Abstract
Functionalized 3-trifluoromethyl-2-isoxazolines and 3-trifluoromethylisoxazoles were easily prepared from trifluoromethyl aldoxime 2 under mild conditions by using DIB as oxidant. Theoretical studies of the reactivity of trifluoroacetonitrile oxide 4 toward olefins and alkynes were carried out. The 3-trifluoromethyl-2-isoxazolines were ring-opened with NaBH4 and NiCl2 to yield the corresponding trifluoromethylated γ-amino alcohols.

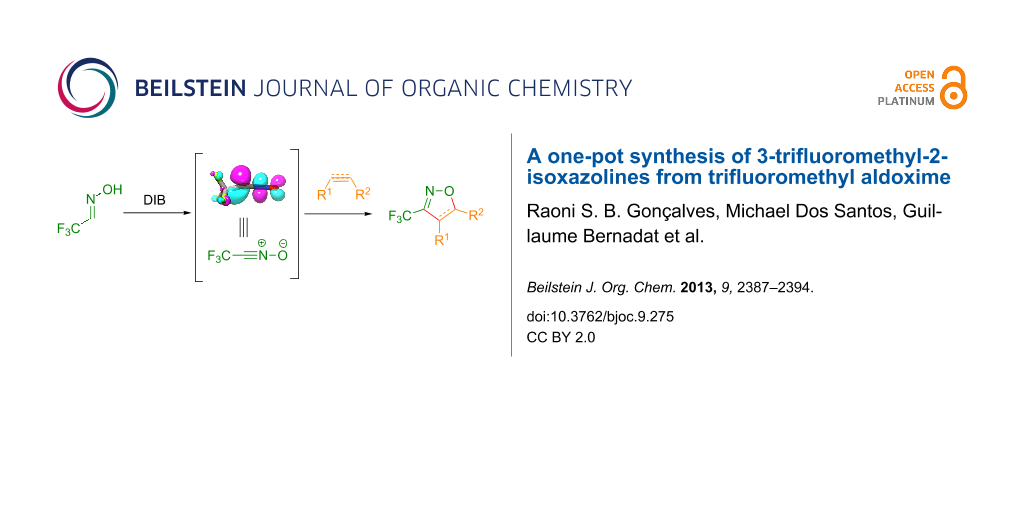
Graphical Abstract
Introduction
2-Isoxazolines are five-membered heterocyclic compounds that have been widely applied in medicinal and organic chemistry. This nucleus is frequently found in natural products [1-4], bioactive molecules [5,6] (Figure 1) and can be used as bioisosteric transformations of amide bonds in order to provide metabolically stable and more active derivatives [7-11]. Moreover, 2-isoxazolines can be cleaved under various conditions to supply a variety of organic functionalities including γ-amino alcohols [12], β-amino acids [13], β-hydroxy ketones [14,15] and β-hydroxy nitriles [14,15].
![[1860-5397-9-275-1]](/bjoc/content/figures/1860-5397-9-275-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Example of bioactive molecules bearing the 2-isoxazoline nucleus.
Figure 1: Example of bioactive molecules bearing the 2-isoxazoline nucleus.
Fluorinated compounds play a central role in different branches of chemistry [16]. The incorporation of a fluorine atom into bioactive molecules causes remarkable changes of their physicochemical properties, which allows the development of substances with improved pharmacological characteristics. Some examples are the synthesis of modified amino acids and peptides, carbohydrates, natural products and the development of more selective enzyme inhibitors [17-21]. Another powerful area, yet a somewhat less utilised role for fluorine is as a tag for 19F NMR that offers several analytical advantages including speed, sensitivity and selectivity [22,23]. Fluorinated molecules have served as valuable 19F NMR probes in high-throughput screening, drug metabolism and protein binding experiments as well as in assessing gene expression [24].
Nevertheless, the preparation of 3-trifluoromethyl-2-isoxazolines 1 has not been extensively studied so far. In the literature only a few examples of the preparation of these derivatives through a multistep procedure are described (Scheme 1) [25]. Initially, trifluoromethyl aldoxime 2 is halogenated to give a volatile trifluoroacetohydroxymoyl chloride or bromide 3, which is usually isolated in low yields. Reaction of intermediate 3 with a base provides trifluoroacetonitrile oxide 4, which can be reacted with olefins (such as styrene, allyl derivatives, etc.) through a 1,3-dipolar cycloaddition to give the desired product. Therefore, the development of a straightforward and mild general procedure to access these valuable derivatives remains of great importance. In the present work, we describe a simple and efficient metal-free protocol for the oxidation of trifluoromethyl aldoxime 2 into trifluoroacetonitrile oxide 4 and a one-pot synthesis of 1 through in situ cyclization of 4 with different dipolarophiles (Scheme 1).
![[1860-5397-9-275-i1]](/bjoc/content/inline/1860-5397-9-275-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of 3-trifluoromethyl-2-isoxazolines.
Scheme 1: Synthesis of 3-trifluoromethyl-2-isoxazolines.
Results and Discussion
Initially, another procedure for the preparation of the trifluoroacetaldehyde oxime 2 was developed. In a previous work [25], 2 was obtained as an etherate complex from the reaction between 2,2,2-trifluoroethane-1,1-diol (TFAL) and hydroxylamine hydrochloride. In our work, reaction of TFAL and an aqueous solution of hydroxylamine (50 wt %) yielded the desired product, which was isolated as a complex of two molecules of aldoxime with one molecule of water after distillation in 70–80% yield (Scheme 2).
