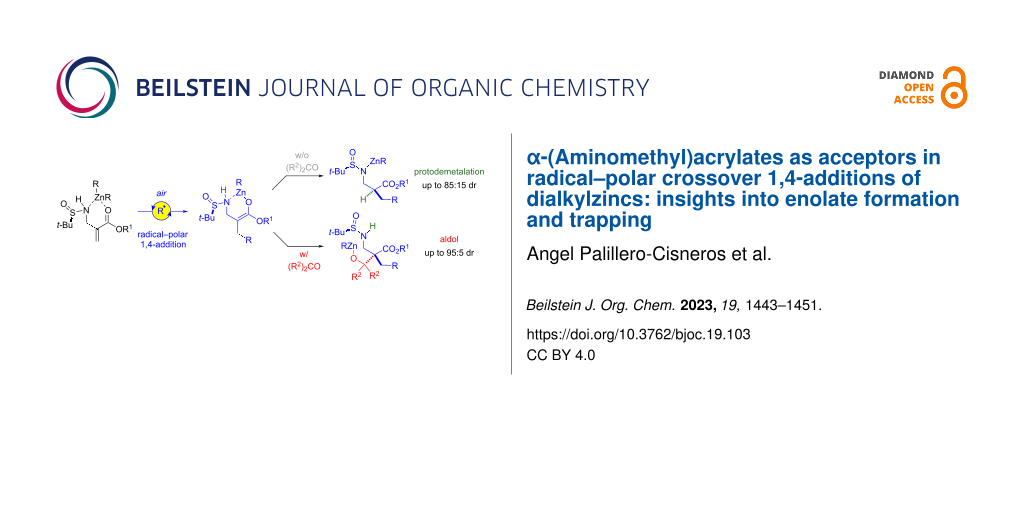
Abstract
We demonstrate that α-(aminomethyl)acrylates are suitable acceptors for 1,4-additions of dialkylzincs in aerobic conditions. The air-promoted radical–polar crossover process involves the 1,4-addition of an alkyl radical followed by homolytic substitution at the zinc atom of dialkylzinc. Coordination of the nitrogen atom to zinc enables this SH2 process which represents a rare example of alkylzinc-group transfer to a tertiary α-carbonyl radical. The zinc enolate thus formed readily undergoes β-fragmentation unless it is trapped by electrophiles in situ. Enolates of substrates having free N–H bonds undergo protodemetalation to provide ultimately the 1,4-addition adduct. In the presence of carbonyl acceptors, aldol condensation occurs providing overall a tandem 1,4-addition–aldol process. When a tert-butanesulfinyl moiety is present on the nitrogen atom, these electrophilic substitution reactions occur with good levels of chiral induction, paving the way to enantioenriched β2-amino acids and β2,2-amino acids.

Graphical Abstract
Introduction
Dialkylzinc reagents react in aerobic medium with a range of α,β-unsaturated carbonyl compounds to provide the corresponding zinc enolates (Scheme 1) [1,2]. While simple, this reaction offers attractive features: 1) it proceeds under mild conditions in the absence of any transition-metal catalyst; 2) the 1,4-addition step can be combined with condensation reactions of the zinc enolate with electrophiles in protocols wherein all the reactive partners can be introduced from the start, given that dialkylzinc reagents offer a large functional group tolerance; and 3) the radical character of the process allows for the use of alkyl iodides as alkyl source in multicomponent reactions. Trialkylboranes can react in a similar way with enones [3] whereas, distinctively, suitable acceptors for the reaction with dialkylzinc reagents also include α,β-unsaturated carboxylic acid derivatives such as α,β-unsaturated (di)esters [4,5], N-enoyloxazolidinones [6,7], N-enoyloxazolidines [8], or alkylidenemalonates [9-11]. These reactions follow a free-radical chain process wherein alkyl radicals (R•) add across the C–C double bond of the 1,4-acceptor, activated by complexation with the dialkylzinc, to deliver an enoxyl radical that undergoes homolytic substitution at zinc (SH2) to produce a zinc enolate and a new R• that propagates the radical chain (Scheme 1). Initiation occurs upon oxidation of the dialkylzinc reagent by oxygen.
![[1860-5397-19-103-i1]](/bjoc/content/inline/1860-5397-19-103-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Air-promoted radical chain reaction of dialkylzinc reagents with α,β-unsaturated carbonyl compounds.
Scheme 1: Air-promoted radical chain reaction of dialkylzinc reagents with α,β-unsaturated carbonyl compounds....
The feasibility of such 1,4-addition reactions is fully reliant on the ease of the intermediate enoxyl radical to undergo alkylzinc-group transfer. Secondary α-carbonyl radicals (Scheme 1, R1 = H) undergo readily homolytic substitution. By contrast, tertiary α-carbonyl radicals (Scheme 1, R1 ≠ H) are less prone, making additions to α-substituted 1,4-acceptors more challenging. Typically, ethyl methacrylate does not react with dialkylzinc reagents [12]. Notwithstanding, 1,4-additions of dialkylzinc reagents have been reported with dehydroamino ester derivatives [13,14] and α-bromoacrylates [15], which both involve an SH2 at zinc of tertiary α-alkoxycarbonyl radicals (Scheme 2, top). Here, the key to unlock the reactivity is the presence of a Lewis-basic substituent coordinated to the zinc atom: this offers a gain in enthalpy associated to the formation of zinc enolates stabilized by chelation and increases the spin density delocalized at the oxygen atom involved in the chelate. Note that the reported 1,4-additions of dialkylzinc reagents to alkylidenemalonates could benefit from a similar effect, even though in this case, the direct formation of an intermediate enolate remains uncertain [11].
