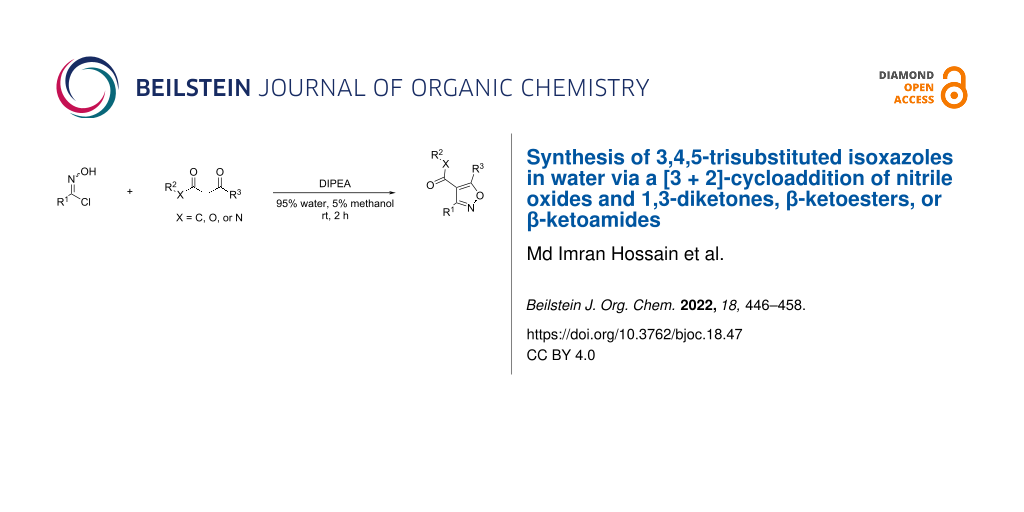
Abstract
Herein we report a method for the synthesis of 3,4,5-trisubstituted isoxazoles in water under mild basic conditions at room temperature via a [3 + 2]-cycloaddition of nitrile oxides and 1,3-diketones, β-ketoesters, or β-ketoamides. We optimized the reaction conditions to control the selectivity of the production of isoxazoles and circumvent other competing reactions, such as O-imidoylation or hetero [3 + 2]-cycloaddition. The reaction happens fast in water and completes within 1–2 hours, which provides an environmentally friendly access to 3,4,5-trisubstituted isoxazoles, an important class of structures found in numerous bioactive natural products and pharmaceuticals. Additionally, we optimized the reaction conditions to produce trifluoromethyl-substituted isoxazoles, a prevalent scaffold in biomedical research and drug discovery programs. We also proposed a plausible mechanism for the selectivity of the [3 + 2]-cycloaddition reaction to produce 3,4,5-trisubstituted isoxazoles. Not to be overlooked are our optimized reaction conditions for the dimerization of hydroximoyl chlorides to form furoxans also known as 1,2,5-oxadiazole 2-oxides, a class of structures with important biological activities due to their unique electronic nature and coordination ability.

Graphical Abstract
Introduction
Isoxazoles are a privileged class of five-membered heterocycles, which are found in numerous bioactive natural products [1,2] and synthetic small molecule drugs [3,4], and are used as important precursors for the synthesis of β-hydroxycarbonyl compounds and γ-amino alcohols [1]. Isoxazoles, appearing in 33 patents from the year 2016 to 2018 [3], are an important drug class due to their wide range of biological activities, such as anticancer [5], antibiotic [6,7], antimicrobial [8], antifungal [9], and anti-inflammatory [10]. Therefore, new methods to develop efficient, high-yielding, and green routes to isoxazoles are always highly desirable.
New approaches to 3,4,5-trisubstituted isoxazoles, particularly to the ones that leverage diverse chemical libraries, are exciting due to a limited number of suitable methods. One current route is the cycloaddition of alkynes and nitrile oxides performed in organic solvents at elevated temperatures (Figure 1) [11-16]. In this method, while 3,5-disubstituted isoxazoles can be accessed from terminal alkynes, 3,4,5-trisubstituted isoxazoles require a high degree of substitution on non-terminal alkynes to activate them for a decent yield of the isoxazole products, thus limiting the scope of the substrates in this method. In addition, this method requires high heat and produces very poor regioselectivity of the products [17,18]. The addition of copper catalysts in this route can help the reaction proceed at room temperature and improve both the regioselectivity and yields of the isoxazoles. However, these catalysts only work for the reaction with terminal alkynes and only produce 3,5-disubstituted isoxazole products (Figure 1) [19,20]. The synthesis of 3,4,5-trisubstituted isoxazoles from highly substituted non-terminal alkynes does not proceed with copper catalysts at room temperature. As an alternative, the usage of ruthenium(II) catalysts enables the reaction to proceed smoothly at room temperature and produces high yields and regioselectivity for both, 3,5-disubstituted and 3,4,5-trisubstituted isoxazoles (Figure 1) [21,22]. Similarly, palladium catalysts were used for the electrophilic intramolecular cyclization of alkynes and aldoximes to produce 3,4,5-trisubstituted isoxazoles, but the scope of the substrates of the method was limited as the substituted 2-alkyne-1-one O-methyl oximes needed to be synthesized independently [23]. While ruthenium(II) and palladium catalysts are useful, they are expensive and environmentally unfriendly.
![[1860-5397-18-47-1]](/bjoc/content/figures/1860-5397-18-47-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
The dehalogenation of hydroximoyl chlorides in the presence of a strong base to generate nitrile oxides and a follow-up cycloaddition with 1,3-diketones, β-ketoesters or β-ketoamides are a commonly used 2-step route to 3,4,5-trisubstituted isoxazoles [24,25]. Xiao Zhou et al. recently reported a direct access to 3,4,5-trisubstituted isoxazoles via an enolate-mediated 1,3-dipolar cycloaddition of β-functionalized ketones with nitrile oxides using organocatalysts (Figure 1) [26,27]. This enolate-mediated cycloaddition, however, requires long reaction time in organic solvents at high temperatures.
Our current work was inspired by a recent report by Kesornpun et al., in which a cycloaddition of nitrile oxides and alkenes or alkynes was carried out in 0.1 M phosphate buffer under weakly acidic conditions (pH 4.0) at room temperature for 15–24 h and generated 3,5-disubstituted isoxazolines and 3,4,5-trisubstituted isoxazoles in good yields (Figure 1) [28]. Notably, no metal catalysts were used in this method. The development of organic reactions in water not only is environmentally friendly, but also finds significant applications in biological systems (e.g., click reactions, bio-conjugation, and bio-orthogonal chemistry). One of such applications is ligation chemistry, in which “click” chemistry through a [3 + 2] biorthogonal cycloaddition between nitrile oxide and strained alkenes has been used to achieve the high-density functionalization of oligodeoxyribonucleotides [29]. However, the alkenes need to be highly strained in order for this reaction to occur without the need for an additional metal catalyst. As an alternative to the mild acidic conditions, we herein report a method for the synthesis of 3,4,5-trisubstituted isoxazoles in water under mild basic conditions at room temperature via a [3 + 2]-cycloaddition of nitrile oxides and 1,3-diketones, β-ketoesters, or β-ketoamides (Figure 1). No additional metal catalyst was used.
