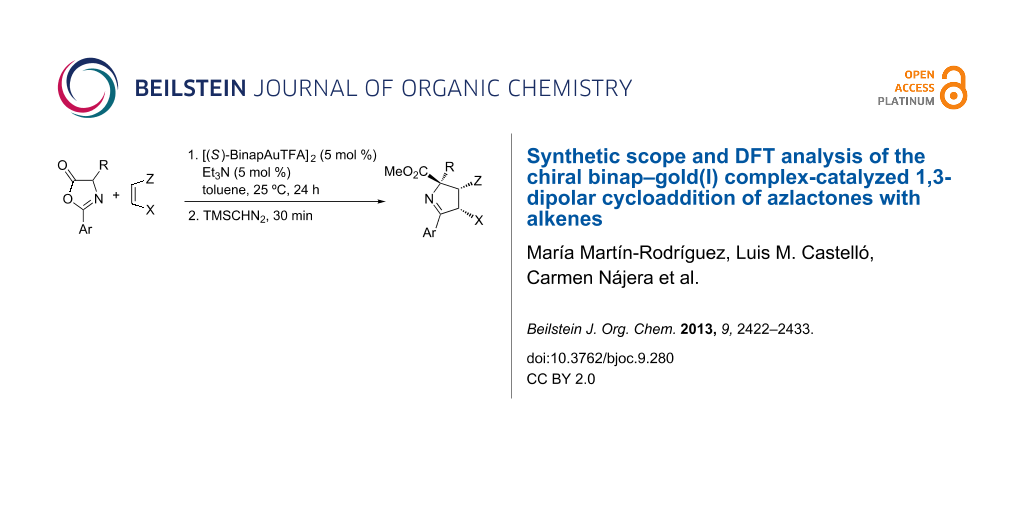
Abstract
The 1,3-dipolar cycloaddition between glycine-derived azlactones with maleimides is efficiently catalyzed by the dimeric chiral complex [(Sa)-Binap·AuTFA]2. The alanine-derived oxazolone only reacts with tert-butyl acrylate giving anomalous regiochemistry, which is explained and supported by Natural Resonance Theory and Nucleus Independent Chemical Shifts calculations. The origin of the high enantiodiscrimination observed with maleimides and tert-butyl acrylate is analyzed using DFT computed at M06/Lanl2dz//ONIOM(b3lyp/Lanl2dz:UFF) level. Several applications of these cycloadducts in the synthesis of new proline derivatives with a 2,5-trans-arrangement and in the preparation of complex fused polycyclic molecules are described.

Graphical Abstract
Introduction
The synthesis of α-amino acids employing an α-amino carbonyl template constitutes the most straightforward route to introduce the α-side chain [1]. As a valid example, oxazol-5-(4H)-ones (azlactones) are suitable heterocycles to perform this C–C bond generation based strategy affording both quaternized and non quaternized α-amino acid derivatives [2-5]. The preparation of azlactones is very simple and their reactivity is very diverse due to their functional groups [2-5]. Many enantioselective and/or diastereoselective processes have been focussed on the elaboration of enantiomerically enriched new non-proteinogenic α-amino acids, such as Michael-type additions [6,7], transition metal-catalyzed allylations [8], Mannich-type additions [9], aldol-type reactions [10], and for other different purposes [11-17]. These substrates can be easily transformed in münchnones, which are potential 1,3-dipoles, after deprotonation and imine-activation with a chiral Lewis acid. Despite of the easy access to this mesoionic heterocycles their enantioselective cycloadditions with electrophilic alkenes have not been exploited. Toste´s group published an efficient 1,3-dipolar cycloaddition (1,3-DC) between alanine, phenylalanine and allylglycine derived azlactones with maleimides and acrylates employing dimetallic (S)-Cy-Segphos(AuOBz)2 complex 1 as a catalyst (2 mol %) in the absence of base (Figure 1) [18,19]. This catalytic system was very effective but the reactions performed with (R)-Binap(AuOBz)2 (Figure 1) as catalyst offered a very low enantioselection, for example, a 8% ee was achieved in the 1,3-DC of alanine derived azlactone and N-phenylmaleimide (NPM).
![[1860-5397-9-280-1]](/bjoc/content/figures/1860-5397-9-280-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Chiral gold(I) complexes employed in 1,3-DC involving azomethine ylides.
Figure 1: Chiral gold(I) complexes employed in 1,3-DC involving azomethine ylides.
Numerous gold-catalyzed transformations employing mild reaction conditions appeared during the last twelve years [20-22]. Initially, coordination arrangements of chiral gold complexes avoided high enantiodiscriminations but, recently, it has been demonstrated that chiral bis-gold complexes type 2 (Figure 1) are very efficient in asymmetric catalysis [23,24]. The high amount of gold per mole of catalyst and the chiral ligand itself make these processes somehow expensive.
The relative lower cost of chiral privileged ligand Binap (versus Cy-Segphos) and the good results obtained in the 1,3-DC of α-imino esters and electrophilic alkenes using the bis-gold(I) complex 3 (where the gold atom:ligand ratio is 1:1, Figure 1) [25-27] inspired us to test it in this azlactone involved cycloaddition. Previous experience in the 1,3-DC between imino esters and electrophilic alkenes revealed that the dimeric chiral gold complex 3 resulted to be unique efficient catalyst in terms of enantioselection rather than the bis-gold complex 4 [25-27]. This data is in a clear contrast to the previously mentioned result for the reactivity of azlactones [18,19]. In this work we describe a more extended study than the analogous one described in a preliminary communication [28] concerning the catalytic activity of complexes 3 and 4 in the 1,3-DC of oxazolones with electrophilic alkenes. Here, a deep DFT analysis and the application of other computational experiments (NRT, NICS) were compared to the experimentally observed results in order to clarify the enantio- and anomalous regioselectivity.
Results and Discussion
Initially, the synthesis of oxazolones 5 was accomplished under mild reaction conditions by mixing N-acyl-α-amino acid derivatives in the presence of dehydrating agents such as carbodiimides [2-5]. Gold(I) complexes 3 and 4, identified and characterized by Puddephatt’s group [X = trifluoroacetate (TFA)] [29-31], were obtained from NaAuCl4 and dimethyl sulfide and the corresponding amount of the chiral Binap ligand. Finally, the anion interchange was promoted by the addition of an equivalent amount of silver(I) salt. These complexes were used immediately after filtration through a celite path. Particularly, complexes 3 and 4 (X = TFA) could be isolated in 96 and 89% yield, respectively, but other gold(I) complexes (see Table 1) with different anions were generated in situ and used as catalysts in the same solution.
Oxazolone derived from glycine 5a was allowed to react with N-phenylmaleimide (NPM) at room temperature (25 °C approx.) using 5 mol % of the chiral catalytic complex and 5 mol % of base (Scheme 1). After completion, a large excess of trimethylsilyldiazomethane was added to obtain the methyl ester of intermediate carboxylic acid 6a (30 min). Compound 7aa was obtained diastereoselectively (>98:2, by 1H NMR spectroscopy) after purification and its absolute configuration was established according to the retention times of signals observed after HPLC analysis employing chiral columns and by comparison with the previously reported data [18,19].
