Abstract
The methodology to prepare 3-substituted 1,5-dibromopentanes I and their immediate precursors, which include 3-substituted 1,5-pentanediols VII or 4-substituted tetrahydropyrans VIII, is surveyed. Such dibromides I are important intermediates in the preparation of liquid crystalline derivatives containing 6-membered heterocyclic rings. Four dibromides 1a–1d containing simple alkyl and more complex fragments at the 3-position were prepared. 3-Propyl- and 3-pentyl-pentane-1,5-diol (2a,b) were prepared starting from either glutaconate or malonate diesters, while tetrahydropyrans 3c and 3d were obtained from tetrahydro-4H-pyran-4-one. The advantages and disadvantages of each route are discussed. Dibromides 1c and 1d were used to prepare sulfonium zwitterions 11c and 11d.

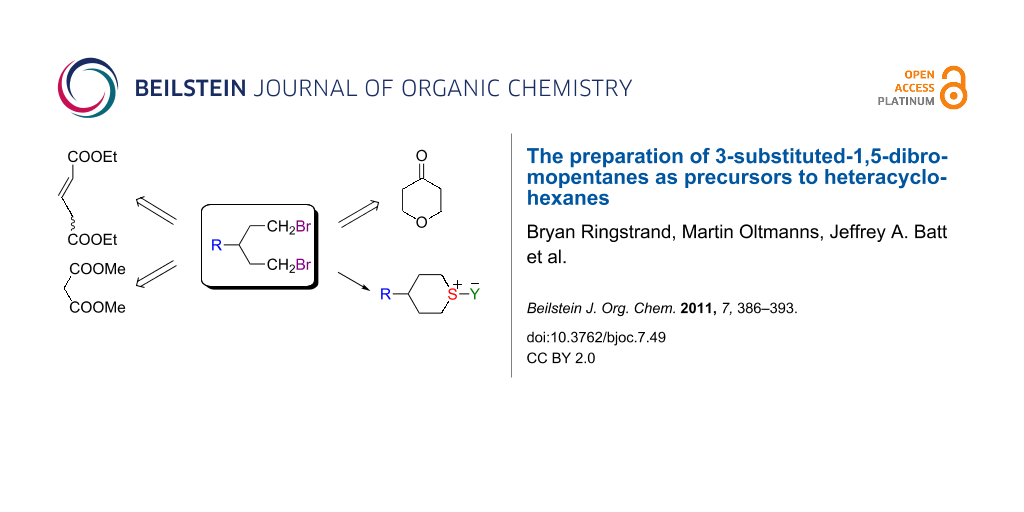
Graphical Abstract
Introduction
3-Substituted 1,5-dibromopentanes I and disulfonates, typically tosylates II, serve as useful intermediates in the preparation of six-membered heterocycles such as 4-substituted-piperidines III, thianes IV, silacyclohexanes V, and phosphorinanes VI (Figure 1). The piperidines III (Y = Ar) have been used as structural elements of liquid crystals [1-6] as well as antithrombotic and neuroprotectant agents [7,8]. Some thianes IV are found in petroleum distillates [9] and several have been used for the preparation of sulfones [7,10,11], sulfoxides [11,12], and sulfonium derivatives [13]. The latter, as well as silacyclohexanes V [14] and phosphorinanes VI [15,16], have been used in conformational and stereochemical studies.
![[1860-5397-7-49-1]](/bjoc/content/figures/1860-5397-7-49-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Methods for synthesis of dibromides I and their use for preparation of 6-membered heterocycles.
Figure 1: Methods for synthesis of dibromides I and their use for preparation of 6-membered heterocycles.
The dibromides I or disulfonates II were prepared from 3-substituted 1,5-pentanediols VII. The former were obtained using HBr and concd sulfuric acid [2], while the latter from p-toluenesulfonyl chloride in the presence of a base such as pyridine [13,17-20]. Dibromides I can also be obtained by the ring-opening of tetrahydropyrans VIII under the same conditions employed for the diols VII [2,12,21,22], or via phase-transfer catalysis conditions with a phosphonium salt [23]. Piperidines III (Y = H) can undergo degradation to form dibromides I although this process is typically less efficient [12,14,15]. The piperidine ring can be reconstituted by ring-closure of I with an appropriate amine or aniline Y–NH2 [2,22]. The thianes IV are typically obtained from I or from II by reaction with Na2S [9,10,12,13], whereas the silacyclohexanes V and phosphorinanes VI are prepared by reacting the Grignard reagent derived from I with dichlorosilanes [14] or phosphonous dichlorides [15], respectively (Figure 1).
Our research program focuses on the development of polar, zwitterionic nematic liquid crystals having large longitudinal dipole moments for formulation of nematic materials with positive dielectric anisotropy [24,25]. The zwitterions consist of six-membered sulfonium rings attached to a boron cluster, either [closo-1-CB9H10]− or [closo-1-CB11H12]−. Therefore, we have interest in cycloalkylating reagents such as dibromides I and disulfonates II containing alkyl or aryl substituents at the 3-position as intermediates for polar nematics. A number of such dibromides I and ditosylates II have been reported in the literature and some are listed in Table 1. For completeness, thianes IV, diols VII, and tetrahydropyrans VIII are also included.
Table 1: Selected compounds reported in the literature.
| R | Literature | |
|---|---|---|
![[Graphic 1]](/bjoc/content/inline/1860-5397-7-49-i11.svg?max-width=637&scale=1.0)
|
Me, n-Pr, n-Bu, C5H11, C6H13, C7H15, C8H17, C9H19, C10H21, Ph, 4-C4H9OPh, 4-ClPh | [2,9,10,12,14,21,22,26] |
![[Graphic 2]](/bjoc/content/inline/1860-5397-7-49-i12.svg?max-width=637&scale=1.0)
|
Me, Et, n-Pr, Ph, 4-CH3OPh, 4-C5H11Ph | [13,17-20] |
![[Graphic 3]](/bjoc/content/inline/1860-5397-7-49-i13.svg?max-width=637&scale=1.0)
|
Me, Et, n-Pr, C6H13, Ph, 4-CH3OPh, 4-C4H9OPh, 4-ClPh, 4-C5H11Ph, PhCH2CH2 | [2,9,17-20,26-28] |
![[Graphic 4]](/bjoc/content/inline/1860-5397-7-49-i14.svg?max-width=637&scale=1.0)
|
Me, Et, n-Pr, n-Bu, C5H11, C6H13, Ph, 4-ClPh, 4-HOPh, 4-NH2Ph | [7,9-13,29,30] |
![[Graphic 5]](/bjoc/content/inline/1860-5397-7-49-i15.svg?max-width=637&scale=1.0)
|
Me, n-Pr, n-Bu, C5H11, C6H13, C7H15, C8H17, C9H19, C10H21, C11H23, Ph, 4-HOPh, 4-C4H9OPh, 4-CF3Ph, 4-NH2Ph | [2,7,12,21,22,31,32] |
Diols VII represent a convenient intermediate for the preparation of dibromides I or ditosylates II, and four routes to VII are presented in Scheme 1. The first route (Method 1A) involves six steps starting from alkylation of malonate diester followed by reduction of the esters, tosylation of the alcohols, carbon homologation with NaCN, hydrolysis of the cyano groups to carboxylic acids, and lastly a second reduction to VII. Overall yields of diols VII using Method 1A are around 10% [27,28]. The second route (Method 1B) consists of a Knoevenagel condensation of malonate diester and an aldehyde followed by conjugate addition of a second equivalent of malonate diester. The tetraester is hydrolyzed, decarboxylated, esterified, and then reduced to give the diol. Yields for tetraesters are in the range of 25–85%, whereas the decarboxylation step gives the corresponding glutaric acids in 60–80% yield [19,33]. Variations of Method 1B involve the use of cyanoacetamide [34-36] or Meldrum’s acid [19,37] instead of malonate diester. For the former, the condensation with aldehyde is efficient and the product is crystalline, which simplifies its purification.
