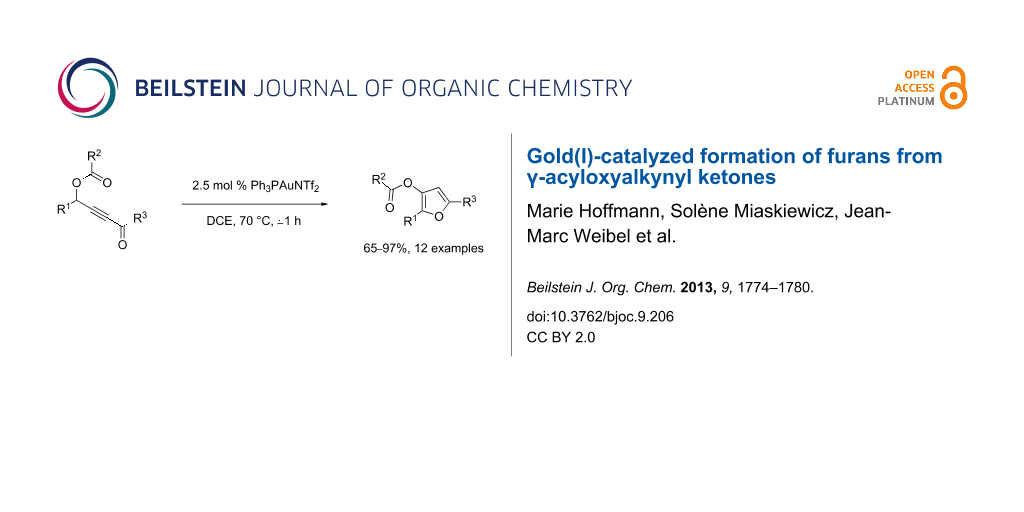
Abstract
Various γ-acyloxyalkynyl ketones were efficiently converted into highly substituted furans with 2.5 mol % of triflimide (triphenylphosphine)gold(I) as a catalyst in dichloroethane at 70 °C.

Graphical Abstract
Introduction
Furans are an important class of aromatic compounds. They are found in many natural products, in pharmaceutical and agrochemical compounds as well as in flavor and fragrance industries [1]. Furans are also routinely used as building blocks in organic synthesis [2,3]. Therefore, a large number of synthetic methods has been developed to construct the furan motif [4,5]. Among them, late transition metal-catalyzed intra- or intermolecular cyclizations of oxygenated functionalities on unsaturated carbon–carbon bonds proved to be powerful synthetic methods due to their mildness, efficiency and diversity [6,7]. In the last decade, gold catalysts with their carbophilic character have emerged as a new tool for furan preparation. As summarized in Scheme 1, furans could now be obtained by either gold(I) or gold(III) catalysis from various types of substrates such as allenyl ketones [8-14], enynes or diynes [15-17], alkynes and sulfur ylides [18,19], alkynyl oxiranes [20-26], alkynyl ketones [27-35], alkynyl alcohols [36-46], and alkynyl ethers [47,48]. Very recently, a three-component coupling reaction toward furans catalyzed by gold(III) has been reported starting from terminal alkenes, glyoxal derivatives and secondary amines [49].
![[1860-5397-9-206-i1]](/bjoc/content/inline/1860-5397-9-206-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Gold(I) or gold(III)-catalyzed furan syntheses with or without nucleophiles.
Scheme 1: Gold(I) or gold(III)-catalyzed furan syntheses with or without nucleophiles.
In this emerging research area, we have been focusing our effort on the development of furan motifs from alkynyl epoxides [21,22,50,51] and new precursors, i.e. γ-acyloxyalkynyl ketones. The latter have already been described to rearrange into furans by using copper catalysts. Indeed, Gevorgyan et al. showed that the combination of copper(I) chloride and triethylamine catalyzed the 1,2-migration/cycloisomerization of γ-acyloxyalkynyl ketones in dimethylacetamide (DMA) at 130 °C (Scheme 2) within 1–46 h [31,52,53]. Despite the relative harsh reaction conditions, furans could be obtained in good to excellent yields. However, one major limitation was ascribed to the types of the employed ketones (R3 in Scheme 2), as only phenyl and tert-butyl alkynyl ketones were able to furnish acceptable yields.
![[1860-5397-9-206-i2]](/bjoc/content/inline/1860-5397-9-206-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Copper(I)-catalyzed 1,2-migration/cycloisomerization of γ-acyloxyalkynyl ketones.
Scheme 2: Copper(I)-catalyzed 1,2-migration/cycloisomerization of γ-acyloxyalkynyl ketones.
We herein report that gold(I) can overcome these limitations, providing a general, fast and very efficient transformation of γ-acyloxyalkynyl ketones into trisubstituted and functionalized furans.