Results and Discussion
The reason why we investigated the reaction under mild basic conditions was because nitrile oxides are often generated by dehalogenation of hydroximoyl chlorides in situ under basic conditions or by dehydration of nitroalkanes [30]. So we were wondering whether we could take advantage of that. There are many possible products formed through the reaction between nitrile oxides and 1,3-diketones (Figure 2), such as the homo-coupled product (dimerization) of the nitrile oxides (Figure 2, path A) and dioxazoles through a hetero [3 + 2] cyclization as another possibility [30] (Figure 2, path B). In addition, there are competing reactions leading to the O-trapping product and the C-trapping product of the nitrile oxides (Figure 2, paths C and D). We were interested in the optimization of the reaction conditions leading to the isoxazole products (path D, Figure 2). For this purpose, we chose 4-fluoro-N-hydroxybenzimidoyl chloride (1a) and 1-phenylbutane-1,3-dione (2a) (Table 1) as the starting materials and a water–methanol mixture as the aqueous reaction medium. A series of organic and inorganic bases was screened at room temperature in various combinations of water–methanol mixtures and other solvents. The results are summarized in Table 1.
![[1860-5397-18-47-2]](/bjoc/content/figures/1860-5397-18-47-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Possible products of the reaction between nitrile oxides and 1,3-diketones. Path D (C-trapping) produces 3,4,5-trisubstituted isoxazoles.
Figure 2: Possible products of the reaction between nitrile oxides and 1,3-diketones. Path D (C-trapping) pro...
Table 1: Optimization of the reaction conditions for the synthesis of 3,4,5-trisubstituted isoxazoles 3.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-18-47-i1.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Base | Solvents | Time (h) | Yield (%) | |
| 3a | 4 | ||||
| 1 | DBU | 5% water, 95% methanol | 18 | – | – |
| 2 | (S)-proline | 5% water, 95% methanol | 18 | – | – |
| 3b | NaHCO3 | 98% water, 2% methanol | 3 | 14 | 55 |
| 4b | Na2CO3 | 98% water, 2% methanol | 3 | 52 | 33 |
| 5 | TEA | 98% water, 2% methanol | 2 | 54 | 22 |
| 6 | TEA | 5% water, 95 % methanol | 2 | 68 | 19 |
| 7 | TEA | CH2Cl2 | 2 | 50 | 28 |
| 8 | TEA | isopropanol | 2 | 57 | 30 |
| 9 | DIPEA | CHCl3 | 1 | 43 | 52 |
| 10 | DIPEA | CH2Cl2 | 1 | 17 | 81 |
| 11 | DIPEA | 5% water, 95 % methanol | 1 | 89 | – |
| 12 | DIPEA | 95% water, 5% methanol | 1 | 98 | – |
| 13c | DIPEA | 2% water, 98 % methanol | 5 | 95 | – |
| 14 | DIPEA (10 mol %) | 95% water, 5% methanol | 2 | – | 70 |
| 15 | – | 5% water, 95 % methanol | 2 | – | 95 |
| 16d | DIPEA (10 mol %) | phosphate buffer (pH 7.4), 5% methanol | 2 | 7 | 76 |
aUnless otherwise noted, the reactions were performed with 0.5 mmol of 1a, 0.5 mmol of 2a, and 3 equivalents of base in 15 mL of the indicated solvents at room temperature for the indicated time. Yields are calculated from NMR spectra of the crude product using acetonitrile as the internal standard. bFour equivalents of NaHCO3 and Na2CO3. cTwo equivalents of DIPEA and 45 mL of the solvent mixture. d10 mol % of DIPEA. TEA = triethylamine, DIPEA = N,N-diisopropylethylamine, DBU = 1,8-diazabicyclo(5.4.0)undec-7-ene.
In our first attempt, DBU or (S)-proline in 5% water, 95% methanol yielded a complex mixture of products that were not easy to distinguish (Table 1, entries 1 and 2). The use of NaHCO3 in 98% water, 2% methanol after 3 hours, produced 14% of the expected 3,4,5-trisubstituted isoxazole 3a and 55% of a compound that we identified as furoxan 4 (a 1,2,5-oxadiazole 2-oxide) which formed through homo-coupling of substrate 1a (Table 1, entry 3). Na2CO3 in 98% water, 2% methanol produced 52% of the isoxazole 3a and 33% of compound 4 after 3 hours (Table 1, entry 4). Triethylamine (TEA) in 98% water, 2% methanol afforded 54% of 3a and 22% of 4 after 2 hours (Table 1, entry 5). However, TEA in 5% water, 95% methanol improved the yield of 3a to 68% as well as the selectivity of 3a (3.6 times) over 4 (Table 1, entry 6). Similar yields and selectivities for 3a were obtained when TEA was used in either dichloromethane (50% yield; 1.8 times selectivity) or isopropanol (57% yield; 1.9 times selectivity) as solvents (Table 1, entries 7 and 8). In contrast, N,N-diisopropylethylamine (DIPEA) in chloroform neither improved the yield (43%) nor the selectivity (0.8 times) of 3a (Table 1, entry 9). In dichloromethane, DIPEA furnished an even lower yield (17%) and selectivity (0.2 times) of 3a (Table 1, entry 10). Interestingly, in 5% water, 95% methanol, DIPEA gave product 3a in 89% yield in 1 hour, while in 95% water, 5% methanol, DIPEA gave 3a in 98% yield in 1 hour, which is the highest yield of our screening in the shortest reaction time (Table 1, entries 11 and 12). The reaction furnished a similar yield (95%) with a reduced amount of DIPEA and reactant concentration (2 equivalents of DIPEA and 1 mM of reactants 1a and 2a). However, these conditions required a longer reaction time (5 hours) to complete (Table 1, entry 13). Additionally, we observed a high selectivity towards compound 4, when we reduced the amount of DIPEA to 10 mol % in 95% water, 5% methanol (Table 1, entry 14) or when the reaction was performed without a base in 5% water, 95% methanol (Table 1, entry 15); in these cases, compound 4 was obtained in excellent yields (70% yield and 95% yield, respectively). Compound 4 was also formed almost exclusively in 76% yield when the reaction was run in phosphate buffer (pH 7.4) in the presence of 5% methanol with 10 mol % of DIPEA (Table 1, entry 16). Notably, no other regioisomers of isoxazole 3a were overserved in any of these reaction conditions.
With the optimized reaction conditions for the formation of 3,4,5-trisubstituted isoxazoles 3a (DIPEA, 95% water, 5% methanol, room temperature, Table 1, entry 12) at hands, we explored the scope of substrates for both the oximes and the β-diketones. First, we carried out the reactions between phenyl hydroximoyl chlorides 1a–c and 1,3-diketones 2b–e (Figure 3). To ensure completion, all reactions were run for 2 hours instead of 1 hour, regardless of substrates having either electron-withdrawing or electron-donating substituents on the benzene rings. The results showed smooth and complete reactions that produced the 3,4,5-trisubstituted isoxazoles 3b–m in good to excellent yields. The overall trend was that electron-donating substituents on the phenyl hydroximoyl chlorides or the phenyl 1,3-diketones produced comparatively lower yields than electron-withdrawing substituents. For example, the reactions afforded a yield from 70% to 80% when one of the reactants contained a p-methoxyphenyl group (compounds 3d–g,j, and m), whereas the reactions afforded a yield from 82% to 95% when one of the reactants contained a Br, F, or CF3 group in the para-position or contained a thiophene ring (compounds 3b,c,h,i,k, and l).