![[1860-5397-9-275-i2]](/bjoc/content/inline/1860-5397-9-275-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
In the recent literature, different conditions have been developed for the direct oxidation of aldoximes [26-31]. Recently, commercially available reagents have been employed under metal-free conditions. A group [27] reported that the hypervalent iodine reagents (diacetoxyiodo)benzene (DIB) and phenyliodine bis(trifluoroacetate) (PIFA) could successfully promote the oxidation of aldoximes to the corresponding nitrile oxide. Those reagents exhibit potent oxidizing properties, comparable to heavy-metal reagents, but with several advantages such as low toxicity, high availability and the possibility to be utilized under mild conditions [32]. Then, we decided to verify their applicability in the oxidation of 2 despite the presence of water. We first screened different oxidative reagents and conditions for the oxidation step and allylbenzene (5a) was chosen as dipolarophile. Our studies for this process are summarized in Table 1.
![[Graphic 1]](/bjoc/content/inline/1860-5397-9-275-i3.svg?max-width=637&scale=1.0)
When DIB was used with triethylamine (TEA) and methanol as solvent, the formation of a complex mixture was observed (Table 1, entry 1). This is probably due to the nucleophilic addition of methanol to the highly electrophilic trifluoroacetonitrile oxide. The utilization of the less nucleophilic alcohol hexafluoroisopropanol (HFIP) [33,34] led to the formation of a complex mixture (Table 1, entry 2). The oxidation of 2 with PIFA in CH2Cl2 afforded the product in only 16% yield (Table 1, entry 4). Better results were obtained by employing DIB in CH2Cl2 as solvent, after which the product could be isolated in an acceptable yield (55%, Table 1, entry 3). [Bis(acetoxy)iodo]benzene (DIB) is a weaker oxidant than PIFA. When the oxidation is carried out with DIB, weak acetic acid instead of strong trifluoroacetic acid is liberated, and the decomposition of the oxazolines is avoided.
Faced with the moderate yield of 1a, we followed the reaction by using 19F NMR. The measurement of the crude mixture with 19F NMR revealed the presence of a side product and despite the total consumption of the aldoxime, a small amount of allylbenzene remained. It is known that nitrile oxides can dimerize or isomerize to yield different products, such as furoxans, isocyanates, 1,2,4-oxadiazoles and 1,4,2,5-dioxadiazines (Figure 2).
![[1860-5397-9-275-2]](/bjoc/content/figures/1860-5397-9-275-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Dimerization and isomerization products from nitrile oxides.
Figure 2: Dimerization and isomerization products from nitrile oxides.
We thus postulated that a competition between the cycloaddition reaction and the dimerization or isomerization pathways could occur. Aiming to confirm our hypothesis we carried out the reaction without the presence of allylbenzene. After 12 h, 2 was completely consumed with the exclusive formation of the previously observed side product. However, attempts to isolate this product failed due to its high volatility. It was therefore co-distilled with CH2Cl2 and the resulted solution was analyzed by 19F NMR coupled and decoupled with proton, 19F,19F-COSY and 19F,19F-NOESY (see Supporting Information File 1). Data confirmed the formation of bis(trifluoromethyl)furoxan 6 (Figure 3) already synthesized by Middleton [35].
![[1860-5397-9-275-3]](/bjoc/content/figures/1860-5397-9-275-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Dimerization of 4 yielding bis(trifluoromethyl)furoxan 6.
Figure 3: Dimerization of 4 yielding bis(trifluoromethyl)furoxan 6.
Considering that the best results were reached utilizing DIB and CH2Cl2 as solvent, these conditions were selected for further optimizations. After several investigations, we could verify that employing two equivalents of aldoxime 2 and two equivalents of DIB led to complete conversion of the starting olefin. The product could be isolated in good yield (76%, see below in Table 2, entry 1). We can note here that water complexed with 2 did not alter the reaction rate.
Table 2: Synthesis of 3-trifluoromethyl-2-isoxazolines and isoxazoles by reaction between aldoxime 2 and olefins or alkynes in the presence of DIB.
![[Graphic 2]](/bjoc/content/inline/1860-5397-9-275-i4.svg?max-width=637&scale=1.0)
|
|||
| entry | substrate | product | isolated yield (%) |
|---|---|---|---|
| 1 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-9-275-i5.svg?max-width=637&scale=1.0)
5a |
![[Graphic 4]](/bjoc/content/inline/1860-5397-9-275-i6.svg?max-width=637&scale=1.0)
1a |
76 |
| 2 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-9-275-i7.svg?max-width=637&scale=1.0)
5b |
![[Graphic 6]](/bjoc/content/inline/1860-5397-9-275-i8.svg?max-width=637&scale=1.0)
1b |
90 |
| 3 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-9-275-i9.svg?max-width=637&scale=1.0)
5c |
![[Graphic 8]](/bjoc/content/inline/1860-5397-9-275-i10.svg?max-width=637&scale=1.0)
1c |
91 |
| 4 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-9-275-i11.svg?max-width=637&scale=1.0)
5d |
![[Graphic 10]](/bjoc/content/inline/1860-5397-9-275-i12.svg?max-width=637&scale=1.0)
1d |
64 |
| 5 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-9-275-i13.svg?max-width=637&scale=1.0)
5e |
