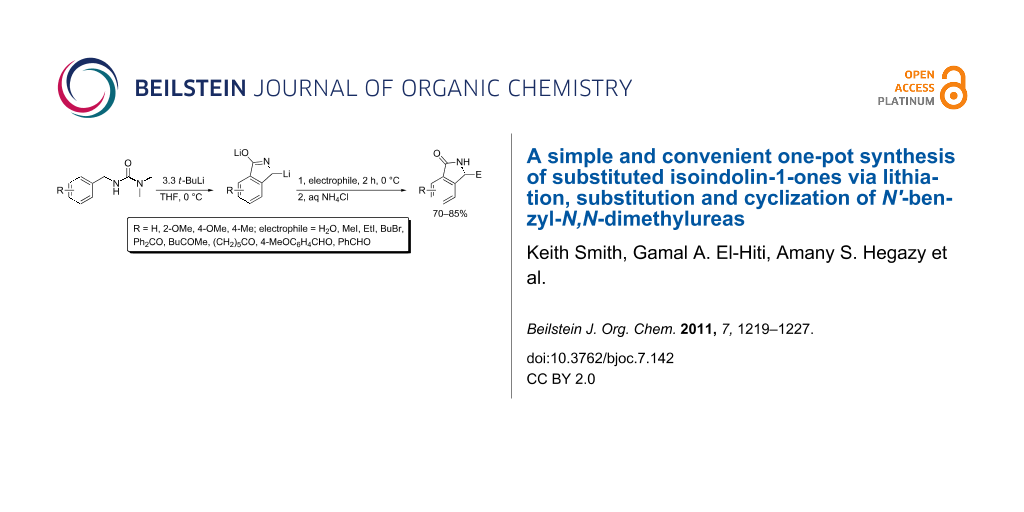
Abstract
Lithiation of N'-benzyl-N,N-dimethylurea and its substituted derivatives with t-BuLi (3.3 equiv) in anhydrous THF at 0 °C followed by reaction with various electrophiles afforded a range of 3-substituted isoindolin-1-ones in high yields.

Graphical Abstract
Introduction
In recent years there has been a great deal of interest in compounds possessing an isoindolinone ring system since it represents the core unit of numerous naturally occurring substances [1-8]. Also, some members that possess this moiety have shown interesting biological properties [9-15].
Several traditional methods are available for the synthesis of isoindolinones [16-25], based on the use of Grignard reagents [26], Diels–Alder reactions [27], Wittig reagents [28], reduction processes [29,30], rearrangement processes [31] and photochemical reactions [32,33]. However, such methods generally require multiple reaction steps and are unsatisfactory both in yield and generality. In recent years several new approaches have been developed for the synthesis of substituted isoindolines, of which the most generally useful involve palladium-catalysed reactions [34-42] or lithiation procedures [43-54].
In particular, among the various lithiation methods two useful approaches to the synthesis of isoindolin-1-ones have been reported (Scheme 1 and Scheme 2) [43,45]. One method simply involves lithiation of a preformed isoindolin-1-one ring system at the 3-position followed by treatment with an electrophile (Scheme 1) [43]. While this approach is straightforward, clearly, its general utility depends on the availability of appropriately substituted analogues of the isoindolin-1-one ring system.
![[1860-5397-7-142-i1]](/bjoc/content/inline/1860-5397-7-142-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Lithiation and substitution of isoindolin-1-ones [43].
Scheme 1: Lithiation and substitution of isoindolin-1-ones [43].
![[1860-5397-7-142-i2]](/bjoc/content/inline/1860-5397-7-142-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Lithiation and cyclization of N-tert-butyl-N-benzylbenzamides [45].
Scheme 2: Lithiation and cyclization of N-tert-butyl-N-benzylbenzamides [45].
The other, potentially more useful approach involves generation of the heterocyclic ring system during the lithiation step. For example, lithiation of N-tert-butyl-N-benzylbenzamides gives intermediates that cyclise to form de-aromatised species. Oxidation to re-aromatise the benzenoid system, followed by treatment with trifluoroacetic acid to remove the tert-butyl group, gives the corresponding isoindolin-1-ones (Scheme 2) [45]. However, this approach gives more modest yields, requires an additional step to remove the tert-butyl group, and also involves incorporating the eventual C-3 substituent into the starting material, which limits the generality. Moreover, the reaction works well only with 3-aryl substituents.
Clayden has improved the yield of isoindolin-1-ones by using 2-methoxybenzamides as the starting materials; in this case the methoxy group acts as a leaving group, which avoids the need for an oxidation step [45]. However, this approach still requires an additional step to remove the tert-butyl group and incorporation of the 3-substituent into the starting material.
As a result of our own interest in the use of lithium reagents in organic synthesis [55-67], we have had occasion to investigate lithiation of various aromatic compounds containing substituted amino groups. Factors that influence the site(s) of lithiation of such compounds can be quite subtle. For example, lithiation of N,N-dimethyl-N'-(substituted phenyl)ureas can occur predominantly on the N,N-dimethyl group (e.g., for the unsubstituted, 4-methyl or 4-methoxy-compounds with t-BuLi at −20 °C) or on the ring next to the urea group (e.g., for the 4-chloro-, 4-fluoro- or 4-trifluoromethyl compounds with either n-BuLi or t-BuLi at 0 °C) [59]. However, a recent report indicates that replacement of the dimethylamino group by a more hindered dialkylamino group in the unsubstituted phenyl derivative results in lithiation on the ring [68]. We have also developed procedures for the lithiation of various substituted benzylamines [69-71]. Similar subtleties over the site(s) of lithiation are also observed for these compounds. The site(s) of lithiation depend on the substituents at nitrogen, the nature and positions of substituents on the aryl ring and/or on the nature of the lithium reagent. For example, lithiation of N-benzylpivalamide with t-BuLi gives a mixture of ring (2-position) and side-chain lithiated species, whereas for N'-benzyl-N,N-dimethylurea side-chain lithiation does not occur [69]. Of particular relevance for the present study was the observation that lithiation of N'-(2-methoxybenzyl)-N,N-dimethylurea (1) with two equivalents of t-BuLi in THF at −20 °C for 2 h followed by reactions with a range of electrophiles gave mixtures of products (Scheme 3) involving ring substitution both next to the urea-containing group (o-substitution; 47–51% yields) and next to the methoxy group (o'-substitution; 38–40% yields) [69]. Formation of 2 and 3 presumably involved lithium intermediates 4 and 5, respectively (Figure 1).
![[1860-5397-7-142-i3]](/bjoc/content/inline/1860-5397-7-142-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Lithiation and substitution of 1 at −20 °C [69].
Scheme 3: Lithiation and substitution of 1 at −20 °C [69].