![[1860-5397-7-49-i1]](/bjoc/content/inline/1860-5397-7-49-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: General methods for preparation of diols VII.
Scheme 1: General methods for preparation of diols VII.
Another route to VII involves a two-step sequence of conjugate addition of an organocopper reagent to glutaconate diester [38] followed by reduction (Method 1C, Scheme 1). Yield of diols from this sequence are typically around 70% [17,38]. The downside to Method 1C is that an excess of Grignard reagent (3–4 equiv) is required; therefore, it is not economical with respect to the alkyl halide. Method 1D also uses glutaconate diester, which is reacted with an aryl iodide in the presence of Pd(0) under Heck conditions. The resulting unsaturated diester is reduced with excess LiAlH4 to give diol VII with an overall yield of about 30% for the two-step process [19].
Tetrahydropyrans VIII represent a second general intermediate for access to dibromides I, and four routes are presented in Scheme 2. The first route involves the addition of a Grignard reagent to tetrahydro-4H-pyran-4-one, elimination of water, and hydrogenation of the olefin (Method 2A). Typical yields for Method 2A range from 20–30% [39,40]. The second route is the Wittig olefination of tetrahydro-4H-pyran-4-one followed by hydrogenation (Method 2B). Yields for the Wittig olefination of tetrahydro-4H-pyran-4-one range from 35–75% [41-43]. The third route (Method 2C) begins from tetrahydropyran-4-carboxylic acid: The acid chloride is reacted with a Grignard reagent, and the resulting ketone reduced under Wolff–Kischner conditions. Typical yields for this sequence are around 40% [21]. A recently described procedure [32] allows for efficient preparation of 4-aryltetrahydropyrans by the coupling of arylboronic acids and 4-chlorotetrahydropyran in the presence of Ni catalyst (Method 2D).
![[1860-5397-7-49-i2]](/bjoc/content/inline/1860-5397-7-49-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: General methods for preparation of tetrahydropyrans VIII.
Scheme 2: General methods for preparation of tetrahydropyrans VIII.
Here, we report the preparation of dibromides 1 having at the 3-position propyl (1a), pentyl (1b), 2-(4-trans-pentylcyclohexyl)ethyl (1c), or 4-propylphenethyl (1d) substituents (Figure 2). We investigate the preparation of appropriate diols 2 starting from malonate (Method 1B, Scheme 1) and glutaconate diesters (Method 1C) as well as appropriate tetrahydropyrans 3 via Wittig olefination (Method 2B, Scheme 2). These routes promise the minimal number of steps and ease of chemical transformations.
![[1860-5397-7-49-2]](/bjoc/content/figures/1860-5397-7-49-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Structures of 1,5-dibromomopentanes 1a–1d.
Figure 2: Structures of 1,5-dibromomopentanes 1a–1d.
Results
Preparation of dibromides
Dibromides 1a–1d were prepared in 67–91% yield in refluxing 48% HBr with tributylhexadecylphosphonium bromide [44] as a phase-transfer catalyst either from diols 2a and 2b or tetrahydropyrans 3a–3d (Scheme 3). When H2SO4 was used instead of the phosphonium salt, the yield of 1b was lower (67%). The dibromides were purified on silica gel and, in addition, 1a and 1b were distilled.
![[1860-5397-7-49-i3]](/bjoc/content/inline/1860-5397-7-49-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Preparation of diols
Diols 2a and 2b, containing simple alkyl chains, were prepared using both the malonate and glutaconate routes, Method 1B and Method 1C, respectively (Scheme 1). The reaction of a 4-fold excess of propylmagnesium chloride with diethyl glutaconate in the presence of TMSCl with a catalytic amount of CuI generated diethyl 3-propylglutarate (4) in 79% yield. Reduction of 4 gave diol 2a in nearly quantitative yield (Scheme 4).
![[1860-5397-7-49-i4]](/bjoc/content/inline/1860-5397-7-49-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Reaction of hexanal with excess dimethyl malonate under Knoevenagel conditions gave dimethyl 3-pentylglutarate (5) in 57% yield after decarboxylation and esterification (Scheme 5). 3-Propylglutarate 4 was obtained in a similar way from butanal in 49% overall yield. Diester 5 was subsequently reduced to give diol 2b in 87% yield.
![[1860-5397-7-49-i5]](/bjoc/content/inline/1860-5397-7-49-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Preparation of tetrahydropyrans
Tetrahydropyrans 3a–3c were prepared in 82%–96% yield by hydrogenation of the corresponding 4-methylenetetrahydropyrans 6a–6c in THF (Scheme 6). Tetrahydropyrans 3a and 3b are volatile so care should be exercised during evaporation of solvent to ensure maximum yields.
![[1860-5397-7-49-i6]](/bjoc/content/inline/1860-5397-7-49-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Preparation of tetrahydropyrans 3a–3c.
Scheme 6: Preparation of tetrahydropyrans 3a–3c.
Negishi coupling of tetrahydropyran 3e with propylzinc chloride following a general literature method [45] using PEPPSI-IR as the Pd(0) source gave tetrahydropyran 3d in 69% yield. Tetrahydropyran 3e was prepared in 86% yield by hydrogenation of the corresponding 4-methylenetetrahydropyran 6e in the presence of ZnBr2 to prevent reductive dechlorination (Scheme 7) [46]. Attempts to use diphenyl sulfide as a catalyst poison [47] resulted only in recovery of the starting material.
![[1860-5397-7-49-i7]](/bjoc/content/inline/1860-5397-7-49-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Preparation of tetrahydropyran 3d.
Scheme 7: Preparation of tetrahydropyran 3d.
Wittig olefination of tetrahydro-4H-pyran-4-one gave substituted 4-methylenetetrahydropyrans 6a–6c and 6e in yields ranging from 20–40% (Scheme 8). Methylenetetrahydropyrans 6a and 6b are volatile compounds so again care should be taken during evaporation of solvent to maximize yields. Phosphoranes were generated using standard conditions by reacting phosphonium salts 7a–7c and 7e with either BuLi in anhydrous THF, or NaHMDS in a 1:1 mixture of anhydrous Et2O and anhydrous CH2Cl2.
![[1860-5397-7-49-i8]](/bjoc/content/inline/1860-5397-7-49-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Preparation of methylenetetrahydropyrans 6.
Scheme 8: Preparation of methylenetetrahydropyrans 6.