![[1860-5397-19-103-i2]](/bjoc/content/inline/1860-5397-19-103-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Enolate formation by zinc radical transfer (SH2 on dialkylzinc reagents).
Scheme 2: Enolate formation by zinc radical transfer (SH2 on dialkylzinc reagents).
With this context in mind, we surmised that β-aminoenoates I could be suitable 1,4-acceptors (Scheme 2, bottom). We previously reported tandem reactions of such substrates wherein the intermediate enoxyl radical II arising from the addition step evolves via intramolecular addition to tethered alkenes [16,17] or alkynes [18]. We wondered if, in the absence of the pending radical acceptor, the presence of the β-nitrogen atom could nevertheless promote zinc enolate formation. Trapping of this enolate would lead to β-amino acid units, a class of compounds that has attracted a great deal of attention [19-24]. An obvious possible shortcoming that had to be considered was still that the generated zinc enolate III having a β-amino group could undergo β-elimination, thereby precluding its synthetic exploitation.
Results and Discussion
Preparation of α-(aminomethyl)acrylates
We commenced our study by preparing a selection of α-(aminomethyl)acrylates with variations of the nitrogen protecting group and the ester substituent. Towards this end, the direct allylation of primary amines 1–3 with methyl α-(bromomethyl)acrylate was contemplated first under several typical conditions that all afforded non-synthetically useful mixtures of mono- and diallylation, even if excess of the nitrogen nucleophiles was used. An alternative strategy was thus developed relying on the allylation of lithium (trimethylsilyl)amides prepared in situ from the parent amines by a lithiation/silylation/lithiation sequence (Table 1). Using this protocol, α-(aminomethyl)acrylates 5 and 6 derived from benzhydrylamine and aniline were prepared in high yields (Table 1, entries 1 and 2). The procedure was poorly efficient with tosylamine, leading to product 7 in low 20% yield [25].
Table 1: Preparation of α-(aminomethyl)acrylates with free N–H bonds.
![[Graphic 1]](/bjoc/content/inline/1860-5397-19-103-i10.svg?max-width=637&scale=1.0)
|
||||
| Entry | Substrate (R) | R1 | Product | Yielda |
| 1 | 1 (CH(Ph)2) | Me | 5 | 81 |
| 2 | 2 (Ph) | Me | 6 | 80 |
| 3 | 3 (Ts) | Me | 7 | 20 |
| 4 | 4 (S(O)t-Bu) | Me | 8a | 69 |
| 5 | 4 (S(O)t-Bu) | t-Bu | 8b | 50 |
| 6 | 4 (S(O)t-Bu) | Bn | 8c | 36 |
aIsolated yield.
With the aim to develop asymmetric variants, we also considered the synthesis of N-(tert-butanesulfinyl) α-(aminomethyl)acrylates 8a–c. For this purpose, the application of the same protocol with (±)-tert-butanesulfinamide (4) and the requisite α-(bromomethyl)acrylates gave satisfactory yields as well. Finally, N-benzyl-N-(tert-butanesulfinyl) α-(aminomethyl)acrylate 10 was prepared by allylation of lithiated N-benzyl tert-butanesulfinamide 9 (Scheme 3).
![[1860-5397-19-103-i3]](/bjoc/content/inline/1860-5397-19-103-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Preparation of α-(aminomethyl)acrylate 10.
Scheme 3: Preparation of α-(aminomethyl)acrylate 10.
1,4-Addition reactions
Having the requisite α-(aminomethyl)acrylates in hands, we carried out an initial survey of their reaction with Et2Zn in CH2Cl2 at −33 °C on addition of air. In these conditions, acrylate 10 led to the recovery (following aqueous work-up) of sulfinamide 9 without traces of formation of the 1,4-adduct (Scheme 4).
![[1860-5397-19-103-i4]](/bjoc/content/inline/1860-5397-19-103-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Reaction of α-(aminomethyl)acrylate 10 with Et2Zn in the presence of air.
Scheme 4: Reaction of α-(aminomethyl)acrylate 10 with Et2Zn in the presence of air.
By contrast, 1,4-addition without subsequent fragmentation was observed starting from α-(aminomethyl)acrylates having free N–H bonds (Table 2). The reaction of Et2Zn with acrylates 5–7 afforded the desired 1,4-addition products 11–13 in 42–55% yield. Better results were obtained starting from 8a, which delivered adduct 14a in 79% yield and 70:30 dr. We also noted that in the absence of deliberately added air, these reactions proceeded only with low conversion. For instance, starting from 8a, product 14a was obtained in only 25% yield along with ≈70% of starting material recovery.