Results and Discussion
In order to find the most appropriate conditions, we applied various gold catalysts in different solvents at different temperatures to the easily available ynone 1a (Table 1), which has been reported to afford furan 2a in 86% yield under Gevorgyan’s conditions (Table 1, entry 1). We started our catalyst screening by using the classical combination of Ph3PAuCl/AgSbF6 and the Gagosz’s catalyst [54], i.e. (triphenylphosphine)gold(I) triflimide, in dichloroethane at room temperature. In both cases, a fast consumption of the starting material 1a was observed compared to the copper(I) catalysis, but lower yields of furans, 44% and 65% respectively, were obtained mostly due to the formation of the hydration product 3 (Table 1, entries 2 and 3 versus entry 1). We found out that running the reaction at 70 °C instead of at room temperature completely prevented the byproduct formation. At this temperature good to quantitative yields of furans 2a were achieved in less than 30 min, and 5 mol % of Ph3PAuNTf2 turned out to be the more efficient catalyst (Table 1, entries 2 and 3 versus entries 4 and 5). We then verified that the hydrate product was not a transient intermediate in this rearrangement by subjecting pure compound 3 to the latter reaction conditions and, even after 3 h at 70 °C, no trace of furan 2a could be detected by 1H NMR analysis. Interestingly, decreasing the catalytic loading from 5 to 1 mol % still provided the furan in less than 1 h and in high yields (Table 1, entries 6 and 7 versus entry 5). However, hydration started to compete again at low loading, as evidenced by tiny amounts of 3 in the NMR spectrum of the crude (Table 1, entry 7). Control experiments revealed that other triflimide salts of coinage metals were not suited for this transformation. Indeed, silver(I) triflimide resulted mainly in degradation (Table 1, entry 8) and tetrakis(acetonitrile)copper(I) triflimide furnished the furan 2a in only modest yield even after prolonged reaction time (Table 1, entry 9).
Table 1: Screening of the catalysts and the conditions.
![[Graphic 1]](/bjoc/content/inline/1860-5397-9-206-i4.svg?max-width=637&scale=1.0)
|
||||||
| Entry | Catalyst (mol %) | Solvent | T (°C) | Time (h) | Yield 2a (%) | Yield 3 (%) |
|---|---|---|---|---|---|---|
| 1 | CuCl (5) | Et3N/DMAa | 130 | 17 | 86b | – |
| 2 | Ph3PAuCl/AgSbF6 (5) | DCEc | rt | 1 | 44 | 22 |
| 3 | Ph3PAuNTf2 (5) | DCE | rt | 2.5 | 65 | 27 |
| 4 | Ph3PAuCl/AgSbF6 (5) | DCE | 70 | 0.1 | 71 | – |
| 5 | Ph3PAuNTf2 (5) | DCE | 70 | 0.25 | 97 | – |
| 6 | Ph3PAuNTf2 (2.5) | DCE | 70 | 0.5 | 95 | – |
| 7 | Ph3PAuNTf2 (1) | DCE | 70 | 0.7 | 91 | <5d |
| 8 | AgNTf2 (5) | DCE | 70 | 16 | –e | 15d |
| 9 | [Cu(MeCN)4]NTf2 (5) | DCE | 70 | 16 | 44 | 9d |
aDimethylacetamide. bReported yield from ref [52]. cDichloroethane. dEstimated yield based on the 1H NMR of the crude mixture. eDegradation occurs.
With these conditions in hand (Table 1, entry 6), we started investigating the scope of the reaction by preparing various acyloxyalkynyl ketones (Table 2). As for the phenyl alkynyl ketone 1a, the corresponding tert-butyl alkynyl ketone 1b turned out to be a good substrate for this transformation confirming Gevorgyan’s results (Table 2, entry 1 versus entry 2). Despite its bulkiness, full conversion was achieved within 30 min and 2.5 mol % of Ph3PAuNTf2, affording furan 2b in 93% yield. We also evaluated the influence of the alkynyl substitution by increasing the size of the R1 group. To implement this, we introduced secondary and tertiary carbon centers next to the acyloxy function by preparing compounds 1c (R1 = 2-decyl) and 1d (R1 = tert-butyl). Compound 1c, similar to 1a, rearranged under these conditions, furnishing the furan 2c in 90% yield (Table 2, entry 3). However, the presence of the sterically demanding tert-butyl group in 1d drastically affected the reaction (Table 2, entry 1 and 3 versus entry 4). Even running the reaction with 5 mol % of catalyst to avoid the hydration product, the reaction took 6 h to reach almost full conversion. Beside the expected furan 2d, its corresponding regioisomer 2d’ arising from 1,3-migration of the pivaloyl group is formed in this reaction and both products were obtained in a combined yield of 68% (Table 2, entry 4). We next varied the nature of the migratory acyloxy group. We synthetized similar substrates 1e–1h bearing pivaloyl, benzoyl, acetyl and 2-phenylacetyl groups, respectively. These compounds were engaged in the gold-catalyzed process affording the furans 2e–2h in the same range of yields (70–80%), suggesting that the nature of the acyloxy group had no crucial influence on the rearrangement. Indeed, the slight differences in terms of yield could be ascribed to the formation of hydration products (5–15%), and the reaction times of each reaction were inferior or equal to 1 h (Table 2, entries 5–8). We then turned our attention to the problematic R3 position in which only phenyl and tert-butyl substituents adjacent to the ketone, i.e. without enolizable position, were tolerated under copper(I)-catalyzed reaction conditions. We were pleased to observe that various other substituents, such as methyl, propyl, 2-phenylethyl, 3-benzyloxypropyl (Table 2, entries 9–12), were fully compatible with our gold-catalysis giving furans 2i–l in good yields.