![[1860-5397-7-142-1]](/bjoc/content/figures/1860-5397-7-142-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
We found that when the lithiation reaction was carried out at 0 °C rather than at −20 °C, followed by reaction with an electrophile, it produced lower yields of the corresponding substituted products 2 and 3, along with some residual 1 and a small amount of a new product of structure 6 (Figure 1). By increasing the amount of lithiating agent and extending the period of the lithiation reaction, we were able to increase the yield of 6, and in a preliminary communication we reported that directed lithiation of various N'-benzyl-N,N-dimethylureas with t-BuLi (3.3 equiv) in anhydrous THF at 0 °C followed by reaction with various electrophiles afforded the corresponding 3-substituted isoindolin-1-ones in high yields [72]. We now report full details of that work, extend the scope of the reaction and examine the diastereoselectivity of the reaction with prochiral electrophiles.
Results and Discussion
As noted above, when lithiation of 1 was carried out at 0 °C rather than at −20 °C prior to reaction with an electrophile, lower yields of 2 and 3 along with some residual 1 and a small amount of 6 were obtained. A possible mechanism for the formation of 6 is shown in Scheme 4. According to this scheme, compounds of the general structure 6 would arise by cyclization of 4 to give 7, followed by further lithiation to give 8, which on reaction with an electrophile would give 6. It appeared likely that the yield of 6 could be increased by use of a larger quantity of t-BuLi and an extended reaction time; therefore, we investigated this possibility.
![[1860-5397-7-142-i4]](/bjoc/content/inline/1860-5397-7-142-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: A possible mechanism for the formation of 6.
Scheme 4: A possible mechanism for the formation of 6.
Indeed, lithiation of 1 with t-BuLi (3.3 equiv) in anhydrous THF at 0 °C for 6 h, followed by treatment with iodomethane, gave 9 (i.e., 6, where E = Me) in 79% yield (Table 1). While an increased yield of 6 (E = Me, i.e., 9, produced from 7) and the disappearance of 2 (produced from 4) were expected, the disappearance of 3 (produced from 5) was a surprise. It would appear that at 0 °C, not only did 4 cyclize to give 7, but 5 was also in equilibrium with 4, allowing its eventual conversion into 7 and then 8.
Table 1: Synthesis of 3-substituted 4-methoxyisoindolin-1-ones 9–18.
![[Graphic 1]](/bjoc/content/inline/1860-5397-7-142-i5.svg?max-width=637&scale=1.0)
|
|||
| Product | Electrophile | E | Yield (%)a |
|---|---|---|---|
| 9 | MeI | Me | 79 |
| 10 | EtI | Et | 84 |
| 11 | BuBr | Bu | 72 |
| 12 | H2O | H | 82 |
| 13 | Ph2CO | Ph2C(OH) | 81 |
| 14 | (CH2)5CO | (CH2)5C(OH) | 78 |
| 15 | BuCOMe | BuC(OH)Me | 78 |
| 16 | PhCOMe | PhC(OH)Me | 80 |
| 17 | 4-MeOC6H4CHO | 4-MeOC6H4CH(OH) | 81 |
| 18 | PhCHO | PhCH(OH) | 80 |
aYield of material isolated by washing or triturating the crude product with diethyl ether.
This fortuitous finding appeared to offer potential as a general synthetic method and the same lithiation procedure was therefore used with a range of other electrophiles including other haloalkanes, water, symmetrical and unsymmetrical ketones, and aldehydes. After work-up of the reaction mixtures, the crude products were triturated and/or washed with diethyl ether to give products 10–18 in high yields (Table 1).
From the results recorded in Table 1, it appeared that the reaction was a general process for production of 3-substituted 4-methoxyisoindolin-1-ones 9–18 in high yields from reactions with a variety of electrophiles. The identity of the ring system was confirmed by the crystallization of 12 and subjecting it to X-ray crystallography (Figure 2). All the products were analysed by standard spectroscopic methods and showed, for example, the expected molecular ions in their mass spectra. Compounds 10–14 were readily identified by their NMR spectra, which showed all the expected signals. In the 1H NMR spectra of compounds 10 and 11, the expected diastereotopicity of the two hydrogens of the CH2 groups closest to the ring was evident. For compound 13 the two phenyl groups appeared as separated signals in the 13C NMR spectrum, verifying that they were also diastereotopic, and similarly, the two sides of the cyclohexane ring in compound 14 also appeared as separated signals. The NMR spectra of products 15–18 were relatively complex. Therefore, in order to get further information about these products, they were each crystallized from ethyl acetate. The 1H NMR spectra of the crystallized materials showed what appeared to be a single component in each case. The crystalline products were therefore subjected to X-ray crystallography. The structures found are illustrated in Figure 2.
![[1860-5397-7-142-2]](/bjoc/content/figures/1860-5397-7-142-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: X-Ray crystal structures of crystallized compounds 12 and 15–18.
Figure 2: X-Ray crystal structures of crystallized compounds 12 and 15–18.
The X-ray crystal structures of the isolated crystals of products 15, 17 and 18 showed that they were α-(R*)-3-(S*)-diastereoisomers, while that of 16 showed that it was the α-(R*)-3-(R*)-diastereoisomer (Figure 2). With this information in hand, the NMR spectra of the initial products obtained by trituration or washing of the original crude products were re-examined. These spectra showed the presence of the crystallized product and a second component, the identifiable resonances of which were consistent with the other diastereoisomer. The two diastereoisomers were present in nearly equal proportions (between 47:53 and 42:58), with the α-(R*)-3-(R*)-diastereoisomer being the somewhat predominant diastereoisomer in all cases.
We have shown previously that lithiation of N'-benzyl-N,N-dimethylurea and N'-(4-substituted benzyl)-N,N-dimethylureas with t-BuLi (two equiv) in THF at −78 °C for 4 h followed by reactions with a variety of electrophiles gave high yields of products involving substitution at the 2-position [69]. Therefore, it was of interest to investigate further the scope of the process represented in Table 1 with other ring-substituted N'-benzyl-N,N-dimethylureas 19 (R = H, 4-OMe, 4-Me; Table 2). Each substrate was lithiated according to the standard procedure with t-BuLi (3.3 equiv) in anhydrous THF at 0 °C for 6 h, and then treated with various electrophiles. After work-up, as described above, the substituted isoindolin-1-ones 20–40 were obtained in high yields (Table 2).
Table 2: Synthesis of various substituted isoindolin-1-ones 20–40.