Table 2: Air-promoted 1,4-addition of Et2Zn onto α-(aminomethyl)acrylates having free N–H bonds.a
![[Graphic 2]](/bjoc/content/inline/1860-5397-19-103-i11.svg?max-width=637&scale=1.0)
|
||||
| Entry | Substrate (R) | Product | Yieldb | drc |
| 1 | 5 (CH(Ph)2) | 11 | 42 | |
| 2 | 6 (Ph) | 12 | 55 | |
| 3 | 7 (Ts) | 13 | 55 | |
| 4 | 8a (S(O)t-Bu) | 14a | 76 | 70:30 |
| 5 | 8a (S(O)t-Bu) | 14a | 25d | 50:50 |
aGeneral conditions: α-(aminomethyl)acrylate (0.2 mmol), Et2Zn (1.0 mmol), CH2Cl2 (6 mL), air (20 mL introduced via syringe at 0.5 mL·min−1 rate). bIsolated yield. cRatio of diastereomers measured by 1H NMR spectroscopy prior to purification. dNo air was added.
These results are relevant in the sense that not only they demonstrate that the oxygen-promoted 1,4-addition of α-(aminomethyl)acrylates with free N–H bonds is a productive process, but also that the tert-butanesulfinyl moiety is well tolerated and that 1,4-stereoinduction can be achieved. Hence, in order to improve the levels of diastereoselectivity, we investigated further the reaction conditions starting with enoate 8a as model substrate (Table 3).
Table 3: Optimization of the air-promoted 1,4-addition of dialkylzinc reagents onto N-(tert-butanesulfinyl) α-(aminomethyl)acrylates.
![[Graphic 3]](/bjoc/content/inline/1860-5397-19-103-i12.svg?max-width=637&scale=1.0)
|
||||||
| Entry | Substrate (R1) | R2 | Product | Variation of conditionsa | Yieldb | drc,d |
| 1 | 8a (Me) | Et | 14a | none | 76 | 70:30 |
| 2 | 8a (Me) | Et | 14a | −78 °C instead of −33 °C | 60 | 57:43 |
| 3 | 8a (Me) | Et | 14a | oxygen (5 mL) was added at once immediately after Et2Zn | 83 | 59:41 |
| 4 | 8a (Me) | Et | 14a | toluene instead of CH2Cl2 | 82 | 75:25 |
| 5 | 8a (Me) | Et | 14a | hexane instead of CH2Cl2 | 82 | 85:15 |
| 6 | 8b (t-Bu) | Et | 14b | hexane instead of CH2Cl2 | 88 | 85:15 |
| 7 | 8c (Bn) | Et | 14c | none | 76 | 70:30 |
| 8 | 8a (Me) | Bu | 15a | none | 71 | 67:33 |
| 9 | 8a (Me) | Me | 16a | none | n.r.e | |
| 10 | 8a (Me) | Me | 16a | hexane instead of CH2Cl2 | n.r.e | |
aAll reactions were conducted at a 0.2 mmol scale using 20 mL of air. bIsolated yield (mixture of diastereoisomers). cMeasured by 1H or 13C NMR spectroscopy prior to purification. dThe relative configuration of the major diastereomer is shown in the scheme; it was determined by chemical correlation for 14b (see below) and inferred by analogy for 14a and 14c. eNo reaction.
Carrying out the reaction at −78 °C instead of −33 °C was deleterious both for the yield and the selectivity (Table 3, entry 2). By contrast, we rapidly learned that leaving diethylzinc in contact with the starting acrylate for 1 h prior to the addition of air had a significant impact on the stereoselectivity. When air was introduced directly after Et2Zn (Table 3, entry 3) a much lower 59:41 dr was observed. This behavior was suggestive of the need for coordination of diethylzinc both to the carbonyl and sulfinyl unit to achieve good levels of selectivity. Hence, to reinforce Lewis pair formation, the reaction was also carried out in apolar solvents such as toluene and hexane (entries 4 and 5 in Table 3). In hexane, an 88% yield with 85:15 dr was obtained, which constituted the best conditions. Importantly, the protocol was found to be similarly applicable with enoates 8b (Table 3, entry 6) and 8c (entry 7) having tert-butyl and benzyl ester groups, which, as the methyl ester unit, are typical in the context of amino acid synthesis. ZnBu2 was also amenable to 1,4-addition (Table 3, entry 8), but not ZnMe2 (entries 9 and 10). This difference can be ascribed to a less favorable homolytic substitution reaction of ZnMe2 in relation to its higher analogues and is in line with previous literature observations [11].
The configuration of the major diastereomer was determined by chemical correlation (Scheme 5). Product (RS)-14b (85:15 dr), i.e., a mixture of two enantiomerically pure diastereomers, was obtained from (RS)-tert-butylsulfinamide upon allylation with tert-butyl α-(bromomethyl)acrylate followed by 1,4-addition with Et2Zn. It was then converted into the known β2-amino acid 17 by TFA-promoted concomitant deprotection of the nitrogen and the ester groups. The sample was found to have a negative optical rotation, thereby indicating that the major enantiomer present had S configuration [26]. This allowed to establish that the configuration of the major diastereomer present in (RS)-14b was (RS,S), and thus the sense of chiral induction for the 1,4-addition reactions reported in Table 2.