Heating the corresponding alkyl halide (Br or I) with PPh3 under reflux in concentrated solutions of toluene, benzene, or acetonitrile gave phosphonium salts 7a–7c and 7e. Starting from 4-chlorophenylacetic acid, 4-chlorophenethyl bromide (8) was prepared in 78% overall yield according to the literature method [48] (Scheme 9). The phosphonium salt 7c, an intermediate to dibromide 1c, was prepared starting from the known 2-(trans-4-pentylcyclohexyl)acetaldehyde (9) [49]. The aldehyde was reduced with NaBH4 and subsequently brominated with PBr3 to give 1-bromo-2-(trans-4-pentylcyclohexyl)ethane (10) in overall yield of 42% (Scheme 9). Bromide 10 has been reported in the literature, but experimental details are lacking [50,51].
![[1860-5397-7-49-i9]](/bjoc/content/inline/1860-5397-7-49-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Preparation of bromides 8 and 10.
Scheme 9: Preparation of bromides 8 and 10.
The utility of dibromides 1 was demonstrated by preparation of sulfonium derivatives 11. Thus, compound 11b was previously obtained in 39% yield (before recrystallization) by alkylative cyclization of masked mercaptan [closo-1-CB9H8-1-SCHNMe2-10-I] 12 with dibromide 1b in the presence of Me4N+OH−·5H2O in MeCN [25]. The same procedure gave 11c in 25% yield after recrystallization, when starting from dibromide 1c. Replacement of Me4N+OH−·5H2O with Cs2CO3 and a catalytic amount of Bu4N+Br− in analogous reactions with dibromides 1c and 1d led to the sulfonium derivatives 11c and 11d in higher isolated yields of about 35% (Scheme 10). The higher yield for the cyclization in the presence of Cs2CO3 is attributed to less dehydrobromination of the electrophile than observed with Me4N+OH−.
![[1860-5397-7-49-i10]](/bjoc/content/inline/1860-5397-7-49-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Preparation of sulfonium derivatives 11.
Scheme 10: Preparation of sulfonium derivatives 11.
Discussion
Evaluation of the syntheses described here reveals advantages and disadvantages of the methods used for preparation of diols VII and tetrahydropyrans VIII. Both VII and VIII can be efficiently converted to dibromides I using HBr and tributylhexadecylphosphonium bromide as the phase-transfer catalyst.
Diols VII are obtained by reduction of a 3-substituted glutaric acid or ester. The latter can be prepared in a single step from glutaconate diester and a Grignard reagent. However, this method is practically limited to simple 3-alkyl-1,5-pentanediols, such as 1a and 1b, since large excess of Grignard reagents is required. The second method, in which glutaric acids are obtained from aldehydes and malonate ester, is more general and suitable for large scale synthesis, but involves an additional step.
Tetrahydropyrans VIII can be prepared in two steps via Wittig olefination. The efficiency of this method is limited by the initial olefination step of tetrahyro-4H-pyran-4-one, where the yields range from 20–40%. This step could potentially be improved by using different reaction conditions or an excess of the ylide. The hydrogenation of the corresponding olefins is straightforward and proceeds in high yield. In the case where a Pd-sensitive group is present, such as a chlorine substituent in 6e, modification of the Pd catalyst with ZnBr2 permits efficient hydrogenation without loss of the halogen. The chlorophenyl derivative 3e is a convenient precursor to a variety of 4-alkylphenyl and 4-arylphenyl derivatives that can be obtained via Negishi or Suzuki coupling methods (i.e., 3d). Another such potentially general intermediate is 4-(4-hydroxyphenyl)tetrahydro-4H-pyran [31], which can be O-alkylated to provide 4-alkoxyphenyl derivatives [2].
Overall, we have demonstrated that dibromides I containing simple groups, such as propyl or pentyl, can be prepared in yields of about 70% starting from glutaconate diester (three steps, entry 2; Table 2), 36–38% from a malonate (four steps, entries 1 and 5), and on average 25% starting from tetrahydro-4H-pyran-4-one (three steps, entries 3 and 5). Dibromides I containing larger fragments such as 3-propylphenethyl or 2-(4-trans-pentylcyclohexyl)ethyl were prepared exclusively starting from tetrahydro-4H-pyran-4-one in overall yields averaging about 20% (three steps, entries 6 and 7). For comparison Table 2 includes the preparation of dibromides I (R = aryl) involving Heck-type arylation (entry 8) and dibromides I (R = alkyl) starting from tetrahydropyran-4-carboxylic acid (entry 9).
Table 2: Summary of dibromide 1 syntheses.
| Entry |
![[Graphic 6]](/bjoc/content/inline/1860-5397-7-49-i16.svg?max-width=637&scale=1.18182)
|
Route (Method) | Number of steps | Overall yield (%) |
|---|---|---|---|---|
| 1 | R = C3H7 (1a) | Malonate (1B) | 4 | 36 |
| 2 | R = C3H7 (1a) | Glutaconate (1C) | 3 | 68 |
| 3 | R = C3H7 (1a) | Pyranone (2B) | 3 | 17 |
| 4 | R = C5H11 (1b) | Malonate (1B) | 4 | 38 |
| 5 | R = C5H11 (1b) | Pyranone (2B) | 3 | 33 |
| 6 | R = C5H11C6H10CH2CH2 (1c) | Pyranone (2B) | 3 | 20 |
| 7 | R = C3H7C6H4CH2CH2 (1d) | Pyranone (2B) | 4 | 18a |
| 8 | R = C5H11C6H4 | Glutaconate (1D) | 3 | 26b |
| 9 | R = C3H7–C10H21 | 4-COOH-pyran (2C) | 4 | 40c |
a4th step includes replacement of Cl in Ar–Cl with propyl by Pd-catalyzed coupling reaction. Excluding this step the yield for the dibromide would have been 17% over three steps. bIsolated as the ditosylate Ref. [19] cRef. [21].
It appears that the most economical way to prepare simple 3-alkyl-1,5-dibromopentanes is by using the aldehyde/malonate or aldehyde/cyanoacetamide method (Method 1B, Scheme 1), while the glutaconate method (Method 1C) is the simplest and most efficient. For the preparation of 3-aryl derivatives, the most direct methods are the Heck-type coupling with glutaconate ester (Method 1D) and Suzuki-type coupling to 4-chlorotetrahydropyran (Method 2D).
The preparation of dibromides I containing larger organic fragments from tetrahydropyrans VIII is inefficient and proceeds with low overall yields (Method 2B, Scheme 2). Attempts at conserving the alkylating reagent by using a stoichiometric Wittig olefination rather than conjugate addition of excess Grignard reagent to glutaconate did not give the expected result. It is possible that the aldehyde/cyanoacetamide method (Method 1B) [36] could prove advantageous and provide a synthesis which is more economical with respect to the alkyl substrate.
Conclusion
Overall, several routes to dibromides I and ditosylates II were reviewed and investigated experimentally. These studies open the way for rational syntheses of other, specifically designed dielectrophiles that are important intermediates in the preparation of polar liquid crystals. In this context, we demonstrated the preparation of sulfonium derivatives 11c and 11d from dibromides 1c and 1d as precursors to such compounds. Other examples will be reported elsewhere.
