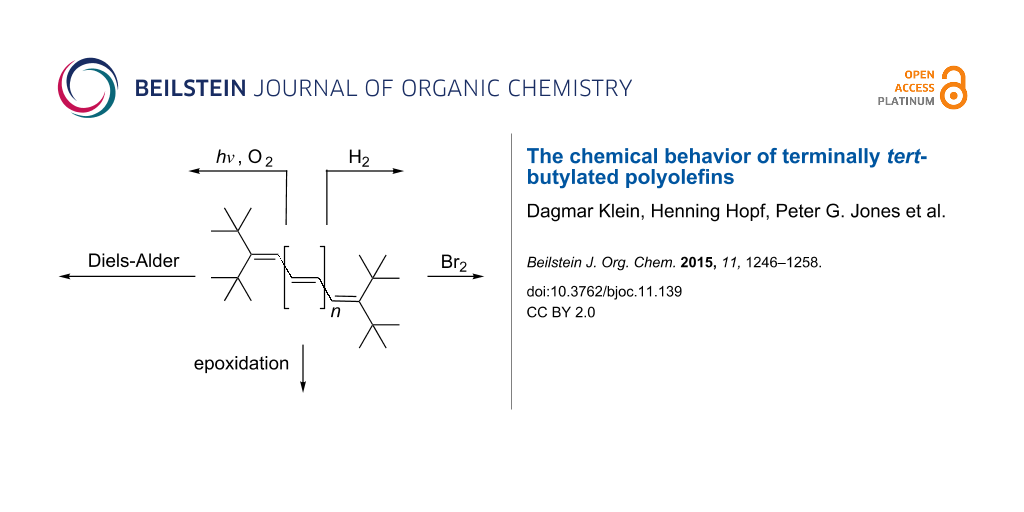
Abstract
The chemical behavior of various oligoenes 2 has been studied. The catalytic hydrogenation of diene 3 yielded monoene 4. Triene 7 was hydrogenated to diene 8, monoene 9 and saturated hydrocarbon 10. Bromine addition to 3 and 7 yielded the dibromides 17 and 18, respectively, i.e., the oligoene system has been attacked at its terminal olefinic carbon atoms. Analogously, the higher vinylogs 19 and 20 yielded the 1,8- and 1,10-bromine adduts 23 and 24, respectively, when less than 1 equivalent of bromine was employed. Treatment of tetraene 19 with excess bromine provided tetrabromide 25. In epoxidation reactions, both with meta-chloroperbenzoic acid (MCPBA) and dimethyldioxirane (DMDO) two model oligoenes were studied: triene 7 and tetraene 19. Whereas 7 furnished the rearrangement product 31 with MCPBA, it yielded the symmetrical epoxide 32 with DMDO. Analogously, 19 was converted to mono-epoxide 33 with MCPBA and to 34 with DMDO. Diels–Alder addition of 7 with N-phenyltriazolinedione (PTAD) did not take place. Extension of the conjugated π-system to the next higher vinylog, 19, caused NPTD-addition to the symmetrical adduct 37 in good yield. Comparable results were observed on adding NPTD (equivalent amount) to pentaene 20 and hexaene 21. Using 36 in excess provided the 2:1-adduct 40 from 21 and led to a complex mixture of adducts from heptaene 22. With tetracyanoethylene (TCNE) as the dienophile, tetraolefin 19 yielded the symmetrical adduct 43, although the reaction temperature had to be increased. Pentaene 20 and hexaene 21 led to corresponding results, adducts 44 and 45 being produced in acceptable yields. With nonaene 42 and TCNE the 2:1-adduct 48 was generated according to its spectroscopic data. Exploratory photochemical studies were carried out with tetraene 19 as the model compound. On irradiation this reacted with oxygen to the stable endo-peroxide 52.

Graphical Abstract
Introduction
Several years ago we described [2] a general synthesis of a series of terminally substituted, conjugated polyenes, 2, beginning with the diene and ending with the decatriene (Scheme 1). We also reported the X-ray structures of compounds with n = 1, 2, 3, 4, 5 and 7 and discussed the structural similarities, in particular the distortions associated with the bulky tert-butyl groups.
![[1860-5397-11-139-i1]](/bjoc/content/inline/1860-5397-11-139-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: The polyenes 2 stabilized by terminal tert-butyl substituents.
Scheme 1: The polyenes 2 stabilized by terminal tert-butyl substituents.
For all these hydrocarbons, di-tert-butylketone (1) served as the starting material, which was chain-elongated by preparative sequences involving standard Wittig, Wittig–Horner and McMurry coupling reactions. Nearly all of the polyenes 2 (as well as many of their precursors) were characterized by X-ray structural analysis. These polyolefins are substructures of the most famous polyene, “polyacetylene”.
Having prepared and unambiguously characterized the hydro-carbons 2, we now turn to their chemical behavior. In the sense that these polyolefins can serve as structural models for “polyacetylene”, their chemical properties should also reflect that of the polymer. Chemical reactions of the higher acyclic polyolefins have scarcely been studied, and we hence decided to carry out typical, textbook olefin reactions of a representative selection of the hydrocarbons 2.