![[Graphic 12]](/bjoc/content/inline/1860-5397-9-275-i14.svg?max-width=637&scale=1.0)
1e |
51 |
| 6 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-9-275-i15.svg?max-width=637&scale=1.0)
5f |
![[Graphic 14]](/bjoc/content/inline/1860-5397-9-275-i16.svg?max-width=637&scale=1.0)
1f |
56 |
| 7 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-9-275-i17.svg?max-width=637&scale=1.0)
5g |
![[Graphic 16]](/bjoc/content/inline/1860-5397-9-275-i18.svg?max-width=637&scale=1.0)
1g |
82 |
| 8 |
![[Graphic 17]](/bjoc/content/inline/1860-5397-9-275-i19.svg?max-width=637&scale=1.0)
5h |
![[Graphic 18]](/bjoc/content/inline/1860-5397-9-275-i20.svg?max-width=637&scale=1.0)
1h |
74 |
| 9 |
![[Graphic 19]](/bjoc/content/inline/1860-5397-9-275-i21.svg?max-width=637&scale=1.0)
5i |
![[Graphic 20]](/bjoc/content/inline/1860-5397-9-275-i22.svg?max-width=637&scale=1.0)
1i |
24 |
| 10 |
![[Graphic 21]](/bjoc/content/inline/1860-5397-9-275-i23.svg?max-width=637&scale=1.0)
5j |
![[Graphic 22]](/bjoc/content/inline/1860-5397-9-275-i24.svg?max-width=637&scale=1.0)
1j |
20 |
| 11 |
![[Graphic 23]](/bjoc/content/inline/1860-5397-9-275-i25.svg?max-width=637&scale=1.0)
5k |
![[Graphic 24]](/bjoc/content/inline/1860-5397-9-275-i26.svg?max-width=637&scale=1.0)
1k |
traces |
| 12 |
![[Graphic 25]](/bjoc/content/inline/1860-5397-9-275-i27.svg?max-width=637&scale=1.0)
5l |
![[Graphic 26]](/bjoc/content/inline/1860-5397-9-275-i28.svg?max-width=637&scale=1.0)
1l |
53 |
| 13 |
![[Graphic 27]](/bjoc/content/inline/1860-5397-9-275-i29.svg?max-width=637&scale=1.0)
5m |
![[Graphic 28]](/bjoc/content/inline/1860-5397-9-275-i30.svg?max-width=637&scale=1.0)
1m |
50 |
| 14 |
![[Graphic 29]](/bjoc/content/inline/1860-5397-9-275-i31.svg?max-width=637&scale=1.0)
5n |
![[Graphic 30]](/bjoc/content/inline/1860-5397-9-275-i32.svg?max-width=637&scale=1.0)
1n |
traces |
Having optimized reagents and conditions, the scope of the reaction was explored with regard to the substrates (Table 2). As observed in the earliest works [11], the cycloaddition of 4 with terminal olefins led to the corresponding 3-trifluoromethyl-5-substituted-2-isoxazoline 1 with complete regioselectivity. No trace of the regioisomer 3-trifluoromethyl-4-substituted-2-isoxazoline could be detected even when yields of cycloadducts were low. From functionalized olefins such as NH–Cbz and NH–Boc allylamines the desired product could be isolated in excellent yields (90% and 91%, respectively, Table 2, entries 2 and 3). Interestingly, the protecting groups were not cleaved, which indicated that reaction conditions are very mild. With allyltrimethylsilane oxazoline 1g was obtained in good yield (82%). From ester and acid derivatives of undecen, isoxazolines were obtained in good yields too (64% and 51%, respectively, Table 2, entries 4 and 5). With para-bromostyrene isoxazoline 1f was obtained in 56% yield. On the other hand, a good yield was reached for disubstituted olefin 5h (Table 2, entry 8). However a complete lack of reactivity was observed for the reaction with the electron-poor olefins 5i, 5j and 5k (Table 2, entries 9–11). The reaction was also carried out with alkynes, providing 3-trifluoromethyl-5-substituted-isoxazoles. Moderate yields were obtained for the reaction with phenylacetylene (5l) and trimethylsilylacetylene (5m, Table 2, entries 12 and 13). However, the electron poor alkyne 5n was unreactive towards trifluoroacetonitrile oxide 4.
1,3-Dipolar cycloaddition reactions have been studied from the theoretical standpoint since the 1970’s onwards [36,37] with an ever-increasing accuracy as computational methods evolved [38]. Assuming that the above-mentioned transformations occur via a concerted mechanism, we decided to perform electronic structure calculations at the B3LYP/6-31G* level in order to further understand the reactivity of the different unsaturated compounds studied in this work against trifluoroacetonitrile oxide 4 (See Supporting Information File 1) [39-43]. Upon energy minimization, a structure with a geometry close to linearity was found for trifluoroacetonitrile oxide (Figure 4), which is consistent with earlier findings [44].
![[1860-5397-9-275-4]](/bjoc/content/figures/1860-5397-9-275-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Depiction of the geometry (left column) and isodensity surface of the reacting frontier molecular orbitals (FMO) at 50% probability (right column) of trifluoroacetonitrile oxide 4 (top row) and protected aminoalkene 5c (bottom row) calculated at the B3LYP/6-31G* level.
Figure 4: Depiction of the geometry (left column) and isodensity surface of the reacting frontier molecular o...
Comparison of the energy gaps between the frontier molecular orbital levels of the nitrile oxide and those of the alkene partners (Figure 5) suggest that type-III cycloaddition reactions (where the dipole reacts via its LUMO and the dipolarophile via its HOMO) take place for every combination of reactants reported herein [45].
![[1860-5397-9-275-5]](/bjoc/content/figures/1860-5397-9-275-5.png?scale=2.5&max-width=1024&background=FFFFFF)
Figure 5: FMO energy levels of dipole 4 and dipolarophiles 5a and 5k calculated at the B3LYP/6-31G* level. Continuous and dotted lines indicate the favored (ΔE = 5.407 eV for 5a and 6.673 eV for 5k) and the disfavored (ΔE = 8.178 eV for 5a and 7.955 eV for 5k) molecular orbital interactions, respectively.