![[Graphic 2]](/bjoc/content/inline/1860-5397-7-142-i6.svg?max-width=637&scale=1.0)
|
||||
| Product | R | Electrophile | E | Yield (%)a |
|---|---|---|---|---|
| 20 | H | H2O | H | 71 |
| 21 | H | MeI | Me | 75 |
| 22 | H | EtI | Et | 77 |
| 23 | H | BuBr | Bu | 76b |
| 24 | H | Ph2CO | Ph2C(OH) | 74 |
| 25c | H | PhCHO | PhCH(OH) | 73 |
| 26d | H | 4-MeOC6H4CHO | 4-MeOC6H4CH(OH) | 78 |
| 27 | OMe | H2O | H | 70 |
| 28 | OMe | MeI | Me | 76 |
| 29 | OMe | EtI | Et | 78 |
| 30 | OMe | BuBr | Bu | 77 |
| 31 | OMe | Ph2CO | Ph2C(OH) | 72 |
| 32c | OMe | 4-MeOC6H4CHO | 4-MeOC6H4CH(OH) | 75 |
| 33 | Me | H2O | H | 75 |
| 34 | Me | MeI | Me | 78 |
| 35 | Me | EtI | Et | 75 |
| 36 | Me | (CH2)5C=O | (CH2)5C(OH) | 72 |
| 37 | Me | Ph2CO | Ph2C(OH) | 85 |
| 38e | Me | MeCOBu | MeC(OH)Bu | 77 |
| 39e | Me | PhCHO | PhCH(OH) | 79 |
| 40d | Me | 4-MeOC6H4CHO | 4-MeOC6H4CH(OH) | 83 |
aYield of product isolated by trituration or washing of the initial reaction mixture with diethyl ether. bCompound 41 (Figure 3) was obtained in 5% yield. cThe 1H NMR spectrum of the solid after crystallization showed the structure to be the α-(R*)-3-(R*)-isomer. dThe 1H NMR spectrum of the solid after crystallization showed the structure to be the α-(R*)-3-(S*)-isomer. eX-Ray crystallography showed the product isolated by crystallization to be the α-(R*)-3-(R*)-isomer (Figure 4).
![[1860-5397-7-142-3]](/bjoc/content/figures/1860-5397-7-142-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-7-142-4]](/bjoc/content/figures/1860-5397-7-142-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: X-ray crystal structures of compounds 38 and 39.
Figure 4: X-ray crystal structures of compounds 38 and 39.
The 1H NMR spectra of compounds 22–24, 29–31 and 35–38 showed diastereotopicity features similar to those for compounds reported in Table 1. The NMR spectra of 25, 26, 32, and 38–40 showed the presence of two racemic diastereoisomers in unequal proportions (up to ca. 38:62). However, the NMR spectra of the products after crystallization from ethyl acetate all exhibited one set of signals, indicating that the isolated crystalline product in each case was a single racemic diastereoisomer. X-ray crystallography of the isolated crystals of compounds 38 and 39 showed them to be the α-(R*)-3-(R*)-diastereoisomers (Figure 4). The 1H NMR spectra of the isolated crystalline products 26, 32 and 40 indicated that the crystals were α-(R*)-3-(S*)-diastereoisomers, while the 1H NMR spectrum of the crystallized product 25 showed it to be α-(R*)-3-(R*)-isomer. With the help of the NMR spectra of the crystallized diastereoisomers, re-examination of the spectra of the materials obtained by simple trituration or washing of the original crude product allowed calculation of the proportions of the two diastereoisomers and in all cases the major diastereoisomer was again the α-(R*)-3-(R*)-isomer, as was the case when 2-methoxybenzyl-N,N-dimethylurea was the starting material.
Compound 41 (Figure 3) was obtained as a side product in 5% yield when 1-bromobutane was used as the electrophile. Compound 41 arose due to further lithiation at position 3 of 42, produced in situ from 43, to generate the lithium intermediate 44 (Figure 3). Reaction of the latter with a further equivalent of 1-bromobutane affords 41.
Conclusion
We have developed a novel, simple, efficient, general and high yielding procedure for synthesis of isoindolin-1-ones in a one-step reaction. It allows easy addition of a range of substituents to the initial benzene ring and incorporation of a range of substituents derived from the electrophiles used. Isolation of the pure products is also extremely easy, involving simple trituration and/or washing of the crude product after work up, except in the cases where two diastereoisomers are produced. In such cases the α-(R*)-3-(R*)-isomer is somewhat predominant (ratios of ca. 47:53 to 38:62); crystallization of these mixtures produces one pure diastereoisomer in all cases. The method promises to be a very useful new approach for synthesis of substituted isoindolin-1-ones.
Experimental
General information
Melting point determinations were performed by the open capillary method with a Gallenkamp melting point apparatus and are uncorrected. 1H and 13C NMR spectra were recorded on a Bruker AV400 or AV500 spectrometer at 400 or 500 MHz for 1H and 100 or 125 MHz for 13C measurements. Chemical shifts δ are reported in parts per million (ppm) relative to TMS and coupling constants J are in Hz and have been rounded to the nearest whole number. 13C multiplicities were revealed by DEPT signals. Assignments of signals are based on integration values, coupling patterns and expected chemical shift values and have not been rigorously confirmed. Signals with similar characteristics might be interchanged. Low-resolution mass spectra (see Supporting Information File 1) were recorded on a Quattro II spectrometer, electron impact (EI) at 70 eV and chemical ionization (CI) at 50 eV by the use of NH3 as ionization gas. Atmospheric pressure chemical ionization (APCI) mass spectra were measured on a Waters LCT Premier XE instrument. Electrospray (ES) analyses were performed on a ZQ4000 spectrometer in positive and negative ionisation modes. Accurate mass data were obtained on a MAT900 instrument. IR spectra (see Supporting Information File 1) were recorded on a Perkin Elmer Spectrum One FT-IR spectrometer or a Perkin Elmer 1600 series FT-IR spectrometer. Microanalyses for representative compounds were performed by Warwick analytical service at the University of Warwick. The X-ray single-crystal diffraction data were collected on a Nonius Kappa CCD diffractometer using graphite-monochromated Mo-Kα, (λ = 0.71073 Å) radiation. Crystal and structure refinement data are shown in the Supporting Information File 1. The structures were solved by direct methods using SHELXS-96 [73] and refined with all data on F2 full-matrix least squares using SHELXL-97 [74]. Non-hydrogen atoms were generally refined anisotropically. Hydrogen atom positions were located from difference Fourier maps and a riding model with atomic displacement parameters 1.2 times (1.5 times for methyl groups) those of the atom to which they are bonded were used for subsequent refinements. Full crystallographic data have been deposited with the CCDC, reference numbers 737411 (compound 12), 762624 (compound 15), 766180 (compound 16), 737415 (compound 17), 762623 (compound 18), 766182 (compound 38) and 766181 (compound 39), and can be obtained free of charge via http://www.ccdc.cam.ac.uk/data_request/cif. Alkyl lithiums were obtained from Aldrich Chemical Company and were estimated prior to use by the method of Watson and Eastham [75]. Other reagents and starting materials were obtained from Aldrich Chemical Company and used without further purification. THF was distilled from sodium benzophenone ketyl.