Supporting Information
| Supporting Information File 1: General methods and synthetic procedures. | ||
| Format: PDF | Size: 309.5 KB | Download |
References
-
Adomenas, P.; Sirutkaitis, R. Mol. Cryst. Liq. Cryst. 1985, 124, 269–275. doi:10.1080/00268948508079482
Return to citation in text: [1] -
Karamysheva, L. A.; Kovshev, E. I.; Pavluchenko, A. I.; Roitman, K. V.; Titov, V. V.; Torgova, S. I.; Grebenkin, M. F. Mol. Cryst. Liq. Cryst. 1981, 67, 241–252. doi:10.1080/00268948108070894
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] -
Sheikh-Ali, B. M.; Weiss, R. G. Liq. Cryst. 1991, 10, 575–580. doi:10.1080/02678299108036444
Return to citation in text: [1] -
Sheikh-Ali, B. M.; Weiss, R. G. Liq. Cryst. 1994, 17, 605–615. doi:10.1080/02678299408037332
Return to citation in text: [1] -
Sucrow, W.; Schatull, W. Z. Naturforsch., B 1982, 37B, 1336–1338.
Return to citation in text: [1] -
Tournilhac, F.; Nicoud, J. F.; Simon, J.; Weber, P.; Guillon, D.; Skoulios, A. Liq. Cryst. 1987, 2, 55–61. doi:10.1080/02678298708086637
Return to citation in text: [1] -
Pfefferkorn, J. A.; Choi, C.; Winters, T.; Kennedy, R.; Chi, L.; Perrin, L. A.; Lu, G.; Ping, Y.-W.; McClanahan, T.; Schroeder, R.; Leininger, M. T.; Geyer, A.; Schefzick, S.; Atherton, J. Bioorg. Med. Chem. Lett. 2008, 18, 3338–3343. doi:10.1016/j.bmcl.2008.04.028
Return to citation in text: [1] [2] [3] [4] -
Mantegani, S.; Arlandini, E.; Brambilla, E.; Cremonesi, P.; Varasi, M. Synth. Commun. 2000, 30, 3543–3553. doi:10.1080/00397910008087268
Return to citation in text: [1] -
Whitehead, E. V.; Dean, R. A.; Fidler, F. A. J. Am. Chem. Soc. 1951, 73, 3632–3635. doi:10.1021/ja01152a022
Return to citation in text: [1] [2] [3] [4] [5] -
Volynskii, N. P.; Shcherbakova, L. P. Bull. Acad. Sci. USSR, Div. Chem. Sci. (Engl. Transl.) 1979, 1006–1009.
Return to citation in text: [1] [2] [3] [4] -
Johnson, C. R. J. Am. Chem. Soc. 1963, 85, 1020–1021. doi:10.1021/ja00890a056
Return to citation in text: [1] [2] [3] -
Johnson, C. R.; McCants, D., Jr. J. Am. Chem. Soc. 1965, 87, 1109–1114. doi:10.1021/ja01083a030
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Halfpenny, P. J.; Johnson, P. J.; Robinson, M. J. T.; Ward, M. G. Tetrahedron 1976, 32, 1873–1879. doi:10.1016/0040-4020(76)85189-7
Return to citation in text: [1] [2] [3] [4] [5] -
Nguyen, B. T.; Cartledge, F. K. J. Org. Chem. 1986, 51, 2206–2210. doi:10.1021/jo00362a009
Return to citation in text: [1] [2] [3] [4] -
Quin, L. D.; Lee, S. O. J. Org. Chem. 1978, 43, 1424–1429. doi:10.1021/jo00401a029
Return to citation in text: [1] [2] [3] -
Marsi, K. L.; Jasperse, J. L.; Llort, F. M.; Kanne, D. B. J. Org. Chem. 1977, 42, 1306–1311. doi:10.1021/jo00428a008
Return to citation in text: [1] -
Chang, C.-S.; Lin, Y.-T.; Shih, S.-R.; Lee, C.-C.; Lee, Y.-C.; Tai, C.-L.; Tseng, S.-N.; Chern, J.-H. J. Med. Chem. 2005, 48, 3522–3535. doi:10.1021/jm050033v
Return to citation in text: [1] [2] [3] [4] -
Cope, A. C.; Cotter, R. J. J. Org. Chem. 1964, 29, 3467–3469. doi:10.1021/jo01035a005
Return to citation in text: [1] [2] [3] -
Kaszynski, P.; Huang, J.; Jenkins, G. S.; Bairamov, K. A.; Lipiak, D. Mol. Cryst. Liq. Cryst. 1995, 260, 315–332. doi:10.1080/10587259508038705
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Allinger, N. L.; Neumann, C. L.; Sugiyama, H. J. Org. Chem. 1971, 36, 1360–1365. doi:10.1021/jo00809a010
Return to citation in text: [1] [2] [3] -
Thomas, J.; Clough, D. J. Pharm. Pharmacol. 1963, 15, 167–177.
Return to citation in text: [1] [2] [3] [4] [5] -
Stapp, P. R.; Drake, C. A. J. Org. Chem. 1971, 36, 522–525. doi:10.1021/jo00803a007
Return to citation in text: [1] [2] [3] [4] -
Bairamov, K. A.; Douglass, A. G.; Kaszynski, P. Synth. Commun. 1998, 28, 527–540. doi:10.1080/00397919808005108
Return to citation in text: [1] -
Ringstrand, B.; Kaszynski, P. J. Mater. Chem. 2011, 21, 90–95. doi:10.1039/c0jm02075c
Return to citation in text: [1] -
Ringstrand, B.; Kaszynski, P.; Januszko, A.; Young, V. G., Jr. J. Mater. Chem. 2009, 19, 9204–9212. doi:10.1039/b913701g
Return to citation in text: [1] [2] -
Allinger, N. L.; Greenberg, S. J. Am. Chem. Soc. 1959, 81, 5733–5736. doi:10.1021/ja01530a050
Return to citation in text: [1] [2] -
Irwin, A. J.; Lok, K. P.; Huang, K. W.-C.; Jones, J. B. J. Chem. Soc., Perkin Trans. 1 1978, 1636–1642. doi:10.1039/P19780001636
Return to citation in text: [1] [2] -
Jones, J. B.; Lok, K. P. Can. J. Chem. 1979, 57, 1025–1032. doi:10.1139/v79-170
Return to citation in text: [1] [2] -
Sarges, R.; Hank, R. F.; Blake, J. F.; Bordner, J.; Bussolotti, D. L.; Hargrove, D. M.; Treadway, J. L.; Gibbs, E. M. J. Med. Chem. 1996, 39, 4783–4803. doi:10.1021/jm950364f
Return to citation in text: [1] -
Onesta, R.; Castelfranchi, G. Gazz. Chim. Ital. 1959, 89, 1127–1138.