![[1860-5397-18-47-3]](/bjoc/content/figures/1860-5397-18-47-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Reactions between various arylhydroximoyl chlorides and 1,3-diketones. The reactions were performed with 0.5 mmol of 1, 0.5 mmol of 2, and 3 equivalents of DIPEA in 15 mL of 95% water, 5% methanol at room temperature for 2 hours. The yields were calculated after the isolation and purification of products.
Figure 3: Reactions between various arylhydroximoyl chlorides and 1,3-diketones. The reactions were performed...
We also explored the reactions between phenyl hydroximoyl chlorides 1a–c and β-ketoesters 2g,h or β-ketoamides 2f,i under the optimized reaction conditions (DIPEA, 95% water, 5% methanol, room temperature, 2 hours). The results showed that phenyl, benzyl, and ethyl β-ketoesters or β-ketoamides reacted smoothly with the phenyl hydroximoyl chlorides and gave the corresponding 3,4,5-trisubstituted isoxazoles 3n–w in good to excellent yields (Figure 4). Generally, the yields of the reactions with β-ketoesters and β-ketoamides were comparable with those obtained with 1,3-diketones.
![[1860-5397-18-47-4]](/bjoc/content/figures/1860-5397-18-47-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Reactions between various phenyl hydroximoyl chlorides and β-ketoesters or β-ketoamides. The reactions were performed with 0.5 mmol of 1, 0.5 mmol of 2, and 3 equivalents of DIPEA in 15 mL of 95% water, 5% methanol at room temperature for 2 hours. The yields were calculated after the isolation and purification of products.
Figure 4: Reactions between various phenyl hydroximoyl chlorides and β-ketoesters or β-ketoamides. The reacti...
We also explored the reactions between 4-fluorophenyl hydroximoyl chloride (1a), which was the phenyl hydroximoyl chloride that gave the best yield, and diethyl malonate (2j) or dibenzyl malonate (2k) under the optimized reaction conditions. However, the reactions did not proceed (Figure 5). We suspected that the increased electron-donating effect on both sides of the 1,3-diketones in 2j or 2k made the methylene group less acidic, thus DIPEA was not able to deprotonate the methylene for the nucleophilic addition and successive cyclization to happen.
![[1860-5397-18-47-5]](/bjoc/content/figures/1860-5397-18-47-5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Reactions between 4-fluorophenyl hydroximoyl chloride (1a) and diethyl malonate (2j) or dibenzyl malonate (2k) did not proceed.
Figure 5: Reactions between 4-fluorophenyl hydroximoyl chloride (1a) and diethyl malonate (2j) or dibenzyl ma...
In addition, we further explored the scope of substrates with a trifluoromethyl group, a strong electron-withdrawing substituent, instead of methyl, an electron-donating group, in the 1,3-diketone starting materials (Table 2). The reaction between 4-fluorophenyl hydroximoyl chloride (1a) and 4,4,4-trifluoro-1-phenyl-1,3-butanedione (2l) under the optimized reaction conditions (Table 2, entry 1) gave a complex mixture of products with only a trace amount of the expected trifluoromethyl-substituted isoxazole 3x being detected. Meanwhile, some of the starting material 2l remained unreacted and we suspected the lower solubility of compound 2l in water was responsible for the low yield of the reaction. Since trifluoromethyl-substituted isoxazoles are an important and prevalent scaffold in biomedical research and drug discovery programs, we decided to optimize the reaction conditions for the synthesis of the trifluoromethyl-substituted isoxazole 3x. We varied the solvent mixtures and the bases, but kept the reaction temperature at room temperature and the reaction time at 2 hours, because these are the highlights of our synthetic method. The results are collected in Table 2. First, we varied the proportion of methanol in the solvent mixture with water to increase the solubility of 4,4,4-trifluoro-1-phenyl-1,3-butanedione (2l). As the percentage of methanol in the solvent mixture increased from 5% to 50%, 75%, and 95%, with DIPEA as the base, the yield of the trifluoromethyl-substituted isoxazole 3x increased from trace amounts to 40% (Table 2, entries 1–4). With dichloromethane as the solvent and DIPEA as the base, the reaction gave 3x in 40% yield (Table 2, entry 5). When isopropanol was used as the solvent and TEA was used as the base, the reaction produced 3x in 40% yield (Table 2, entry 9). When benzene, dichloromethane, or a 5% water, 95% methanol mixture was used as the solvent and TEA was used as the base, the reaction gave a yield from 5% to 22% (Table 2, entries 6–8). Overall, the best results, i.e., 40% yield of the trifluoromethyl-substituted isoxazole 3x, were obtained with DIPEA, 5% water, 95% methanol, room temperature, 2 hours, or with DIPEA, dichloromethane, room temperature, 2 hours, or with TEA, isopropanol, room temperature, 2 hours (Table 2, entries 4, 5, and 9).
Table 2: Optimization of the reaction conditions to synthesize the trifluoromethyl-substituted isoxazole 3x.a
![[Graphic 2]](/bjoc/content/inline/1860-5397-18-47-i2.svg?max-width=637&scale=1.0)
|
|||
| Entry | Base | Solvents | Yield of 3x (%) |
| 1 | DIPEA | 95% water, 5% methanol | traces |
| 2 | DIPEA | 50% water, 50% methanol | traces |
| 3 | DIPEA | 25% water, 75% methanol | 30 |
| 4 | DIPEA | 5% water, 95% methanol | 40 |
| 5 | DIPEA | dichloromethane | 40 |
| 6 | TEA | 5% water, 95% methanol | 22 |
| 7 | TEA | dichloromethane | 20 |
| 8 | TEA | benzene | 5 |
| 9 | TEA | isopropanol | 40 |
aThe reactions were performed with 0.5 mmol of 4-fluorophenyl hydroximoyl chloride (1a), 0.5 mmol of 4,4,4-trifluoro-1-phenyl-1,3-butanedione (2l), and 3 equivalents of base in 15 mL of the indicated solvent at room temperature for 2 hours.
We then applied the reaction conditions DIPEA, 5% water, 95% methanol, room temperature, 2 hours (Table 2, entry 4) to the reactions between phenyl hydroximoyl chlorides 1a,c and 4,4,4-trifluoro-1-phenyl- (2l) and 4,4,4-trifluoro-1-naphthylbutane-1,3-dione (2m) to synthesize the trifluoromethyl-substituted isoxazoles 3y–aa (Figure 6). The reactions proceeded smoothly and produced the expected trifluoromethyl-substituted isoxazole products. Similar to the yield of compound 3x, the yields of 3y–aa were between 35–40%. Overall, the reactions to synthesize trifluoromethyl-substituted isoxazoles produced lower yields than those of methyl-substituted isoxazoles.
![[1860-5397-18-47-6]](/bjoc/content/figures/1860-5397-18-47-6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Reactions between phenyl hydroximoyl chlorides 1a,c and 4,4,4-trifluoro-1-phenyl- (2l) and 4,4,4-trifluoro-1-naphthylbutane-1,3-dione (2m) to synthesize trifluoromethyl-substituted isoxazoles 3y–aa.