![[1860-5397-9-280-i1]](/bjoc/content/inline/1860-5397-9-280-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: 1,3-DC of azlactone 5a and NPM.
Scheme 1: 1,3-DC of azlactone 5a and NPM.
Using this model reaction (Scheme 1), we tested the dimeric gold complex [(Sa)-Binap·AuTFA]2 according to the previous experience obtained in the 1,3-DC involving imino esters and electrophilic alkenes and the reaction conditions employed by Toste’s group [18,19]. The use of fluorobenzene as solvent or co-solvent did not afford neither good conversions nor enantioselectivities, even working with the dimetallic complex 4 (X = TFA) (Table 1, entries 1–4). After the evaluation of the influence of the solvent, we concluded that toluene was the most appropriate solvent for these reactions (Table 1, entries 5–9), being the chemical yield high (90%) and the enantiodiscrimination excellent (99% ee). The presence of triethylamine as base is crucial for this transformation, it ensures both of the high conversions and enantioselections (Table 1, entries 11–14). Other different bases such as DBU, and DIPEA did not improve the result achieved by the analogous reaction carried out with triethylamine (Table 1, entries 12 and 13). Again, the presence of the chiral catalytic complex 4 (X = TFA) did not give the expected results (Table 1, entries 6 and 10). The enantiomerically pure form of 7aa with opposite absolute configuration was isolated by working in the presence of [(Ra)-Binap·AuTFA]2 complex (Table 1, entry 11). Surprisingly, no reaction was observed in the presence of silver(I) complex (Sa)-Binap·AgTFA (Table 1, entry 15). In this section the effect of different anions of the metal complex was studied as well. In contrast with the negligible reaction observed when poor basic anion, such as perchlorate, was essayed (Table 1, entry 16), anions with basic character such as acetate or benzoate, incorporated to the chemical structure of the gold(I) catalyst, promoted the enantioselective reaction although with lower efficiency (Table 1, entries 17 and 18) [32].
Table 1: Optimization of the 1,3-dipolar cycloaddition of 5a and NPM using chiral complexes.
| Entry | Catalyst/Xa | Solvent | Base | Yieldb (%) | eec (%) |
|---|---|---|---|---|---|
| 1 | (Sa)-3/TFA | PhF | Et3N | <50 | 7 |
| 2 | (Sa)-4/TFA | PhF | Et3N | ___d | <5 |
| 3 | (Sa)-3/TFA | PhF-THF | Et3N | ___d | ___d |
| 4 | (Sa)-4/TFA | PhF-THF | Et3N | ___d | <5 |
| 5 | (Sa)-3/TFA | THF | Et3N | 76 | 49 |
| 6 | (Sa)-4/TFA | THF | Et3N | ___d | nd |
| 7 | (Sa)-3/TFA | DCM | Et3N | 88 | 80 |
| 8 | (Sa)-3/TFA | Et2O | Et3N | 85 | 76 |
| 9 | (Sa)-3/TFA | PhMe | Et3N | 90 | 99 |
| 10 | (Sa)-4/TFA | PhMe | Et3N | ___d | ___d |
| 11 | (Ra)-3/TFA | PhMe | Et3N | 90 | –99 |
| 12 | (Sa)-3/TFA | PhMe | DBU | 70e | 80 |
| 13 | (Sa)-3/TFA | PhMe | DIPEA | 90 | 98 |
| 14 | (Sa)-3/TFA | PhMe | none | ___d | ___d |
| 15 | (Sa)-Binap·AgTFA | PhMe | Et3N | ___d | ___d |
| 16 | (Sa)-3/ClO4 | PhMe | Et3N | ___d | ___d |
| 17 | (Sa)-3/OAc | PhMe | Et3N | 90 | 64 |
| 18 | (Sa)-3/OBz | PhMe | Et3N | 91 | 74 |
aThe gold catalysts were freshly generated in situ. bAfter flash chromatography (silica gel). The observed exo:endo ratio was always >98:2 (1H NMR). cDetermined by using analytical chiral HPLC columns (Daicel, Chiralpak AS). dNot determined.
The scope of the reaction was next surveyed. Firstly, azlactone 5a was allowed to react with several maleimides (Scheme 2, and Table 2, entries 1–10). NPM and 4-acetoxyphenylmaleimide were the best entries of this series affording almost enantiomerically pure bicyclic products 7aa and 7ae, respectively (Table 2, entries 1 and 8). N-Substituted methyl, ethyl and benzylmaleimides did not afford compounds 7 with so high enantioselections. Then, a lower temperature (−20 °C) was attempted but the increment of ee for N-methyl- and N-ethylmaleimides was not very noticeable (Table 2, entries 2, 3 and 4, 5, respectively). Nevertheless, a gap of 21 units of ee was achieved in the case of the reaction involving N-benzylmaleimide (Table 2, compare entries 6 and 7). In the case of N-(4-bromophenyl)maleimide a good enantioselection was observed when the reaction was run at −20 °C furnishing enantiomerically pure 7af in good chemical yields (Table 2, entries 9 and 10). The variation of the arene substituent of the azlactones promoted also excellent to good enantioselections in compounds 7ba and 7ca (Table 2, entries 11 and 12). Even working with an heteroaromatic substituent, such as 2-thienyl, compound 7da was isolated in 95% ee (Table 2, entry 13).
![[1860-5397-9-280-i2]](/bjoc/content/inline/1860-5397-9-280-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: General 1,3-DC between azlactones 5 with maleimides.
Scheme 2: General 1,3-DC between azlactones 5 with maleimides.
Table 2: 1,3-Dipolar cycloaddition of azlactones 5a with maleimides.
| Entry | Ar, 5a | R | Product 7 | Yieldb (%) | eec (%) |
|---|---|---|---|---|---|
| 1 | Ph, 5a | Ph | 7aa | 90 | 99 |
| 2 | Ph, 5a | Me | 7ab | 90 | 54 |
| 3 | Ph, 5a | Med | 7ab | 79 | 60 |
| 4 | Ph, 5a | Et | 7ac | 87 | 62 |
| 5 | Ph, 5a | Etd | 7ac | 70 | 70 |
| 6 | Ph, 5a | Bn | 7ad | 90 | 50 |
| 7 | Ph, 5a | Bnd | 7ad | 83 | 71 |
| 8 | Ph, 5a | 4-(AcO)C6H4 | 7ae | 90 | 99 |
| 9 | Ph, 5a | 4-BrC6H4 | 7af | 82 | 91 |
| 10 | Ph, 5a | 4-BrC6H4d | 7af | 84 | 99 |
| 11 | 4-MeC6H4, 5b | Ph | 7ba | 78 | 99 |
| 12 | 4-ClC6H4, 5c | Ph | 7ca | 83 | 98 |
| 13 | 2-Thienyl, 5d | Ph | 7da | 80 | 95 |
aThe gold catalyst was freshly generated in situ. bAfter flash chromatography (silica gel). The observed exo:endo ratio was always >98:2 (1H NMR). cDetermined by using analytical chiral HPLC columns (Daicel, Chiralpak AS). dReaction run at −20 °C.