Table 2: Scope of the gold(I)-catalyzed formation of furans from γ-acyloxyalkynyl ketones.
![[Graphic 2]](/bjoc/content/inline/1860-5397-9-206-i5.svg?max-width=637&scale=1.0)
|
||||
| Entry | Substrates 1 | Time (h) | Furans 2 | Yield (%) |
|---|---|---|---|---|
| 1 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-9-206-i6.svg?max-width=637&scale=1.0)
1a |
0.5 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-9-206-i7.svg?max-width=637&scale=1.0)
2a |
95 |
| 2 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-9-206-i8.svg?max-width=637&scale=1.0)
1b |
0.5 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-9-206-i9.svg?max-width=637&scale=1.0)
2b |
93 |
| 3 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-9-206-i10.svg?max-width=637&scale=1.0)
1c |
0.75 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-9-206-i11.svg?max-width=637&scale=1.0)
2c |
90 |
| 4 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-9-206-i12.svg?max-width=637&scale=1.0)
1d |
6 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-9-206-i13.svg?max-width=637&scale=1.0)
2d/2d’ |
68a |
| 5 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-9-206-i14.svg?max-width=637&scale=1.0)
1e |
1 |
![[Graphic 12]](/bjoc/content/inline/1860-5397-9-206-i15.svg?max-width=637&scale=1.0)
2e |
78 |
| 6 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-9-206-i16.svg?max-width=637&scale=1.0)
1f |
0.5 |
![[Graphic 14]](/bjoc/content/inline/1860-5397-9-206-i17.svg?max-width=637&scale=1.0)
2f |
81 |
| 7 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-9-206-i18.svg?max-width=637&scale=1.0)
1g |
0.75 |
![[Graphic 16]](/bjoc/content/inline/1860-5397-9-206-i19.svg?max-width=637&scale=1.0)
2g |
70 |
| 8 |
![[Graphic 17]](/bjoc/content/inline/1860-5397-9-206-i20.svg?max-width=637&scale=1.0)
1h |
1 |
![[Graphic 18]](/bjoc/content/inline/1860-5397-9-206-i21.svg?max-width=637&scale=1.0)
2h |
75 |
| 9 |
![[Graphic 19]](/bjoc/content/inline/1860-5397-9-206-i22.svg?max-width=637&scale=1.0)
1i |
0.33 |
![[Graphic 20]](/bjoc/content/inline/1860-5397-9-206-i23.svg?max-width=637&scale=1.0)
2i |
77 |
| 10 |
![[Graphic 21]](/bjoc/content/inline/1860-5397-9-206-i24.svg?max-width=637&scale=1.0)
1j |
0.5 |
![[Graphic 22]](/bjoc/content/inline/1860-5397-9-206-i25.svg?max-width=637&scale=1.0)
2j |
68 |
| 11 |
![[Graphic 23]](/bjoc/content/inline/1860-5397-9-206-i26.svg?max-width=637&scale=1.0)
1k |
0.33 |
![[Graphic 24]](/bjoc/content/inline/1860-5397-9-206-i27.svg?max-width=637&scale=1.0)
2k |
74 |
| 12 |
![[Graphic 25]](/bjoc/content/inline/1860-5397-9-206-i28.svg?max-width=637&scale=1.0)
1l |
0.33 |
![[Graphic 26]](/bjoc/content/inline/1860-5397-9-206-i29.svg?max-width=637&scale=1.0)
2l |
65 |
aCumulative yield of furans 2d and 2d’; reaction performed with 5 mol % of catalyst.
Two different mechanistic hypotheses could be envisaged in the rearrangement of γ-acyloxyalkynyl ketones into furans based on multifaceted gold-catalyst properties, i.e. the ability of gold cations to act as π or σ Lewis acids (Scheme 3) [21,55,56]. Intramolecular [1,4]-addition of the acyloxy function by oxophilic activation of γ-acyloxyalkynyl ketones could lead to the formation of gold allenolate A, which is in equilibrium with both Z or E vinylgold B and C [57]. Intermediate B, which could also be generated by carbophilic gold activation followed by nucleophilic addition of the acyloxy part, could evolve into the gold carbenoid species D [31]. Intermediate C, possessing the correct stereochemistry, and D might then cyclize by an attack of the carbonyl function on the carbon bearing the R1 substituent to afford the oxygenated five-membered ring E. Furan would finally be formed after tautomerization and protodemetalation of intermediate E.
![[1860-5397-9-206-i3]](/bjoc/content/inline/1860-5397-9-206-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Mechanistic hypothesis for gold(I)-catalyzed conversion of γ-acyloxyalkynyl ketones into furans.
Scheme 3: Mechanistic hypothesis for gold(I)-catalyzed conversion of γ-acyloxyalkynyl ketones into furans.
Conclusion
We have reported an efficient, very general and regioselective preparation of functionalized furans through a gold(I)-catalyzed rearrangement of γ-acyloxyalkynyl ketones under mild conditions. Further work is currently underway in our laboratory to fully understand this novel rearrangement.