Results and Discussion
Catalytic hydrogenation
We started our studies on the reactive behavior of polyolefins 2 with one of the formally simplest alkene reactions: catalytic hydrogenation.
When diene 3 was hydrogenated under relatively mild conditions (Pd/C, EtOH, room temp.), the only product that we could isolate was mono-olefin 4 (Scheme 2), a formal 1,4-addition product of hydrogen to 3.
![[1860-5397-11-139-i2]](/bjoc/content/inline/1860-5397-11-139-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: The catalytic hydrogenation of diene 3.
Scheme 2: The catalytic hydrogenation of diene 3.
Its structure was established by the usual spectroscopic and analytical data (see Supporting Information File 1). To determine the configuration of the double bond, an X-ray structural investigation was carried out. Although the quality of the structure determination was disappointing because of high residual electron density, it sufficed to determine the connectivity of the atoms in the compound and to demonstrate that the double bond configuration was E (Figure 1). The molecule possesses no crystallographic symmetry, but displays twofold symmetry to a good approximation (rms deviation 0.015 Å). The torsion angles across the central C1…C4 moiety are C5–C1…C4–C7 53.4° and C6–C1…C4–C8 −14.6°. Distortions from "normal" dimensions may be attributed to the steric effects of the tert-butyl groups; thus the C–CMe3 bonds are long (1.58 Å), the sp3 angles Me3C–C–CMe3 are wide (120°), and the sp2 C–C=C angles in the central chain are also wide (127°). For individual values, the deposited material should be consulted. The molecules pack end-to-end to form chains parallel to the b axis; neighboring chains extend the packing, again by translation, to layers parallel to the bc plane at z ≈ ¼ and z ≈ ¾.
![[1860-5397-11-139-1]](/bjoc/content/figures/1860-5397-11-139-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: The structure of compound 4 in the crystal. Ellipsoids correspond to 30% probability levels.
Figure 1: The structure of compound 4 in the crystal. Ellipsoids correspond to 30% probability levels.
The hydrogenation mixture yielded no evidence for the formation of a 1,2-adduct (5) or of the fully saturated hydrocarbon 6. In fact, the primary adduct 4 was inert towards hydrogen even under very harsh hydrogenation conditions (Pt, 100 °C, acetic acid). We assume that the spatial shielding of the double bond, as indicated by the X-ray structure, is responsible for this. Furthermore, we were also unable to epoxidize or brominate 4.
The next higher vinylog of 3, the triene 7, is much more easily hydrogenated. Even under mild conditions (Pd/C, EtOH/hexane, room temp.) it is readily reduced (Scheme 3).
![[1860-5397-11-139-i3]](/bjoc/content/inline/1860-5397-11-139-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: The catalytic hydrogenation of triene 7.
Scheme 3: The catalytic hydrogenation of triene 7.
After 5 minutes of hydrogenation, three products were detected: the 1,6-adduct 8, a diene (main product); the mono olefin 9; and the fully saturated hydrocarbon 10. When the reaction time was increased, the amounts of 9 and 10 grew at the cost of 8, and when the hydrogenation was run for two days, alkane 10 was the only reduction product. It seems safe to assume that we are dealing with a stepwise process, with 8 and 9 serving as intermediates en route to 10. Comparing the two experiments starting from 3 and 7, it is obvious that two vicinal (t-Bu)2CH-substituents are required to shield a double bond from hydrogenation. The spectroscopic data of 8–10 can be found in Supporting Information File 1. Since we did not expect to obtain fundamentally new results with the oligoenes beyond 7, we stopped the hydrogenation experiments at this stage.
Our hydrogenation results with α,ω-fully tert-butylated oligoenes are similar to the studies of Kuhn and Winterstein with several α,ω-diphenylpolyenes [3]. These authors hydrogenated their aromatic derivatives under different condi-tions; results comparable to ours were obtained with aluminum amalgam in moist ether. The primary product with the hexatriene, the octatetraene and the decapentaene derivatives were always the 1,ω-adducts. Although more highly reduced derivatives – similar to, e.g., 9 – were not obtained, exhaustive hydrogenation of the 1,8-hydrogenated products always provided the α,ω-diphenylalkanes.
Brominations
The behavior of dienes towards bromine has been studied extensively and, in particular, the addition of bromine to buta-1,3-diene is discussed in most textbooks on organic chemistry [4]. Elementary bromine adds to buta-1,3-diene in halogenated solvents to give mostly the 1,4-addition product (ratio 1,4-/1,2-product: 7:1) [5]. Turning to substituted dienes, 2,3-dimethylbuta-1,3-diene (11) initially also provides the 1,4-adduct 12, which subsequently is saturated to the tetrabromide 13 by reaction with a second equivalent of bromine (Scheme 4) [6-8].
![[1860-5397-11-139-i4]](/bjoc/content/inline/1860-5397-11-139-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Addition of bromine to model dienes.