Figure 6: Reactions between phenyl hydroximoyl chlorides 1a,c and 4,4,4-trifluoro-1-phenyl- (2l) and 4,4,4-tr...
Our observed results with 1,3-diketones with substituents of different electronic nature and the variety of the polarities of the solvents can be explained by the enolization of the 1,3-diketones as previously was described in the literature [31,32]. Strongly electron-withdrawing substituents present in the 1,3-diketones would increase the degree of enolization of the 1,3-diketones [30,33], and the polarity of the solvent would have only a little effect on the keto–enol tautomerization of such 1,3-diketones [30]. During our studies, we also observed the effect of solvent polarity on the keto–enol equilibrium of the 1,3-diketones, which influences the results of the [3 + 2] cycloaddition reactions under mild basic conditions, via nuclear magnetic resonance (NMR) spectroscopy. The 1H NMR spectra of 1-phenyl-1,3-butanedione (2a) in CDCl3 and in methanol-d4 indicated that the enol tautomer of the 1,3-diketone was predominant in CDCl3, while the keto tautomer was predominant in methanol-d4, which is a more polar solvent than CDCl3 (Figure 7). Our observations thus reinforce Meyer’s rule [34,35], which states that the keto tautomer is favored as the solvent polarity increases. This also corroborates our observations and explains why under mild basic conditions, the 3,4,5-trisubstituted isoxazole 3a was formed exclusively in polar solvents like water or methanol (Table 1, entries 11–13), whereas the furoxan 4 was formed predominantly in nonpolar solvents such as chloroform or dichloromethane (Table 1, entries 9 and 10).
![[1860-5397-18-47-7]](/bjoc/content/figures/1860-5397-18-47-7.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: 1H NMR spectra of 1-phenyl-1,3-butanedione (2a) in methanol-d4 (top) and in CDCl3 (bottom).
Figure 7: 1H NMR spectra of 1-phenyl-1,3-butanedione (2a) in methanol-d4 (top) and in CDCl3 (bottom).
A plausible mechanism for the formation of the 3,4,5-trisubstituted isoxazoles 3a (the only regioisomer that was formed) is shown in Figure 8. In a polar solvent like water or methanol, the 1,3-diketone is deprotonated by the base DIPEA to form the carbanion I that is solvated by the polar solvent. The carbanion I then adds to the carbon (carbocation) of the nitrile oxide, to give the intermediate II. Due to steric effects, the intermediate II undergoes bond rotation to the lowest-energy conformation II-D in which the methyl group is at a close distance to the nitrile oxide. Then, cyclization followed by the elimination of water (formation of the aromatic ring is the driving force), produces the 3,4,5-trisubstituted isoxazole 3a (Figure 8). Of note, II-A–II-D are conformers, not resonance structures, and the isoxazole 3a is likely the thermodynamical product. The solvent polarity also affects the keto–enol equilibrium of the intermediate II-D. In polar solvents, the keto tautomer is predominant as an electrophilic group for the intramolecular cyclization, while in nonpolar solvents, the enol tautomer could not accept a nucleophilic attack for a further cycloaddition. In addition, the conjugation between the new C=N bond and the enol double bond promotes the enol tautomer formation. This may also explain why the trifluoromethylated 1,3-diketones produced the trifluoromethyl-substituted isoxazoles with lower yields than the methyl-substituted isoxazoles. Significant enolization of the corresponding trifluoromethyl intermediate II-D could not transfer into the target sufficiently. Furthermore, when the amount of DIPEA was reduced to 0–10 mol % (Table 1, entries 14–16), or when the reaction was carried out in a nonpolar solvent like chloroform or dichloromethane, the amount of carbanion I in the reaction medium is much smaller. Therefore, the formation of the homo-coupled product (furoxan 4) outcompeted the [3 + 2]-cycloaddition.
![[1860-5397-18-47-8]](/bjoc/content/figures/1860-5397-18-47-8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: A plausible mechanism for the formation of the 3,4,5-trisubstituted isoxazoles 3 in the presence of DIPEA in polar solvents like water or methanol.
Figure 8: A plausible mechanism for the formation of the 3,4,5-trisubstituted isoxazoles 3 in the presence of...
Not to be overlooked is our discovery of the optimized reaction conditions for the formation of furoxan 4 with 95% yield (5% water, 95% methanol, room temperature, 2 hours; see Table 1, entry 15), which adds another method for the synthesis of furoxans to the current literature. The synthesis of furoxans or 1,2,5-oxadiazole-2-oxides was first reported by Kekulé in 1857 [36]. Since then, these nitric oxide donors have shown important biological activities due to their unique electronic and coordination ability [37,38], such as antitumor [39] and antiparasitic [40,41]. Previously, furoxans were synthesized via dimerization of hydroximoyl chlorides in the presence of a base, such as trimethylamine in ether [42], or Na2CO3 in DME [43]. Our results showed that when 5% water, 95% methanol was used as a solvent mixture, the dimerization of hydroximoyl chlorides happened fast at room temperature without the need of a base. The obtained furoxan 4 was found to be a stable product; when stirring it in a 5% water, 95% methanol mixture in the presence of DIPEA overnight, no degradation was observed.
Conclusion
We have developed a method for the synthesis of 3,4,5-trisubstituted isoxazoles in aqueous medium under mild basic conditions at room temperature. The reaction proceeds via a [3 + 2]-cycloaddition of nitrile oxides and 1,3-diketones, β-ketoesters, or β-ketoamides. When DIPEA is used as the base and 95% water, 5% methanol is used as the solvent mixture, the reaction is complete within 1–2 hours at room temperature. Our method provides a fast and environmentally friendly access to 3,4,5-trisubstituted isoxazoles, an important class of structures found in numerous bioactive natural products and pharmaceuticals. Producing good to excellent yields in short reaction time in aqueous media, our method has the potential for significant applications in biological systems (e.g., click reactions, bioconjugation, and bio-orthogonal chemistry). Our method could also find potential applications in the production of important 3,4,5-trisubstituted isoxazoles, such as the precursors of many β-lactamase-resistant antibiotics like oxacillin, cloxacillin, dicloxacillin, and flucloxacillin (Figure 9), which share a similar 3,4,5-trisubstituted isoxazole structure to the synthesized compounds in this report. In addition, we optimized the reaction conditions to produce trifluoromethyl-substituted isoxazoles, a prevalent scaffold in biomedical research and drug discovery programs. We also proposed a plausible mechanism for the selectivity of the [3 + 2]-cycloaddition reaction to produce 3,4,5-trisubstituted isoxazoles. Not to be overlooked is our optimized reaction conditions for the dimerization of hydroximoyl chlorides to form furoxans or 1,2,5-oxadiazole 2-oxides, a class of structures with important biological activities due to their unique electronic and coordination ability.
![[1860-5397-18-47-9]](/bjoc/content/figures/1860-5397-18-47-9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 9: Structures of β-lactamase-resistant antibiotics oxacillin, cloxacillin, dicloxacillin, and flucloxacillin.
Figure 9: Structures of β-lactamase-resistant antibiotics oxacillin, cloxacillin, dicloxacillin, and flucloxa...