Experimental
General procedure for gold(I)-catalyzed formation of furans from γ-acyloxyalkynyl ketones. In an oven-dried flask, γ-acyloxyalkynyl ketone (0.4 mmol) was dissolved in dry dichloroethane (0.1 M) and heated to 70 °C under an argon atmosphere. Ph3PAuNTf2 (2.5 mol %) was then added to the stirred solution at 70 °C. The reaction was monitored by thin-layer chromatography until completion. The solvent was then removed in vacuo, and the crude residue was purified by silica gel flash chromatography (pentane/Et2O).
Supporting Information
| Supporting Information File 1: General procedures, characterization data and NMR spectra for compounds 1a–l, 2a–l and 3. | ||
| Format: PDF | Size: 7.7 MB | Download |
References
-
Keay, B. A.; Hopkins, J. M.; Dibble, P. W. Furans and their Benzo Derivatives: Applications. Comprehensive Heterocyclic Chemistry III; Elsevier: Amsterdam, The Netherlands, 2008; pp 571–623. doi:10.1016/B978-008044992-0.00308-4
Return to citation in text: [1] -
Lipshutz, B. H. Chem. Rev. 1986, 86, 795–819. doi:10.1021/cr00075a005
Return to citation in text: [1] -
Balme, G.; Bouyssi, D.; Monteiro, N. Heterocycles 2007, 73, 87–124. doi:10.3987/REV-07-SR(U)2
Return to citation in text: [1] -
Hou, X. L.; Cheung, H. Y.; Hon, T. Y.; Kwan, P. L.; Lo, T. H.; Tong, S. Y.; Wong, H. N. C. Tetrahedron 1998, 54, 1955–2020. doi:10.1016/S0040-4020(97)10303-9
Return to citation in text: [1] -
Kirsch, S. F. Org. Biomol. Chem. 2006, 4, 2076–2080. doi:10.1039/b602596j
Return to citation in text: [1] -
Brown, R. C. D. Angew. Chem., Int. Ed. 2005, 44, 850–852. doi:10.1002/anie.200461668
Return to citation in text: [1] -
Patil, N. T.; Yamamoto, Y. Chem. Rev. 2008, 108, 3395–3442. doi:10.1021/cr050041j
Return to citation in text: [1] -
Hashmi, A. S. K.; Schwarz, L.; Choi, J.-H.; Frost, T. M. Angew. Chem., Int. Ed. 2000, 39, 2285–2288. doi:10.1002/1521-3773(20000703)39:13<2285::AID-ANIE2285>3.0.CO;2-F
Return to citation in text: [1] -
Zhou, C.-Y.; Chan, P. W. H.; Che, C.-M. Org. Lett. 2006, 8, 325–328. doi:10.1021/ol052696c
Return to citation in text: [1] -
Sromek, A. W.; Rubina, M.; Gevorgyan, V. J. Am. Chem. Soc. 2005, 127, 10500–10501. doi:10.1021/ja053290y
Return to citation in text: [1] -
Dudnik, A. S.; Xia, Y.; Li, Y.; Gevorgyan, V. J. Am. Chem. Soc. 2010, 132, 7645–7655. doi:10.1021/ja910290c
Return to citation in text: [1] -
Dudnik, A. S.; Sromek, A. W.; Rubina, M.; Kim, J. T.; Kel'in, A. V.; Gevorgyan, V. J. Am. Chem. Soc. 2008, 130, 1440–1452. doi:10.1021/ja0773507
Return to citation in text: [1] -
Dudnik, A. S.; Gevorgyan, V. Angew. Chem., Int. Ed. 2007, 46, 5195–5197. doi:10.1002/anie.200701128
Return to citation in text: [1] -
Wang, E.; Fu, X.; Xie, X.; Chen, J.; Gao, H.; Liu, Y. Tetrahedron Lett. 2011, 52, 1968–1972. doi:10.1016/j.tetlet.2011.02.057
Return to citation in text: [1] -
Li, E.; Yao, W.; Xie, X.; Wang, C.; Shao, Y.; Li, Y. Org. Biomol. Chem. 2012, 10, 2960–2965. doi:10.1039/c2ob07173h
Return to citation in text: [1] -
Kramer, S.; Madsen, J. L. H.; Rottländer, M.; Skrydstrup, T. Org. Lett. 2010, 12, 2758–2761. doi:10.1021/ol1008685
Return to citation in text: [1] -
Nun, P.; Dupuy, S.; Gaillard, S.; Poater, A.; Cavallo, L.; Nolan, S. P. Catal. Sci. Technol. 2011, 1, 58–61. doi:10.1039/c0cy00055h
Return to citation in text: [1] -