Scheme 4: Addition of bromine to model dienes.
Whereas an isomer of 11, hexa-2,5-diene, behaves similarly to 11 [9], the terminally fully methylated diene 14 does not react further than 15 on bromine addition, no tetrabromide 16 being formed [10].
An increase in the steric bulk of the (terminal) substituents should lead to comparable results; and this is indeed the case (Scheme 5), as shown by diene 3.
![[1860-5397-11-139-i5]](/bjoc/content/inline/1860-5397-11-139-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Bromine addition to diene 3 and triene 7.
Scheme 5: Bromine addition to diene 3 and triene 7.
The 1,4-addition product 17, a colorless solid, is produced in near quantitative yield (93%). It is easily characterized by its spectroscopic data, which are listed in Supporting Information File 1. Unfortunately, we were unable to prepare crystals of 17 suitable for an X-ray structural analysis.
Interestingly, Kuhn and Winterstein in their study on the chemical behavior of α,ω-diphenyloligoenes obtained different results: for 1,4-diphenylbuta-1,3-diene, bromine addition furnished the 1,2-adduct in 95% yield [3]. As in the case of the hydrogenation of 3, 1,4-adduct 17 is inert to further bromine addition; the steric bulk of the substituents at the remaining double bond has increased even further.
When excess bromine is added to triene 7, we also obtained only the 1,6-addition product 18. This is inferred from the NMR spectra of the raw product mixture. The 13C NMR spectrum only displays 5 signals (see Supporting Information File 1), half the number of the signals expected for a less symmetrical 1,6-adduct than the one shown in Scheme 5. Again, no further adducts were detected, a fact which we ascribe to the “protection” of the double bonds by the (t-Bu)2CBr-substituents. Unfortunately, 18 is unstable. Even measurement of its NMR spectra is accompanied by decomposition: the color of the CDCl3 solution rapidly changed to brown, and then black. Again, this result is in marked contrast to Kuhn´s experiment with the 1,6-diphenylhexatriene. In this latter case tetra- and even hexabromo adducts were also obtained [3].
Proceeding to the next higher vinylogs, 19 and 20, an analogous reactivity pattern to that above is observed when less than an equivalent of bromine is employed. However, in the case of 19 only a product mixture containing the 1,8-adduct 23 as the minor component (19:23 2.5:1) was obtained, from which the (unstable) 23 could not be isolated in pure form (Scheme 6). In the case of 20 we were more successful: the dibromide 24 was not only obtained in analytically pure form, but also as colorless needles suitable for an X-ray investigation. Whereas the spectroscopic data of 23 and 24 are given in Supporting Information File 1, the structure of 24 in the solid state is discussed here. The molecule (Figure 2a) possesses exact inversion symmetry, but its non-crystallographic symmetry is close to 2/m (C2h), with an rms deviation of 0.21 Å. Distortions associated with the tert-butyl groups are largely similar to those discussed above for 4, but the angle C1–C2=C3 is wider still at 131° (other chain angles are 122–125°). The bromine atom is approximately synperiplanar to C3 across the C1–C2 bond (torsion angle 12.2°). The molecular packing (Figure 2b) involves herringbone-type layers perpendicular to (![[Graphic 1]](/bjoc/content/inline/1860-5397-11-139-i14.svg?max-width=637&scale=1.18182) ). The shortest H…Br contacts of 3.05 Å, probably of only marginal significance, are formed between layers.
). The shortest H…Br contacts of 3.05 Å, probably of only marginal significance, are formed between layers.
![[1860-5397-11-139-i6]](/bjoc/content/inline/1860-5397-11-139-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Bromine addition to the higher oligoenes 19–22.
Scheme 6: Bromine addition to the higher oligoenes 19–22.
![[1860-5397-11-139-2]](/bjoc/content/figures/1860-5397-11-139-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2:
(a) The structure of compound 24 in the crystal. Ellipsoids correspond to 50% probability levels. (b) Packing diagram of compound 24 viewed perpendicular to (![[Graphic 2]](/bjoc/content/inline/1860-5397-11-139-i15.svg?max-width=637&scale=1.18182) ). Hydrogen atoms are omitted.
). Hydrogen atoms are omitted.
Figure 2: (a) The structure of compound 24 in the crystal. Ellipsoids correspond to 50% probability levels. (...
Having unambiguously established that the mono adduct of bromine to pentaene 20 has the structure of a 1,10-addition product with all-E-configuration of the double bonds, i.e., 24, we postulate that the corresponding adducts, 17, 18 and 23, have analogous structures, as shown in Scheme 5 and Scheme 6, respectively.