When benzylamine was employed as alternative quenching reagent to trimethylsilyldiazomethane, the generation of the corresponding N-benzylamide in 76% yield and 96% ee was achieved after 17 h at 25 °C (Scheme 3) [18,19].
![[1860-5397-9-280-i3]](/bjoc/content/inline/1860-5397-9-280-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
The study of the key points of the enantiodiscrimination step and mechanism for the 1,3-DC of azlactone 7aa and NPM can be originated by the presence of a more active homochiral dimer catalyst (Sa,Sa)-3 (X = TFA) with a lower TS energy with all the reaction components, rather than the corresponding heterochiral ones and even lower than homochiral dimer catalyst (Ra,Ra)-3 (X = TFA). The clear positive non-linear effects (NLE) described in Figure 2 supported this hypothesis [33].
![[1860-5397-9-280-2]](/bjoc/content/figures/1860-5397-9-280-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Positive non-linear effects (NLE) observed in 1,3-DC of azlactone 7aa and NPM.
Figure 2: Positive non-linear effects (NLE) observed in 1,3-DC of azlactone 7aa and NPM.
Next, we studied the reaction between the oxazolone 5aa and NPM catalyzed by [(Sa)-Binap-AuTFA]2. In previous works, we have demonstrated that the stereoselectivity of the 1,3-DC employing chiral metallic Lewis acids arises from the blockage of one of the prochiral faces [34]. Starting from this selected conformation of the catalyst, our results show that the (2Re,5Re) prochiral face is less hindered than the other prochiral face in the most stable conformation of [{(Sa)-Binap-Au}2]-5aa complex (Figure 3). As expected, the existence of dimeric gold units is crucial in the blockage of one of the prochiral faces, and therefore, in the stereochemical outcome of the final cycloadducts [26,27].
![[1860-5397-9-280-3]](/bjoc/content/figures/1860-5397-9-280-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Main geometrical features and relative Gibbs free energies (in kcal mol−1 at 298 K) of complexes [(Sa)-Binap-Au]2-5aa and [(Sa)-Binap-Au]2-5aa-b computed at M06/Lanl2dz//ONIOM (b3lyp/Lanl2dz:UFF). High-level and low-level layers are represented as ball and stick and wireframe models, respectively. Blue surface represents the solvent-accessible surface with a probe radius of 1.9 Å.
Figure 3: Main geometrical features and relative Gibbs free energies (in kcal mol−1 at 298 K) of complexes [(S...
Refined computational results showed the exo-approach [35] is the preferred one. In this analysis, only that approach was considered. The less energetic computed TS are depicted on Figure 4 (see Supporting Information File 1 for further information of additional TS’s).
![[1860-5397-9-280-4]](/bjoc/content/figures/1860-5397-9-280-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Main geometrical features and relative Gibbs free energies (in kcal mol−1) of the less energetic transition states associated with the 1,3-DC of 5aa and NPM catalyzed by (Sa)-Binap gold dimers computed at M06/Lanl2dz//ONIOM(b3lyp/Lanl2dz:UFF) level of theory. High-level and low level layers are represented as ball and stick and wireframe models, respectively. Distances are in Å. Blue and purple surfaces represent the solvent-accessible surface of the catalyst and NPM with a probe radius of 1.9 Å.
Figure 4: Main geometrical features and relative Gibbs free energies (in kcal mol−1) of the less energetic tr...
The computed transition structures correspond to concerted but highly asynchronous cycloadditions (Figure 4). Our calculations show that there is a different overlap between the accessible-solvent surface of the catalyst and the one of the incoming dipolarophile. That implies an increase of the 4e− Pauli repulsion between the reactives in TSNPMdown compared to TSNPMup, and thus an increase of the activation barrier. Moreover, lower energy to deform the initial ylide (strain energy) is required in the latter TS. With that energetic diference, the computed ee is about 99%, in good agreement with the experimental results (Table 2, entry 1).
The complete reaction path of the cycloaddition process is shown in Scheme 4. We do not study computationaly the second synthetic step, namely the ring-opening of the tricyclic-cycloadduct, because that step has no relevance in the stereochemical outcome of the reaction.
![[1860-5397-9-280-i4]](/bjoc/content/inline/1860-5397-9-280-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Reaction Gibbs free energy associated with the 1,3-DC of 5aa and NPM catalyzed by (Sa)-Binap gold dimers computed at M06/Lanl2dz//ONIOM (b3lyp/Lanl2dz:UFF) level of theory.
Scheme 4: Reaction Gibbs free energy associated with the 1,3-DC of 5aa and NPM catalyzed by (Sa)-Binap gold d...
We also studied the last step of the catalytic cycle that ensures the recovery of the catalyst obtaining a favourable Gibbs energy of −55.3 kcal mol−1 (Scheme 5).
![[1860-5397-9-280-i5]](/bjoc/content/inline/1860-5397-9-280-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: ΔG calculation for the recovery of the catalytic active species.
Scheme 5: ΔG calculation for the recovery of the catalytic active species.
No chemical reaction occurred when 5a was combined with other dipolarophiles such as fumarates, maleates, vinyl phenyl sulfone, trans-1,2-bis(phenylsulfonyl)ethylene, chalcone, crotonaldehyde and cinnamaldehyde at the same reaction conditions [36]. Another drawback was the poor reactivity observed when α-substituted azlactones were used as starting material in the named reaction with NPM. However, the alanine-derived 4-methyloxazole-5-one 10, surprisingly, reacted at 25 and at 0 °C with tert-butyl acrylate yielding cycloadduct 11 in good yields and moderate to good enantioselections (Scheme 6).
![[1860-5397-9-280-i6]](/bjoc/content/inline/1860-5397-9-280-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: 1,3-DC of azlactone 10 and tert-butyl acrylate.
Scheme 6: 1,3-DC of azlactone 10 and tert-butyl acrylate.
If we compare this result with previous ones obtained using α-imino esters, this last diastereoselective cycloaddition exhibited an opposite regioselection. Besides, the resulting relative configuration of Δ1-pyrroline 11 is equivalent to the exo-approach of the dipolarophile when an endo-transition state was the most favourable in the gold(I)-catalyzed 1,3-DC with α-imino esters and alkenes [37].