Kramer, S.; Skrydstrup, T. Angew. Chem., Int. Ed. 2012, 51, 4681–4684. doi:10.1002/anie.201200307
Return to citation in text: [1] -
Huang, X.; Peng, B.; Luparia, M.; Gomes, L. F. R.; Veiros, L. F.; Maulide, N. Angew. Chem., Int. Ed. 2012, 51, 8886–8890. doi:10.1002/anie.201203637
Return to citation in text: [1] -
Hashmi, A. S. K.; Sinha, P. Adv. Synth. Catal. 2004, 346, 432–438. doi:10.1002/adsc.200303201
Return to citation in text: [1] -
Blanc, A.; Tenbrink, K.; Weibel, J.-M.; Pale, P. J. Org. Chem. 2009, 74, 5342–5348. doi:10.1021/jo9008172
Return to citation in text: [1] [2] [3] -
Blanc, A.; Alix, A.; Weibel, J.-M.; Pale, P. Eur. J. Org. Chem. 2010, 1644–1647. doi:10.1002/ejoc.200901331
Return to citation in text: [1] [2] -
Shu, X.-Z.; Liu, X.-Y.; Xiao, H.-Q.; Ji, K.-G.; Guo, L.-N.; Qi, C.-Z.; Liang, Y.-M. Adv. Synth. Catal. 2007, 349, 2493–2498. doi:10.1002/adsc.200700319
Return to citation in text: [1] -
Ji, K.-G.; Shen, Y.-W.; Shu, X.-Z.; Xiao, H.-Q.; Bian, Y.-J.; Liang, Y.-M. Adv. Synth. Catal. 2008, 350, 1275–1280. doi:10.1002/adsc.200800130
Return to citation in text: [1] -
Ji, K.-G.; Shu, X.-Z.; Chen, J.; Zhao, S.-C.; Zheng, Z.-J.; Liu, X.-Y.; Liang, Y.-M. Org. Biomol. Chem. 2009, 7, 2501–2505. doi:10.1039/b905332h
Return to citation in text: [1] -
Dai, L.-Z.; Shi, M. Tetrahedron Lett. 2008, 49, 6437–6439. doi:10.1016/j.tetlet.2008.08.093
Return to citation in text: [1] -
Yao, T.; Zhang, X.; Larock, R. C. J. Am. Chem. Soc. 2004, 126, 11164–11165. doi:10.1021/ja0466964
Return to citation in text: [1] -
Yao, T.; Zhang, X.; Larock, R. C. J. Org. Chem. 2005, 70, 7679–7685. doi:10.1021/jo0510585
Return to citation in text: [1] -
Liu, X.; Pan, Z.; Shu, X.; Duan, X.; Liang, Y. Synlett 2006, 1962–1964. doi:10.1055/s-2006-947363
Return to citation in text: [1] -
Zhang, J.; Schmalz, H.-G. Angew. Chem., Int. Ed. 2006, 45, 6704–6707. doi:10.1002/anie.200601252
Return to citation in text: [1] -
Schwier, T.; Sromek, A. W.; Yap, D. M. L.; Chernyak, D.; Gevorgyan, V. J. Am. Chem. Soc. 2007, 129, 9868–9878. doi:10.1021/ja072446m
Return to citation in text: [1] [2] [3] -
Oh, C. H.; Lee, S. J.; Lee, J. H.; Na, Y. J. Chem. Commun. 2008, 5794–5796. doi:10.1039/b812077c
Return to citation in text: [1] -
Belting, V.; Krause, N. Org. Biomol. Chem. 2009, 7, 1221–1225. doi:10.1039/b819704k
Return to citation in text: [1] -
Rodríguez, A.; Moran, W. J. Tetrahedron Lett. 2011, 52, 2605–2607. doi:10.1016/j.tetlet.2011.03.086
Return to citation in text: [1] -
Li, Y.; Wheeler, K. A.; Dembinski, R. Adv. Synth. Catal. 2010, 352, 2761–2766. doi:10.1002/adsc.201000411
Return to citation in text: [1] -
Liu, Y.; Song, F.; Song, Z.; Liu, M.; Yan, B. Org. Lett. 2005, 7, 5409–5412. doi:10.1021/ol052160r
Return to citation in text: [1] -
Du, X.; Song, F.; Lu, Y.; Chen, H.; Liu, Y. Tetrahedron 2009, 65, 1839–1845. doi:10.1016/j.tet.2008.11.109
Return to citation in text: [1] -
Zhang, X.; Lu, Z.; Fu, C.; Ma, S. J. Org. Chem. 2010, 75, 2589–2598. doi:10.1021/jo100146p
Return to citation in text: [1] -
Kim, S.; Lee, P. H. Adv. Synth. Catal. 2008, 350, 547–551. doi:10.1002/adsc.200700471
Return to citation in text: [1] -
Kim, S.; Kang, D.; Shin, S.; Lee, P. H. Tetrahedron Lett. 2010, 51, 1899–1901. doi:10.1016/j.tetlet.2010.02.026
Return to citation in text: [1] -