![[1860-5397-19-103-i5]](/bjoc/content/inline/1860-5397-19-103-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Chemical correlation to determine the configuration of the major diastereomer of (RS)-14b.
Scheme 5: Chemical correlation to determine the configuration of the major diastereomer of (RS)-14b.
Tandem 1,4-addition–aldol condensation reactions
We then went on to consider tandem 1,4-addition–aldol condensation reactions (Scheme 6), which offer the interesting prospect of generating an all-carbon quaternary stereocenter. α-(Aminomethyl)acrylates 5–7 reacted smoothly at −33 °C within 2 h with Et2Zn in the presence of cyclohexanone to afford amino alcohols 18–20 in quite good yields (63–68%). Even better yields were obtained with enoates 8a and 8b both with cyclohexanone and acetone as carbonyl partners. Starting from 8a and carrying out the reaction in CH2Cl2, product 21a was obtained in 86% yield with 75:25 dr. Alike for the 1,4-addition protocol, better stereoinduction was obtained by performing the reaction in hexane: 8b was converted into 21b and 22 in 77–84% yield with higher than 90:10 dr. It is also interesting to note that the levels of induction for the 1,4-addition–aldol condensations are somewhat higher than those obtained for the 1,4-additions. Aldehydes also proved competent terminal electrophiles for the tandem sequence. Illustratively, adducts 23 and 24 were obtained from α-(aminomethyl)acrylates 5 and 8a in 77–88% yields, albeit as poorly selective mixtures of diastereoisomers. This lack of stereocontrol is not surprising, given the well-known difficulty to control the relative configuration between the two adjacent stereocenters created during aldol condensations with zinc enolates.
![[1860-5397-19-103-i6]](/bjoc/content/inline/1860-5397-19-103-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Air-promoted tandem 1,4-addition–aldol condensation reactions of Et2Zn with α-(aminomethyl)acrylates.
Scheme 6: Air-promoted tandem 1,4-addition–aldol condensation reactions of Et2Zn with α-(aminomethyl)acrylate...
Mechanistic insights
The last part of our work was devoted to gain mechanistic insight for the developed reaction protocols through several diagnostic experiments. Regarding the 1,4-addition process, the lower reactivity noted in the absence of air (Table 2, entry 5) represents already a strong indication for a radical addition mechanism. This is further supported by the result of an I-atom transfer experiment (Scheme 7, top). In the presence of two equivalents of iPrI, the reaction of 8a with Et2Zn leads to a mixture of product 14a and product 25a, incorporating an iPr moiety, in a 14a/25a 30:70 mixture. Product 25a is formed on addition of an iPr radical generated by I-atom transfer from iPrI to the Et radical, and is diagnostic for the formation of the latter in the reaction medium.
![[1860-5397-19-103-i7]](/bjoc/content/inline/1860-5397-19-103-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Diagnostic experiments for a radical mechanism and for enolate formation.
Scheme 7: Diagnostic experiments for a radical mechanism and for enolate formation.
Deuterium labeling experiments were then performed to substantiate the formation of a zinc enolate following radical addition (Scheme 7, bottom). Much to our surprise however, no deuterium incorporation is observed on quenching with ND4Cl/D2O the reaction between 8a and Et2Zn. By contrast, a significant deuterium incorporation is obtained when deuterated starting material (8a-d) is engaged. The combination of these two results is in agreement with the formation of a zinc enolate that undergoes proto- (or deuterio)demetalation with the N–H (or N–D) as proton (or deuterium) source.
To further analyze the influence of the presence of an N–H function, we performed other reactions with N-benzyl enoate 10 which proved highly informative. As discussed previously (Scheme 4), application of the developed protocol for 1,4-addition to 10 only yields N-benzyl-N-tert-butylsulfinamide following β-elimination. By contrast, in the presence of benzaldehyde, 1,4-addition–aldol condensation is predominant, yielding 26 in 56% yield as a 49:25:23:3 diastereomeric mixture (Scheme 8). When 10 is exposed to Et3B in the presence of iPrI, benzaldehyde, and O2, which are conditions known to promote radical 1,4-addition, only formation of telomers [7] is noted. This lends clear evidence that the intermediate enoxyl radical does not intervene neither in the β-fragmentation (Scheme 4) nor in the addition across the carbonyl bonds.
![[1860-5397-19-103-i8]](/bjoc/content/inline/1860-5397-19-103-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Diagnostic experiments with N-benzyl enoate 10.
Scheme 8: Diagnostic experiments with N-benzyl enoate 10.
Overall, the mechanistic investigations support the scenario depicted in Scheme 9. Oxygen (in air) triggers a free-radical chain reaction between α-(aminomethyl)acrylates and dialkylzinc reagents that entails 1,4-addition and SH2 of the formed enoxyl radical facilitated by coordination of nitrogen to zinc. The zinc enolate thus formed evolves following different pathways according to the type of substrate and reaction conditions. In the absence of a carbonyl electrophile, enolates of substrates with trisubstituted nitrogen groups undergo β-fragmentation. By contrast, those derived from substrates having N–H bonds undergo protodemetalation to provide ultimately the 1,4-addition adduct. In the presence of carbonyl acceptors, these two competitive reactions are superseded and the enolate engages in aldol condensation regardless of its nitrogen substitution; the outcome of the reaction is a tandem 1,4-addition–aldol process. When the tert-butanesulfinyl moiety is present on the nitrogen atom, electrophilic substitution of the intermediate enolates (protodemetalation or aldol condensation) occurs with decent levels of chiral induction. It should be mentioned here that our attempts to trap the intermediate enolate with a carbon electrophile other than carbonyl acceptors (i.e., iodomethane) were not successful and protodemetalation of the enolate outcompeted methylation.