To gain more insight into the unexpected regioselectivity of the 1,3-DC depicted in Scheme 6, calculations within the DFT framework were performed. In the accepted mechanism of the metal catalyzed 1,3-DC of azomethine ylides and acrylates, the α-carbon atom of the azomethine ylide (C2 in Figure 5) reacts with the β-carbon of the acrylate moiety, independently of the mechanism (concerted fashion or via Michael-like transition state followed by a Mannich-like ring closure in a stepwise mechanism yields the same cycloadduct) [38]. This fact is assumed to be a consequence of the unsymmetrical electron density in the 1,3-dipole moiety, being higher in the carbon in α-position to the carboxy group (C2).
![[1860-5397-9-280-5]](/bjoc/content/figures/1860-5397-9-280-5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: (A) Schematic representation of the model gold(I) ylides. (B) HOMO of the ylides and expansion orbital coefficient values of carbon atoms 2 and 5 computed at HF/Lanl2dz level of theory. Hydrogen atoms are omitted for clarity. (C) Most stable Lewis structures of the ylides obtained with the Natural Resonance Theory (NRT) analysis.
Figure 5: (A) Schematic representation of the model gold(I) ylides. (B) HOMO of the ylides and expansion orbi...
Initially, a model azomethine ylide derived from oxazolone 10 was considered (Figure 5). Moreover, an acyclic w-shaped ylide analogue (Ylide-II) was also studied as a reference. We chose this latter 1,3-dipole because it is known that with this kind of reactive species, the reaction yields cycloadducts possessing a standard regioselectivity in 1,3-DC with acrylates [38]. Since our goal was to understand the origins of the unusual regioselectivity observed in the reaction between dipoles of type Ylide-I with acrylates, trimethylphosphine was coordinated directly to the gold(I) atom in our model (Figure 5).
Analysis of atomic expansion coefficients of the HOMO of Ylide I reveal no significant difference between the azomethine ylides reported in Figure 5. However, Natural Resonance Theory Analysis (NRT) [39-41] shows that the negative charge in the Lewis structure of Ylide I is mainly placed on C5. In the case of Ylide II, this negative charge is placed on the oxygen of the carboxy group instead. The importance of these electronic distributions was verified by Nucleus Independent Chemical Shifts (NICS) calculations in the ring point of the oxazoline [42]. The NICS value of −7.3 ppm pointed to the aromaticity of that ring in Ylide I. These results explain the existence of different regioselectivities for both ylides.
Following the same calculation patterns previously shown for the reaction with NPM, the results of the main geometrical features an relative Gibbs free energies were determined for the approach of the gold(I) complex·azlactone 10 to tert-butyl acrylate (Figure 6).
![[1860-5397-9-280-6]](/bjoc/content/figures/1860-5397-9-280-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Main geometrical features and relative Gibbs free energies (in kcal mol−1 at 298 K) of complexes [{(Sa)-Binap-Au}2]-I computed at M06/Lanl2dz//ONIOM (b3lyp/Lanl2dz:UFF). High-level and low-level layers are represented as ball and stick and wireframe models, respectively. Blue surface represents the solvent-accessible surface with a probe radius of 1.9 Å.
Figure 6: Main geometrical features and relative Gibbs free energies (in kcal mol−1 at 298 K) of complexes [{(...
In order to have a complete view of the reaction mechanism, all transition structures corresponding to the endo- or exo-approaches of the acrylate moiety as well as possible regiochemistry of the selected 1,3-DC, were considered. The main geometrical features of the less energetic transition structures are depicted in Figure 7.
![[1860-5397-9-280-7]](/bjoc/content/figures/1860-5397-9-280-7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: Main geometrical features and relative Gibbs free energies (in kcal mol−1) of the less energetic transition states associated with the 1,3-DC of 10 and tert-butyl acrylate catalyzed by (Sa)-Binap gold dimers computed at M06/Lanl2dz//ONIOM(b3lyp/Lanl2dz:UFF). High-level and low level layers are represented as ball and stick and wireframe models, respectively. Distances are in Å. Hydrogen atoms are omitted for clarity.
Figure 7: Main geometrical features and relative Gibbs free energies (in kcal mol−1) of the less energetic tr...
Our calculations show that the less energetic transition structure associated with the 1,3-DC of 10 and tert-butyl acrylate is TS11exo (Figure 7), is in good agreement with the experimental results in which a high ee of the corresponding stereoisomer was observed. The formation of the enantiomer (TS11ent) was found to have an activation barrier of 4.5 kcal mol−1 higher in energy. That difference can be a consequence of the higher strain energy necessary to deform the initial ylide. Our calculations also pointed out the stabilizing interaction of the carboxy group of the incoming acrylate and the gold atom closest to the ylide moiety, despite the long distance (dAu-C=O = 2.8 Å). In fact, the exo-approach is ca. 11 kcal mol−1 lower in energy than the endo analogue (TS11exo vs TS11endo in Figure 7). Moreover, the a priori expected regiochemistry of the cycloaduct, in which C2–Cβ and C5–Cα are new bonds (12), was considered. In this case, TS12 is 12.1 kcal mol−1 higher in energy than TS11exo. It is noticeable that transition structures associated with the formation of C2–Cα and C5–Cβ bonds (TS11exo, TS11ent and TS11endo) correspond to concerted but highly asynchronous cycloadditions. On the other hand, TS12 is associated with a stepwise mechanism.
As possible applications of the resulting pyrrolines 7aa, it was submitted to different transformations. For example, it could be reduced to the corresponding pyrrolidines employing sodium cyanoborohydride in acidic media. In this reaction, a 1:1 mixture of 2,5-cis-pyrrolidine 13 and its 5-epimer 14 (2,5-trans) was isolated in good chemical yield (71%) (Scheme 7, reaction a). Fortunately, 5-epimer 14 (2,5-trans) was diastereoselectively generated through a 10% Pd/C-catalyzed hydrogenation using 4 atmospheres of hydrogen during three days at 25 °C (Scheme 7, reaction b). This trans- arrangement in molecule 14 is not very easy to built because several steps were needed using other synthetic strategies [43].
![[1860-5397-9-280-i7]](/bjoc/content/inline/1860-5397-9-280-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Reduction of heterocycle 7aa under different conditions.
Scheme 7: Reduction of heterocycle 7aa under different conditions.
Pyrrolines also possess a typical 1,3-dipole precursor structure (azomethine ylide), so a second cycloaddition was attempted with a new equivalent of N-methylmaleimide. The reaction took place under microwave assisted heating (1 h, 75 W) using triethylamine as base and toluene as solvent at 120 °C. Polycyclic compound 15 was finally obtained in 50% yield as single diastereoisomer (Scheme 8). Despite being a solid product it was not possible to perform an X-ray diffraction analysis. Positive (CH derived from NPM with the CH derived from NMM) nOe experiments supported the drawn absolute configuration of 15.
![[1860-5397-9-280-i8]](/bjoc/content/inline/1860-5397-9-280-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Double 1,3-DC to give polycycle 15.
Scheme 8: Double 1,3-DC to give polycycle 15.