General procedure for the synthesis of 3-substituted isoindolin-1-ones 9–18 and 20–40. A solution of t-BuLi in heptane (3.9 mL, 1.7 M, 6.6 mmol) was added to a cold (0 °C), stirred solution of N'-(substituted benzyl)-N,N-dimethylurea (1 or 19; 2.0 mmol) in anhydrous THF (20 mL) under a N2 atmosphere. Formation of the monolithium reagent was observed as a yellow solution and the dilithium reagent was observed as a reddish orange solution, after which the colour changed to deep red. The mixture was stirred at 0 °C for 6 h after which an electrophile (2.2 mmol), in anhydrous THF (8 mL) if solid, otherwise neat, was added. The mixture was stirred for 2 h at 0 °C then the cooling bath was removed and the mixture allowed to warm to room temperature. It was then diluted with Et2O (10 mL) and quenched with aq sat. NH4Cl (10 mL). The organic layer was separated, washed with H2O (2 × 10 mL), dried (MgSO4), and evaporated under reduced pressure. The residue obtained was triturated with diethyl ether (20 mL) to give a white solid which was filtered and washed with diethyl ether (20 mL) to give the pure product. The solid obtained was recrystallized from ethyl acetate. Some of the products were previously reported and in these cases the melting point and the spectral data were in agreement with reported values. Of those compounds that had not been reported previously, representative examples were subjected to microanalysis, and in all cases gave analyses consistent with the assigned structures. For other compounds, therefore, proof of purity was established by a combination of tlc (single spot), sharp melting point and clean NMR spectra. Elemental composition of a molecular ion or pseudo molecular ion confirmed the formula.
Supporting Information
| Supporting Information File 1: Characterization data of all compounds given in the article and NMR spectra and X-ray information for representative compounds. CCDC 737411, 737415, 762623, 762624, 766180, 766181 and 766182. | ||
| Format: PDF | Size: 7.2 MB | Download |
References
-
Couture, A.; Deniau, E.; Grandclaudon, P.; Hoarau, C.; Rys, V. Tetrahedron Lett. 2002, 43, 2207–2210. doi:10.1016/S0040-4039(02)00236-8
Return to citation in text: [1] -
Lawrence, N. J.; Liddle, J.; Bushell, S. M.; Jackson, D. A. Z. J. Org. Chem. 2002, 67, 457–464. doi:10.1021/jo0159901
Return to citation in text: [1] -
Fuchs, J. R.; Funk, R. L. Org. Lett. 2001, 3, 3923–3925. doi:10.1021/ol016795b
Return to citation in text: [1] -
Abramovitch, R. A.; Shinkai, I.; Mavunkel, B. J.; More, K. M.; O'Connor, S.; Ooi, G. H.; Pennington, W. T.; Srinivasan, P. C.; Stowers, J. R. Tetrahedron 1996, 52, 3339–3354. doi:10.1016/0040-4020(95)01115-3
Return to citation in text: [1] -
Wood, J. L.; Stoltz, B. M.; Goodman, S. N. J. Am. Chem. Soc. 1996, 118, 10656–10657. doi:10.1021/ja9626143
Return to citation in text: [1] -
Fang, F. G.; Danishefsky, S. J. Tetrahedron Lett. 1989, 30, 2747–2750. doi:10.1016/S0040-4039(00)99115-9
Return to citation in text: [1] -
Valencia, E.; Freyer, A. J.; Shamma, M.; Fajardo, V. Tetrahedron Lett. 1984, 25, 599–602. doi:10.1016/S0040-4039(00)99948-9
Return to citation in text: [1] -
Fajardo, V.; Elango, V.; Cassels, B. K.; Shamma, M. Tetrahedron Lett. 1982, 23, 39–42. doi:10.1016/S0040-4039(00)97526-9
Return to citation in text: [1] -
Moon, E.-Y.; Oh, J.-M.; Kim, Y.-H.; Ryoo, I.-J.; Yoo, I.-D. Biol. Pharm. Bull. 2009, 32, 1689–1694. doi:10.1248/bpb.32.1689
Return to citation in text: [1] -
Lawson, E. C.; Luci, D. K.; Ghosh, S.; Kinney, W. A.; Reynolds, C. H.; Qi, J.; Smith, C. E.; Wang, Y.; Minor, L. K.; Haertlein, B. J.; Parry, T. J.; Damiano, B. P.; Maryanoff, B. E. J. Med. Chem. 2009, 52, 7432–7445. doi:10.1021/jm900683d
Return to citation in text: [1] -
Maugeri, C.; Alisi, M. A.; Apicella, C.; Cellai, L.; Dragone, P.; Fioravanzo, E.; Florio, S.; Furlotti, G.; Mangano, G.; Ombrato, R.; Luisi, R.; Pompei, R.; Rincicotti, V.; Russo, V.; Vitiello, M.; Cazzolla, N. Bioorg. Med. Chem. 2008, 16, 3091–3107. doi:10.1016/j.bmc.2007.12.030
Return to citation in text: [1] -
Cid, H. M. B.; Tränkle, C.; Baumann, K.; Pick, R.; Mies-Klomfass, E.; Kostenis, E.; Mohr, K.; Holzgrabe, U. J. Med. Chem. 2000, 43, 2155–2164. doi:10.1021/jm991136e
Return to citation in text: [1] -
Zhuang, Z.-P.; Kung, M.-P.; Mu, M.; Kung, H. F. J. Med. Chem. 1998, 41, 157–166. doi:10.1021/jm970296s
Return to citation in text: [1] -
Taylor, E. C.; Zhou, P.; Jennings, L. D.; Mao, Z.; Hu, B.; Jun, J.-G. Tetrahedron Lett. 1997, 38, 521–524. doi:10.1016/S0040-4039(96)02397-0
Return to citation in text: [1] -
De Clercq, E. J. Med. Chem. 1995, 38, 2491–2517. doi:10.1021/jm00014a001
Return to citation in text: [1] -
Marinicheva, G. E.; Gubina, T. I. Chem. Heterocycl. Compd. 2004, 40, 1517–1535. doi:10.1007/s10593-005-0094-7
Return to citation in text: [1] -