![[1860-5397-19-103-i9]](/bjoc/content/inline/1860-5397-19-103-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Reactivity manifolds for the air-promoted tandem 1,4-addition–electrophilic substitution reaction between dialkylzinc reagents and α-(aminomethyl)acrylates (N-(tert-butanesulfinyl) derivatives shown).
Scheme 9: Reactivity manifolds for the air-promoted tandem 1,4-addition–electrophilic substitution reaction b...
Conclusion
In conclusion, we have demonstrated that α-(aminomethyl)acrylates are suitable acceptors for 1,4-additions with dialkylzincs in aerobic conditions. Coordination of the nitrogen atom to zinc is crucial to enable the SH2 step of the tertiary α-carbonyl radical that follows radical 1,4-addition in order to deliver a zinc enolate. The latter is poised to undergo β-fragmentation, but this process can be outcompeted by in situ electrophilic substitution reactions which offer synthetically useful procedures: 1,4-addition (for substrates having N–H bonds) or tandem 1,4-addition–aldol reactions (in the presence of carbonyl electrophiles). Asymmetric variants of these transformations are possible using the tert-butanesulfinyl chiral auxiliary on the nitrogen atom. The levels of 1,4-stereoinduction are significant but a convincing model to account for it cannot be put forward at this point. Nonetheless, from a synthetic methodology point of view, the reported protocols are relevant as they offer a new, direct and modular route to enantioenriched α-mono- and α,α-disubstituted β-amino acids (β2-amino acids and β2,2-amino acids), with, for the latter, the noteworthy stereocontrolled construction of an all-carbon quaternary stereocenter. Furthermore, our protocol provides a complement to existing literature, as none of the previously reported methods to convert α-(aminomethyl)acrylates into enantioenriched β-amino acids is applicable for the preparation of β2,2-amino acids [27-31].
Experimental
1. Procedure for the monoallylation of primary amines and tert-butylsulfinamide (preparation of compounds 5–7 and 8a–c). In a round-bottomed flask under argon, n-BuLi (1.0 equiv, soln. in heptane) was added dropwise to a THF (0.2 mol·L−1) solution of the appropriate primary amine or tert-butylsulfinamide (1.0 equiv) at −55 °C. The mixture was then stirred at rt for 30 min, cooled to −55 °C, and trimethylsilyl chloride (1.0 equiv) was added. The mixture was then stirred at rt for 30 min, cooled to −55 °C, and n-BuLi (1.0 equiv, soln. in heptane) was added dropwise. The mixture was stirred at rt for 30 min, cooled to −78 °C, and the corresponding α-(bromomethyl)acrylate (1.0 equiv) was added. The reaction mixture was then stirred for 2 h letting the temperature rise to rt and quenched with aq 1 M NH4Cl. The aqueous layer was extracted with EtOAc (3×) and the combined organic layer was washed (brine), dried (MgSO4), and concentrated under reduced pressure to provide the crude product which was then purified by column chromatography on silica gel.
2. Procedure for the air-promoted 1,4-addition of dialkylzinc reagents to α-(aminomethyl)acrylates (preparation of compounds 11–13, 14a–c, and 15a). In a Schlenk-tube under argon, the appropriate α-(aminomethyl)acrylate (0.2 mmol) was dissolved in the indicated reaction solvent (3 mL) and the solution was cooled to −33 °C. Then, Et2Zn (1 M in hexanes, 1.0 mL, 1.0 mmol) was added dropwise and the solution was stirred for 1 h. Air (20 mL) was introduced directly into the solution via a syringe fitted with a CaCl2 pad at a 0.5 mL/min rate (syringe pump). After the end of the air addition, the mixture was stirred for an additional 80 min at −33 °C and then quenched with aq NH4Cl (5 mL) at 0 °C. The aqueous layer was extracted with CH2Cl2 (2×). The combined organic layer was washed (brine), dried (MgSO4), and concentrated under reduced pressure to provide the crude product which was then purified by column chromatography on silica gel.
3. Procedure for the air-promoted tandem 1,4-addition–aldol reaction between dialkylzinc reagents, α-(aminomethyl)acrylates and carbonyl derivatives (preparation of compounds 18–20, 21a–b, 22–24). In a Schlenk-tube under argon, the appropriate α-(aminomethyl)acrylate (0.2 mmol) was dissolved in the indicated reaction solvent (3 mL) and the solution was cooled to −33 °C. The carbonyl electrophile (1.0 mmol) and then Et2Zn (1 M in hexanes, 1.0 mL, 1.0 mmol) were added dropwise and the solution was stirred for 1 h. Air (20 mL) was introduced directly into the solution via a syringe fitted with a CaCl2 pad at a 0.5 mL/min rate (syringe pump). After the end of the air addition, the mixture was stirred for an additional 80 min at −33 °C and then quenched with aq NH4Cl (5 mL) at 0 °C. The aqueous layer was extracted with CH2Cl2 (2×). The combined organic layer was washed (brine), dried (MgSO4), and concentrated under reduced pressure to provide the crude product which was then purified by column chromatography on silica gel.