Other different dipolarophiles were attempted to react with starting 7aa obtaining very complex mixtures including decomposed materials. In the most cases, reactions had to be refluxed for 24 h (110 °C, toluene) because microwave assisted irradiation was not as effective as occurred in the reaction with NMM. For example, the purification of the crude reaction mixture of the cycloaddition of 7aa with β-nitrostyrene afforded an overall poor yield (~28%) of a complex 4:15:10 mixture of three compounds (16, 17, and 18) (Scheme 9) [44]. The desired compound 16 was identified (almost as unique diastereoisomer) in low chemical yield (<5%) together with two pyrrole derivatives 17 (only one stereoisomer), and 18. The last compound was formed by a retro-cycloaddition of the pyrroline 7aa with elimination of NMM, which was favoured by a prolonged heating [45].
![[1860-5397-9-280-i9]](/bjoc/content/inline/1860-5397-9-280-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Reaction between 7aa and nitrostyrene.
Scheme 9: Reaction between 7aa and nitrostyrene.
Conclusion
In this work it has been demonstrated the efficiency of the chiral [BinapAuTFA]2 complexes in the enantioselective 1,3-DC between azlactone derived from glycine and maleimides, especially those containing a N-aromatic substituent, and between alanine derived oxazolone with tert-butyl acrylate. In the last example the regiochemistry was totally opposite to the common trend of these cycloadditions. This behaviour has been explained for the first time using NRT, NICS, whilst DFT calculations served to justify the elevated enantioselection observed in the 1,3-DC between azlactones and maleimides. The general scope is not very wide but enantioselections obtained are quite good. Very interesting pyrrolidines with a trans-arrangement were obtained after hydrogenation of the pyrroline precursor.
Supporting Information
Description of all procedures and characterization of all new compounds, as well as computational details and coordinate tables are reported in the Supporting Information.
| Supporting Information File 1: Experimental and analytical data. | ||
| Format: PDF | Size: 741.6 KB | Download |
Acknowledgements
This work has been supported by the DGES of the Spanish Ministerio de Ciencia e Innovación (MICINN) (Consolider INGENIO 2010 CSD2007-00006, FEDER-CTQ2007-62771/BQU, CTQ2010-20387 and by the Hispano-Brazilian project PHB2008-0037-PC), Generalitat Valenciana (PROMETEO/ 2009/039), the Basque government (Grant IT-324-07) and by the University of Alicante. M. M.-R. and L. C. also thank DGES for grants. The authors also thank the SGI/IZO-SGIker of UPV/EHU for allocation of computational resources.
References
-
Nájera, C.; Sansano, J. M. Chem. Rev. 2007, 107, 4584–4671. doi:10.1021/cr050580o
Return to citation in text: [1] -
Fisk, J. S.; Mosey, R. A.; Tepe, J. J. Chem. Soc. Rev. 2007, 36, 1432–1440. doi:10.1039/b511113g
Return to citation in text: [1] [2] [3] -
Hewlett, N. M.; Hupp, C. D.; Tepe, J. J. Synthesis 2009, 2825–2839. doi:10.1055/s-0029-1216924
Return to citation in text: [1] [2] [3] -
Alba, A.-N. R.; Ríos, R. Chem.–Asian J. 2011, 6, 720–734. doi:10.1002/asia.201000636
Return to citation in text: [1] [2] [3] -
El-Mekabaty, A. Int. J. Mod. Org. Chem. 2013, 2, 40–66.
Return to citation in text: [1] [2] [3] -
Ávila, E. P.; de Mello, A. C.; Diniz, R.; Amarante, G. W. Eur. J. Org. Chem. 2013, 1881–1883.
See for a recent contribution.
Return to citation in text: [1] -
Uraguchi, D.; Ueki, Y.; Ooi, T. Chem. Sci. 2012, 3, 842–845. doi:10.1039/c1sc00678a
See for a recent contribution.
Return to citation in text: [1] -
Chen, W.; Hartwig, J. F. J. Am. Chem. Soc. 2013, 135, 2068–2071. doi:10.1021/ja311363a
Return to citation in text: [1] -
Zhang, W.-Q.; Cheng, L.-F.; Yu, J.; Gong, L.-Z. Angew. Chem., Int. Ed. 2012, 51, 4085–4088. doi:10.1002/anie.201107741
Return to citation in text: [1] -
Terada, M.; Tanaka, H.; Sorimachi, K. J. Am. Chem. Soc. 2009, 131, 3430–3431. doi:10.1021/ja8090643
Return to citation in text: [1] -
Weber, M.; Frey, W.; Peters, R. Adv. Synth. Catal. 2012, 354, 1443–1449. doi:10.1002/adsc.201200085
See for alkylations at multiple positions.
Return to citation in text: [1] -
Trost, B. M.; Czabaniuk, L. C. J. Am. Chem. Soc. 2012, 134, 5778–5781. doi:10.1021/ja301461p
See for alkylations at multiple positions.
Return to citation in text: [1] -
Dell’Amico, L.; Albrecht, L.; Naicker, T.; Poulsen, P. H.; Jørgensen, K. A. J. Am. Chem. Soc. 2013, 135, 8063–8070. doi:10.1021/ja4029928
See for alkylations at multiple positions.
Return to citation in text: [1] -
Breman, A. C.; Smits, J. M. M.; de Gelder, R.; van Maarseveen, J. H.; Ingemann, S.; Hiemstra, H. Synlett 2012, 23, 2195–2200. doi:10.1055/s-0032-1317081
See for participation of α-alkylidene azlactones in 1,3-DC as dipolarophiles.
Return to citation in text: [1] -
González-Esguevillas, M.; Adrio, J.; Carretero, J. C. Chem. Commun. 2013, 49, 4649–4651. doi:10.1039/c3cc41663a
See for participation of α-alkylidene azlactones in 1,3-DC as dipolarophiles.
Return to citation in text: [1] -
Ho, H. T.; Levere, M. E.; Fournier, D.; Montembault, V.; Pascual, S.; Fontaine, L. Aust. J. Chem. 2012, 65, 970–977. doi:10.1071/CH12192
See for the synthesis of new polymers.
Return to citation in text: [1] -
Horlacher, O. P.; Hartkoorn, R. C.; Cole, S. T.; Altmann, K.-H. ACS Med. Chem. Lett. 2013, 4, 264–268. doi:10.1021/ml300385q
See for the preparation of natural products.