Figure 5: FMO energy levels of dipole 4 and dipolarophiles 5a and 5k calculated at the B3LYP/6-31G* level. Co...
In this scenario, with the same dipole, the reactivity is expected to increase with the HOMO energy level of the dipolarophile. A simple scatter plot of yield versus the latter variable confirms and illustrates this trend (Figure 6), which can be interpreted the following way: when the gap between the LUMO energy level of the nitrile oxide and the HOMO energy level of the alkene becomes too high (relatively to 7.139 eV, which is the energy difference for the self-cycloaddition process), the dimerization pathway is favored and the yield of isoxazole drops.
![[1860-5397-9-275-6]](/bjoc/content/figures/1860-5397-9-275-6.png?scale=2.5&max-width=1024&background=FFFFFF)
Figure 6: Yields of the cycloaddition reaction plotted against the HOMO energy levels of the dipolarophile partner among 5a–c,f–n.
Figure 6: Yields of the cycloaddition reaction plotted against the HOMO energy levels of the dipolarophile pa...
With only a single regioisomer being isolated, we also considered the coefficients (Table 3) and shapes (Figure 4) of frontier molecular orbitals. This data is compatible with the observed regioselectivity [37]. Steric factors can also exert notable influence, but they would guide regioselectivity in the same direction.
Table 3: FMO coefficients of the 1,3-dipole 4 and representative dipolarophiles (atomic orbital is indicated in parentheses).
| coefficients | ||
|---|---|---|
| reactant | Ca | Cb |
| 5c | 0.180 (2py) | 0.133 (2px) |
| 5g | 0.321 (2pz) | 0.246 (2pz) |
| 4a | 0.434 (2py) | 0.299 (2py) |
aColumns Ca and Cb contain coefficients for C and O.
In a subsequent step, we decided to preliminarily study the ring opening reaction of the 3-trifluoromethyl-2-isoxazolines in order to prepare the corresponding trifluoromethylated γ-amino alcohols. The major and almost the only route to synthesize these amino alcohols is the reduction of β-aminocarbonyl compounds prepared from Mannich-type reactions [46-48]. The ring opening of 2-methyl-3-trifluoromethylisoxazolines by utilizing H2 and Raney-Ni as catalysts was described by Tanaka and co-workers [49]. However, this methodology is restricted to the synthesis of N-methylated amino alcohols. Instead we investigated the reduction and the ring opening of 3-trifluoromethyl-2-isoxazolines 1a and 1b in the presence of NaBH4 and NiCl2 as additives [50]. Under these conditions, a total conversion of the starting material was observed by 19F NMR, and products 7a and 7b were obtained in moderate yields. However the diastereoisomeric excess was very poor (10% de). The results are reported in Table 4.
![[Graphic 31]](/bjoc/content/inline/1860-5397-9-275-i33.svg?max-width=637&scale=1.0)
![[Graphic 32]](/bjoc/content/inline/1860-5397-9-275-i34.svg?max-width=637&scale=1.0)
![[Graphic 33]](/bjoc/content/inline/1860-5397-9-275-i35.svg?max-width=637&scale=1.0)
![[Graphic 34]](/bjoc/content/inline/1860-5397-9-275-i36.svg?max-width=637&scale=1.0)
![[Graphic 35]](/bjoc/content/inline/1860-5397-9-275-i37.svg?max-width=637&scale=1.0)
Conclusion
In conclusion, we have developed a simple, mild and efficient one-step procedure for the synthesis of functionalized 3-trifluoromethyl-2-isoxazolines and 3-trifluoromethyl-2-isoxazoles from trifluoromethyl aldoxime 2 by utilizing DIB as oxidant. The applicability of the 3-trifluoromethyl-2-isoxazolines to supply different fluorinated building blocks was demonstrated by the easy ring opening of these intermediates with NaBH4 and NiCl2, yielding the corresponding trifluoromethylated γ-amino alcohol.
Supporting Information
| Supporting Information File 1: General methods, synthetic procedure, spectroscopic data, 1H NMR, 13C NMR and 19F NMR of compounds of 2, 1a-1j, 1l-1n, 7a, 7b, 6 and computational results. | ||
| Format: PDF | Size: 2.8 MB | Download |
Acknowledgements
R.S.B.G thanks the FAPERJ, CNPq, the Marie Curie program, the Farmaguinhos/FioCruz, and PGQu for financial support. Central Glass Co. Ltd. is gratefully acknowledged for kindly providing fluoral hydrate and HFIP, as well as the IT department from Université Paris-Sud for providing computing resources. The authors would like thank M’bokeno Gomes Lowa, Erasmus student. We also thank the French Fluorine Network (GIS Fluor).
References
-
Feuer, H., Ed. Nitrile oxides, nitrones, and nitronates in organic synthesis, 2nd ed.; John Wiley & Sons: Hoboken, NJ, 2008.
Return to citation in text: [1] -
Cicchi, S.; Cordero, F. M.; Giomi, D. Five-membered ring systems: With O&N atoms. In Progress in Heterocyclic Chemistry; Gribble, G. W.; Joule, J. A., Eds.; Pergamon: Oxford, 2003; Vol. 15, pp 261–283.