Praveen, C.; Kiruthiga, P.; Perumal, P. T. Synlett 2009, 1990–1996. doi:10.1055/s-0029-1217517
Return to citation in text: [1] -
Hashmi, A. S. K.; Häffner, T.; Rudolph, M.; Rominger, F. Eur. J. Org. Chem. 2011, 667–671. doi:10.1002/ejoc.201001479
Return to citation in text: [1] -
Kotikalapudi, R.; Swamy, K. C. K. Tetrahedron Lett. 2012, 53, 3831–3834. doi:10.1016/j.tetlet.2012.04.060
Return to citation in text: [1] -
Aponick, A.; Li, C.-Y.; Malinge, J.; Marques, E. F. Org. Lett. 2009, 11, 4624–4627. doi:10.1021/ol901901m
Return to citation in text: [1] -
Egi, M.; Azechi, K.; Akai, S. Org. Lett. 2009, 11, 5002–5005. doi:10.1021/ol901942t
Return to citation in text: [1] -
Arcadi, A.; Alfonsi, M.; Chiarini, M.; Marinelli, F. J. Organomet. Chem. 2009, 694, 576–582. doi:10.1016/j.jorganchem.2008.12.013
Return to citation in text: [1] -
Istrate, F. M.; Gagosz, F. Beilstein J. Org. Chem. 2011, 7, 878–885. doi:10.3762/bjoc.7.100
Return to citation in text: [1] -
Suhre, M. H.; Reif, M.; Kirsch, S. F. Org. Lett. 2005, 7, 3925–3927. doi:10.1021/ol0514101
Return to citation in text: [1] -
Li, J.; Liu, L.; Ding, D.; Sun, J.; Ji, Y.; Dong, J. Org. Lett. 2013, 15, 2884–2887. doi:10.1021/ol401239j
Return to citation in text: [1] -
Blanc, A.; Tenbrink, K.; Weibel, J.-M.; Pale, P. J. Org. Chem. 2009, 74, 4360–4363. doi:10.1021/jo900483m
Return to citation in text: [1] -
Borghèse, S.; Louis, B.; Blanc, A.; Pale, P. Catal. Sci. Technol. 2011, 1, 981–986. doi:10.1039/c1cy00154j
Return to citation in text: [1] -
Sromek, A. W.; Kel’in, A. V.; Gevorgyan, V. Angew. Chem., Int. Ed. 2004, 43, 2280–2282. doi:10.1002/anie.200353535
Return to citation in text: [1] [2] -
Kim, J. T.; Kel'in, A. V.; Gevorgyan, V. Angew. Chem., Int. Ed. 2003, 42, 98–101. doi:10.1002/anie.200390064
Return to citation in text: [1] -
Mézailles, N.; Ricard, L.; Gagosz, F. Org. Lett. 2005, 7, 4133–4136. doi:10.1021/ol0515917
Return to citation in text: [1] -
Yamamoto, Y. J. Org. Chem. 2007, 72, 7817–7831. doi:10.1021/jo070579k
Return to citation in text: [1] -
Ngwerume, S.; Lewis, W.; Camp, J. E. J. Org. Chem. 2013, 78, 920–934. doi:10.1021/jo302349k
Return to citation in text: [1] -
Egi, M.; Azechi, K.; Saneto, M.; Shimizu, K.; Akai, S. J. Org. Chem. 2010, 75, 2123–2126. doi:10.1021/jo100048j
Return to citation in text: [1]
| 31. | Schwier, T.; Sromek, A. W.; Yap, D. M. L.; Chernyak, D.; Gevorgyan, V. J. Am. Chem. Soc. 2007, 129, 9868–9878. doi:10.1021/ja072446m |
| 21. | Blanc, A.; Tenbrink, K.; Weibel, J.-M.; Pale, P. J. Org. Chem. 2009, 74, 5342–5348. doi:10.1021/jo9008172 |
| 55. | Yamamoto, Y. J. Org. Chem. 2007, 72, 7817–7831. doi:10.1021/jo070579k |
| 56. | Ngwerume, S.; Lewis, W.; Camp, J. E. J. Org. Chem. 2013, 78, 920–934. doi:10.1021/jo302349k |
| 57. | Egi, M.; Azechi, K.; Saneto, M.; Shimizu, K.; Akai, S. J. Org. Chem. 2010, 75, 2123–2126. doi:10.1021/jo100048j |
| 1. | Keay, B. A.; Hopkins, J. M.; Dibble, P. W. Furans and their Benzo Derivatives: Applications. Comprehensive Heterocyclic Chemistry III; Elsevier: Amsterdam, The Netherlands, 2008; pp 571–623. doi:10.1016/B978-008044992-0.00308-4 |
| 8. | Hashmi, A. S. K.; Schwarz, L.; Choi, J.-H.; Frost, T. M. Angew. Chem., Int. Ed. 2000, 39, 2285–2288. doi:10.1002/1521-3773(20000703)39:13<2285::AID-ANIE2285>3.0.CO;2-F |