When the tetraene 19 was treated with a six-fold excess of bromine, a tetrabromide 25 was produced in good yield (73%). The compound could be kept at −10 °C without decomposition for considerable times and was recrystallized from pentane to provide needles "suitable" for X-ray structural analysis; its qualitative structure in the solid state is reproduced in Figure 3, but should be interpreted with great caution because of unsatisfactory refinement. The compound appeared to crystallize in P21/c with a = 10.243, b = 10.467, c = 13.222 Å, β = 96.39° and Z = 2 (at −130 °C). The refinement was unsatisfactory because the carbon atoms could not be refined anisotropically, and several bond lengths and angles were unrealistic. Possible sources of error would include disorder, unidentified weak reflections corresponding to a larger cell (the data were recorded on a serial diffractometer), or an incorrect space group. Refinements in lower symmetry space groups were, however, not better. We believe that the chemical nature of compound 25 has nevertheless been qualitatively confirmed.
![[1860-5397-11-139-3]](/bjoc/content/figures/1860-5397-11-139-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: The structure of compound 25 in the crystal. This was a structure of poor quality and served only to determine the connectivity.
Figure 3: The structure of compound 25 in the crystal. This was a structure of poor quality and served only t...
The spectroscopic data of 25 (see Supporting Information File 1) are consistent with its solid state structure. There are several plausible mechanisms that would explain the formation of 25. This bis-bromine adduct could be formed from 19 either by two consecutive 1,4-bromine addition processes or by a 1,8-addition followed by trans-bromination of its central double bond. This bond should be favored for steric reasons over the two terminal double bonds of the triene intermediate 23. Since this dibromide was not available in pure form, we did not carry out the control experiment required to distinguish experimentally between the first and the second path.
The higher oligoenes 20, 21, and 22 react similarly to 19 with excess bromine and provide the corresponding polybromides 26–28 in the yields shown in Scheme 6. Since we were not able to obtain X-ray data for these adducts, their exact stereostructures must be left open for the time being. It is quite clear, though, that the terminal, highly substituted quaternary carbon atoms protect their neighboring double bond from further attack. The attack by “positive” bromine at the di-tert-butylated carbon atoms in the first step of the addition process is probably associated with the better stabilization by resonance of the cationic intermediate thus generated compared to the alternative positively charged intermediate produced by initial bromine attack at the penultimate carbon atom. In the former case the positive charge can shift to the other end of the polyolefin system, where it is stabilized by the combined hyperconjugative effect of two tert-butyl groups. It is this other terminal carbon atom that is attacked by the bromide ion in the termination step, resulting in the overall formation of 1,x-bromine adducts.
Epoxidations
Epoxidation reactions of highly substituted dienes have been carried out by us and by others, and it has been demonstrated that whenever competing reaction pathways exist, it is usually the more highly substituted double bond that is preferentially attacked, regardless of the steric bulk of the substituents [11]. As far as the higher vinylogs are concerned, we decided to study an odd oligoene, the triene 7 (Scheme 7), and an even representative, the tetraene 19 (Scheme 8).
![[1860-5397-11-139-i7]](/bjoc/content/inline/1860-5397-11-139-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Epoxidation of triene 7 with MCPBA and DMDO.
Scheme 7: Epoxidation of triene 7 with MCPBA and DMDO.
In the first epoxidation experiment, 7 was treated with m-chloroperbenzoic acid (MCPBA) in chloroform at room temperature overnight. Surprisingly, it was neither the mono- epoxide 29 nor its non-terminal isomer 32 but the ketone 31 that was obtained in low yield (ca. 25%). The structure follows from the spectroscopic data (see Supporting Information File 1); the carbonyl group is readily seen in the IR spectrum (νmax = 1702 cm−1) and the carbonyl carbon signal in the 13C NMR spectrum at δ = 217 ppm is also of particular diagnostic value. We propose that 31 is produced from epoxide 29 by initial protonation to the oxonium ion 30, which then undergoes a Wagner–Meerwein rearrangement followed by deprotonation. Under non-acidic conditions, this process would not be expected; and indeed, when 7 was oxidized with dimethyldioxirane (DMDO) in acetone at room temperature, epoxide 32 with a central oxirane ring is produced in acceptable yield (59%). Since we were unable to obtain single crystals of this derivative, the assignment of its exact stereostructure (syn- or anti-epoxide) must remain tentative. Since many other epoxidations take place with retention of the original double bond configuration, we assume that the anti-configuration is more probable in the present case as well. The spectroscopic data (see Supporting Information File 1) also support the structure shown in Scheme 7. Of particular value is the 13C NMR spectrum, which shows a halved set of signals, as expected for a symmetrical structure. The completely substituted olefinic carbon atoms display a signal at δ = 161 ppm, whereas the –CH= carbon atoms absorb at 122 ppm. The oxirane carbon atoms appear at 59 ppm.
When the double bond chain is extended by one –CH=CH– group, results were obtained comparable to those observed for 7 (Scheme 8). Thus, tetraene 19 yielded mono-epoxide 33 with MCPBA in chloroform in fair yield (36%). With DMDO in sub-stoichiometric amounts, mono-epoxide 34 was produced in acceptable yield (49%). Employing excess DMDO yields a complex product mixture in which we could identify 34 as well as more highly epoxidized products (by mass spectrometric analysis) to which we tentatively assign structures 35. Our attempts to obtain pure products by chromatographic separation have in this latter case so far failed.