Return to citation in text: [1] -
Melhado, A. D.; Luparia, M.; Toste, F. D. J. Am. Chem. Soc. 2007, 129, 12638–12639. doi:10.1021/ja074824t
Return to citation in text: [1] [2] [3] [4] [5] -
Melhado, A. D.; Amarante, G. W.; Wang, Z. J.; Luparia, M.; Toste, F. D. J. Am. Chem. Soc. 2011, 133, 3517–3527. doi:10.1021/ja1095045
Return to citation in text: [1] [2] [3] [4] [5] -
Hasmi, A. S. K.; Toste, F. D., Eds. Modern Gold Catalyzed Synthesis; Wiley-VCH: Weinheim, Germany, 2012. doi:10.1002/9783527646869
Return to citation in text: [1] -
Rudolph, M.; Hashmi, A. S. K. Chem. Soc. Rev. 2012, 41, 2448–2462. doi:10.1039/c1cs15279c
Return to citation in text: [1] -
Peixoto de Almeida, M.; Carabineiro, S. A. C. ChemCatChem 2012, 4, 18–29. doi:10.1002/cctc.201100288
Return to citation in text: [1] -
Bandini, M.; Bottoni, A.; Chiarucci, M.; Cera, G.; Miscione, G. P. J. Am. Chem. Soc. 2012, 134, 20690–20700. doi:10.1021/ja3086774
Return to citation in text: [1] -
Brazeau, J.-F.; Zhang, S.; Colomer, I.; Corkey, B. K.; Toste, F. D. J. Am. Chem. Soc. 2012, 134, 2742–2749. doi:10.1021/ja210388g
And references cited therein.
Return to citation in text: [1] -
Martín-Rodríguez, M.; Nájera, C.; Sansano, J. M.; Wu, F.-L. Tetrahedron: Asymmetry 2010, 21, 1184–1186. doi:10.1016/j.tetasy.2010.06.011
And see corrigendum Tetrahedron: Asymmetry 2010, 21, 2559–2560.
Return to citation in text: [1] [2] -
Martín-Rodríguez, M.; Nájera, C.; Sansano, J. M.; de Cózar, A.; Cossío, F. P. Chem.–Eur. J. 2011, 17, 14224–14233. doi:10.1002/chem.201101606
Return to citation in text: [1] [2] [3] -
Martín-Rodríguez, M.; Nájera, C.; Sansano, J. M.; de Cózar, A.; Cossío, F. P. Beilstein J. Org. Chem. 2011, 7, 988–996. doi:10.3762/bjoc.7.111
Return to citation in text: [1] [2] [3] -
Martín-Rodríguez, M.; Nájera, C.; Sansano, J. M. Synlett 2012, 62–65. doi:10.1055/s-0030-1260334
Return to citation in text: [1] -
Wheaton, C. A.; Jennings, M. C.; Puddephatt, R. J. J. Am. Chem. Soc. 2006, 128, 15370–15371. doi:10.1021/ja066450u
Return to citation in text: [1] -
Wheaton, C. A.; Puddephatt, R. J. Angew. Chem., Int. Ed. 2007, 46, 4461–4463. doi:10.1002/anie.200701325
Return to citation in text: [1] -
Wheaton, C. A.; Jennings, M. C.; Puddephatt, R. J. Z. Naturforsch. 2009, 64b, 1569–1577.
Return to citation in text: [1] -
Gorin, D. J.; Sherry, B. D.; Toste, F. D. Chem. Rev. 2008, 108, 3351–3378. doi:10.1021/cr068430g
See for a comprehensive study of the ligand effects in homogeneous gold(I) catalysis.
Return to citation in text: [1] -
Satyanarayana, T.; Abraham, S.; Kagan, H. B. Angew. Chem., Int. Ed. 2009, 48, 456–494. doi:10.1002/anie.200705241
Return to citation in text: [1] -
de Cózar, A.; Cossío, F. P. Phys. Chem. Chem. Phys. 2011, 13, 10858–10868. doi:10.1039/c1cp20682f
Return to citation in text: [1] -
The exo descriptor is referred to the approach of the two reaction components where the tert-butyl ester and carbonyl group of the azlactone are placed in oposite direction.
Return to citation in text: [1] -
Terada, M.; Nii, H. Chem.–Eur. J. 2011, 17, 1760–1763. doi:10.1002/chem.201003015
The published [4 + 2] cycloaddition of azlactones to β,γ-unsaturated α-ketoesters was unsuccessfully attempted. Only a low yield of O-alkylation addition product to the ketone group was detected.
Return to citation in text: [1] -
In ref. [18] and [19] the same result was obtained, but no explanation to this anomalous addition was given.
Return to citation in text: [1] -
Nájera, C.; de Gracia Retamosa, M.; Sansano, J. M.; de Cózar, A.; Cossío, F. P. Eur. J. Org. Chem. 2007, 5038–5049. doi:10.1002/ejoc.200700267
Return to citation in text: [1] [2] -
Glendening, E. D.; Weinhold, F. J. Comput. Chem. 1998, 19, 593–609. doi:10.1002/(SICI)1096-987X(19980430)19:6<593::AID-JCC3>3.0.CO;2-M
Return to citation in text: [1] -
Glendening, E. D.; Weinhold, F. J. Comput. Chem. 1998, 19, 610–627. doi:10.1002/(SICI)1096-987X(19980430)19:6<610::AID-JCC4>3.0.CO;2-U
Return to citation in text: [1] -
Glendening, E. D.; Badenhoop, J. K.; Weinhold, F. J. Comput. Chem. 1998, 19, 628–646. doi:10.1002/(SICI)1096-987X(19980430)19:6<628::AID-JCC5>3.0.CO;2-T
Return to citation in text: [1] -
Bader, R. F. W. Atoms in Molecules-A Quantum Theory; Clarendon Press: Oxford, 1900.
Return to citation in text: [1] -
Lee, M.; Lee, Y.-J.; Park, E.; Park, Y.; Ha, M. W.; Hong, S.; Lee, Y.-J.; Kim, T.-S.; Kim, M.; Park, H. Org. Biomol. Chem. 2013, 11, 2039–2046. doi:10.1039/c3ob27089k
See for an example.
Return to citation in text: [1] -
Kim, Y.; Kim, J.; Park, S. B. Org. Lett. 2009, 11, 17–20. doi:10.1021/ol8022193
See for a regioselective synthesis of tetrasubstituted pyrroles by 1,3-DC from azlactones and spontaneous decarboxylation.
Return to citation in text: [1] -
Ortiz, A. L.; Echegoyen, L.; Delgado, J. L.; Martin, N. Handb. Carbon Nano Mater. 2011, 1, 325–373.
See for retro 1,3-DC, that has also been observed in many thermal processes.