Return to citation in text: [1] -
Koyama, H.; Boueres, J. K.; Miller, D. J.; Berger, J. P.; MacNaul, K. L.; Wang, P.-R.; Ippolito, M. C.; Wright, S. D.; Agrawal, A. K.; Moller, D. E.; Sahoo, S. P. Bioorg. Med. Chem. Lett. 2005, 15, 3347–3351. doi:10.1016/j.bmcl.2005.05.028
Return to citation in text: [1] [2] -
González-Bobes, F.; Fu, G. C. J. Am. Chem. Soc. 2006, 128, 5360–5361. doi:10.1021/ja0613761
Return to citation in text: [1] [2] -
Boots, M. R.; Yeh, Y.-M.; Boots, S. G. J. Pharm. Sci. 1980, 69, 506–509. doi:10.1002/jps.2600690507
Return to citation in text: [1] -
Farmer, E. H.; Martin, S. R. W. J. Chem. Soc. 1933, 960–962. doi:10.1039/jr9330000960
Return to citation in text: [1] -
Curtis, R. H.; Day, J. N. E.; Kimmins, L. G. J. Chem. Soc., Trans. 1923, 123, 3131–3140. doi:10.1039/CT9232303131
Return to citation in text: [1] -
Day, J. N. E.; Thorpe, J. F. J. Chem. Soc., Trans. 1920, 117, 1465–1474. doi:10.1039/CT9201701465
Return to citation in text: [1] [2] -
Hedge, J. A.; Kruse, C. W.; Snyder, H. R. J. Org. Chem. 1961, 26, 3166–3170. doi:10.1021/jo01067a032
Return to citation in text: [1] -
Leotta, G. J., III; Overman, L. E.; Welmaker, G. S. J. Org. Chem. 1994, 59, 1946. doi:10.1021/jo00086a065
Return to citation in text: [1] [2] -
Booth, H.; Khedhair, K. A.; Readshaw, S. A. Tetrahedron 1987, 43, 4699–4723. doi:10.1016/S0040-4020(01)86912-X
Return to citation in text: [1] -
Chini, M.; Crotti, P.; Gardelli, C.; Macchia, F. Tetrahedron 1994, 50, 1261–1274. doi:10.1016/S0040-4020(01)80836-X
Return to citation in text: [1] -
Margot, C.; Rizzolio, M.; Schlosser, M. Tetrahedron 1990, 46, 2411–2424. doi:10.1016/S0040-4020(01)82022-6
Return to citation in text: [1] -
Kabat, M. M.; Lange, M.; Wovkulich, P. M.; Usokovic, M. R. Tetrahedron Lett. 1992, 33, 7701–7704. doi:10.1016/0040-4039(93)88021-A
Return to citation in text: [1] -
Angelastro, M. R.; Baugh, L. E.; Bey, P.; Burkhart, J. P.; Chen, T.-M.; Durham, S. L.; Hare, C. M.; Huber, E. W.; Janusz, M. J.; Koehl, J. R.; Marquart, A. L.; Mehdi, S.; Peet, N. P. J. Med. Chem. 1994, 37, 4538–4553. doi:10.1021/jm00052a013
Return to citation in text: [1] -
Landini, D.; Montanari, F.; Rolla, F. Synthesis 1978, 771–773. doi:10.1055/s-1978-24888
Return to citation in text: [1] -
Organ, M. G.; Avola, S.; Dubovyk, I.; Hadei, N.; Kantchev, E. A. B.; O'Brien, C. J.; Valente, C. Chem.–Eur. J. 2006, 12, 4749–4755. doi:10.1002/chem.200600206
Return to citation in text: [1] -
Wu, G.; Huang, M.; Richards, M.; Poirier, M.; Wen, X.; Draper, R. W. Synthesis 2003, 1657–1660. doi:10.1055/s-2003-40878
Return to citation in text: [1] -
Mori, A.; Mizusaki, T.; Kawase, M.; Maegawa, T.; Monguchi, Y.; Takao, S.; Takagi, Y.; Sajiki, H. Adv. Synth. Catal. 2008, 350, 406–410. doi:10.1002/adsc.200700571
Return to citation in text: [1] -
Lambert, J. B.; Mark, H. W.; Magyar, E. S. J. Am. Chem. Soc. 1977, 99, 3059–3067. doi:10.1021/ja00451a035
Return to citation in text: [1] -
Jankowiak, A.; Januszko, A.; Ringstrand, B.; Kaszynski, P. Liq. Cryst. 2008, 35, 65–77. doi:10.1080/02678290701744561
Return to citation in text: [1] -
Dąbrowski, R.; Dziaduszek, J.; Szczuciński, T.; Parka, J. Mol. Cryst. Liq. Cryst. 1995, 260, 201–215. doi:10.1080/10587259508038692
Return to citation in text: [1] -
Fearon, J. E.; Gray, G. W.; Ifill, A. D.; Toyne, K. J. Mol. Cryst. Liq. Cryst. 1985, 124, 89–103. doi:10.1080/00268948508079467
Return to citation in text: [1]
| 38. | Leotta, G. J., III; Overman, L. E.; Welmaker, G. S. J. Org. Chem. 1994, 59, 1946. doi:10.1021/jo00086a065 |
| 17. | Chang, C.-S.; Lin, Y.-T.; Shih, S.-R.; Lee, C.-C.; Lee, Y.-C.; Tai, C.-L.; Tseng, S.-N.; Chern, J.-H. J. Med. Chem. 2005, 48, 3522–3535. doi:10.1021/jm050033v |
| 38. | Leotta, G. J., III; Overman, L. E.; Welmaker, G. S. J. Org. Chem. 1994, 59, 1946. doi:10.1021/jo00086a065 |
| 19. | Kaszynski, P.; Huang, J.; Jenkins, G. S.; Bairamov, K. A.; Lipiak, D. Mol. Cryst. Liq. Cryst. 1995, 260, 315–332. doi:10.1080/10587259508038705 |
| 1. | Adomenas, P.; Sirutkaitis, R. Mol. Cryst. Liq. Cryst. 1985, 124, 269–275. doi:10.1080/00268948508079482 |
| 2. | Karamysheva, L. A.; Kovshev, E. I.; Pavluchenko, A. I.; Roitman, K. V.; Titov, V. V.; Torgova, S. I.; Grebenkin, M. F. Mol. Cryst. Liq. Cryst. 1981, 67, 241–252. doi:10.1080/00268948108070894 |
| 3. | Sheikh-Ali, B. M.; Weiss, R. G. Liq. Cryst. 1991, 10, 575–580. doi:10.1080/02678299108036444 |
| 4. | Sheikh-Ali, B. M.; Weiss, R. G. Liq. Cryst. 1994, 17, 605–615. doi:10.1080/02678299408037332 |
| 5. | Sucrow, W.; Schatull, W. Z. Naturforsch., B 1982, 37B, 1336–1338. |