![[1860-5397-11-139-i8]](/bjoc/content/inline/1860-5397-11-139-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Epoxidation of tetraene 19 with MCPBA and DMDO.
Scheme 8: Epoxidation of tetraene 19 with MCPBA and DMDO.
To summarize the epoxidation experiments, it appears that MCPBA prefers to attack the terminal double bonds of our sterically shielded oligoenes, whereas there are indications that DMDO oxidizes “inner” double bonds preferentially. In principle, heterocycles with larger rings could also be produced in these experiments; however, at present we have no experimental evidence for these alternative routes.
Diels–Alder reactions
Since we expected steric hindrance effects to play a pronounced role in the Diels–Alder additions of the shielded oligoene 3, we decided to begin our experiments with one of the most reactive dienophiles, N-phenyltriazolinedione (36, PTAD, Scheme 9).
![[1860-5397-11-139-i9]](/bjoc/content/inline/1860-5397-11-139-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Diels–Alder addition of PTAD (36) to triene 7 and tetraene 19.
Scheme 9: Diels–Alder addition of PTAD (36) to triene 7 and tetraene 19.
No reaction took place between triene 7 and 36, even if the reaction mixture was heated to reflux. Clearly, the steric influence of the four bulky substituents is too great, and it either prevents the population of a cisoid conformation of diene 7 or it causes too much steric hindrance between the two cycloaddition partners en route to the transition state. This situation changes when the polyene chain is elongated by one double bond: tetraene 19 and dienophile 36 provide cycloadduct 37 in good yield (75%) under mild reaction conditions. Note that only this symmetrical regioisomer is produced – the alternative involving a terminal diene unit is not observed. The spectroscopic data (see Supporting Information File 1) already hinted that the cycloadduct 37 had been produced; the structure assignment was confirmed by X-ray structural analysis.
The structure of 37 involves two independent molecules, one of which is shown in Figure 4. The molecules are closely similar, with an rms deviation of only 0.09 Å for all non-H atoms. The angles at C2 and C7 are greatly widened (to 132–135°). The central six-membered ring displays a 1,2-diplanar ("sofa") conformation, whereby the atom N1 lies 0.5 Å out of the plane of the other five atoms.
![[1860-5397-11-139-4]](/bjoc/content/figures/1860-5397-11-139-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: The structure of compound 37 in the crystal. Only one of two independent molecules is shown. Ellipsoids correspond to 30% probability levels.
Figure 4: The structure of compound 37 in the crystal. Only one of two independent molecules is shown. Ellips...
The next two higher vinylogs, pentaene 20 and hexaene 21, reacted similarly with PTAD (Scheme 10).
![[1860-5397-11-139-i10]](/bjoc/content/inline/1860-5397-11-139-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Diels-Alder addition of oligoenes 20 and 21 with PTAD (36).
Scheme 10: Diels-Alder addition of oligoenes 20 and 21 with PTAD (36).
In neither case are the terminal double bonds involved in the cycloaddition process. In the case of pentaene 20, the unsymmetrical adduct 38 was isolated in 66% yield, and with hexaene 21 the symmetrical adduct 39 was obtained. The lower yield (24%) in this case is misleading, since we also re-isolated 50% of the starting oligoolefin 21 in this experiment with just one equivalent of PTAD. The course of the reaction in the latter case is more difficult to follow. Whereas 37 and 38 are colorless, cycloadduct 39 is yellow. In the former cases the reactions can be followed visually analogous to a titration, with the intensely colored PTAD serving as the indicator. In case of the transformation 21→38, the color changes from red to yellow and is hence less readily monitored.
When an excess of 36 is employed in the Diels–Alder reaction, the overall picture changes (Scheme 11).
![[1860-5397-11-139-i11]](/bjoc/content/inline/1860-5397-11-139-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 11: Addition of excess PTAD (36) to hexaene 21 and heptaene 22.
Scheme 11: Addition of excess PTAD (36) to hexaene 21 and heptaene 22.
With 21 (and extended reaction times) the 2:1-adduct 40 is produced in acceptable yield (47%) as the main product. In other words, both penultimate double bonds have also participated in the addition process (again the terminal double bonds remain untouched). Very probably this process also occurs stereospecifically, but since we could not obtain single crystals of X-ray quality in this case we refrain from stereochemical assignments (note also that the added heterocycles could in principle be in syn- or anti-orientation).
Chemically and stereochemically the situation becomes even more intricate on further extension of the polyene chain. When heptaene 22 is treated with an equimolar amount of 36, a complex mixture is obtained consisting of starting material, mono-adducts (25%, MS analysis) and bis-adducts (12%). Chromatographic separation of the products turned out to be impossible, but we believe that compound 41 shown in Scheme 11 is among them; as in all other experiments the terminal double bonds remained untouched.