Return to citation in text: [1] -
Namboothiri, I. N. N.; Rastogi, N. Isoxazolines from Nitro Compounds: Synthesis and Applications. In Synthesis of Heterocycles via Cycloadditions I; Gupta, R. R.; Hassner, A., Eds.; Springer: Berlin, 2008; Vol. 12, pp 1–44. doi:10.1007/7081_2007_101
Return to citation in text: [1] -
Savage, G. P. Curr. Org. Chem. 2010, 14, 1478–1499. doi:10.2174/138527210791616812
Return to citation in text: [1] -
El Sayed, K. A.; Bartyzel, P.; Shen, X.; Perry, T. L.; Zjawiony, J. K.; Hamann, M. T. Tetrahedron 2000, 56, 949–953. doi:10.1016/S0040-4020(99)01093-5
Return to citation in text: [1] -
Proksch, P.; Putz, A.; Ortlepp, S.; Kjer, J.; Bayer, M. Phytochem. Rev. 2010, 9, 475–489. doi:10.1007/s11101-010-9178-9
Return to citation in text: [1] -
Castellano, S.; Kuck, D.; Viviano, M.; Yoo, J.; López-Vallejo, F.; Conti, P.; Tamborini, L.; Pinto, A.; Medina-Franco, J. L.; Sbardella, G. J. Med. Chem. 2011, 54, 7663–7677. doi:10.1021/jm2010404
Return to citation in text: [1] -
Dallanoce, C.; Frigerio, F.; Martelli, G.; Grazioso, G.; Matera, C.; Pomè, D. Y.; Pucci, L.; Clementi, F.; Gotti, C.; De Amici, M. Bioorg. Med. Chem. 2010, 18, 4498–4508. doi:10.1016/j.bmc.2010.04.065
Return to citation in text: [1] -
Cheng, J.-F.; Huang, Y.; Penuliar, R.; Nishimoto, M.; Liu, L.; Arrhenius, T.; Yang, G.; O'Leary, E.; Barbosa, M.; Barr, R.; Dyck, J. R. B.; Lopaschuk, G. D.; Nadzan, A. M. J. Med. Chem. 2006, 49, 4055–4058. doi:10.1021/jm0605029
Return to citation in text: [1] -
Benltifa, M.; Hayes, J. M.; Vidal, S.; Gueyrard, D.; Goekjian, P. G.; Praly, J.-P.; Kizilis, G.; Tiraidis, C.; Alexacou, K.-M.; Chrysina, E. D.; Zographos, S. E.; Leonidas, D. D.; Archontis, G.; Oikonomakos, N. G. Bioorg. Med. Chem. 2009, 17, 7368–7380. doi:10.1016/j.bmc.2009.08.060
Return to citation in text: [1] -
Rakesh; Sun, D.; Lee, R. B.; Tangallapally, R. P.; Lee, R. E. Eur. J. Med. Chem. 2009, 44, 460–472. doi:10.1016/j.ejmech.2008.04.007
Return to citation in text: [1] [2] -
Tangallapally, R. P.; Sun, D.; Rakesh; Budha, N.; Lee, R. E. B.; Lenaerts, A. J.; Meibohm, B.; Lee, R. E. Bioorg. Med. Chem. Lett. 2007, 17, 6638–6642. doi:10.1016/j.bmcl.2007.09.048
Return to citation in text: [1] -
Tang, S.; He, J.; Sun, Y.; He, L.; She, X. J. Org. Chem. 2010, 75, 1961–1966. doi:10.1021/jo1000065
Return to citation in text: [1] -
Jiang, D.; Chen, Y. J. Org. Chem. 2008, 73, 9181–9183. doi:10.1021/jo801831c
Return to citation in text: [1] [2] -
Maimone, T. J.; Shi, J.; Ashida, S.; Baran, P. S. J. Am. Chem. Soc. 2009, 131, 17066–17067. doi:10.1021/ja908194b
Return to citation in text: [1] [2] -
Chambers, R. D. Fluorine in Organic Chemistry; Blackwell Publishing Ltd.: Oxford, 2004. doi:10.1002/9781444305371
Return to citation in text: [1] -
O'Hagan, D. Chem. Soc. Rev. 2008, 37, 308–319. doi:10.1039/b711844a
Return to citation in text: [1] -
Bégué, J.-P.; Bonnet-Delpon, D. Bioorganic and Medicinal Chemistry of Fluorine; Wiley: Hoboken, 2008. doi:10.1002/9780470281895
Return to citation in text: [1] -
Uneyama, K. Organofluorine Chemistry; Blackwell Publishing: Oxford, 2006. doi:10.1002/9780470988589
Return to citation in text: [1] -
Kirsch, P. Modern Fluoroorganic Chemistry; Wiley: Weinheim, Germany, 2004. doi:10.1002/352760393X
Return to citation in text: [1] -
Banks, R. E.; Smart, B. E.; Tatlow, J. C. Organofluorine Chemistry, Principles and Commercial Applications; Plenum: New York, 1994.