Return to citation in text: [1]
| 38. | Nájera, C.; de Gracia Retamosa, M.; Sansano, J. M.; de Cózar, A.; Cossío, F. P. Eur. J. Org. Chem. 2007, 5038–5049. doi:10.1002/ejoc.200700267 |
| 38. | Nájera, C.; de Gracia Retamosa, M.; Sansano, J. M.; de Cózar, A.; Cossío, F. P. Eur. J. Org. Chem. 2007, 5038–5049. doi:10.1002/ejoc.200700267 |
| 39. | Glendening, E. D.; Weinhold, F. J. Comput. Chem. 1998, 19, 593–609. doi:10.1002/(SICI)1096-987X(19980430)19:6<593::AID-JCC3>3.0.CO;2-M |
| 40. | Glendening, E. D.; Weinhold, F. J. Comput. Chem. 1998, 19, 610–627. doi:10.1002/(SICI)1096-987X(19980430)19:6<610::AID-JCC4>3.0.CO;2-U |
| 41. | Glendening, E. D.; Badenhoop, J. K.; Weinhold, F. J. Comput. Chem. 1998, 19, 628–646. doi:10.1002/(SICI)1096-987X(19980430)19:6<628::AID-JCC5>3.0.CO;2-T |
| 1. | Nájera, C.; Sansano, J. M. Chem. Rev. 2007, 107, 4584–4671. doi:10.1021/cr050580o |
| 8. | Chen, W.; Hartwig, J. F. J. Am. Chem. Soc. 2013, 135, 2068–2071. doi:10.1021/ja311363a |
| 28. | Martín-Rodríguez, M.; Nájera, C.; Sansano, J. M. Synlett 2012, 62–65. doi:10.1055/s-0030-1260334 |
| 6. |
Ávila, E. P.; de Mello, A. C.; Diniz, R.; Amarante, G. W. Eur. J. Org. Chem. 2013, 1881–1883.
See for a recent contribution. |
| 7. |
Uraguchi, D.; Ueki, Y.; Ooi, T. Chem. Sci. 2012, 3, 842–845. doi:10.1039/c1sc00678a
See for a recent contribution. |
| 2. | Fisk, J. S.; Mosey, R. A.; Tepe, J. J. Chem. Soc. Rev. 2007, 36, 1432–1440. doi:10.1039/b511113g |
| 3. | Hewlett, N. M.; Hupp, C. D.; Tepe, J. J. Synthesis 2009, 2825–2839. doi:10.1055/s-0029-1216924 |
| 4. | Alba, A.-N. R.; Ríos, R. Chem.–Asian J. 2011, 6, 720–734. doi:10.1002/asia.201000636 |
| 5. | El-Mekabaty, A. Int. J. Mod. Org. Chem. 2013, 2, 40–66. |
| 2. | Fisk, J. S.; Mosey, R. A.; Tepe, J. J. Chem. Soc. Rev. 2007, 36, 1432–1440. doi:10.1039/b511113g |
| 3. | Hewlett, N. M.; Hupp, C. D.; Tepe, J. J. Synthesis 2009, 2825–2839. doi:10.1055/s-0029-1216924 |
| 4. | Alba, A.-N. R.; Ríos, R. Chem.–Asian J. 2011, 6, 720–734. doi:10.1002/asia.201000636 |
| 5. | El-Mekabaty, A. Int. J. Mod. Org. Chem. 2013, 2, 40–66. |
| 25. |
Martín-Rodríguez, M.; Nájera, C.; Sansano, J. M.; Wu, F.-L. Tetrahedron: Asymmetry 2010, 21, 1184–1186. doi:10.1016/j.tetasy.2010.06.011
And see corrigendum Tetrahedron: Asymmetry 2010, 21, 2559–2560. |
| 26. | Martín-Rodríguez, M.; Nájera, C.; Sansano, J. M.; de Cózar, A.; Cossío, F. P. Chem.–Eur. J. 2011, 17, 14224–14233. doi:10.1002/chem.201101606 |
| 27. | Martín-Rodríguez, M.; Nájera, C.; Sansano, J. M.; de Cózar, A.; Cossío, F. P. Beilstein J. Org. Chem. 2011, 7, 988–996. doi:10.3762/bjoc.7.111 |
| 18. | Melhado, A. D.; Luparia, M.; Toste, F. D. J. Am. Chem. Soc. 2007, 129, 12638–12639. doi:10.1021/ja074824t |
| 2. | Fisk, J. S.; Mosey, R. A.; Tepe, J. J. Chem. Soc. Rev. 2007, 36, 1432–1440. doi:10.1039/b511113g |
| 3. | Hewlett, N. M.; Hupp, C. D.; Tepe, J. J. Synthesis 2009, 2825–2839. doi:10.1055/s-0029-1216924 |
| 4. | Alba, A.-N. R.; Ríos, R. Chem.–Asian J. 2011, 6, 720–734. doi:10.1002/asia.201000636 |
| 5. | El-Mekabaty, A. Int. J. Mod. Org. Chem. 2013, 2, 40–66. |
| 18. | Melhado, A. D.; Luparia, M.; Toste, F. D. J. Am. Chem. Soc. 2007, 129, 12638–12639. doi:10.1021/ja074824t |
| 19. | Melhado, A. D.; Amarante, G. W.; Wang, Z. J.; Luparia, M.; Toste, F. D. J. Am. Chem. Soc. 2011, 133, 3517–3527. doi:10.1021/ja1095045 |
| 19. | Melhado, A. D.; Amarante, G. W.; Wang, Z. J.; Luparia, M.; Toste, F. D. J. Am. Chem. Soc. 2011, 133, 3517–3527. doi:10.1021/ja1095045 |
| 18. | Melhado, A. D.; Luparia, M.; Toste, F. D. J. Am. Chem. Soc. 2007, 129, 12638–12639. doi:10.1021/ja074824t |
| 19. | Melhado, A. D.; Amarante, G. W.; Wang, Z. J.; Luparia, M.; Toste, F. D. J. Am. Chem. Soc. 2011, 133, 3517–3527. doi:10.1021/ja1095045 |
| 23. | Bandini, M.; Bottoni, A.; Chiarucci, M.; Cera, G.; Miscione, G. P. J. Am. Chem. Soc. 2012, 134, 20690–20700. doi:10.1021/ja3086774 |
| 24. |
Brazeau, J.-F.; Zhang, S.; Colomer, I.; Corkey, B. K.; Toste, F. D. J. Am. Chem. Soc. 2012, 134, 2742–2749. doi:10.1021/ja210388g
And references cited therein. |
| 44. |
Kim, Y.; Kim, J.; Park, S. B. Org. Lett. 2009, 11, 17–20. doi:10.1021/ol8022193
See for a regioselective synthesis of tetrasubstituted pyrroles by 1,3-DC from azlactones and spontaneous decarboxylation. |
| 11. |
Weber, M.; Frey, W.; Peters, R. Adv. Synth. Catal. 2012, 354, 1443–1449. doi:10.1002/adsc.201200085
See for alkylations at multiple positions. |
| 12. |
Trost, B. M.; Czabaniuk, L. C. J. Am. Chem. Soc. 2012, 134, 5778–5781. doi:10.1021/ja301461p
See for alkylations at multiple positions. |
| 13. |
Dell’Amico, L.; Albrecht, L.; Naicker, T.; Poulsen, P. H.; Jørgensen, K. A. J. Am. Chem. Soc. 2013, 135, 8063–8070. doi:10.1021/ja4029928
See for alkylations at multiple positions. |
| 14. |
Breman, A. C.; Smits, J. M. M.; de Gelder, R.; van Maarseveen, J. H.; Ingemann, S.; Hiemstra, H. Synlett 2012, 23, 2195–2200. doi:10.1055/s-0032-1317081
See for participation of α-alkylidene azlactones in 1,3-DC as dipolarophiles. |
| 15. |
González-Esguevillas, M.; Adrio, J.; Carretero, J. C. Chem. Commun. 2013, 49, 4649–4651. doi:10.1039/c3cc41663a
See for participation of α-alkylidene azlactones in 1,3-DC as dipolarophiles. |
| 16. |
Ho, H. T.; Levere, M. E.; Fournier, D.; Montembault, V.; Pascual, S.; Fontaine, L. Aust. J. Chem. 2012, 65, 970–977. doi:10.1071/CH12192
See for the synthesis of new polymers. |
| 17. |
Horlacher, O. P.; Hartkoorn, R. C.; Cole, S. T.; Altmann, K.-H. ACS Med. Chem. Lett. 2013, 4, 264–268. doi:10.1021/ml300385q
See for the preparation of natural products. |
| 25. |
Martín-Rodríguez, M.; Nájera, C.; Sansano, J. M.; Wu, F.-L. Tetrahedron: Asymmetry 2010, 21, 1184–1186. doi:10.1016/j.tetasy.2010.06.011
And see corrigendum Tetrahedron: Asymmetry 2010, 21, 2559–2560. |
| 26. | Martín-Rodríguez, M.; Nájera, C.; Sansano, J. M.; de Cózar, A.; Cossío, F. P. Chem.–Eur. J. 2011, 17, 14224–14233. doi:10.1002/chem.201101606 |
| 27. | Martín-Rodríguez, M.; Nájera, C.; Sansano, J. M.; de Cózar, A.; Cossío, F. P. Beilstein J. Org. Chem. 2011, 7, 988–996. doi:10.3762/bjoc.7.111 |
| 45. |
Ortiz, A. L.; Echegoyen, L.; Delgado, J. L.; Martin, N. Handb. Carbon Nano Mater. 2011, 1, 325–373.