| 6. | Tournilhac, F.; Nicoud, J. F.; Simon, J.; Weber, P.; Guillon, D.; Skoulios, A. Liq. Cryst. 1987, 2, 55–61. doi:10.1080/02678298708086637 |
| 11. | Johnson, C. R. J. Am. Chem. Soc. 1963, 85, 1020–1021. doi:10.1021/ja00890a056 |
| 12. | Johnson, C. R.; McCants, D., Jr. J. Am. Chem. Soc. 1965, 87, 1109–1114. doi:10.1021/ja01083a030 |
| 9. | Whitehead, E. V.; Dean, R. A.; Fidler, F. A. J. Am. Chem. Soc. 1951, 73, 3632–3635. doi:10.1021/ja01152a022 |
| 10. | Volynskii, N. P.; Shcherbakova, L. P. Bull. Acad. Sci. USSR, Div. Chem. Sci. (Engl. Transl.) 1979, 1006–1009. |
| 12. | Johnson, C. R.; McCants, D., Jr. J. Am. Chem. Soc. 1965, 87, 1109–1114. doi:10.1021/ja01083a030 |
| 13. | Halfpenny, P. J.; Johnson, P. J.; Robinson, M. J. T.; Ward, M. G. Tetrahedron 1976, 32, 1873–1879. doi:10.1016/0040-4020(76)85189-7 |
| 46. | Wu, G.; Huang, M.; Richards, M.; Poirier, M.; Wen, X.; Draper, R. W. Synthesis 2003, 1657–1660. doi:10.1055/s-2003-40878 |
| 7. | Pfefferkorn, J. A.; Choi, C.; Winters, T.; Kennedy, R.; Chi, L.; Perrin, L. A.; Lu, G.; Ping, Y.-W.; McClanahan, T.; Schroeder, R.; Leininger, M. T.; Geyer, A.; Schefzick, S.; Atherton, J. Bioorg. Med. Chem. Lett. 2008, 18, 3338–3343. doi:10.1016/j.bmcl.2008.04.028 |
| 10. | Volynskii, N. P.; Shcherbakova, L. P. Bull. Acad. Sci. USSR, Div. Chem. Sci. (Engl. Transl.) 1979, 1006–1009. |
| 11. | Johnson, C. R. J. Am. Chem. Soc. 1963, 85, 1020–1021. doi:10.1021/ja00890a056 |
| 14. | Nguyen, B. T.; Cartledge, F. K. J. Org. Chem. 1986, 51, 2206–2210. doi:10.1021/jo00362a009 |
| 47. | Mori, A.; Mizusaki, T.; Kawase, M.; Maegawa, T.; Monguchi, Y.; Takao, S.; Takagi, Y.; Sajiki, H. Adv. Synth. Catal. 2008, 350, 406–410. doi:10.1002/adsc.200700571 |
| 9. | Whitehead, E. V.; Dean, R. A.; Fidler, F. A. J. Am. Chem. Soc. 1951, 73, 3632–3635. doi:10.1021/ja01152a022 |
| 12. | Johnson, C. R.; McCants, D., Jr. J. Am. Chem. Soc. 1965, 87, 1109–1114. doi:10.1021/ja01083a030 |
| 14. | Nguyen, B. T.; Cartledge, F. K. J. Org. Chem. 1986, 51, 2206–2210. doi:10.1021/jo00362a009 |
| 15. | Quin, L. D.; Lee, S. O. J. Org. Chem. 1978, 43, 1424–1429. doi:10.1021/jo00401a029 |
| 44. | Landini, D.; Montanari, F.; Rolla, F. Synthesis 1978, 771–773. doi:10.1055/s-1978-24888 |
| 7. | Pfefferkorn, J. A.; Choi, C.; Winters, T.; Kennedy, R.; Chi, L.; Perrin, L. A.; Lu, G.; Ping, Y.-W.; McClanahan, T.; Schroeder, R.; Leininger, M. T.; Geyer, A.; Schefzick, S.; Atherton, J. Bioorg. Med. Chem. Lett. 2008, 18, 3338–3343. doi:10.1016/j.bmcl.2008.04.028 |
| 8. | Mantegani, S.; Arlandini, E.; Brambilla, E.; Cremonesi, P.; Varasi, M. Synth. Commun. 2000, 30, 3543–3553. doi:10.1080/00397910008087268 |
| 2. | Karamysheva, L. A.; Kovshev, E. I.; Pavluchenko, A. I.; Roitman, K. V.; Titov, V. V.; Torgova, S. I.; Grebenkin, M. F. Mol. Cryst. Liq. Cryst. 1981, 67, 241–252. doi:10.1080/00268948108070894 |
| 22. | Stapp, P. R.; Drake, C. A. J. Org. Chem. 1971, 36, 522–525. doi:10.1021/jo00803a007 |
| 45. | Organ, M. G.; Avola, S.; Dubovyk, I.; Hadei, N.; Kantchev, E. A. B.; O'Brien, C. J.; Valente, C. Chem.–Eur. J. 2006, 12, 4749–4755. doi:10.1002/chem.200600206 |
| 2. | Karamysheva, L. A.; Kovshev, E. I.; Pavluchenko, A. I.; Roitman, K. V.; Titov, V. V.; Torgova, S. I.; Grebenkin, M. F. Mol. Cryst. Liq. Cryst. 1981, 67, 241–252. doi:10.1080/00268948108070894 |
| 2. | Karamysheva, L. A.; Kovshev, E. I.; Pavluchenko, A. I.; Roitman, K. V.; Titov, V. V.; Torgova, S. I.; Grebenkin, M. F. Mol. Cryst. Liq. Cryst. 1981, 67, 241–252. doi:10.1080/00268948108070894 |
| 12. | Johnson, C. R.; McCants, D., Jr. J. Am. Chem. Soc. 1965, 87, 1109–1114. doi:10.1021/ja01083a030 |
| 21. | Thomas, J.; Clough, D. J. Pharm. Pharmacol. 1963, 15, 167–177. |
| 22. | Stapp, P. R.; Drake, C. A. J. Org. Chem. 1971, 36, 522–525. doi:10.1021/jo00803a007 |
| 15. | Quin, L. D.; Lee, S. O. J. Org. Chem. 1978, 43, 1424–1429. doi:10.1021/jo00401a029 |
| 16. | Marsi, K. L.; Jasperse, J. L.; Llort, F. M.; Kanne, D. B. J. Org. Chem. 1977, 42, 1306–1311. doi:10.1021/jo00428a008 |
| 23. | Bairamov, K. A.; Douglass, A. G.; Kaszynski, P. Synth. Commun. 1998, 28, 527–540. doi:10.1080/00397919808005108 |
| 32. | González-Bobes, F.; Fu, G. C. J. Am. Chem. Soc. 2006, 128, 5360–5361. doi:10.1021/ja0613761 |
| 14. | Nguyen, B. T.; Cartledge, F. K. J. Org. Chem. 1986, 51, 2206–2210. doi:10.1021/jo00362a009 |
| 39. | Booth, H.; Khedhair, K. A.; Readshaw, S. A. Tetrahedron 1987, 43, 4699–4723. doi:10.1016/S0040-4020(01)86912-X |
| 40. | Chini, M.; Crotti, P.; Gardelli, C.; Macchia, F. Tetrahedron 1994, 50, 1261–1274. doi:10.1016/S0040-4020(01)80836-X |