Return to citation in text: [1] -
Woods, J. R.; Mo, H.; Bieberich, A. A.; Alavanja, T.; Colby, D. A. J. Med. Chem. 2011, 54, 7934–7941. doi:10.1021/jm201114t
Return to citation in text: [1] -
Jordan, J. B.; Poppe, L.; Xia, X.; Cheng, A. C.; Sun, Y.; Michelsen, K.; Eastwood, H.; Schnier, P. D.; Nixey, T.; Zhong, W. J. Med. Chem. 2012, 55, 678–687. doi:10.1021/jm201441k
Return to citation in text: [1] -
Dalvit, C. Prog. Nucl. Magn. Reson. Spectrosc. 2007, 51, 243–271. doi:10.1016/j.pnmrs.2007.07.002
Return to citation in text: [1] -
Tanaka, K.; Masuda, H.; Mitsuhashi, K. Bull. Chem. Soc. Jpn. 1984, 57, 2184–2187. doi:10.1246/bcsj.57.2184
Return to citation in text: [1] [2] -
Yoshimura, A.; Zhu, C.; Middleton, K. R.; Todora, A. D.; Kastern, B. J.; Maskaev, A. V.; Zhdankin, V. V. Chem. Commun. 2013, 49, 4800–4802. doi:10.1039/c3cc41164h
Return to citation in text: [1] -
Mendelsohn, B. A.; Lee, S.; Kim, S.; Teyssier, F.; Aulakh, V. S.; Ciufolini, M. A. Org. Lett. 2009, 11, 1539–1542. doi:10.1021/ol900194v
Return to citation in text: [1] [2] -
Shing, T. K. M.; Wong, W. F.; Cheng, H. M.; Kwok, W. S.; So, K. H. Org. Lett. 2007, 9, 753–756. doi:10.1021/ol062873p
Return to citation in text: [1] -
Singh, V.; Hutait, S.; Biswas, S.; Batra, S. Eur. J. Org. Chem. 2010, 531–539. doi:10.1002/ejoc.200901166
Return to citation in text: [1] -
Pinto, A.; Conti, P.; Grazioso, G.; Tamborini, L.; Madsen, U.; Nielsen, B.; De Micheli, C. Eur. J. Med. Chem. 2011, 46, 787–793. doi:10.1016/j.ejmech.2010.12.020
Return to citation in text: [1] -
Minakata, S.; Okumura, S.; Nagamachi, T.; Takeda, Y. Org. Lett. 2011, 13, 2966–2969. doi:10.1021/ol2010616
Return to citation in text: [1] -
Zhdankin, V. V.; Stang, P. J. Chem. Rev. 2008, 108, 5299–5358. doi:10.1021/cr800332c
Return to citation in text: [1] -
Bégué, J.-P.; Bonnet-Delpon, D.; Crousse, B. Synlett 2004, 18–29. doi:10.1055/s-2003-44973
Return to citation in text: [1] -
Shuklov, I. A.; Dubrovina, N. V.; Börner, A. Synthesis 2007, 2925–2943. doi:10.1055/s-2007-983902
Return to citation in text: [1] -
Middleton, W. J. J. Org. Chem. 1984, 49, 919–922. doi:10.1021/jo00179a031
Return to citation in text: [1] -
Houk, K. N.; Sims, J.; Duke, R. E.; Strozier, R. W.; George, J. K. J. Am. Chem. Soc. 1973, 95, 7287–7301. doi:10.1021/ja00803a017
Return to citation in text: [1] -
Houk, K. N.; Sims, J.; Watts, C. R.; Luskus, L. J. J. Am. Chem. Soc. 1973, 95, 7301–7315. doi:10.1021/ja00803a018
Return to citation in text: [1] [2] -
Lan, Y.; Zou, L.; Cao, Y.; Houk, K. N. J. Phys. Chem. A 2011, 115, 13906–13920. doi:10.1021/jp207563h
Return to citation in text: [1] -
Kohn, W.; Sham, L. J. Phys. Rev. 1965, 140, A1133–A1138. doi:10.1103/PhysRev.140.A1133
Return to citation in text: [1] -
Hohenberg, P.; Kohn, W. Phys. Rev. 1964, 136, B864–B871. doi:10.1103/PhysRev.136.B864
Return to citation in text: [1] -
Becke, A. D. J. Chem. Phys. 1993, 98, 5648–5652. doi:10.1063/1.464913
Return to citation in text: [1] -
Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. B 1988, 37, 785–789. doi:10.1103/PhysRevB.37.785
Return to citation in text: [1] -
Hehre, W. J.; Radom, L.; Schleyer, P. V. R.; Pople, J. Ab Initio Molecular Orbital Theory; Wiley: New York, 1986.
Return to citation in text: [1] -
Havasi, B.; Pasinszki, T.; Westwood, N. P. C. J. Phys. Chem. A 2005, 109, 3864–3874. doi:10.1021/jp040725f
Return to citation in text: [1] -
Sustmann, R. Pure Appl. Chem. 1974, 40, 569–593. doi:10.1351/pac197440040569
Return to citation in text: [1] -
Michaut, V.; Metz, F.; Paris, J.-M.; Plaquevent, J.-C. J. Fluorine Chem. 2007, 128, 889–895. doi:10.1016/j.jfluchem.2007.03.007
Return to citation in text: [1] -
Sukach, V. A.; Golovach, N. M.; Pirozhenko, V. V.; Rusanov, E. B.; Vovk, M. V. Tetrahedron: Asymmetry 2008, 19, 761–764. doi:10.1016/j.tetasy.2008.02.023
Return to citation in text: [1] -
Huguenot, F.; Brigaud, T. J. Org. Chem. 2006, 71, 2159–2162. doi:10.1021/jo052323p