Tetracyanoethylene (TCNE) is usually less reactive as a dienophile than PTAD; this is also the case when the above mentioned oligoenes are employed as the diene components (Scheme 12). As expected, triene 7 did not react with TCNE, neither at room temperature nor at elevated temperatures. Tetraene 19 gives the expected (see Supporting Information File 1 for experimental details) symmetrical adduct 43, but only under reflux conditions (THF, 65 °C). In addition to the spectroscopic data, the solid state structure of 43 was determined by crystal structure analysis.
![[1860-5397-11-139-i12]](/bjoc/content/inline/1860-5397-11-139-i12.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 12: TCNE addition to oligoolefins: from tetraene 19 to nonaene 42.
Scheme 12: TCNE addition to oligoolefins: from tetraene 19 to nonaene 42.
The structure of this adduct is in many ways similar to that of the related PTAD-adduct 37. The ring conformation is again a "sofa", whereby C26 lies 0.7 Å out of the plane of the other five atoms (Figure 5). The angles at C2 and C7 are again wide at 131–132°, although not quite as wide as for 37. By chance, there are again two independent molecules in the asymmetric unit that are closely similar (the rms deviation for all non-H atoms except methyl C is 0.08 Å).
![[1860-5397-11-139-5]](/bjoc/content/figures/1860-5397-11-139-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: The structure of compound 43 in the crystal. Only one of two independent molecules is shown. Ellipsoids correspond to 30% probability levels.
Figure 5: The structure of compound 43 in the crystal. Only one of two independent molecules is shown. Ellips...
As expected, from pentaene 20 the Diels–Alder adduct 44 is produced in 77% yield, and from hexaene 21 the mono-adduct 45 (49%). Finally, with heptaene 22 and nonaene 42 the adducts 46 and 47 were obtained in varying yields. In the latter case some decomposition of the product was noted during work-up and we isolated a TCNE bis-adduct, 48, for the first time in this series (Scheme 12). Again, we do not attempt to describe the exact stereochemical outcome of the addition, since the experimental evidence is too meagre.
Exploratory cycloaddition experiments were carried out with dimethyl acetylenedicarboxylate and maleic anhydride, but none of these dienophiles reacted with, e.g., heptaene 22 up to 75 °C.
In summary, the tert-butyl protected oligoenes participate in Diels−Alder reactions, as diene components with up to nine consecutive double bonds, with very reactive dienophiles (PTAD, TCNE). Only in the most extended cases are 2:1 adducts produced, and in all cases the terminal double bonds survive the cycloaddition process.
Photochemical behavior
Although oligo- and polyene substructures are present as chromophores in, inter alia, the visual pigments [12], the carotenoid antennae of photosynthesis [13,14], and vitamins A [15] and D [16], relatively little is known about their basic photochemical reactions, such as photoisomerizations and/or photoadditions. For the unsubstituted hydrocarbons, this is not surprising in view of their general instability and the difficulty of obtaining pure diastereomers.
With our stabilized oligoenes in hand, we started an exploratory study to investigate typical photoreactions of unsaturated systems. As illustrated in Scheme 13, we selected tetraene 19 as a model compound, since the previous investigations had shown (see above) that in many cases four consecutive double bonds are required to observe chemical transformations.
![[1860-5397-11-139-i13]](/bjoc/content/inline/1860-5397-11-139-i13.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 13: Photochemical experiments with tetraene 19.
Scheme 13: Photochemical experiments with tetraene 19.
The first two processes involved photoisomerizations. It is well known that E/Z-isomerizations take place readily in oligoenes, even when daylight is used for photoexcitation. However, rather than isomerizing to 49, a mixture of diastereomers, 19 remained unchanged when irradiated with a daylight lamp in deuteriochloroform. The only process that we could observe was a “polymerization” reaction, which slowly destroyed the substrate. Likewise, no photocyclization to the divinylcyclobutene derivate 51 was noted. [2 + 2] Photodimerizations of olefins have often been described, whether in solution or in the solid state. For 19, this reaction, which would lead to the photodimer 50 – or any other cycloadduct – was not observed. It is known from the solid state structures of the tert-butylated oligoenes that the distance between two polyolefin chains is markedly larger than the intermolecular distance between two double bonds that successfully undergo a [2 + 2] cycloaddition (between 3.5 and 4 Å, the so-called “topochemical reaction control”). Furthermore, in derivatives 2 two adjacent polyolefin chains are orthogonal to each other, because of the steric bulk of the tert-butyl moieties. The p-orbitals of the double bonds are hence prevented from overlap [1,2].
So far we have only been successful in observing a single photochemical reaction between 19 and an “external” reagent: the photoaddition of oxygen to its unhindered, inner diene unit. After 20 h irradiation with a daylight lamp in deuteriochloroform solution in the presence of air, endo-peroxide 52 was isolated in 46% yield. Its structure follows from its spectroscopic data (see Supporting Information File 1) and, in particular, an X-ray structural investigation of single crystals obtained from a petrol ether solution.