| 9. | Zhou, C.-Y.; Chan, P. W. H.; Che, C.-M. Org. Lett. 2006, 8, 325–328. doi:10.1021/ol052696c |
| 10. | Sromek, A. W.; Rubina, M.; Gevorgyan, V. J. Am. Chem. Soc. 2005, 127, 10500–10501. doi:10.1021/ja053290y |
| 11. | Dudnik, A. S.; Xia, Y.; Li, Y.; Gevorgyan, V. J. Am. Chem. Soc. 2010, 132, 7645–7655. doi:10.1021/ja910290c |
| 12. | Dudnik, A. S.; Sromek, A. W.; Rubina, M.; Kim, J. T.; Kel'in, A. V.; Gevorgyan, V. J. Am. Chem. Soc. 2008, 130, 1440–1452. doi:10.1021/ja0773507 |
| 13. | Dudnik, A. S.; Gevorgyan, V. Angew. Chem., Int. Ed. 2007, 46, 5195–5197. doi:10.1002/anie.200701128 |
| 14. | Wang, E.; Fu, X.; Xie, X.; Chen, J.; Gao, H.; Liu, Y. Tetrahedron Lett. 2011, 52, 1968–1972. doi:10.1016/j.tetlet.2011.02.057 |
| 54. | Mézailles, N.; Ricard, L.; Gagosz, F. Org. Lett. 2005, 7, 4133–4136. doi:10.1021/ol0515917 |
| 6. | Brown, R. C. D. Angew. Chem., Int. Ed. 2005, 44, 850–852. doi:10.1002/anie.200461668 |
| 7. | Patil, N. T.; Yamamoto, Y. Chem. Rev. 2008, 108, 3395–3442. doi:10.1021/cr050041j |
| 52. | Sromek, A. W.; Kel’in, A. V.; Gevorgyan, V. Angew. Chem., Int. Ed. 2004, 43, 2280–2282. doi:10.1002/anie.200353535 |
| 4. | Hou, X. L.; Cheung, H. Y.; Hon, T. Y.; Kwan, P. L.; Lo, T. H.; Tong, S. Y.; Wong, H. N. C. Tetrahedron 1998, 54, 1955–2020. doi:10.1016/S0040-4020(97)10303-9 |
| 5. | Kirsch, S. F. Org. Biomol. Chem. 2006, 4, 2076–2080. doi:10.1039/b602596j |
| 21. | Blanc, A.; Tenbrink, K.; Weibel, J.-M.; Pale, P. J. Org. Chem. 2009, 74, 5342–5348. doi:10.1021/jo9008172 |
| 22. | Blanc, A.; Alix, A.; Weibel, J.-M.; Pale, P. Eur. J. Org. Chem. 2010, 1644–1647. doi:10.1002/ejoc.200901331 |
| 50. | Blanc, A.; Tenbrink, K.; Weibel, J.-M.; Pale, P. J. Org. Chem. 2009, 74, 4360–4363. doi:10.1021/jo900483m |
| 51. | Borghèse, S.; Louis, B.; Blanc, A.; Pale, P. Catal. Sci. Technol. 2011, 1, 981–986. doi:10.1039/c1cy00154j |
| 2. | Lipshutz, B. H. Chem. Rev. 1986, 86, 795–819. doi:10.1021/cr00075a005 |
| 3. | Balme, G.; Bouyssi, D.; Monteiro, N. Heterocycles 2007, 73, 87–124. doi:10.3987/REV-07-SR(U)2 |
| 31. | Schwier, T.; Sromek, A. W.; Yap, D. M. L.; Chernyak, D.; Gevorgyan, V. J. Am. Chem. Soc. 2007, 129, 9868–9878. doi:10.1021/ja072446m |
| 52. | Sromek, A. W.; Kel’in, A. V.; Gevorgyan, V. Angew. Chem., Int. Ed. 2004, 43, 2280–2282. doi:10.1002/anie.200353535 |
| 53. | Kim, J. T.; Kel'in, A. V.; Gevorgyan, V. Angew. Chem., Int. Ed. 2003, 42, 98–101. doi:10.1002/anie.200390064 |
| 27. | Yao, T.; Zhang, X.; Larock, R. C. J. Am. Chem. Soc. 2004, 126, 11164–11165. doi:10.1021/ja0466964 |
| 28. | Yao, T.; Zhang, X.; Larock, R. C. J. Org. Chem. 2005, 70, 7679–7685. doi:10.1021/jo0510585 |
| 29. | Liu, X.; Pan, Z.; Shu, X.; Duan, X.; Liang, Y. Synlett 2006, 1962–1964. doi:10.1055/s-2006-947363 |
| 30. | Zhang, J.; Schmalz, H.-G. Angew. Chem., Int. Ed. 2006, 45, 6704–6707. doi:10.1002/anie.200601252 |
| 31. | Schwier, T.; Sromek, A. W.; Yap, D. M. L.; Chernyak, D.; Gevorgyan, V. J. Am. Chem. Soc. 2007, 129, 9868–9878. doi:10.1021/ja072446m |
| 32. | Oh, C. H.; Lee, S. J.; Lee, J. H.; Na, Y. J. Chem. Commun. 2008, 5794–5796. doi:10.1039/b812077c |
| 33. | Belting, V.; Krause, N. Org. Biomol. Chem. 2009, 7, 1221–1225. doi:10.1039/b819704k |
| 34. | Rodríguez, A.; Moran, W. J. Tetrahedron Lett. 2011, 52, 2605–2607. doi:10.1016/j.tetlet.2011.03.086 |