Supporting Information
| Supporting Information File 1: General information, characterization data, chemical correlation, and copies of NMR spectra. | ||
| Format: PDF | Size: 5.4 MB | Download |
References
-
Bazin, S.; Feray, L.; Bertrand, M. P. Chimia 2006, 60, 260–265. doi:10.2533/000942906777674679
Return to citation in text: [1] -
Chemla, F.; Pérez-Luna, A. Radical-Polar Crossover Reactions. In Free Radicals: Fundamentals and Applications in Organic Synthesis 2; Fensterbank, L.; Ollivier, C., Eds.; Science of Synthesis; Thieme: Stuttgart, Germany, 2021; pp 209–357. doi:10.1055/sos-sd-233-00075
Return to citation in text: [1] -
Brown, H. C.; Midland, M. M. Angew. Chem., Int. Ed. Engl. 1972, 11, 692–700. doi:10.1002/anie.197206921
Return to citation in text: [1] -
Bazin, S.; Feray, L.; Vanthuyne, N.; Siri, D.; Bertrand, M. P. Tetrahedron 2007, 63, 77–85. doi:10.1016/j.tet.2006.10.049
Return to citation in text: [1] -
Maury, J.; Mouysset, D.; Feray, L.; Marque, S. R. A.; Siri, D.; Bertrand, M. P. Chem. – Eur. J. 2012, 18, 3241–3247. doi:10.1002/chem.201102366
Return to citation in text: [1] -
Bazin, S.; Feray, L.; Vanthuyne, N.; Bertrand, M. P. Tetrahedron 2005, 61, 4261–4274. doi:10.1016/j.tet.2005.02.042
Return to citation in text: [1] -
Bazin, S.; Feray, L.; Siri, D.; Naubron, J.-V.; Bertrand, M. P. Chem. Commun. 2002, 2506–2507. doi:10.1039/b206695e
Return to citation in text: [1] [2] -
Zelocualtecatl-Montiel, I.; García-Álvarez, F.; Juárez, J. R.; Orea, L.; Gnecco, D.; Mendoza, A.; Chemla, F.; Ferreira, F.; Jackowski, O.; Aparicio, D. M.; Perez-Luna, A.; Terán, J. L. Asian J. Org. Chem. 2017, 6, 67–70. doi:10.1002/ajoc.201600501
Return to citation in text: [1] -
Yamada, K.-i.; Konishi, T.; Nakano, M.; Fujii, S.; Cadou, R.; Yamamoto, Y.; Tomioka, K. J. Org. Chem. 2012, 77, 5775–5780. doi:10.1021/jo300944f
Return to citation in text: [1] -
Yamada, K.-i.; Matsumoto, Y.; Fujii, S.; Konishi, T.; Yamaoka, Y.; Takasu, K. J. Org. Chem. 2016, 81, 3809–3817. doi:10.1021/acs.joc.6b00485
Return to citation in text: [1] -
Lingua, H.; Dwadnia, N.; Siri, D.; Bertrand, M. P.; Feray, L. Tetrahedron 2018, 74, 7507–7515. doi:10.1016/j.tet.2018.11.029
Return to citation in text: [1] [2] [3] -
Bertrand, M. P.; Feray, L.; Nouguier, R.; Perfetti, P. J. Org. Chem. 1999, 64, 9189–9193. doi:10.1021/jo9912404
Return to citation in text: [1] -
Miyabe, H.; Asada, R.; Yoshida, K.; Takemoto, Y. Synlett 2004, 540–542. doi:10.1055/s-2004-815407
Return to citation in text: [1] -
Miyabe, H.; Asada, R.; Takemoto, Y. Tetrahedron 2005, 61, 385–393. doi:10.1016/j.tet.2004.10.104
Return to citation in text: [1] -
Vibert, F.; Maury, J.; Lingua, H.; Besson, E.; Siri, D.; Bertrand, M. P.; Feray, L. Tetrahedron 2015, 71, 8991–9002. doi:10.1016/j.tet.2015.09.045
Return to citation in text: [1] -
Denes, F.; Chemla, F.; Normant, J. F. Angew. Chem., Int. Ed. 2003, 42, 4043–4046. doi:10.1002/anie.200250474
Return to citation in text: [1] -
Denes, F.; Cutri, S.; Pérez-Luna, A.; Chemla, F. Chem. – Eur. J. 2006, 12, 6506–6513. doi:10.1002/chem.200600334
Return to citation in text: [1] -
Beniazza, R.; Romain, E.; Chemla, F.; Ferreira, F.; Jackowski, O.; Perez-Luna, A. Eur. J. Org. Chem. 2015, 7661–7665. doi:10.1002/ejoc.201501173
Return to citation in text: [1] -