Return to citation in text: [1] -
Tanaka, K.; Sugimoto, Y.; Okafuji, Y.; Tachikawa, M.; Mitsuhashi, K. J. Heterocycl. Chem. 1989, 26, 381–385. doi:10.1002/jhet.5570260221
Return to citation in text: [1] -
Nonn, M.; Kiss, L.; Sillanpää, R.; Fülöp, F. Beilstein J. Org. Chem. 2012, 8, 100–106. doi:10.3762/bjoc.8.10
Return to citation in text: [1]
| 50. | Nonn, M.; Kiss, L.; Sillanpää, R.; Fülöp, F. Beilstein J. Org. Chem. 2012, 8, 100–106. doi:10.3762/bjoc.8.10 |
| 1. | Feuer, H., Ed. Nitrile oxides, nitrones, and nitronates in organic synthesis, 2nd ed.; John Wiley & Sons: Hoboken, NJ, 2008. |
| 2. | Cicchi, S.; Cordero, F. M.; Giomi, D. Five-membered ring systems: With O&N atoms. In Progress in Heterocyclic Chemistry; Gribble, G. W.; Joule, J. A., Eds.; Pergamon: Oxford, 2003; Vol. 15, pp 261–283. |
| 3. | Namboothiri, I. N. N.; Rastogi, N. Isoxazolines from Nitro Compounds: Synthesis and Applications. In Synthesis of Heterocycles via Cycloadditions I; Gupta, R. R.; Hassner, A., Eds.; Springer: Berlin, 2008; Vol. 12, pp 1–44. doi:10.1007/7081_2007_101 |
| 4. | Savage, G. P. Curr. Org. Chem. 2010, 14, 1478–1499. doi:10.2174/138527210791616812 |
| 13. | Tang, S.; He, J.; Sun, Y.; He, L.; She, X. J. Org. Chem. 2010, 75, 1961–1966. doi:10.1021/jo1000065 |
| 27. | Mendelsohn, B. A.; Lee, S.; Kim, S.; Teyssier, F.; Aulakh, V. S.; Ciufolini, M. A. Org. Lett. 2009, 11, 1539–1542. doi:10.1021/ol900194v |
| 12. | Tangallapally, R. P.; Sun, D.; Rakesh; Budha, N.; Lee, R. E. B.; Lenaerts, A. J.; Meibohm, B.; Lee, R. E. Bioorg. Med. Chem. Lett. 2007, 17, 6638–6642. doi:10.1016/j.bmcl.2007.09.048 |
| 32. | Zhdankin, V. V.; Stang, P. J. Chem. Rev. 2008, 108, 5299–5358. doi:10.1021/cr800332c |
| 7. | Castellano, S.; Kuck, D.; Viviano, M.; Yoo, J.; López-Vallejo, F.; Conti, P.; Tamborini, L.; Pinto, A.; Medina-Franco, J. L.; Sbardella, G. J. Med. Chem. 2011, 54, 7663–7677. doi:10.1021/jm2010404 |
| 8. | Dallanoce, C.; Frigerio, F.; Martelli, G.; Grazioso, G.; Matera, C.; Pomè, D. Y.; Pucci, L.; Clementi, F.; Gotti, C.; De Amici, M. Bioorg. Med. Chem. 2010, 18, 4498–4508. doi:10.1016/j.bmc.2010.04.065 |
| 9. | Cheng, J.-F.; Huang, Y.; Penuliar, R.; Nishimoto, M.; Liu, L.; Arrhenius, T.; Yang, G.; O'Leary, E.; Barbosa, M.; Barr, R.; Dyck, J. R. B.; Lopaschuk, G. D.; Nadzan, A. M. J. Med. Chem. 2006, 49, 4055–4058. doi:10.1021/jm0605029 |
| 10. | Benltifa, M.; Hayes, J. M.; Vidal, S.; Gueyrard, D.; Goekjian, P. G.; Praly, J.-P.; Kizilis, G.; Tiraidis, C.; Alexacou, K.-M.; Chrysina, E. D.; Zographos, S. E.; Leonidas, D. D.; Archontis, G.; Oikonomakos, N. G. Bioorg. Med. Chem. 2009, 17, 7368–7380. doi:10.1016/j.bmc.2009.08.060 |
| 11. | Rakesh; Sun, D.; Lee, R. B.; Tangallapally, R. P.; Lee, R. E. Eur. J. Med. Chem. 2009, 44, 460–472. doi:10.1016/j.ejmech.2008.04.007 |
| 25. | Tanaka, K.; Masuda, H.; Mitsuhashi, K. Bull. Chem. Soc. Jpn. 1984, 57, 2184–2187. doi:10.1246/bcsj.57.2184 |
| 5. | El Sayed, K. A.; Bartyzel, P.; Shen, X.; Perry, T. L.; Zjawiony, J. K.; Hamann, M. T. Tetrahedron 2000, 56, 949–953. doi:10.1016/S0040-4020(99)01093-5 |
| 6. | Proksch, P.; Putz, A.; Ortlepp, S.; Kjer, J.; Bayer, M. Phytochem. Rev. 2010, 9, 475–489. doi:10.1007/s11101-010-9178-9 |
| 26. | Yoshimura, A.; Zhu, C.; Middleton, K. R.; Todora, A. D.; Kastern, B. J.; Maskaev, A. V.; Zhdankin, V. V. Chem. Commun. 2013, 49, 4800–4802. doi:10.1039/c3cc41164h |
| 27. | Mendelsohn, B. A.; Lee, S.; Kim, S.; Teyssier, F.; Aulakh, V. S.; Ciufolini, M. A. Org. Lett. 2009, 11, 1539–1542. doi:10.1021/ol900194v |
| 28. | Shing, T. K. M.; Wong, W. F.; Cheng, H. M.; Kwok, W. S.; So, K. H. Org. Lett. 2007, 9, 753–756. doi:10.1021/ol062873p |
| 29. | Singh, V.; Hutait, S.; Biswas, S.; Batra, S. Eur. J. Org. Chem. 2010, 531–539. doi:10.1002/ejoc.200901166 |
| 30. | Pinto, A.; Conti, P.; Grazioso, G.; Tamborini, L.; Madsen, U.; Nielsen, B.; De Micheli, C. Eur. J. Med. Chem. 2011, 46, 787–793. doi:10.1016/j.ejmech.2010.12.020 |