| 35. | Li, Y.; Wheeler, K. A.; Dembinski, R. Adv. Synth. Catal. 2010, 352, 2761–2766. doi:10.1002/adsc.201000411 |
| 47. | Istrate, F. M.; Gagosz, F. Beilstein J. Org. Chem. 2011, 7, 878–885. doi:10.3762/bjoc.7.100 |
| 48. | Suhre, M. H.; Reif, M.; Kirsch, S. F. Org. Lett. 2005, 7, 3925–3927. doi:10.1021/ol0514101 |
| 20. | Hashmi, A. S. K.; Sinha, P. Adv. Synth. Catal. 2004, 346, 432–438. doi:10.1002/adsc.200303201 |
| 21. | Blanc, A.; Tenbrink, K.; Weibel, J.-M.; Pale, P. J. Org. Chem. 2009, 74, 5342–5348. doi:10.1021/jo9008172 |
| 22. | Blanc, A.; Alix, A.; Weibel, J.-M.; Pale, P. Eur. J. Org. Chem. 2010, 1644–1647. doi:10.1002/ejoc.200901331 |
| 23. | Shu, X.-Z.; Liu, X.-Y.; Xiao, H.-Q.; Ji, K.-G.; Guo, L.-N.; Qi, C.-Z.; Liang, Y.-M. Adv. Synth. Catal. 2007, 349, 2493–2498. doi:10.1002/adsc.200700319 |
| 24. | Ji, K.-G.; Shen, Y.-W.; Shu, X.-Z.; Xiao, H.-Q.; Bian, Y.-J.; Liang, Y.-M. Adv. Synth. Catal. 2008, 350, 1275–1280. doi:10.1002/adsc.200800130 |
| 25. | Ji, K.-G.; Shu, X.-Z.; Chen, J.; Zhao, S.-C.; Zheng, Z.-J.; Liu, X.-Y.; Liang, Y.-M. Org. Biomol. Chem. 2009, 7, 2501–2505. doi:10.1039/b905332h |
| 26. | Dai, L.-Z.; Shi, M. Tetrahedron Lett. 2008, 49, 6437–6439. doi:10.1016/j.tetlet.2008.08.093 |
| 49. | Li, J.; Liu, L.; Ding, D.; Sun, J.; Ji, Y.; Dong, J. Org. Lett. 2013, 15, 2884–2887. doi:10.1021/ol401239j |
| 18. | Kramer, S.; Skrydstrup, T. Angew. Chem., Int. Ed. 2012, 51, 4681–4684. doi:10.1002/anie.201200307 |
| 19. | Huang, X.; Peng, B.; Luparia, M.; Gomes, L. F. R.; Veiros, L. F.; Maulide, N. Angew. Chem., Int. Ed. 2012, 51, 8886–8890. doi:10.1002/anie.201203637 |
| 15. | Li, E.; Yao, W.; Xie, X.; Wang, C.; Shao, Y.; Li, Y. Org. Biomol. Chem. 2012, 10, 2960–2965. doi:10.1039/c2ob07173h |
| 16. | Kramer, S.; Madsen, J. L. H.; Rottländer, M.; Skrydstrup, T. Org. Lett. 2010, 12, 2758–2761. doi:10.1021/ol1008685 |
| 17. | Nun, P.; Dupuy, S.; Gaillard, S.; Poater, A.; Cavallo, L.; Nolan, S. P. Catal. Sci. Technol. 2011, 1, 58–61. doi:10.1039/c0cy00055h |
| 36. | Liu, Y.; Song, F.; Song, Z.; Liu, M.; Yan, B. Org. Lett. 2005, 7, 5409–5412. doi:10.1021/ol052160r |
| 37. | Du, X.; Song, F.; Lu, Y.; Chen, H.; Liu, Y. Tetrahedron 2009, 65, 1839–1845. doi:10.1016/j.tet.2008.11.109 |
| 38. | Zhang, X.; Lu, Z.; Fu, C.; Ma, S. J. Org. Chem. 2010, 75, 2589–2598. doi:10.1021/jo100146p |
| 39. | Kim, S.; Lee, P. H. Adv. Synth. Catal. 2008, 350, 547–551. doi:10.1002/adsc.200700471 |
| 40. | Kim, S.; Kang, D.; Shin, S.; Lee, P. H. Tetrahedron Lett. 2010, 51, 1899–1901. doi:10.1016/j.tetlet.2010.02.026 |
| 41. | Praveen, C.; Kiruthiga, P.; Perumal, P. T. Synlett 2009, 1990–1996. doi:10.1055/s-0029-1217517 |
| 42. | Hashmi, A. S. K.; Häffner, T.; Rudolph, M.; Rominger, F. Eur. J. Org. Chem. 2011, 667–671. doi:10.1002/ejoc.201001479 |
| 43. | Kotikalapudi, R.; Swamy, K. C. K. Tetrahedron Lett. 2012, 53, 3831–3834. doi:10.1016/j.tetlet.2012.04.060 |
| 44. | Aponick, A.; Li, C.-Y.; Malinge, J.; Marques, E. F. Org. Lett. 2009, 11, 4624–4627. doi:10.1021/ol901901m |
| 45. | Egi, M.; Azechi, K.; Akai, S. Org. Lett. 2009, 11, 5002–5005. doi:10.1021/ol901942t |
| 46. | Arcadi, A.; Alfonsi, M.; Chiarini, M.; Marinelli, F. J. Organomet. Chem. 2009, 694, 576–582. doi:10.1016/j.jorganchem.2008.12.013 |
© 2013 Hoffmann et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)