The molecule of 52 is shown in Figure 6. The O–O bond length is 1.4755(12) Å, which corresponds well to the mean value of 1.480 Å obtained from the Cambridge Crystallographic Database [17] for similar ring systems (69 hits, 81 values; one severe outlier omitted). The six-membered ring has an approximate “sofa” conformation, whereby O1 lies 0.75 Å out of the plane of the other five atoms. The angles at C2 and C7 are 134, 132°. Despite the steric shielding provided by the tert-butyl groups, the molecules associate in pairs via a weak hydrogen bond H3…O1, 2.57 Å.
![[1860-5397-11-139-6]](/bjoc/content/figures/1860-5397-11-139-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: The structure of compound 52 in the crystal. Ellipsoids correspond to 50% probability levels.
Figure 6: The structure of compound 52 in the crystal. Ellipsoids correspond to 50% probability levels.
The photochemical addition of oxygen to numerous diene systems has been investigated by many authors [18-20]. In most cases this photooxidation involves singlet oxygen that is generated from triplet oxygen by irradiation in the presence of a sensitizer such as chlorophyll. Since the above experiment was carried out in the absence of a sensitizer, the formation of 52 must be explained by a different mechanism. One alternative could be the photochemical generation of a diradical from the conjugated oligoene 19 and interception of the former by the oxygen present in the reaction solution. Interestingly, when the solution is degassed before irradiation and the photolysis is carried out under argon, only polymeric material is produced from 19 after extended irradiation (20 h).
Supporting Information
| Supporting Information File 1: Experimental part. | ||
| Format: PDF | Size: 488.3 KB | Download |
References
-
Betz, M.; Hopf, H.; Ernst, L.; Jones, P. G.; Okamoto, Y. Chem. – Eur. J. 2011, 17, 231–247. doi:10.1002/chem.201001250
Return to citation in text: [1] -
Klein, D.; Kiliçkiran, P.; Mlynek, C.; Hopf, H.; Dix, I.; Jones, P. G. Chem. – Eur. J. 2010, 16, 10507–10522. doi:10.1002/chem.201000019
Return to citation in text: [1] [2] -
Kuhn, R.; Winterstein, A. Helv. Chim. Acta 1928, 11, 123–144. doi:10.1002/hlca.19280110109
Return to citation in text: [1] [2] [3] -
Streitwieser, A.; Heathcock, C. H.; Kosower, E. M. Introduction to Organic Chemistry, 4th ed.; McMillan: London, 1992.
Return to citation in text: [1] -
Bellucci, G.; Berti, G.; Bioanchini, R.; Ingrosso, G.; Yates, K. J. Org. Chem. 1981, 46, 2315–2323. doi:10.1021/jo00324a021
Return to citation in text: [1] -
Sweeting, O. J.; Johnson, J. R. J. Am. Chem. Soc. 1946, 68, 1057–1061. doi:10.1021/ja01210a042
Return to citation in text: [1] -
Said, E. Z.; Tipping, A. E. J. Chem. Soc., Perkin Trans. 1 1972, 1399–1403. doi:10.1039/P19720001399
Return to citation in text: [1] -
Collier, W. L.; Macomber, R. S. J. Org. Chem. 1973, 38, 1367–1369. doi:10.1021/jo00947a027
Return to citation in text: [1] -
Heasley, G. E.; Heasley, V. L.; Manatt, S. L.; Day, H. A.; Hodges, R. V.; Kroon, P. A.; Redfield, D. A.; Rold, T. L.; Williamson, D. E. J. Org. Chem. 1973, 38, 4109–4117. doi:10.1021/jo00963a004
Return to citation in text: [1] -
Korte, F.; Scharf, D.; Büchel, K.-H. Justus Liebigs Ann. Chem. 1963, 664, 97–113. doi:10.1002/jlac.19636640107
Return to citation in text: [1] -
Hänel, R. Ph.D. Thesis, Braunschweig, 1995.
unpublished work.
Return to citation in text: [1] -
Abrahamson, E. W.; Ostroy, S. E. Prog. Biophys. Mol. Biol. 1967, 7, 179–215. doi:10.1016/0079-6107(67)90007-7
Return to citation in text: [1] -
Cogdell, R. J.; Frank, H. A. Biochim. Biophys. Acta 1987, 895, 63–79. doi:10.1016/S0304-4173(87)80008-3
Return to citation in text: [1] -
Siefermann-Harms, D. Biochim. Biophys. Acta 1985, 811, 325–355. doi:10.1016/0304-4173(85)90006-0
Return to citation in text: [1] -
Chichester, C. O.; Nakayama, T. O. M. The biosynthesis of carotinoids and vitamine A. In Biogenesis of Natural Products; Bernfeld, P., Ed.; Pergamon Press: Oxford, 1963.