Bonnett, R.; North, S. A. Adv. Heterocycl. Chem. 1981, 29, 341–399. doi:10.1016/S0065-2725(08)60790-4
Return to citation in text: [1] -
White, J. D.; Mann, M. E. Adv. Heterocycl. Chem. 1969, 10, 113–147. doi:10.1016/S0065-2725(08)60496-1
Return to citation in text: [1] -
Medimagh, R.; Marque, S.; Prim, D.; Marrot, J.; Chatti, S. Org. Lett. 2009, 11, 1817–1820. doi:10.1021/ol9003965
Return to citation in text: [1] -
Li, L.; Wang, M.; Zhang, X.; Jiang, Y.; Ma, D. Org. Lett. 2009, 11, 1309–1312. doi:10.1021/ol9000922
Return to citation in text: [1] -
Marcos, C. F.; Marcaccini, S.; Menchi, G.; Pepino, R.; Torroba, T. Tetrahedron Lett. 2008, 49, 149–152. doi:10.1016/j.tetlet.2007.10.154
Return to citation in text: [1] -
López-Valdez, G.; Olguín-Uribe, S.; Miranda, L. D. Tetrahedron Lett. 2007, 48, 8285–8289. doi:10.1016/j.tetlet.2007.09.142
Return to citation in text: [1] -
Lamblin, M.; Couture, A.; Deniau, E.; Grandclaudon, P. Tetrahedron 2007, 63, 2664–2669. doi:10.1016/j.tet.2007.01.021
Return to citation in text: [1] -
Pérard-Viret, J.; Prangé, T.; Tomas, A.; Royer, J. Tetrahedron 2002, 58, 5103–5108. doi:10.1016/S0040-4020(02)00462-3
Return to citation in text: [1] -
Yao, T.; Larock, R. C. J. Org. Chem. 2005, 70, 1432–1437. doi:10.1021/jo048007c
Return to citation in text: [1] -
Ang, W. S.; Halton, B. Aust. J. Chem. 1971, 24, 851–856. doi:10.1071/CH9710851
Return to citation in text: [1] -
Gutierrez, A. J.; Shea, K. J.; Svoboda, J. J. J. Org. Chem. 1989, 54, 4335–4344. doi:10.1021/jo00279a021
Return to citation in text: [1] -
Epsztajn, J.; Grzelak, R.; Jóźwiak, A. Synthesis 1996, 1212–1216. doi:10.1055/s-1996-4366
Return to citation in text: [1] -
Luzzio, F. A.; Zacherl, D. P. Tetrahedron Lett. 1998, 39, 2285–2288. doi:10.1016/S0040-4039(98)00293-7
Return to citation in text: [1] -
Milewska, M. J.; Bytner, T.; Połoński, T. Synthesis 1996, 1485–1488. doi:10.1055/s-1996-4405
Return to citation in text: [1] -
Guillaumel, J.; Boccara, N.; Demersemann, P.; Royer, R. J. Chem. Soc., Chem. Commun. 1988, 1604–1605. doi:10.1039/C39880001604
Return to citation in text: [1] -
Griesbeck, A. G.; Hirt, J.; Kramer, W.; Dallakian, P. Tetrahedron 1998, 54, 3169–3180. doi:10.1016/S0040-4020(98)00063-5
Return to citation in text: [1] -
Weidner-Wells, M. A.; Oda, K.; Mazzocchi, P. H. Tetrahedron 1997, 53, 3475–3486. doi:10.1016/S0040-4020(97)00108-7
Return to citation in text: [1] -
Cho, C. S.; Ren, W. X. Tetrahedron Lett. 2009, 50, 2097–2099. doi:10.1016/j.tetlet.2009.02.109
Return to citation in text: [1] -
Sun, C.; Xu, B. J. Org. Chem. 2008, 73, 7361–7364. doi:10.1021/jo801219j
Return to citation in text: [1] -
Cao, H.; McNamee, L.; Alper, H. Org. Lett. 2008, 10, 5281–5284. doi:10.1021/ol8021403
Return to citation in text: [1] -
Orito, K.; Miyazawa, M.; Nakamura, T.; Horibata, A.; Ushito, H.; Nagasaki, H.; Yuguchi, M.; Yamashita, S.; Yamazaki, T.; Tokuda, M. J. Org. Chem. 2006, 71, 5951–5958. doi:10.1021/jo060612n
Return to citation in text: [1] -
Couty, S.; Liégault, B.; Meyer, C.; Cossy, J. Org. Lett. 2004, 6, 2511–2514. doi:10.1021/ol049302m
Return to citation in text: [1] -
Cho, C. S.; Shim, H. S.; Choi, H.-J.; Kim, T.-J.; Shim, S. C.; Kim, M. C. Tetrahedron Lett. 2000, 41, 3891–3893. doi:10.1016/S0040-4039(00)00495-0
Return to citation in text: [1] -
Cho, C. S.; Jiang, L. H.; Shim, S. C. Synth. Commun. 1998, 28, 849–857. doi:10.1080/00032719808006483
Return to citation in text: [1] -
Cho, C. S.; Lee, J. W.; Lee, D. Y.; Shim, S. C.; Kim, T. J. Chem. Commun. 1996, 2115–2116. doi:10.1039/cc9960002115
Return to citation in text: [1] -
Grigg, R.; Loganathan, V.; Santhakumar, V.; Sridharan, V.; Teasdale, A. Tetrahedron Lett. 1991, 32, 687–690. doi:10.1016/S0040-4039(00)74860-X
Return to citation in text: [1] -
Couture, A.; Deniau, E.; Ionescu, D.; Grandclaudon, P. Tetrahedron Lett. 1998, 39, 2319–2320. doi:10.1016/S0040-4039(98)00136-1
Return to citation in text: [1] [2] [3] [4] -
Couture, A.; Deniau, E.; Grandclaudon, P. Tetrahedron 1997, 53, 10313–10330. doi:10.1016/S0040-4020(97)00680-7
Return to citation in text: [1] -
Clayden, J.; Menet, C. J. Tetrahedron Lett. 2003, 44, 3059–3062. doi:10.1016/S0040-4039(03)00545-8
Return to citation in text: [1] [2] [3] [4] [5] -
Clayden, J.; Menet, C. J.; Mansfield, D. J. Org. Lett. 2000, 2, 4229–4232. doi:10.1021/ol006786n
Return to citation in text: [1] -
Clayden, J.; Tchabanenko, K. Chem. Commun. 2000, 317–318. doi:10.1039/a909325g
Return to citation in text: [1] -
Ahmed, A.; Clayden, J.; Yasin, S. A. Chem. Commun. 1999, 231–232. doi:10.1039/a808218i
Return to citation in text: [1] -
Clayden, J.; Knowles, F. E.; Baldwin, I. R. J. Am. Chem. Soc. 2005, 127, 2412–2413. doi:10.1021/ja042415g
Return to citation in text: [1] -
Epsztajn, J.; Jóźwiak, A.; Kołuda, P.; Sadokierska, I.; Wilkowska, I. D. Tetrahedron 2000, 56, 4837–4844. doi:10.1016/S0040-4020(00)00403-8