Experimental
General information
All chemicals and solvents were obtained from Sigma-Aldrich or Fisher Scientific and were used as received unless specified. 1H NMR and 13C NMR spectra were recorded on a Bruker-400 and or a Bruker-500 spectrometer using CDCl3, methanol-d4, or DMSO-d6 as the solvent. Chemical shifts (δ) were recorded in parts per million and referenced to CDCl3 (7.24 ppm for 1H NMR and 77.23 ppm for 13C NMR), methanol-d4 (3.31 ppm for 1H NMR and 49.15 ppm for 13C NMR), or DMSO-d6 (2.50 ppm for 1H NMR and 39.52 ppm for 13C NMR). 19F NMR spectra were recorded on a Bruker-400 spectrometer. Coupling constants (J) are given in Hz. The following abbreviations were used to designate the multiplicities: s = singlet, d = doublet, t = triplet, q = quartet, quint = quintet, m = multiplet, br = broad. Melting points were measured using an OptiMelt automated melting point system. Exact high-resolution mass determinations were analyzed on a JEOL AccuToF 4G LCplus atmospheric pressure ionization time-of-flight mass spectrometer (Jeol, Tokyo, Japan) fitted with direct analysis in real-time (DART) ion source (IonSense DART controller, Saugus, MA, USA). The DART ion source was operated with helium gas (approximately 4.0 L/min flow rate), the gas heater (350 °C), and the source grid (350 V). The data acquisition range was from m/z 50 to 1000. Polyethylene glycol (PEG 600) was used for the exact mass calibration.
General synthetic procedure for phenyl hydroximoyl chlorides
The phenyl hydroximoyl chlorides (Figure S1 in Supporting Information File 1) were synthesized by following literature procedures [44,45]. To a solution of the corresponding benzaldehyde (1 mmol) in a 1:1 EtOH/H2O mixture (1 mL) was added sodium acetate (1.1 mmol) and hydroxylamine hydrochloride (1.1 mmol). The reaction mixture was stirred for 4 h at rt. After the reaction was complete, as indicated by thin-layer chromatography (TLC), the mixture was extracted with ethyl acetate. The organic phase was dried with Na2SO4 and concentrated under reduced pressure. Purification via column chromatography (ethyl acetate/hexanes) resulted in the aldoxime product.
To a solution of the aldoxime (1 mmol) in DMF was added N-chlorosuccinimide and the mixture stirred for 18 h at rt. After the reaction was complete, as indicated by TLC, the reaction mixture was poured into water and extracted with ethyl acetate. The organic phase was washed with brine and dried with Na2SO4. The solvent was then evaporated under reduced pressure to produce the desired phenyl hydroximoyl chloride, which was used immediately for the [3 + 2]-cycloaddition reaction without further purification.
General synthetic procedure for 1,3-diketones
The 1,3-diketones (Figure S2 in Supporting Information File 1) were synthesized by following a literature procedure [46]. To a suspension of NaH (1.60 g of dispersion in oil, 40 mmol) in ethyl acetate (20 mL) at 0 °C was added slowly a solution of the corresponding ketone (10 mmol) in ethyl acetate (20 mL). The reaction mixture was stirred at rt for 12 h. After the reaction was complete, as indicated by TLC, the mixture was carefully treated with 10% aqueous NH4Cl (30 mL) and the pH adjusted to 5 with a solution of hydrochloric acid (3 M). The aqueous phase was then separated and extracted with ethyl acetate. The organic phase was dried with Na2SO4 and concentrated under reduced pressure. Purification via column chromatography (20:1 hexanes/ethyl acetate) resulted in the desired 1,3-diketone.
General synthetic procedure for the [3 + 2]-cycloaddition reaction
To a solution of the 1,3-diketone, β-ketoester, or β-ketoamide (0.5 mmol, 1 equiv) in methanol was added water, phenyl hydroximoyl chloride (1 equiv), and DIPEA (3 equiv) at room temperature (total volume of methanol and water = 15 mL; 95% water, 5% methanol). The reaction mixture was stirred for 1–2 h until all of the starting materials were consumed (TLC, 10% ethyl acetate in hexanes). After the reaction was complete, the product was extracted with ethyl acetate, dried with Na2SO4, concentrated under reduced pressure, and purified via column chromatography (ethyl acetate/hexanes).
Supporting Information
| Supporting Information File 1: Synthetic schemes for phenyl hydroximoyl chlorides and 1,3-diketones, characterization data, and copies of 1H, 13C, and 19F NMR spectra. | ||
| Format: PDF | Size: 7.7 MB | Download |
Funding
This work was supported by the American Association of Colleges of Pharmacy (2018 New Investigator Award to H. V. L.), the National Institute of General Medical Sciences (P30GM122733 pilot project award to H. V. L.), and funds from the Department of BioMolecular Sciences at the University of Mississippi, School of Pharmacy. The content is solely the responsibility of the authors and does not necessarily represent the official views of these funders.
References
-
Baraldi, P. G.; Barco, A.; Benetti, S.; Pollini, G. P.; Simoni, D. Synthesis 1987, 857–869. doi:10.1055/s-1987-28105
Return to citation in text: [1] [2] -
Rahbœk, L.; Christophersen, C. The Isoxazole Alkaloids. The Alkaloids: Chemistry and Biology; Elsevier, 2001; Vol. 57, pp 185–233. doi:10.1016/s0099-9598(01)57004-2
Return to citation in text: [1] -
Zhu, J.; Mo, J.; Lin, H.-Z.; Chen, Y.; Sun, H. Bioorg. Med. Chem. 2018, 26, 3065–3075. doi:10.1016/j.bmc.2018.05.013
Return to citation in text: [1] [2] -
Sysak, A.; Obmińska-Mrukowicz, B. Eur. J. Med. Chem. 2017, 137, 292–309. doi:10.1016/j.ejmech.2017.06.002
Return to citation in text: [1] -
Jensen, M. R.; Schoepfer, J.; Radimerski, T.; Massey, A.; Guy, C. T.; Brueggen, J.; Quadt, C.; Buckler, A.; Cozens, R.; Drysdale, M. J.; Garcia-Echeverria, C.; Chène, P. Breast Cancer Res. 2008, 10, R33. doi:10.1186/bcr1996
Return to citation in text: [1] -
Hong, Y. L.; Hossler, P. A.; Calhoun, D. H.; Meshnick, S. R. Antimicrob. Agents Chemother. 1995, 39, 1756–1763. doi:10.1128/aac.39.8.1756
Return to citation in text: [1] -
Luckner, P.; Brandsch, M. Eur. J. Pharm. Biopharm. 2005, 59, 17–24. doi:10.1016/j.ejpb.2004.07.008
Return to citation in text: [1] -
Ali, K. A.; Hosni, H. M.; Ragab, E. A.; El-Moez, S. I. A. Arch. Pharm. (Weinheim, Ger.) 2012, 345, 231–239. doi:10.1002/ardp.201100186
Return to citation in text: [1] -
Brown, G. D.; Denning, D. W.; Gow, N. A. R.; Levitz, S. M.; Netea, M. G.; White, T. C. Sci. Transl. Med. 2012, 4, 165rv13. doi:10.1126/scitranslmed.3004404
Return to citation in text: [1] -
Talley, J. J.; Malecha, J. W.; Bertenshaw, S.; Graneto, M. J.; Carter, J. S.; Li, J.; Nagarajan, S.; Brown, D. L.; Rogier, D. J., Jr.; Penning, T. D.; Khanna, I. K.; Xu, X.; Weier, R. M. Substituted Benzenesulfonamide Derivatives as Prodrug of COX-2 Inhibitors. WO Pat. Appl. WO1997038986A1, Oct 23, 1997.