Return to citation in text: [1] -
Jacobs, H. J. C.; Havinga, E. Photochemistry of Vitamin D and its Isomers and of Simple Trienes. In Advances in Photochemistry; Pitts, J. N.; Hammond, G. S.; Gollnick, K.; Grosjean, D., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, 1979; Vol. 11, pp 305–374. doi:10.1002/9780470133415.ch4
Return to citation in text: [1] -
Groom, C. R.; Allen, F. H. Angew. Chem., Int. Ed. 2014, 53, 662–671. doi:10.1002/anie.201306438
Return to citation in text: [1] -
Schenck, G. O.; Ziegler, K. Naturwissenschaften 1944, 32, 157. doi:10.1007/BF01467891
Return to citation in text: [1] -
Denny, R. W.; Nickon, A. Org. React. 1973, 20, 133–336. doi:10.1002/0471264180.or020.02
Return to citation in text: [1] -
Frimer, A. A. Chem. Rev. 1979, 79, 359–387. doi:10.1021/cr60321a001
Return to citation in text: [1]
| 18. | Schenck, G. O.; Ziegler, K. Naturwissenschaften 1944, 32, 157. doi:10.1007/BF01467891 |
| 19. | Denny, R. W.; Nickon, A. Org. React. 1973, 20, 133–336. doi:10.1002/0471264180.or020.02 |
| 20. | Frimer, A. A. Chem. Rev. 1979, 79, 359–387. doi:10.1021/cr60321a001 |
| 2. | Klein, D.; Kiliçkiran, P.; Mlynek, C.; Hopf, H.; Dix, I.; Jones, P. G. Chem. – Eur. J. 2010, 16, 10507–10522. doi:10.1002/chem.201000019 |
| 6. | Sweeting, O. J.; Johnson, J. R. J. Am. Chem. Soc. 1946, 68, 1057–1061. doi:10.1021/ja01210a042 |
| 7. | Said, E. Z.; Tipping, A. E. J. Chem. Soc., Perkin Trans. 1 1972, 1399–1403. doi:10.1039/P19720001399 |
| 8. | Collier, W. L.; Macomber, R. S. J. Org. Chem. 1973, 38, 1367–1369. doi:10.1021/jo00947a027 |
| 1. | Betz, M.; Hopf, H.; Ernst, L.; Jones, P. G.; Okamoto, Y. Chem. – Eur. J. 2011, 17, 231–247. doi:10.1002/chem.201001250 |
| 2. | Klein, D.; Kiliçkiran, P.; Mlynek, C.; Hopf, H.; Dix, I.; Jones, P. G. Chem. – Eur. J. 2010, 16, 10507–10522. doi:10.1002/chem.201000019 |
| 5. | Bellucci, G.; Berti, G.; Bioanchini, R.; Ingrosso, G.; Yates, K. J. Org. Chem. 1981, 46, 2315–2323. doi:10.1021/jo00324a021 |
| 17. | Groom, C. R.; Allen, F. H. Angew. Chem., Int. Ed. 2014, 53, 662–671. doi:10.1002/anie.201306438 |
| 4. | Streitwieser, A.; Heathcock, C. H.; Kosower, E. M. Introduction to Organic Chemistry, 4th ed.; McMillan: London, 1992. |
| 15. | Chichester, C. O.; Nakayama, T. O. M. The biosynthesis of carotinoids and vitamine A. In Biogenesis of Natural Products; Bernfeld, P., Ed.; Pergamon Press: Oxford, 1963. |
| 3. | Kuhn, R.; Winterstein, A. Helv. Chim. Acta 1928, 11, 123–144. doi:10.1002/hlca.19280110109 |
| 16. | Jacobs, H. J. C.; Havinga, E. Photochemistry of Vitamin D and its Isomers and of Simple Trienes. In Advances in Photochemistry; Pitts, J. N.; Hammond, G. S.; Gollnick, K.; Grosjean, D., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, 1979; Vol. 11, pp 305–374. doi:10.1002/9780470133415.ch4 |
| 3. | Kuhn, R.; Winterstein, A. Helv. Chim. Acta 1928, 11, 123–144. doi:10.1002/hlca.19280110109 |
| 12. | Abrahamson, E. W.; Ostroy, S. E. Prog. Biophys. Mol. Biol. 1967, 7, 179–215. doi:10.1016/0079-6107(67)90007-7 |
| 3. | Kuhn, R.; Winterstein, A. Helv. Chim. Acta 1928, 11, 123–144. doi:10.1002/hlca.19280110109 |
| 13. | Cogdell, R. J.; Frank, H. A. Biochim. Biophys. Acta 1987, 895, 63–79. doi:10.1016/S0304-4173(87)80008-3 |
| 14. | Siefermann-Harms, D. Biochim. Biophys. Acta 1985, 811, 325–355. doi:10.1016/0304-4173(85)90006-0 |
| 10. | Korte, F.; Scharf, D.; Büchel, K.-H. Justus Liebigs Ann. Chem. 1963, 664, 97–113. doi:10.1002/jlac.19636640107 |
| 9. | Heasley, G. E.; Heasley, V. L.; Manatt, S. L.; Day, H. A.; Hodges, R. V.; Kroon, P. A.; Redfield, D. A.; Rold, T. L.; Williamson, D. E. J. Org. Chem. 1973, 38, 4109–4117. doi:10.1021/jo00963a004 |
© 2015 Klein et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)