Return to citation in text: [1] -
Epsztajn, J.; Jóźwiak, A.; Szcześniak, A. K. Synth. Commun. 1994, 24, 1789–1798. doi:10.1080/00397919408010186
Return to citation in text: [1] -
Epsztajn, J.; Jóźwiak, A.; Szcześniak, A. K. Tetrahedron 1993, 49, 929–938. doi:10.1016/S0040-4020(01)80334-3
Return to citation in text: [1] -
Epsztajn, J.; Jóźwiak, A.; Czech, K.; Szcześniak, A. K. Monatsh. Chem. 1990, 121, 909–921. doi:10.1007/BF00808955
Return to citation in text: [1] -
Lamblin, M.; Couture, A.; Deniau, E.; Grandclaudon, P. Tetrahedron: Asymmetry 2008, 19, 111–123. doi:10.1016/j.tetasy.2007.11.014
Return to citation in text: [1] -
Smith, K.; El-Hiti, G. A.; Abdo, M. A.; Abdel-Megeed, M. F. J. Chem. Soc., Perkin Trans. 1 1995, 1029–1033. doi:10.1039/P19950001029
Return to citation in text: [1] -
Smith, K.; El-Hiti, G. A.; Abdel-Megeed, M. F.; Abdo, M. A. J. Org. Chem. 1996, 61, 647–655. doi:10.1021/jo950988t
Return to citation in text: [1] -
Smith, K.; El-Hiti, G. A.; Abdel-Megeed, M. F.; Abdo, M. A. J. Org. Chem. 1996, 61, 656–661. doi:10.1021/jo950989l
Return to citation in text: [1] -
Smith, K.; El-Hiti, G. A.; Pritchard, G. J.; Hamilton, A. J. Chem. Soc., Perkin Trans. 1 1999, 2299–2303. doi:10.1039/A903467F
Return to citation in text: [1] -
Smith, K.; El-Hiti, G. A.; Shukla, A. P. J. Chem. Soc., Perkin Trans. 1 1999, 2305–2313. doi:10.1039/A903464A
Return to citation in text: [1] [2] -
Smith, K.; El-Hiti, G. A.; Hawes, A. C. Synthesis 2003, 2047–2052. doi:10.1055/s-2003-41019
Return to citation in text: [1] -
Smith, K.; El-Hiti, G. A.; Mahgoub, S. A. Synthesis 2003, 2345–2348. doi:10.1055/s-2003-41064
Return to citation in text: [1] -
El-Hiti, G. A. Synthesis 2003, 2799–2804. doi:10.1055/s-2003-42486
Return to citation in text: [1] -
Smith, K.; El-Hiti, G. A.; Abdel-Megeed, M. F. Synthesis 2004, 2121–2130. doi:10.1055/s-2004-829169
Return to citation in text: [1] -
El-Hiti, G. A. Synthesis 2004, 363–368. doi:10.1055/s-2004-815923
Return to citation in text: [1] -
Smith, K.; El-Hiti, G. A.; Hegazy, A. S. J. Sulfur Chem. 2005, 26, 121–131. doi:10.1080/17415990500151472
Return to citation in text: [1] -
Smith, K.; El-Hiti, G. A.; Hegazy, A. S. Synthesis 2005, 2951–2961. doi:10.1055/s-2005-916036
Return to citation in text: [1] -
Smith, K.; Barratt, M. L. J. Org. Chem. 2007, 72, 1031–1034. doi:10.1021/jo062024f
Return to citation in text: [1] -
Houlden, C. E.; Lloyd-Jones, G. C.; Booker-Milburn, K. I. Org. Lett. 2010, 12, 3090–3092. doi:10.1021/ol101102y
Return to citation in text: [1] -
Smith, K.; El-Hiti, G. A.; Hegazy, A. S.; Fekri, A.; Kariuki, B. M. ARKIVOC 2009, (xiv), 266–300.
http://www.arkat-usa.org/get-file/32973/
Return to citation in text: [1] [2] [3] [4] [5] -
Smith, K.; El-Hiti, G. A.; Hegazy, A. S. Synlett 2009, 2242–2244. doi:10.1055/s-0029-1217722
Return to citation in text: [1] -
Smith, K.; El-Hiti, G. A.; Hegazy, A. S. Synthesis 2010, 1371–1380. doi:10.1055/s-0029-1219277
Return to citation in text: [1] -
Smith, K.; El-Hiti, G. A.; Hegazy, A. S. Chem. Commun. 2010, 46, 2790–2792. doi:10.1039/b926983e
Return to citation in text: [1] -
Sheldrick, G. M. Acta Crystallogr., Sect. A 1990, 46, 467–473. doi:10.1107/S0108767390000277
Return to citation in text: [1] -
SHELXS-97, Program for Crystal Structure Refinement; Universität Göttingen: Germany, 1997.
Return to citation in text: [1] -
Watson, S. C.; Eastham, J. F. J. Organomet. Chem. 1967, 9, 165–168. doi:10.1016/S0022-328X(00)92418-5
Return to citation in text: [1]
| 74. | SHELXS-97, Program for Crystal Structure Refinement; Universität Göttingen: Germany, 1997. |
| 75. | Watson, S. C.; Eastham, J. F. J. Organomet. Chem. 1967, 9, 165–168. doi:10.1016/S0022-328X(00)92418-5 |
| 1. | Couture, A.; Deniau, E.; Grandclaudon, P.; Hoarau, C.; Rys, V. Tetrahedron Lett. 2002, 43, 2207–2210. doi:10.1016/S0040-4039(02)00236-8 |
| 2. | Lawrence, N. J.; Liddle, J.; Bushell, S. M.; Jackson, D. A. Z. J. Org. Chem. 2002, 67, 457–464. doi:10.1021/jo0159901 |
| 3. | Fuchs, J. R.; Funk, R. L. Org. Lett. 2001, 3, 3923–3925. doi:10.1021/ol016795b |
| 4. | Abramovitch, R. A.; Shinkai, I.; Mavunkel, B. J.; More, K. M.; O'Connor, S.; Ooi, G. H.; Pennington, W. T.; Srinivasan, P. C.; Stowers, J. R. Tetrahedron 1996, 52, 3339–3354. doi:10.1016/0040-4020(95)01115-3 |
| 5. | Wood, J. L.; Stoltz, B. M.; Goodman, S. N. J. Am. Chem. Soc. 1996, 118, 10656–10657. doi:10.1021/ja9626143 |
| 6. | Fang, F. G.; Danishefsky, S. J. Tetrahedron Lett. 1989, 30, 2747–2750. doi:10.1016/S0040-4039(00)99115-9 |
| 7. | Valencia, E.; Freyer, A. J.; Shamma, M.; Fajardo, V. Tetrahedron Lett. 1984, 25, 599–602. doi:10.1016/S0040-4039(00)99948-9 |
| 8. | Fajardo, V.; Elango, V.; Cassels, B. K.; Shamma, M. Tetrahedron Lett. 1982, 23, 39–42. doi:10.1016/S0040-4039(00)97526-9 |