| 13. | Halfpenny, P. J.; Johnson, P. J.; Robinson, M. J. T.; Ward, M. G. Tetrahedron 1976, 32, 1873–1879. doi:10.1016/0040-4020(76)85189-7 |
| 13. | Halfpenny, P. J.; Johnson, P. J.; Robinson, M. J. T.; Ward, M. G. Tetrahedron 1976, 32, 1873–1879. doi:10.1016/0040-4020(76)85189-7 |
| 17. | Chang, C.-S.; Lin, Y.-T.; Shih, S.-R.; Lee, C.-C.; Lee, Y.-C.; Tai, C.-L.; Tseng, S.-N.; Chern, J.-H. J. Med. Chem. 2005, 48, 3522–3535. doi:10.1021/jm050033v |
| 18. | Cope, A. C.; Cotter, R. J. J. Org. Chem. 1964, 29, 3467–3469. doi:10.1021/jo01035a005 |
| 19. | Kaszynski, P.; Huang, J.; Jenkins, G. S.; Bairamov, K. A.; Lipiak, D. Mol. Cryst. Liq. Cryst. 1995, 260, 315–332. doi:10.1080/10587259508038705 |
| 20. | Allinger, N. L.; Neumann, C. L.; Sugiyama, H. J. Org. Chem. 1971, 36, 1360–1365. doi:10.1021/jo00809a010 |
| 41. | Margot, C.; Rizzolio, M.; Schlosser, M. Tetrahedron 1990, 46, 2411–2424. doi:10.1016/S0040-4020(01)82022-6 |
| 42. | Kabat, M. M.; Lange, M.; Wovkulich, P. M.; Usokovic, M. R. Tetrahedron Lett. 1992, 33, 7701–7704. doi:10.1016/0040-4039(93)88021-A |
| 43. | Angelastro, M. R.; Baugh, L. E.; Bey, P.; Burkhart, J. P.; Chen, T.-M.; Durham, S. L.; Hare, C. M.; Huber, E. W.; Janusz, M. J.; Koehl, J. R.; Marquart, A. L.; Mehdi, S.; Peet, N. P. J. Med. Chem. 1994, 37, 4538–4553. doi:10.1021/jm00052a013 |
| 2. | Karamysheva, L. A.; Kovshev, E. I.; Pavluchenko, A. I.; Roitman, K. V.; Titov, V. V.; Torgova, S. I.; Grebenkin, M. F. Mol. Cryst. Liq. Cryst. 1981, 67, 241–252. doi:10.1080/00268948108070894 |
| 9. | Whitehead, E. V.; Dean, R. A.; Fidler, F. A. J. Am. Chem. Soc. 1951, 73, 3632–3635. doi:10.1021/ja01152a022 |
| 10. | Volynskii, N. P.; Shcherbakova, L. P. Bull. Acad. Sci. USSR, Div. Chem. Sci. (Engl. Transl.) 1979, 1006–1009. |
| 12. | Johnson, C. R.; McCants, D., Jr. J. Am. Chem. Soc. 1965, 87, 1109–1114. doi:10.1021/ja01083a030 |
| 14. | Nguyen, B. T.; Cartledge, F. K. J. Org. Chem. 1986, 51, 2206–2210. doi:10.1021/jo00362a009 |
| 21. | Thomas, J.; Clough, D. J. Pharm. Pharmacol. 1963, 15, 167–177. |
| 22. | Stapp, P. R.; Drake, C. A. J. Org. Chem. 1971, 36, 522–525. doi:10.1021/jo00803a007 |
| 26. | Allinger, N. L.; Greenberg, S. J. Am. Chem. Soc. 1959, 81, 5733–5736. doi:10.1021/ja01530a050 |
| 15. | Quin, L. D.; Lee, S. O. J. Org. Chem. 1978, 43, 1424–1429. doi:10.1021/jo00401a029 |
| 48. | Lambert, J. B.; Mark, H. W.; Magyar, E. S. J. Am. Chem. Soc. 1977, 99, 3059–3067. doi:10.1021/ja00451a035 |
| 24. | Ringstrand, B.; Kaszynski, P. J. Mater. Chem. 2011, 21, 90–95. doi:10.1039/c0jm02075c |
| 25. | Ringstrand, B.; Kaszynski, P.; Januszko, A.; Young, V. G., Jr. J. Mater. Chem. 2009, 19, 9204–9212. doi:10.1039/b913701g |
| 49. | Jankowiak, A.; Januszko, A.; Ringstrand, B.; Kaszynski, P. Liq. Cryst. 2008, 35, 65–77. doi:10.1080/02678290701744561 |
| 50. | Dąbrowski, R.; Dziaduszek, J.; Szczuciński, T.; Parka, J. Mol. Cryst. Liq. Cryst. 1995, 260, 201–215. doi:10.1080/10587259508038692 |
| 51. | Fearon, J. E.; Gray, G. W.; Ifill, A. D.; Toyne, K. J. Mol. Cryst. Liq. Cryst. 1985, 124, 89–103. doi:10.1080/00268948508079467 |
| 34. | Farmer, E. H.; Martin, S. R. W. J. Chem. Soc. 1933, 960–962. doi:10.1039/jr9330000960 |
| 35. | Curtis, R. H.; Day, J. N. E.; Kimmins, L. G. J. Chem. Soc., Trans. 1923, 123, 3131–3140. doi:10.1039/CT9232303131 |
| 36. | Day, J. N. E.; Thorpe, J. F. J. Chem. Soc., Trans. 1920, 117, 1465–1474. doi:10.1039/CT9201701465 |
| 19. | Kaszynski, P.; Huang, J.; Jenkins, G. S.; Bairamov, K. A.; Lipiak, D. Mol. Cryst. Liq. Cryst. 1995, 260, 315–332. doi:10.1080/10587259508038705 |
| 37. | Hedge, J. A.; Kruse, C. W.; Snyder, H. R. J. Org. Chem. 1961, 26, 3166–3170. doi:10.1021/jo01067a032 |
| 27. | Irwin, A. J.; Lok, K. P.; Huang, K. W.-C.; Jones, J. B. J. Chem. Soc., Perkin Trans. 1 1978, 1636–1642. doi:10.1039/P19780001636 |
| 28. | Jones, J. B.; Lok, K. P. Can. J. Chem. 1979, 57, 1025–1032. doi:10.1139/v79-170 |
| 19. | Kaszynski, P.; Huang, J.; Jenkins, G. S.; Bairamov, K. A.; Lipiak, D. Mol. Cryst. Liq. Cryst. 1995, 260, 315–332. doi:10.1080/10587259508038705 |
| 33. | Boots, M. R.; Yeh, Y.-M.; Boots, S. G. J. Pharm. Sci. 1980, 69, 506–509. doi:10.1002/jps.2600690507 |
| 36. | Day, J. N. E.; Thorpe, J. F. J. Chem. Soc., Trans. 1920, 117, 1465–1474. doi:10.1039/CT9201701465 |
| 7. | Pfefferkorn, J. A.; Choi, C.; Winters, T.; Kennedy, R.; Chi, L.; Perrin, L. A.; Lu, G.; Ping, Y.-W.; McClanahan, T.; Schroeder, R.; Leininger, M. T.; Geyer, A.; Schefzick, S.; Atherton, J. Bioorg. Med. Chem. Lett. 2008, 18, 3338–3343. doi:10.1016/j.bmcl.2008.04.028 |