See for retro 1,3-DC, that has also been observed in many thermal processes. |
| 10. | Terada, M.; Tanaka, H.; Sorimachi, K. J. Am. Chem. Soc. 2009, 131, 3430–3431. doi:10.1021/ja8090643 |
| 42. | Bader, R. F. W. Atoms in Molecules-A Quantum Theory; Clarendon Press: Oxford, 1900. |
| 9. | Zhang, W.-Q.; Cheng, L.-F.; Yu, J.; Gong, L.-Z. Angew. Chem., Int. Ed. 2012, 51, 4085–4088. doi:10.1002/anie.201107741 |
| 20. | Hasmi, A. S. K.; Toste, F. D., Eds. Modern Gold Catalyzed Synthesis; Wiley-VCH: Weinheim, Germany, 2012. doi:10.1002/9783527646869 |
| 21. | Rudolph, M.; Hashmi, A. S. K. Chem. Soc. Rev. 2012, 41, 2448–2462. doi:10.1039/c1cs15279c |
| 22. | Peixoto de Almeida, M.; Carabineiro, S. A. C. ChemCatChem 2012, 4, 18–29. doi:10.1002/cctc.201100288 |
| 43. |
Lee, M.; Lee, Y.-J.; Park, E.; Park, Y.; Ha, M. W.; Hong, S.; Lee, Y.-J.; Kim, T.-S.; Kim, M.; Park, H. Org. Biomol. Chem. 2013, 11, 2039–2046. doi:10.1039/c3ob27089k
See for an example. |
| 18. | Melhado, A. D.; Luparia, M.; Toste, F. D. J. Am. Chem. Soc. 2007, 129, 12638–12639. doi:10.1021/ja074824t |
| 19. | Melhado, A. D.; Amarante, G. W.; Wang, Z. J.; Luparia, M.; Toste, F. D. J. Am. Chem. Soc. 2011, 133, 3517–3527. doi:10.1021/ja1095045 |
| 29. | Wheaton, C. A.; Jennings, M. C.; Puddephatt, R. J. J. Am. Chem. Soc. 2006, 128, 15370–15371. doi:10.1021/ja066450u |
| 30. | Wheaton, C. A.; Puddephatt, R. J. Angew. Chem., Int. Ed. 2007, 46, 4461–4463. doi:10.1002/anie.200701325 |
| 31. | Wheaton, C. A.; Jennings, M. C.; Puddephatt, R. J. Z. Naturforsch. 2009, 64b, 1569–1577. |
| 18. | Melhado, A. D.; Luparia, M.; Toste, F. D. J. Am. Chem. Soc. 2007, 129, 12638–12639. doi:10.1021/ja074824t |
| 19. | Melhado, A. D.; Amarante, G. W.; Wang, Z. J.; Luparia, M.; Toste, F. D. J. Am. Chem. Soc. 2011, 133, 3517–3527. doi:10.1021/ja1095045 |
| 36. |
Terada, M.; Nii, H. Chem.–Eur. J. 2011, 17, 1760–1763. doi:10.1002/chem.201003015
The published [4 + 2] cycloaddition of azlactones to β,γ-unsaturated α-ketoesters was unsuccessfully attempted. Only a low yield of O-alkylation addition product to the ketone group was detected. |
| 37. | In ref. [18] and [19] the same result was obtained, but no explanation to this anomalous addition was given. |
| 26. | Martín-Rodríguez, M.; Nájera, C.; Sansano, J. M.; de Cózar, A.; Cossío, F. P. Chem.–Eur. J. 2011, 17, 14224–14233. doi:10.1002/chem.201101606 |
| 27. | Martín-Rodríguez, M.; Nájera, C.; Sansano, J. M.; de Cózar, A.; Cossío, F. P. Beilstein J. Org. Chem. 2011, 7, 988–996. doi:10.3762/bjoc.7.111 |
| 35. | The exo descriptor is referred to the approach of the two reaction components where the tert-butyl ester and carbonyl group of the azlactone are placed in oposite direction. |
| 33. | Satyanarayana, T.; Abraham, S.; Kagan, H. B. Angew. Chem., Int. Ed. 2009, 48, 456–494. doi:10.1002/anie.200705241 |
| 34. | de Cózar, A.; Cossío, F. P. Phys. Chem. Chem. Phys. 2011, 13, 10858–10868. doi:10.1039/c1cp20682f |
| 32. |
Gorin, D. J.; Sherry, B. D.; Toste, F. D. Chem. Rev. 2008, 108, 3351–3378. doi:10.1021/cr068430g
See for a comprehensive study of the ligand effects in homogeneous gold(I) catalysis. |
| 18. | Melhado, A. D.; Luparia, M.; Toste, F. D. J. Am. Chem. Soc. 2007, 129, 12638–12639. doi:10.1021/ja074824t |
| 19. | Melhado, A. D.; Amarante, G. W.; Wang, Z. J.; Luparia, M.; Toste, F. D. J. Am. Chem. Soc. 2011, 133, 3517–3527. doi:10.1021/ja1095045 |
© 2013 Martín-Rodríguez et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)