Return to citation in text: [1] -
Radhakrishna, A. S.; Sivaprakash, K.; Singh, B. B. Synth. Commun. 1991, 21, 1625–1629. doi:10.1080/00397919108021062
Return to citation in text: [1] -
Moriya, O.; Nakamura, H.; Kageyama, T.; Urata, Y. Tetrahedron Lett. 1989, 30, 3987–3990. doi:10.1016/s0040-4039(00)99302-x
Return to citation in text: [1] -
Minakata, S.; Okumura, S.; Nagamachi, T.; Takeda, Y. Org. Lett. 2011, 13, 2966–2969. doi:10.1021/ol2010616
Return to citation in text: [1] -
Mendelsohn, B. A.; Lee, S.; Kim, S.; Teyssier, F.; Aulakh, V. S.; Ciufolini, M. A. Org. Lett. 2009, 11, 1539–1542. doi:10.1021/ol900194v
Return to citation in text: [1] -
Zhao, G.; Liang, L.; Wen, C. H. E.; Tong, R. Org. Lett. 2019, 21, 315–319. doi:10.1021/acs.orglett.8b03829
Return to citation in text: [1] -
Bourbeau, M. P.; Rider, J. T. Org. Lett. 2006, 8, 3679–3680. doi:10.1021/ol061260+
Return to citation in text: [1] -
Fouli, F. A.; Habashy, M. M.; El-Kafrawy, A. F.; Youseef, A. S. A.; El-Adly, M. M. J. Prakt. Chem. 1987, 329, 1116–1122. doi:10.1002/prac.19873290622
Return to citation in text: [1] -
Jäger, V.; Colinas, P. A. Nitrile Oxides. In Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry Toward Heterocycles and Natural Products; Padwa, A.; Pearson, W. H., Eds.; John Wiley & Sons: New York, NY, USA, 2002; Vol. 59, pp 363–472. doi:10.1002/0471221902.ch6
Return to citation in text: [1] -
Himo, F.; Lovell, T.; Hilgraf, R.; Rostovtsev, V. V.; Noodleman, L.; Sharpless, K. B.; Fokin, V. V. J. Am. Chem. Soc. 2005, 127, 210–216. doi:10.1021/ja0471525
Return to citation in text: [1] -
Hansen, T. V.; Wu, P.; Fokin, V. V. J. Org. Chem. 2005, 70, 7761–7764. doi:10.1021/jo050163b
Return to citation in text: [1] -
Oakdale, J. S.; Sit, R. K.; Fokin, V. V. Chem. – Eur. J. 2014, 20, 11101–11110. doi:10.1002/chem.201402559
Return to citation in text: [1] -
Grecian, S.; Fokin, V. V. Angew. Chem., Int. Ed. 2008, 47, 8285–8287. doi:10.1002/anie.200801920
Return to citation in text: [1] -
Waldo, J. P.; Larock, R. C. J. Org. Chem. 2007, 72, 9643–9647. doi:10.1021/jo701942e
Return to citation in text: [1] -
Mirzaei, Y. R.; Weaver, M. J.; Steiger, S. A.; Kearns, A. K.; Gajewski, M. P.; Rider, K. C.; Beall, H. D.; Natale, N. R. Tetrahedron 2012, 68, 10360–10364. doi:10.1016/j.tet.2012.09.084
Return to citation in text: [1] -
Liu, B.; Liu, G.; Xin, Z.; Serby, M. D.; Zhao, H.; Schaefer, V. G.; Falls, H. D.; Kaszubska, W.; Collins, C. A.; Sham, H. L. Bioorg. Med. Chem. Lett. 2004, 14, 5223–5226. doi:10.1016/j.bmcl.2004.06.060
Return to citation in text: [1] -
Zhou, X.; Xu, X.; Shi, Z.; Liu, K.; Gao, H.; Li, W. Org. Biomol. Chem. 2016, 14, 5246–5250. doi:10.1039/c6ob00717a
Return to citation in text: [1] -
Li, W.; Zhou, X.; Shi, Z.; Liu, Y.; Liu, Z.; Gao, H. Org. Biomol. Chem. 2016, 14, 9985–9988. doi:10.1039/c6ob02025a
Return to citation in text: [1] -
Kesornpun, C.; Aree, T.; Mahidol, C.; Ruchirawat, S.; Kittakoop, P. Angew. Chem., Int. Ed. 2016, 55, 3997–4001. doi:10.1002/anie.201511730
Return to citation in text: [1] -
Gutsmiedl, K.; Wirges, C. T.; Ehmke, V.; Carell, T. Org. Lett. 2009, 11, 2405–2408. doi:10.1021/ol9005322
Return to citation in text: [1] -
Sloop, J. C.; Bumgardner, C. L.; Washington, G.; Loehle, W. D.; Sankar, S. S.; Lewis, A. B. J. Fluorine Chem. 2006, 127, 780–786. doi:10.1016/j.jfluchem.2006.02.012
Return to citation in text: [1] [2] [3] [4] -
Arslan, N. B.; Özdemir, N. J. Mol. Model. 2015, 21, 19. doi:10.1007/s00894-015-2574-8
Return to citation in text: [1] -
Cook, A. G.; Feltman, P. M. J. Chem. Educ. 2010, 87, 678–679. doi:10.1021/ed1000813
Return to citation in text: [1] -
Geraldes, C. F. G. C.; Barros, M. T.; Maycock, C. D.; Silva, M. I. J. Mol. Struct. 1990, 238, 335–346. doi:10.1016/0022-2860(90)85025-e
Return to citation in text: [1] -
Meyer, K. H. Justus Liebigs Ann. Chem. 1911, 380, 212–220. doi:10.1002/jlac.19113800206
Return to citation in text: [1] -
Cortney, C. H.; Krishnan, V. V. J. Chem. Educ. 2020, 97, 825–830. doi:10.1021/acs.jchemed.9b00737
Return to citation in text: [1] -
Kekulé, A. Ann. Chem. Pharm. 1857, 101, 200–213. doi:10.1002/jlac.18571010208
Return to citation in text: [1] -
Medana, C.; Ermondi, G.; Fruttero, R.; Di Stilo, A.; Ferretti, C.; Gasco, A. J. Med. Chem. 1994, 37, 4412–4416. doi:10.1021/jm00051a020
Return to citation in text: [1] -
Seymour, C. P.; Tohda, R.; Tsubaki, M.; Hayashi, M.; Matsubara, R. J. Org. Chem. 2017, 82, 9647–9654. doi:10.1021/acs.joc.7b01709
Return to citation in text: [1] -
Ramazani, A.; Karimi, M.; Hosseinzadeh, Z.; Rezayati, S.; Hanifehpour, Y.; Joo, S. W. Curr. Org. Chem. 2021, 25, 757–778. doi:10.2174/1385272825666210208183751