| 27. | Gutierrez, A. J.; Shea, K. J.; Svoboda, J. J. J. Org. Chem. 1989, 54, 4335–4344. doi:10.1021/jo00279a021 |
| 45. | Clayden, J.; Menet, C. J. Tetrahedron Lett. 2003, 44, 3059–3062. doi:10.1016/S0040-4039(03)00545-8 |
| 26. | Ang, W. S.; Halton, B. Aust. J. Chem. 1971, 24, 851–856. doi:10.1071/CH9710851 |
| 45. | Clayden, J.; Menet, C. J. Tetrahedron Lett. 2003, 44, 3059–3062. doi:10.1016/S0040-4039(03)00545-8 |
| 16. | Marinicheva, G. E.; Gubina, T. I. Chem. Heterocycl. Compd. 2004, 40, 1517–1535. doi:10.1007/s10593-005-0094-7 |
| 17. | Bonnett, R.; North, S. A. Adv. Heterocycl. Chem. 1981, 29, 341–399. doi:10.1016/S0065-2725(08)60790-4 |
| 18. | White, J. D.; Mann, M. E. Adv. Heterocycl. Chem. 1969, 10, 113–147. doi:10.1016/S0065-2725(08)60496-1 |
| 19. | Medimagh, R.; Marque, S.; Prim, D.; Marrot, J.; Chatti, S. Org. Lett. 2009, 11, 1817–1820. doi:10.1021/ol9003965 |
| 20. | Li, L.; Wang, M.; Zhang, X.; Jiang, Y.; Ma, D. Org. Lett. 2009, 11, 1309–1312. doi:10.1021/ol9000922 |
| 21. | Marcos, C. F.; Marcaccini, S.; Menchi, G.; Pepino, R.; Torroba, T. Tetrahedron Lett. 2008, 49, 149–152. doi:10.1016/j.tetlet.2007.10.154 |
| 22. | López-Valdez, G.; Olguín-Uribe, S.; Miranda, L. D. Tetrahedron Lett. 2007, 48, 8285–8289. doi:10.1016/j.tetlet.2007.09.142 |
| 23. | Lamblin, M.; Couture, A.; Deniau, E.; Grandclaudon, P. Tetrahedron 2007, 63, 2664–2669. doi:10.1016/j.tet.2007.01.021 |
| 24. | Pérard-Viret, J.; Prangé, T.; Tomas, A.; Royer, J. Tetrahedron 2002, 58, 5103–5108. doi:10.1016/S0040-4020(02)00462-3 |
| 25. | Yao, T.; Larock, R. C. J. Org. Chem. 2005, 70, 1432–1437. doi:10.1021/jo048007c |
| 43. | Couture, A.; Deniau, E.; Ionescu, D.; Grandclaudon, P. Tetrahedron Lett. 1998, 39, 2319–2320. doi:10.1016/S0040-4039(98)00136-1 |
| 9. | Moon, E.-Y.; Oh, J.-M.; Kim, Y.-H.; Ryoo, I.-J.; Yoo, I.-D. Biol. Pharm. Bull. 2009, 32, 1689–1694. doi:10.1248/bpb.32.1689 |
| 10. | Lawson, E. C.; Luci, D. K.; Ghosh, S.; Kinney, W. A.; Reynolds, C. H.; Qi, J.; Smith, C. E.; Wang, Y.; Minor, L. K.; Haertlein, B. J.; Parry, T. J.; Damiano, B. P.; Maryanoff, B. E. J. Med. Chem. 2009, 52, 7432–7445. doi:10.1021/jm900683d |
| 11. | Maugeri, C.; Alisi, M. A.; Apicella, C.; Cellai, L.; Dragone, P.; Fioravanzo, E.; Florio, S.; Furlotti, G.; Mangano, G.; Ombrato, R.; Luisi, R.; Pompei, R.; Rincicotti, V.; Russo, V.; Vitiello, M.; Cazzolla, N. Bioorg. Med. Chem. 2008, 16, 3091–3107. doi:10.1016/j.bmc.2007.12.030 |
| 12. | Cid, H. M. B.; Tränkle, C.; Baumann, K.; Pick, R.; Mies-Klomfass, E.; Kostenis, E.; Mohr, K.; Holzgrabe, U. J. Med. Chem. 2000, 43, 2155–2164. doi:10.1021/jm991136e |
| 13. | Zhuang, Z.-P.; Kung, M.-P.; Mu, M.; Kung, H. F. J. Med. Chem. 1998, 41, 157–166. doi:10.1021/jm970296s |
| 14. | Taylor, E. C.; Zhou, P.; Jennings, L. D.; Mao, Z.; Hu, B.; Jun, J.-G. Tetrahedron Lett. 1997, 38, 521–524. doi:10.1016/S0040-4039(96)02397-0 |
| 15. | De Clercq, E. J. Med. Chem. 1995, 38, 2491–2517. doi:10.1021/jm00014a001 |
| 43. | Couture, A.; Deniau, E.; Ionescu, D.; Grandclaudon, P. Tetrahedron Lett. 1998, 39, 2319–2320. doi:10.1016/S0040-4039(98)00136-1 |
| 32. | Griesbeck, A. G.; Hirt, J.; Kramer, W.; Dallakian, P. Tetrahedron 1998, 54, 3169–3180. doi:10.1016/S0040-4020(98)00063-5 |
| 33. | Weidner-Wells, M. A.; Oda, K.; Mazzocchi, P. H. Tetrahedron 1997, 53, 3475–3486. doi:10.1016/S0040-4020(97)00108-7 |
| 43. | Couture, A.; Deniau, E.; Ionescu, D.; Grandclaudon, P. Tetrahedron Lett. 1998, 39, 2319–2320. doi:10.1016/S0040-4039(98)00136-1 |
| 44. | Couture, A.; Deniau, E.; Grandclaudon, P. Tetrahedron 1997, 53, 10313–10330. doi:10.1016/S0040-4020(97)00680-7 |
| 45. | Clayden, J.; Menet, C. J. Tetrahedron Lett. 2003, 44, 3059–3062. doi:10.1016/S0040-4039(03)00545-8 |
| 46. | Clayden, J.; Menet, C. J.; Mansfield, D. J. Org. Lett. 2000, 2, 4229–4232. doi:10.1021/ol006786n |
| 47. | Clayden, J.; Tchabanenko, K. Chem. Commun. 2000, 317–318. doi:10.1039/a909325g |
| 48. | Ahmed, A.; Clayden, J.; Yasin, S. A. Chem. Commun. 1999, 231–232. doi:10.1039/a808218i |
| 49. | Clayden, J.; Knowles, F. E.; Baldwin, I. R. J. Am. Chem. Soc. 2005, 127, 2412–2413. doi:10.1021/ja042415g |
| 50. | Epsztajn, J.; Jóźwiak, A.; Kołuda, P.; Sadokierska, I.; Wilkowska, I. D. Tetrahedron 2000, 56, 4837–4844. doi:10.1016/S0040-4020(00)00403-8 |
| 51. | Epsztajn, J.; Jóźwiak, A.; Szcześniak, A. K. Synth. Commun. 1994, 24, 1789–1798. doi:10.1080/00397919408010186 |
| 52. | Epsztajn, J.; Jóźwiak, A.; Szcześniak, A. K. Tetrahedron 1993, 49, 929–938. doi:10.1016/S0040-4020(01)80334-3 |
| 53. | Epsztajn, J.; Jóźwiak, A.; Czech, K.; Szcześniak, A. K. Monatsh. Chem. 1990, 121, 909–921. doi:10.1007/BF00808955 |
| 54. | Lamblin, M.; Couture, A.; Deniau, E.; Grandclaudon, P. Tetrahedron: Asymmetry 2008, 19, 111–123. doi:10.1016/j.tetasy.2007.11.014 |