| 9. | Whitehead, E. V.; Dean, R. A.; Fidler, F. A. J. Am. Chem. Soc. 1951, 73, 3632–3635. doi:10.1021/ja01152a022 |
| 10. | Volynskii, N. P.; Shcherbakova, L. P. Bull. Acad. Sci. USSR, Div. Chem. Sci. (Engl. Transl.) 1979, 1006–1009. |
| 11. | Johnson, C. R. J. Am. Chem. Soc. 1963, 85, 1020–1021. doi:10.1021/ja00890a056 |
| 12. | Johnson, C. R.; McCants, D., Jr. J. Am. Chem. Soc. 1965, 87, 1109–1114. doi:10.1021/ja01083a030 |
| 13. | Halfpenny, P. J.; Johnson, P. J.; Robinson, M. J. T.; Ward, M. G. Tetrahedron 1976, 32, 1873–1879. doi:10.1016/0040-4020(76)85189-7 |
| 29. | Sarges, R.; Hank, R. F.; Blake, J. F.; Bordner, J.; Bussolotti, D. L.; Hargrove, D. M.; Treadway, J. L.; Gibbs, E. M. J. Med. Chem. 1996, 39, 4783–4803. doi:10.1021/jm950364f |
| 30. | Onesta, R.; Castelfranchi, G. Gazz. Chim. Ital. 1959, 89, 1127–1138. |
| 2. | Karamysheva, L. A.; Kovshev, E. I.; Pavluchenko, A. I.; Roitman, K. V.; Titov, V. V.; Torgova, S. I.; Grebenkin, M. F. Mol. Cryst. Liq. Cryst. 1981, 67, 241–252. doi:10.1080/00268948108070894 |
| 2. | Karamysheva, L. A.; Kovshev, E. I.; Pavluchenko, A. I.; Roitman, K. V.; Titov, V. V.; Torgova, S. I.; Grebenkin, M. F. Mol. Cryst. Liq. Cryst. 1981, 67, 241–252. doi:10.1080/00268948108070894 |
| 7. | Pfefferkorn, J. A.; Choi, C.; Winters, T.; Kennedy, R.; Chi, L.; Perrin, L. A.; Lu, G.; Ping, Y.-W.; McClanahan, T.; Schroeder, R.; Leininger, M. T.; Geyer, A.; Schefzick, S.; Atherton, J. Bioorg. Med. Chem. Lett. 2008, 18, 3338–3343. doi:10.1016/j.bmcl.2008.04.028 |
| 12. | Johnson, C. R.; McCants, D., Jr. J. Am. Chem. Soc. 1965, 87, 1109–1114. doi:10.1021/ja01083a030 |
| 21. | Thomas, J.; Clough, D. J. Pharm. Pharmacol. 1963, 15, 167–177. |
| 22. | Stapp, P. R.; Drake, C. A. J. Org. Chem. 1971, 36, 522–525. doi:10.1021/jo00803a007 |
| 31. | Koyama, H.; Boueres, J. K.; Miller, D. J.; Berger, J. P.; MacNaul, K. L.; Wang, P.-R.; Ippolito, M. C.; Wright, S. D.; Agrawal, A. K.; Moller, D. E.; Sahoo, S. P. Bioorg. Med. Chem. Lett. 2005, 15, 3347–3351. doi:10.1016/j.bmcl.2005.05.028 |
| 32. | González-Bobes, F.; Fu, G. C. J. Am. Chem. Soc. 2006, 128, 5360–5361. doi:10.1021/ja0613761 |
| 19. | Kaszynski, P.; Huang, J.; Jenkins, G. S.; Bairamov, K. A.; Lipiak, D. Mol. Cryst. Liq. Cryst. 1995, 260, 315–332. doi:10.1080/10587259508038705 |
| 13. | Halfpenny, P. J.; Johnson, P. J.; Robinson, M. J. T.; Ward, M. G. Tetrahedron 1976, 32, 1873–1879. doi:10.1016/0040-4020(76)85189-7 |
| 17. | Chang, C.-S.; Lin, Y.-T.; Shih, S.-R.; Lee, C.-C.; Lee, Y.-C.; Tai, C.-L.; Tseng, S.-N.; Chern, J.-H. J. Med. Chem. 2005, 48, 3522–3535. doi:10.1021/jm050033v |
| 18. | Cope, A. C.; Cotter, R. J. J. Org. Chem. 1964, 29, 3467–3469. doi:10.1021/jo01035a005 |
| 19. | Kaszynski, P.; Huang, J.; Jenkins, G. S.; Bairamov, K. A.; Lipiak, D. Mol. Cryst. Liq. Cryst. 1995, 260, 315–332. doi:10.1080/10587259508038705 |
| 20. | Allinger, N. L.; Neumann, C. L.; Sugiyama, H. J. Org. Chem. 1971, 36, 1360–1365. doi:10.1021/jo00809a010 |
| 25. | Ringstrand, B.; Kaszynski, P.; Januszko, A.; Young, V. G., Jr. J. Mater. Chem. 2009, 19, 9204–9212. doi:10.1039/b913701g |
| 2. | Karamysheva, L. A.; Kovshev, E. I.; Pavluchenko, A. I.; Roitman, K. V.; Titov, V. V.; Torgova, S. I.; Grebenkin, M. F. Mol. Cryst. Liq. Cryst. 1981, 67, 241–252. doi:10.1080/00268948108070894 |
| 9. | Whitehead, E. V.; Dean, R. A.; Fidler, F. A. J. Am. Chem. Soc. 1951, 73, 3632–3635. doi:10.1021/ja01152a022 |
| 17. | Chang, C.-S.; Lin, Y.-T.; Shih, S.-R.; Lee, C.-C.; Lee, Y.-C.; Tai, C.-L.; Tseng, S.-N.; Chern, J.-H. J. Med. Chem. 2005, 48, 3522–3535. doi:10.1021/jm050033v |
| 18. | Cope, A. C.; Cotter, R. J. J. Org. Chem. 1964, 29, 3467–3469. doi:10.1021/jo01035a005 |
| 19. | Kaszynski, P.; Huang, J.; Jenkins, G. S.; Bairamov, K. A.; Lipiak, D. Mol. Cryst. Liq. Cryst. 1995, 260, 315–332. doi:10.1080/10587259508038705 |
| 20. | Allinger, N. L.; Neumann, C. L.; Sugiyama, H. J. Org. Chem. 1971, 36, 1360–1365. doi:10.1021/jo00809a010 |
| 26. | Allinger, N. L.; Greenberg, S. J. Am. Chem. Soc. 1959, 81, 5733–5736. doi:10.1021/ja01530a050 |
| 27. | Irwin, A. J.; Lok, K. P.; Huang, K. W.-C.; Jones, J. B. J. Chem. Soc., Perkin Trans. 1 1978, 1636–1642. doi:10.1039/P19780001636 |
| 28. | Jones, J. B.; Lok, K. P. Can. J. Chem. 1979, 57, 1025–1032. doi:10.1139/v79-170 |
| 31. | Koyama, H.; Boueres, J. K.; Miller, D. J.; Berger, J. P.; MacNaul, K. L.; Wang, P.-R.; Ippolito, M. C.; Wright, S. D.; Agrawal, A. K.; Moller, D. E.; Sahoo, S. P. Bioorg. Med. Chem. Lett. 2005, 15, 3347–3351. doi:10.1016/j.bmcl.2005.05.028 |
© 2011 Ringstrand et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)