Return to citation in text: [1] -
dos Santos Fernandes, G. F.; de Souza, P. C.; Marino, L. B.; Chegaev, K.; Guglielmo, S.; Lazzarato, L.; Fruttero, R.; Chung, M. C.; Pavan, F. R.; dos Santos, J. L. Eur. J. Med. Chem. 2016, 123, 523–531. doi:10.1016/j.ejmech.2016.07.039
Return to citation in text: [1] -
Mott, B. T.; Cheng, K. C.-C.; Guha, R.; Kommer, V. P.; Williams, D. L.; Vermeire, J. J.; Cappello, M.; Maloney, D. J.; Rai, G.; Jadhav, A.; Simeonov, A.; Inglese, J.; Posner, G. H.; Thomas, C. J. MedChemComm 2012, 3, 1505–1511. doi:10.1039/c2md20238g
Return to citation in text: [1] -
Hwang, K.-J.; Kang, H. Bull. Korean Chem. Soc. 1998, 19, 506–507. doi:10.5012/bkcs.1998.19.5.506
Return to citation in text: [1] -
Matsubara, R.; Ando, A.; Hasebe, H.; Kim, H.; Tsuneda, T.; Hayashi, M. J. Org. Chem. 2020, 85, 5959–5972. doi:10.1021/acs.joc.0c00326
Return to citation in text: [1] -
Senadi, G. C.; Mutra, M. R.; Lu, T.-Y.; Wang, J.-J. Green Chem. 2017, 19, 4272–4277. doi:10.1039/c7gc01449j
Return to citation in text: [1] -
Tsai, Y.-h.; Borini Etichetti, C. M.; Cicetti, S.; Girardini, J. E.; Spanevello, R. A.; Suárez, A. G.; Sarotti, A. M. Bioorg. Med. Chem. Lett. 2020, 30, 127247. doi:10.1016/j.bmcl.2020.127247
Return to citation in text: [1] -
Inagaki, S.; Saito, K.; Suto, S.; Aihara, H.; Sugawara, A.; Tamura, S.; Kawano, T. J. Org. Chem. 2018, 83, 13834–13846. doi:10.1021/acs.joc.8b02153
Return to citation in text: [1]
| 40. | dos Santos Fernandes, G. F.; de Souza, P. C.; Marino, L. B.; Chegaev, K.; Guglielmo, S.; Lazzarato, L.; Fruttero, R.; Chung, M. C.; Pavan, F. R.; dos Santos, J. L. Eur. J. Med. Chem. 2016, 123, 523–531. doi:10.1016/j.ejmech.2016.07.039 |
| 41. | Mott, B. T.; Cheng, K. C.-C.; Guha, R.; Kommer, V. P.; Williams, D. L.; Vermeire, J. J.; Cappello, M.; Maloney, D. J.; Rai, G.; Jadhav, A.; Simeonov, A.; Inglese, J.; Posner, G. H.; Thomas, C. J. MedChemComm 2012, 3, 1505–1511. doi:10.1039/c2md20238g |
| 42. | Hwang, K.-J.; Kang, H. Bull. Korean Chem. Soc. 1998, 19, 506–507. doi:10.5012/bkcs.1998.19.5.506 |
| 43. | Matsubara, R.; Ando, A.; Hasebe, H.; Kim, H.; Tsuneda, T.; Hayashi, M. J. Org. Chem. 2020, 85, 5959–5972. doi:10.1021/acs.joc.0c00326 |
| 1. | Baraldi, P. G.; Barco, A.; Benetti, S.; Pollini, G. P.; Simoni, D. Synthesis 1987, 857–869. doi:10.1055/s-1987-28105 |
| 2. | Rahbœk, L.; Christophersen, C. The Isoxazole Alkaloids. The Alkaloids: Chemistry and Biology; Elsevier, 2001; Vol. 57, pp 185–233. doi:10.1016/s0099-9598(01)57004-2 |
| 5. | Jensen, M. R.; Schoepfer, J.; Radimerski, T.; Massey, A.; Guy, C. T.; Brueggen, J.; Quadt, C.; Buckler, A.; Cozens, R.; Drysdale, M. J.; Garcia-Echeverria, C.; Chène, P. Breast Cancer Res. 2008, 10, R33. doi:10.1186/bcr1996 |
| 24. | Mirzaei, Y. R.; Weaver, M. J.; Steiger, S. A.; Kearns, A. K.; Gajewski, M. P.; Rider, K. C.; Beall, H. D.; Natale, N. R. Tetrahedron 2012, 68, 10360–10364. doi:10.1016/j.tet.2012.09.084 |
| 25. | Liu, B.; Liu, G.; Xin, Z.; Serby, M. D.; Zhao, H.; Schaefer, V. G.; Falls, H. D.; Kaszubska, W.; Collins, C. A.; Sham, H. L. Bioorg. Med. Chem. Lett. 2004, 14, 5223–5226. doi:10.1016/j.bmcl.2004.06.060 |
| 3. | Zhu, J.; Mo, J.; Lin, H.-Z.; Chen, Y.; Sun, H. Bioorg. Med. Chem. 2018, 26, 3065–3075. doi:10.1016/j.bmc.2018.05.013 |
| 26. | Zhou, X.; Xu, X.; Shi, Z.; Liu, K.; Gao, H.; Li, W. Org. Biomol. Chem. 2016, 14, 5246–5250. doi:10.1039/c6ob00717a |
| 27. | Li, W.; Zhou, X.; Shi, Z.; Liu, Y.; Liu, Z.; Gao, H. Org. Biomol. Chem. 2016, 14, 9985–9988. doi:10.1039/c6ob02025a |
| 1. | Baraldi, P. G.; Barco, A.; Benetti, S.; Pollini, G. P.; Simoni, D. Synthesis 1987, 857–869. doi:10.1055/s-1987-28105 |
| 21. | Oakdale, J. S.; Sit, R. K.; Fokin, V. V. Chem. – Eur. J. 2014, 20, 11101–11110. doi:10.1002/chem.201402559 |
| 22. | Grecian, S.; Fokin, V. V. Angew. Chem., Int. Ed. 2008, 47, 8285–8287. doi:10.1002/anie.200801920 |
| 3. | Zhu, J.; Mo, J.; Lin, H.-Z.; Chen, Y.; Sun, H. Bioorg. Med. Chem. 2018, 26, 3065–3075. doi:10.1016/j.bmc.2018.05.013 |
| 4. | Sysak, A.; Obmińska-Mrukowicz, B. Eur. J. Med. Chem. 2017, 137, 292–309. doi:10.1016/j.ejmech.2017.06.002 |
| 23. | Waldo, J. P.; Larock, R. C. J. Org. Chem. 2007, 72, 9643–9647. doi:10.1021/jo701942e |
| 10. | Talley, J. J.; Malecha, J. W.; Bertenshaw, S.; Graneto, M. J.; Carter, J. S.; Li, J.; Nagarajan, S.; Brown, D. L.; Rogier, D. J., Jr.; Penning, T. D.; Khanna, I. K.; Xu, X.; Weier, R. M. Substituted Benzenesulfonamide Derivatives as Prodrug of COX-2 Inhibitors. WO Pat. Appl. WO1997038986A1, Oct 23, 1997. |