Cheng, R. P.; Gellman, S. H.; DeGrado, W. F. Chem. Rev. 2001, 101, 3219–3232. doi:10.1021/cr000045i
Return to citation in text: [1] -
Liu, M.; Sibi, M. P. Tetrahedron 2002, 58, 7991–8035. doi:10.1016/s0040-4020(02)00991-2
Return to citation in text: [1] -
Lelais, G.; Seebach, D. Biopolymers 2004, 76, 206–243. doi:10.1002/bip.20088
Return to citation in text: [1] -
Seebach, D.; Gardiner, J. Acc. Chem. Res. 2008, 41, 1366–1375. doi:10.1021/ar700263g
Return to citation in text: [1] -
Seebach, D.; Beck, A. K.; Capone, S.; Deniau, G.; Groselj, U.; Zass, E. Synthesis 2009, 1–32. doi:10.1055/s-0028-1087490
Return to citation in text: [1] -
Noda, H.; Shibasaki, M. Eur. J. Org. Chem. 2020, 2350–2361. doi:10.1002/ejoc.201901596
Return to citation in text: [1] -
Xuan, J.; Daniliuc, C. G.; Studer, A. Org. Lett. 2016, 18, 6372–6375. doi:10.1021/acs.orglett.6b03267
Return to citation in text: [1] -
Gutiérrez-García, V. M.; Reyes-Rangel, G.; Muñoz-Muñiz, O.; Juaristi, E. Helv. Chim. Acta 2002, 85, 4189–4199. doi:10.1002/hlca.200290004
Return to citation in text: [1] -
Sibi, M. P.; Tatamidani, H.; Patil, K. Org. Lett. 2005, 7, 2571–2573. doi:10.1021/ol050630b
Return to citation in text: [1] -
Qiu, L.; Prashad, M.; Hu, B.; Prasad, K.; Repič, O.; Blacklock, T. J.; Kwong, F. Y.; Kok, S. H. L.; Lee, H. W.; Chan, A. S. C. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 16787–16792. doi:10.1073/pnas.0704461104
Return to citation in text: [1] -
Guo, Y.; Shao, G.; Li, L.; Wu, W.; Li, R.; Li, J.; Song, J.; Qiu, L.; Prashad, M.; Kwong, F. Y. Adv. Synth. Catal. 2010, 352, 1539–1553. doi:10.1002/adsc.201000122
Return to citation in text: [1] -
Li, L.; Chen, B.; Ke, Y.; Li, Q.; Zhuang, Y.; Duan, K.; Huang, Y.; Pang, J.; Qiu, L. Chem. – Asian J. 2013, 8, 2167–2174. doi:10.1002/asia.201300339
Return to citation in text: [1] -
Sibi, M. P.; Patil, K. Angew. Chem., Int. Ed. 2004, 43, 1235–1238. doi:10.1002/anie.200353000
Return to citation in text: [1]
| 7. | Bazin, S.; Feray, L.; Siri, D.; Naubron, J.-V.; Bertrand, M. P. Chem. Commun. 2002, 2506–2507. doi:10.1039/b206695e |
| 27. | Sibi, M. P.; Tatamidani, H.; Patil, K. Org. Lett. 2005, 7, 2571–2573. doi:10.1021/ol050630b |
| 28. | Qiu, L.; Prashad, M.; Hu, B.; Prasad, K.; Repič, O.; Blacklock, T. J.; Kwong, F. Y.; Kok, S. H. L.; Lee, H. W.; Chan, A. S. C. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 16787–16792. doi:10.1073/pnas.0704461104 |
| 29. | Guo, Y.; Shao, G.; Li, L.; Wu, W.; Li, R.; Li, J.; Song, J.; Qiu, L.; Prashad, M.; Kwong, F. Y. Adv. Synth. Catal. 2010, 352, 1539–1553. doi:10.1002/adsc.201000122 |
| 30. | Li, L.; Chen, B.; Ke, Y.; Li, Q.; Zhuang, Y.; Duan, K.; Huang, Y.; Pang, J.; Qiu, L. Chem. – Asian J. 2013, 8, 2167–2174. doi:10.1002/asia.201300339 |
| 31. | Sibi, M. P.; Patil, K. Angew. Chem., Int. Ed. 2004, 43, 1235–1238. doi:10.1002/anie.200353000 |
| 1. | Bazin, S.; Feray, L.; Bertrand, M. P. Chimia 2006, 60, 260–265. doi:10.2533/000942906777674679 |
| 2. | Chemla, F.; Pérez-Luna, A. Radical-Polar Crossover Reactions. In Free Radicals: Fundamentals and Applications in Organic Synthesis 2; Fensterbank, L.; Ollivier, C., Eds.; Science of Synthesis; Thieme: Stuttgart, Germany, 2021; pp 209–357. doi:10.1055/sos-sd-233-00075 |
| 8. | Zelocualtecatl-Montiel, I.; García-Álvarez, F.; Juárez, J. R.; Orea, L.; Gnecco, D.; Mendoza, A.; Chemla, F.; Ferreira, F.; Jackowski, O.; Aparicio, D. M.; Perez-Luna, A.; Terán, J. L. Asian J. Org. Chem. 2017, 6, 67–70. doi:10.1002/ajoc.201600501 |