| 31. | Minakata, S.; Okumura, S.; Nagamachi, T.; Takeda, Y. Org. Lett. 2011, 13, 2966–2969. doi:10.1021/ol2010616 |
| 17. | O'Hagan, D. Chem. Soc. Rev. 2008, 37, 308–319. doi:10.1039/b711844a |
| 18. | Bégué, J.-P.; Bonnet-Delpon, D. Bioorganic and Medicinal Chemistry of Fluorine; Wiley: Hoboken, 2008. doi:10.1002/9780470281895 |
| 19. | Uneyama, K. Organofluorine Chemistry; Blackwell Publishing: Oxford, 2006. doi:10.1002/9780470988589 |
| 20. | Kirsch, P. Modern Fluoroorganic Chemistry; Wiley: Weinheim, Germany, 2004. doi:10.1002/352760393X |
| 21. | Banks, R. E.; Smart, B. E.; Tatlow, J. C. Organofluorine Chemistry, Principles and Commercial Applications; Plenum: New York, 1994. |
| 24. | Dalvit, C. Prog. Nucl. Magn. Reson. Spectrosc. 2007, 51, 243–271. doi:10.1016/j.pnmrs.2007.07.002 |
| 16. | Chambers, R. D. Fluorine in Organic Chemistry; Blackwell Publishing Ltd.: Oxford, 2004. doi:10.1002/9781444305371 |
| 25. | Tanaka, K.; Masuda, H.; Mitsuhashi, K. Bull. Chem. Soc. Jpn. 1984, 57, 2184–2187. doi:10.1246/bcsj.57.2184 |
| 14. | Jiang, D.; Chen, Y. J. Org. Chem. 2008, 73, 9181–9183. doi:10.1021/jo801831c |
| 15. | Maimone, T. J.; Shi, J.; Ashida, S.; Baran, P. S. J. Am. Chem. Soc. 2009, 131, 17066–17067. doi:10.1021/ja908194b |
| 14. | Jiang, D.; Chen, Y. J. Org. Chem. 2008, 73, 9181–9183. doi:10.1021/jo801831c |
| 15. | Maimone, T. J.; Shi, J.; Ashida, S.; Baran, P. S. J. Am. Chem. Soc. 2009, 131, 17066–17067. doi:10.1021/ja908194b |
| 22. | Woods, J. R.; Mo, H.; Bieberich, A. A.; Alavanja, T.; Colby, D. A. J. Med. Chem. 2011, 54, 7934–7941. doi:10.1021/jm201114t |
| 23. | Jordan, J. B.; Poppe, L.; Xia, X.; Cheng, A. C.; Sun, Y.; Michelsen, K.; Eastwood, H.; Schnier, P. D.; Nixey, T.; Zhong, W. J. Med. Chem. 2012, 55, 678–687. doi:10.1021/jm201441k |
| 11. | Rakesh; Sun, D.; Lee, R. B.; Tangallapally, R. P.; Lee, R. E. Eur. J. Med. Chem. 2009, 44, 460–472. doi:10.1016/j.ejmech.2008.04.007 |
| 33. | Bégué, J.-P.; Bonnet-Delpon, D.; Crousse, B. Synlett 2004, 18–29. doi:10.1055/s-2003-44973 |
| 34. | Shuklov, I. A.; Dubrovina, N. V.; Börner, A. Synthesis 2007, 2925–2943. doi:10.1055/s-2007-983902 |
| 46. | Michaut, V.; Metz, F.; Paris, J.-M.; Plaquevent, J.-C. J. Fluorine Chem. 2007, 128, 889–895. doi:10.1016/j.jfluchem.2007.03.007 |
| 47. | Sukach, V. A.; Golovach, N. M.; Pirozhenko, V. V.; Rusanov, E. B.; Vovk, M. V. Tetrahedron: Asymmetry 2008, 19, 761–764. doi:10.1016/j.tetasy.2008.02.023 |
| 48. | Huguenot, F.; Brigaud, T. J. Org. Chem. 2006, 71, 2159–2162. doi:10.1021/jo052323p |
| 49. | Tanaka, K.; Sugimoto, Y.; Okafuji, Y.; Tachikawa, M.; Mitsuhashi, K. J. Heterocycl. Chem. 1989, 26, 381–385. doi:10.1002/jhet.5570260221 |
| 37. | Houk, K. N.; Sims, J.; Watts, C. R.; Luskus, L. J. J. Am. Chem. Soc. 1973, 95, 7301–7315. doi:10.1021/ja00803a018 |
| 39. | Kohn, W.; Sham, L. J. Phys. Rev. 1965, 140, A1133–A1138. doi:10.1103/PhysRev.140.A1133 |
| 40. | Hohenberg, P.; Kohn, W. Phys. Rev. 1964, 136, B864–B871. doi:10.1103/PhysRev.136.B864 |
| 41. | Becke, A. D. J. Chem. Phys. 1993, 98, 5648–5652. doi:10.1063/1.464913 |
| 42. | Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. B 1988, 37, 785–789. doi:10.1103/PhysRevB.37.785 |
| 43. | Hehre, W. J.; Radom, L.; Schleyer, P. V. R.; Pople, J. Ab Initio Molecular Orbital Theory; Wiley: New York, 1986. |
| 44. | Havasi, B.; Pasinszki, T.; Westwood, N. P. C. J. Phys. Chem. A 2005, 109, 3864–3874. doi:10.1021/jp040725f |
| 36. | Houk, K. N.; Sims, J.; Duke, R. E.; Strozier, R. W.; George, J. K. J. Am. Chem. Soc. 1973, 95, 7287–7301. doi:10.1021/ja00803a017 |
| 37. | Houk, K. N.; Sims, J.; Watts, C. R.; Luskus, L. J. J. Am. Chem. Soc. 1973, 95, 7301–7315. doi:10.1021/ja00803a018 |
| 38. | Lan, Y.; Zou, L.; Cao, Y.; Houk, K. N. J. Phys. Chem. A 2011, 115, 13906–13920. doi:10.1021/jp207563h |
© 2013 Gonçalves et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)