| 17. | Fouli, F. A.; Habashy, M. M.; El-Kafrawy, A. F.; Youseef, A. S. A.; El-Adly, M. M. J. Prakt. Chem. 1987, 329, 1116–1122. doi:10.1002/prac.19873290622 |
| 18. | Jäger, V.; Colinas, P. A. Nitrile Oxides. In Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry Toward Heterocycles and Natural Products; Padwa, A.; Pearson, W. H., Eds.; John Wiley & Sons: New York, NY, USA, 2002; Vol. 59, pp 363–472. doi:10.1002/0471221902.ch6 |
| 9. | Brown, G. D.; Denning, D. W.; Gow, N. A. R.; Levitz, S. M.; Netea, M. G.; White, T. C. Sci. Transl. Med. 2012, 4, 165rv13. doi:10.1126/scitranslmed.3004404 |
| 19. | Himo, F.; Lovell, T.; Hilgraf, R.; Rostovtsev, V. V.; Noodleman, L.; Sharpless, K. B.; Fokin, V. V. J. Am. Chem. Soc. 2005, 127, 210–216. doi:10.1021/ja0471525 |
| 20. | Hansen, T. V.; Wu, P.; Fokin, V. V. J. Org. Chem. 2005, 70, 7761–7764. doi:10.1021/jo050163b |
| 8. | Ali, K. A.; Hosni, H. M.; Ragab, E. A.; El-Moez, S. I. A. Arch. Pharm. (Weinheim, Ger.) 2012, 345, 231–239. doi:10.1002/ardp.201100186 |
| 44. | Senadi, G. C.; Mutra, M. R.; Lu, T.-Y.; Wang, J.-J. Green Chem. 2017, 19, 4272–4277. doi:10.1039/c7gc01449j |
| 45. | Tsai, Y.-h.; Borini Etichetti, C. M.; Cicetti, S.; Girardini, J. E.; Spanevello, R. A.; Suárez, A. G.; Sarotti, A. M. Bioorg. Med. Chem. Lett. 2020, 30, 127247. doi:10.1016/j.bmcl.2020.127247 |
| 6. | Hong, Y. L.; Hossler, P. A.; Calhoun, D. H.; Meshnick, S. R. Antimicrob. Agents Chemother. 1995, 39, 1756–1763. doi:10.1128/aac.39.8.1756 |
| 7. | Luckner, P.; Brandsch, M. Eur. J. Pharm. Biopharm. 2005, 59, 17–24. doi:10.1016/j.ejpb.2004.07.008 |
| 11. | Radhakrishna, A. S.; Sivaprakash, K.; Singh, B. B. Synth. Commun. 1991, 21, 1625–1629. doi:10.1080/00397919108021062 |
| 12. | Moriya, O.; Nakamura, H.; Kageyama, T.; Urata, Y. Tetrahedron Lett. 1989, 30, 3987–3990. doi:10.1016/s0040-4039(00)99302-x |
| 13. | Minakata, S.; Okumura, S.; Nagamachi, T.; Takeda, Y. Org. Lett. 2011, 13, 2966–2969. doi:10.1021/ol2010616 |
| 14. | Mendelsohn, B. A.; Lee, S.; Kim, S.; Teyssier, F.; Aulakh, V. S.; Ciufolini, M. A. Org. Lett. 2009, 11, 1539–1542. doi:10.1021/ol900194v |
| 15. | Zhao, G.; Liang, L.; Wen, C. H. E.; Tong, R. Org. Lett. 2019, 21, 315–319. doi:10.1021/acs.orglett.8b03829 |
| 16. | Bourbeau, M. P.; Rider, J. T. Org. Lett. 2006, 8, 3679–3680. doi:10.1021/ol061260+ |
| 46. | Inagaki, S.; Saito, K.; Suto, S.; Aihara, H.; Sugawara, A.; Tamura, S.; Kawano, T. J. Org. Chem. 2018, 83, 13834–13846. doi:10.1021/acs.joc.8b02153 |
| 30. | Sloop, J. C.; Bumgardner, C. L.; Washington, G.; Loehle, W. D.; Sankar, S. S.; Lewis, A. B. J. Fluorine Chem. 2006, 127, 780–786. doi:10.1016/j.jfluchem.2006.02.012 |
| 28. | Kesornpun, C.; Aree, T.; Mahidol, C.; Ruchirawat, S.; Kittakoop, P. Angew. Chem., Int. Ed. 2016, 55, 3997–4001. doi:10.1002/anie.201511730 |
| 29. | Gutsmiedl, K.; Wirges, C. T.; Ehmke, V.; Carell, T. Org. Lett. 2009, 11, 2405–2408. doi:10.1021/ol9005322 |
| 37. | Medana, C.; Ermondi, G.; Fruttero, R.; Di Stilo, A.; Ferretti, C.; Gasco, A. J. Med. Chem. 1994, 37, 4412–4416. doi:10.1021/jm00051a020 |
| 38. | Seymour, C. P.; Tohda, R.; Tsubaki, M.; Hayashi, M.; Matsubara, R. J. Org. Chem. 2017, 82, 9647–9654. doi:10.1021/acs.joc.7b01709 |
| 39. | Ramazani, A.; Karimi, M.; Hosseinzadeh, Z.; Rezayati, S.; Hanifehpour, Y.; Joo, S. W. Curr. Org. Chem. 2021, 25, 757–778. doi:10.2174/1385272825666210208183751 |
| 34. | Meyer, K. H. Justus Liebigs Ann. Chem. 1911, 380, 212–220. doi:10.1002/jlac.19113800206 |
| 35. | Cortney, C. H.; Krishnan, V. V. J. Chem. Educ. 2020, 97, 825–830. doi:10.1021/acs.jchemed.9b00737 |
| 36. | Kekulé, A. Ann. Chem. Pharm. 1857, 101, 200–213. doi:10.1002/jlac.18571010208 |
| 30. | Sloop, J. C.; Bumgardner, C. L.; Washington, G.; Loehle, W. D.; Sankar, S. S.; Lewis, A. B. J. Fluorine Chem. 2006, 127, 780–786. doi:10.1016/j.jfluchem.2006.02.012 |
| 33. | Geraldes, C. F. G. C.; Barros, M. T.; Maycock, C. D.; Silva, M. I. J. Mol. Struct. 1990, 238, 335–346. doi:10.1016/0022-2860(90)85025-e |
| 30. | Sloop, J. C.; Bumgardner, C. L.; Washington, G.; Loehle, W. D.; Sankar, S. S.; Lewis, A. B. J. Fluorine Chem. 2006, 127, 780–786. doi:10.1016/j.jfluchem.2006.02.012 |
| 30. | Sloop, J. C.; Bumgardner, C. L.; Washington, G.; Loehle, W. D.; Sankar, S. S.; Lewis, A. B. J. Fluorine Chem. 2006, 127, 780–786. doi:10.1016/j.jfluchem.2006.02.012 |
| 31. | Arslan, N. B.; Özdemir, N. J. Mol. Model. 2015, 21, 19. doi:10.1007/s00894-015-2574-8 |
| 32. | Cook, A. G.; Feltman, P. M. J. Chem. Educ. 2010, 87, 678–679. doi:10.1021/ed1000813 |
© 2022 Hossain et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.