| 11. | Lingua, H.; Dwadnia, N.; Siri, D.; Bertrand, M. P.; Feray, L. Tetrahedron 2018, 74, 7507–7515. doi:10.1016/j.tet.2018.11.029 |
| 6. | Bazin, S.; Feray, L.; Vanthuyne, N.; Bertrand, M. P. Tetrahedron 2005, 61, 4261–4274. doi:10.1016/j.tet.2005.02.042 |
| 7. | Bazin, S.; Feray, L.; Siri, D.; Naubron, J.-V.; Bertrand, M. P. Chem. Commun. 2002, 2506–2507. doi:10.1039/b206695e |
| 26. | Gutiérrez-García, V. M.; Reyes-Rangel, G.; Muñoz-Muñiz, O.; Juaristi, E. Helv. Chim. Acta 2002, 85, 4189–4199. doi:10.1002/hlca.200290004 |
| 4. | Bazin, S.; Feray, L.; Vanthuyne, N.; Siri, D.; Bertrand, M. P. Tetrahedron 2007, 63, 77–85. doi:10.1016/j.tet.2006.10.049 |
| 5. | Maury, J.; Mouysset, D.; Feray, L.; Marque, S. R. A.; Siri, D.; Bertrand, M. P. Chem. – Eur. J. 2012, 18, 3241–3247. doi:10.1002/chem.201102366 |
| 19. | Cheng, R. P.; Gellman, S. H.; DeGrado, W. F. Chem. Rev. 2001, 101, 3219–3232. doi:10.1021/cr000045i |
| 20. | Liu, M.; Sibi, M. P. Tetrahedron 2002, 58, 7991–8035. doi:10.1016/s0040-4020(02)00991-2 |
| 21. | Lelais, G.; Seebach, D. Biopolymers 2004, 76, 206–243. doi:10.1002/bip.20088 |
| 22. | Seebach, D.; Gardiner, J. Acc. Chem. Res. 2008, 41, 1366–1375. doi:10.1021/ar700263g |
| 23. | Seebach, D.; Beck, A. K.; Capone, S.; Deniau, G.; Groselj, U.; Zass, E. Synthesis 2009, 1–32. doi:10.1055/s-0028-1087490 |
| 24. | Noda, H.; Shibasaki, M. Eur. J. Org. Chem. 2020, 2350–2361. doi:10.1002/ejoc.201901596 |
| 3. | Brown, H. C.; Midland, M. M. Angew. Chem., Int. Ed. Engl. 1972, 11, 692–700. doi:10.1002/anie.197206921 |
| 25. | Xuan, J.; Daniliuc, C. G.; Studer, A. Org. Lett. 2016, 18, 6372–6375. doi:10.1021/acs.orglett.6b03267 |
| 15. | Vibert, F.; Maury, J.; Lingua, H.; Besson, E.; Siri, D.; Bertrand, M. P.; Feray, L. Tetrahedron 2015, 71, 8991–9002. doi:10.1016/j.tet.2015.09.045 |
| 16. | Denes, F.; Chemla, F.; Normant, J. F. Angew. Chem., Int. Ed. 2003, 42, 4043–4046. doi:10.1002/anie.200250474 |
| 17. | Denes, F.; Cutri, S.; Pérez-Luna, A.; Chemla, F. Chem. – Eur. J. 2006, 12, 6506–6513. doi:10.1002/chem.200600334 |
| 13. | Miyabe, H.; Asada, R.; Yoshida, K.; Takemoto, Y. Synlett 2004, 540–542. doi:10.1055/s-2004-815407 |
| 14. | Miyabe, H.; Asada, R.; Takemoto, Y. Tetrahedron 2005, 61, 385–393. doi:10.1016/j.tet.2004.10.104 |
| 18. | Beniazza, R.; Romain, E.; Chemla, F.; Ferreira, F.; Jackowski, O.; Perez-Luna, A. Eur. J. Org. Chem. 2015, 7661–7665. doi:10.1002/ejoc.201501173 |
| 12. | Bertrand, M. P.; Feray, L.; Nouguier, R.; Perfetti, P. J. Org. Chem. 1999, 64, 9189–9193. doi:10.1021/jo9912404 |
| 9. | Yamada, K.-i.; Konishi, T.; Nakano, M.; Fujii, S.; Cadou, R.; Yamamoto, Y.; Tomioka, K. J. Org. Chem. 2012, 77, 5775–5780. doi:10.1021/jo300944f |
| 10. | Yamada, K.-i.; Matsumoto, Y.; Fujii, S.; Konishi, T.; Yamaoka, Y.; Takasu, K. J. Org. Chem. 2016, 81, 3809–3817. doi:10.1021/acs.joc.6b00485 |
| 11. | Lingua, H.; Dwadnia, N.; Siri, D.; Bertrand, M. P.; Feray, L. Tetrahedron 2018, 74, 7507–7515. doi:10.1016/j.tet.2018.11.029 |
| 11. | Lingua, H.; Dwadnia, N.; Siri, D.; Bertrand, M. P.; Feray, L. Tetrahedron 2018, 74, 7507–7515. doi:10.1016/j.tet.2018.11.029 |
© 2023 Palillero-Cisneros et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.