| 31. | Guillaumel, J.; Boccara, N.; Demersemann, P.; Royer, R. J. Chem. Soc., Chem. Commun. 1988, 1604–1605. doi:10.1039/C39880001604 |
| 43. | Couture, A.; Deniau, E.; Ionescu, D.; Grandclaudon, P. Tetrahedron Lett. 1998, 39, 2319–2320. doi:10.1016/S0040-4039(98)00136-1 |
| 45. | Clayden, J.; Menet, C. J. Tetrahedron Lett. 2003, 44, 3059–3062. doi:10.1016/S0040-4039(03)00545-8 |
| 29. | Luzzio, F. A.; Zacherl, D. P. Tetrahedron Lett. 1998, 39, 2285–2288. doi:10.1016/S0040-4039(98)00293-7 |
| 30. | Milewska, M. J.; Bytner, T.; Połoński, T. Synthesis 1996, 1485–1488. doi:10.1055/s-1996-4405 |
| 28. | Epsztajn, J.; Grzelak, R.; Jóźwiak, A. Synthesis 1996, 1212–1216. doi:10.1055/s-1996-4366 |
| 34. | Cho, C. S.; Ren, W. X. Tetrahedron Lett. 2009, 50, 2097–2099. doi:10.1016/j.tetlet.2009.02.109 |
| 35. | Sun, C.; Xu, B. J. Org. Chem. 2008, 73, 7361–7364. doi:10.1021/jo801219j |
| 36. | Cao, H.; McNamee, L.; Alper, H. Org. Lett. 2008, 10, 5281–5284. doi:10.1021/ol8021403 |
| 37. | Orito, K.; Miyazawa, M.; Nakamura, T.; Horibata, A.; Ushito, H.; Nagasaki, H.; Yuguchi, M.; Yamashita, S.; Yamazaki, T.; Tokuda, M. J. Org. Chem. 2006, 71, 5951–5958. doi:10.1021/jo060612n |
| 38. | Couty, S.; Liégault, B.; Meyer, C.; Cossy, J. Org. Lett. 2004, 6, 2511–2514. doi:10.1021/ol049302m |
| 39. | Cho, C. S.; Shim, H. S.; Choi, H.-J.; Kim, T.-J.; Shim, S. C.; Kim, M. C. Tetrahedron Lett. 2000, 41, 3891–3893. doi:10.1016/S0040-4039(00)00495-0 |
| 40. | Cho, C. S.; Jiang, L. H.; Shim, S. C. Synth. Commun. 1998, 28, 849–857. doi:10.1080/00032719808006483 |
| 41. | Cho, C. S.; Lee, J. W.; Lee, D. Y.; Shim, S. C.; Kim, T. J. Chem. Commun. 1996, 2115–2116. doi:10.1039/cc9960002115 |
| 42. | Grigg, R.; Loganathan, V.; Santhakumar, V.; Sridharan, V.; Teasdale, A. Tetrahedron Lett. 1991, 32, 687–690. doi:10.1016/S0040-4039(00)74860-X |
| 59. | Smith, K.; El-Hiti, G. A.; Shukla, A. P. J. Chem. Soc., Perkin Trans. 1 1999, 2305–2313. doi:10.1039/A903464A |
| 45. | Clayden, J.; Menet, C. J. Tetrahedron Lett. 2003, 44, 3059–3062. doi:10.1016/S0040-4039(03)00545-8 |
| 55. | Smith, K.; El-Hiti, G. A.; Abdo, M. A.; Abdel-Megeed, M. F. J. Chem. Soc., Perkin Trans. 1 1995, 1029–1033. doi:10.1039/P19950001029 |
| 56. | Smith, K.; El-Hiti, G. A.; Abdel-Megeed, M. F.; Abdo, M. A. J. Org. Chem. 1996, 61, 647–655. doi:10.1021/jo950988t |
| 57. | Smith, K.; El-Hiti, G. A.; Abdel-Megeed, M. F.; Abdo, M. A. J. Org. Chem. 1996, 61, 656–661. doi:10.1021/jo950989l |
| 58. | Smith, K.; El-Hiti, G. A.; Pritchard, G. J.; Hamilton, A. J. Chem. Soc., Perkin Trans. 1 1999, 2299–2303. doi:10.1039/A903467F |
| 59. | Smith, K.; El-Hiti, G. A.; Shukla, A. P. J. Chem. Soc., Perkin Trans. 1 1999, 2305–2313. doi:10.1039/A903464A |
| 60. | Smith, K.; El-Hiti, G. A.; Hawes, A. C. Synthesis 2003, 2047–2052. doi:10.1055/s-2003-41019 |
| 61. | Smith, K.; El-Hiti, G. A.; Mahgoub, S. A. Synthesis 2003, 2345–2348. doi:10.1055/s-2003-41064 |
| 62. | El-Hiti, G. A. Synthesis 2003, 2799–2804. doi:10.1055/s-2003-42486 |
| 63. | Smith, K.; El-Hiti, G. A.; Abdel-Megeed, M. F. Synthesis 2004, 2121–2130. doi:10.1055/s-2004-829169 |
| 64. | El-Hiti, G. A. Synthesis 2004, 363–368. doi:10.1055/s-2004-815923 |
| 65. | Smith, K.; El-Hiti, G. A.; Hegazy, A. S. J. Sulfur Chem. 2005, 26, 121–131. doi:10.1080/17415990500151472 |
| 66. | Smith, K.; El-Hiti, G. A.; Hegazy, A. S. Synthesis 2005, 2951–2961. doi:10.1055/s-2005-916036 |
| 67. | Smith, K.; Barratt, M. L. J. Org. Chem. 2007, 72, 1031–1034. doi:10.1021/jo062024f |
| 69. |
Smith, K.; El-Hiti, G. A.; Hegazy, A. S.; Fekri, A.; Kariuki, B. M. ARKIVOC 2009, (xiv), 266–300.
http://www.arkat-usa.org/get-file/32973/ |
| 73. | Sheldrick, G. M. Acta Crystallogr., Sect. A 1990, 46, 467–473. doi:10.1107/S0108767390000277 |
| 69. |
Smith, K.; El-Hiti, G. A.; Hegazy, A. S.; Fekri, A.; Kariuki, B. M. ARKIVOC 2009, (xiv), 266–300.
http://www.arkat-usa.org/get-file/32973/ |
| 72. | Smith, K.; El-Hiti, G. A.; Hegazy, A. S. Chem. Commun. 2010, 46, 2790–2792. doi:10.1039/b926983e |
| 69. |
Smith, K.; El-Hiti, G. A.; Hegazy, A. S.; Fekri, A.; Kariuki, B. M. ARKIVOC 2009, (xiv), 266–300.
http://www.arkat-usa.org/get-file/32973/ |
| 69. |
Smith, K.; El-Hiti, G. A.; Hegazy, A. S.; Fekri, A.; Kariuki, B. M. ARKIVOC 2009, (xiv), 266–300.
http://www.arkat-usa.org/get-file/32973/ |
| 68. | Houlden, C. E.; Lloyd-Jones, G. C.; Booker-Milburn, K. I. Org. Lett. 2010, 12, 3090–3092. doi:10.1021/ol101102y |
| 69. |
Smith, K.; El-Hiti, G. A.; Hegazy, A. S.; Fekri, A.; Kariuki, B. M. ARKIVOC 2009, (xiv), 266–300.
http://www.arkat-usa.org/get-file/32973/ |
| 70. | Smith, K.; El-Hiti, G. A.; Hegazy, A. S. Synlett 2009, 2242–2244. doi:10.1055/s-0029-1217722 |
| 71. | Smith, K.; El-Hiti, G. A.; Hegazy, A. S. Synthesis 2010, 1371–1380. doi:10.1055/s-0029-1219277 |
© 2011 Smith et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)