Abstract
Triaryl-2-pyridylidene effectively facilitates the gold-catalyzed oxidative C–H arylation of heteroarenes with arylsilanes as a unique electron-donating ligand on gold. The employment of the 2-pyridylidene ligand, which is one of the strongest electron-donating N-heterocyclic carbenes, resulted in the rate acceleration of the C–H arylation reaction of heterocycles over conventional ligands such as triphenylphosphine and a classical N-heterocyclic carbene. In situ observation and isolation of the 2-pyridylidene-gold(III) species, as well as a DFT study, indicated unusual stability of gold(III) species stabilized by strong electron donation from the 2-pyridylidene ligand. Thus, the gold(I)-to-gold(III) oxidation process is thought to be facilitated by the highly electron-donating 2-pyridylidene ligand.
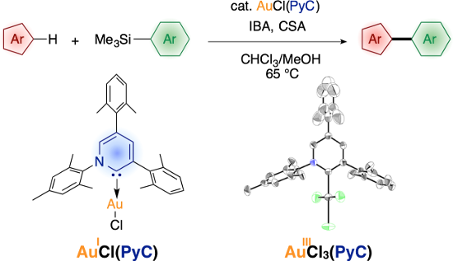
Graphical Abstract
Introduction
Over the past decade, gold salts and complexes have emerged as unique catalysts for the transformation of alkynes, alkenes and allenes [1-30]. In most of the gold-catalyzed reactions, phosphines, N-heterocyclic carbenes, pyridines and salen ligands have been applied as ligands for controlling the stability of catalysts, and chemo-, regio- and enantioselectivities of the reactions [31-36]. Recent advances in the gold-catalyzed reactions are represented by oxidative coupling that is expected to proceed through a gold(I)/gold(III) catalytic cycle [37-81]. In particular, the elegant works of Lloyd-Jones and Russell on gold-catalyzed oxidative C–H arylation of simple arenes with arylsilanes have led the way to novel gold-catalyzed reactions that could not be achieved with other transition metals [68,69]. In these reactions, the oxidation of gold(I) to gold(III) is thought to be a key step in the catalytic cycle consisting of transmetalation with arylsilane, C–H activation and reductive elimination [69]. While gold(I) complexes bearing various ligands are used as gold(III) precursors, it remains unclear whether ligands can still coordinate to the gold center or not under such oxidative reaction conditions. For example, triphenylphosphine is easily oxidized to triphenylphosphine oxide by a hypervalent iodine reagent that has been used as an oxidant for gold-catalyzed C–H arylation [69]. Appropriate ligands that are tolerant to the oxidative conditions would offer numerous benefits such as high activity and stability of gold catalyst, thereby achieving otherwise-difficult oxidative transformations [37-40].
Recently, we have introduced highly electron-donating triaryl-2-pyridylidene (PyC: pyridine-based carbene) [82-84] as a new type of nonclassical N-heterocyclic carbene [85-102]. We demonstrated that the PyC ligand is one of the strongest electron-donating carbene ligands to a gold(I) species (Figure 1) [83]. The AuCl(PyC) complex is very stable, even in air and moisture, and isolable by column chromatography on silica gel. Thus we envisioned that a gold complex with strongly electron-donating PyC would promote the gold(I)-to-gold(III) oxidation process, facilitating oxidative coupling reactions. Herein we report that the PyC ligand facilitates gold-catalyzed oxidative C–H arylation of hereroarenes that has been known to be very sluggish with typical ligand systems [68-72]. In this paper, the C–H arylation reactions of isoxazole, indole, and benzothiophene are presented. In addition, direct observation and isolation of PyC-gold(III) complexes are described.
![[1860-5397-11-295-1]](/bjoc/content/figures/1860-5397-11-295-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Triaryl-2-pyridylidene (PyC) and PyC-gold(I) complex (AuCl(PyC)).
Figure 1: Triaryl-2-pyridylidene (PyC) and PyC-gold(I) complex (AuCl(PyC)).
Results and Discussion
Ligand effect of PyC in gold-catalyzed aromatic C–H arylation
In this study, we selected the gold-catalyzed oxidative C–H arylation of arenes with arylsilanes [68,69], reported by Lloyd-Jones and Russell, to test the ligand effect of PyC (Table 1). Likely due to the low stability of electron-rich heteroarene substrates toward oxidative conditions [103-107], their original conditions usually do not work well for these substrates. For example, when isoxazole (1a: 1 equiv) [108-111] was treated with 1-bromo-4-(trimethylsilyl)benzene (2a: 1 equiv) in chloroform/methanol solution at 65 °C in the presence of AuCl(PPh3) (5 mol %), iodosobenzoic acid (IBA: 1 equiv) and (+)-10-camphorsulfonic acid (CSA: 1 equiv), the corresponding C–H arylation product 3aa was obtained in only 10% yield (Table 1, entry 1). Although the application of IPr, a conventional NHC ligand, to the reaction did not afford 3aa at all (Table 1, entry 2), PyC promoted the reaction with higher yield of 4-arylisoxazole 3aa under these conditions (30%, Table 1, entry 3). In the AuCl(PyC)-catalyzed reaction, 1a was fully consumed, and 4,4’-dibromobiphenyl (4a) derived from the homocoupling of arylsilane 2a was also detected. Furthermore, a significant amount of methyl 2-iodobenzoate (5) was generated through the esterification of a co-product (2-iodobenzoic acid) with methanol. We also tested other iodine(III) reagents such as PhI(OAc)2, PhI(OCOCF3)2 and PhI(OH)(OTs), but they all resulted in lower yields than IBA mainly due to the formation of diaryliodonium PhI(4-BrC6H4)+ produced by the reaction with arylsilane 2a (Table 1, entries 4–6) [69]. Using p-toluenesulfonic acid (TsOH) instead of CSA was less effective (Table 1, entry 7). It was clearly seen that both CSA and methanol had a significant effect on the reaction progress (Table 1, entries 8 and 9). Nevertheless, the highest yield achieved by the use of AuCl(PyC) may be attributed to the highly electron-donating nature of the PyC ligand.
Table 1: Effect of ligand and oxidant in gold-catalyzed oxidative C–H arylation of isoxazole 1a.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-11-295-i3.svg?max-width=637&scale=1.0)
|
||||
| Entry | Au catalyst | Oxidant | Yield [%]b | |
|---|---|---|---|---|
| 3aa | 4a | |||
| 1 | AuCl(PPh3) | IBA | 10 | 7 |
| 2 | AuCl(IPr) | IBA | 0 | 0 |
| 3 | AuCl(PyC) | IBA | 30 | 12 |
| 4 | AuCl(PyC) | PhI(OAc)2 | 4 | 5 |
| 5 | AuCl(PyC) | PhI(OCOCF3)2 | 3 | 4 |
| 6c | AuCl(PyC) | PhI(OH)(OTs) | 9 | 5 |
| 7d | AuCl(PyC) | IBA | 13 | 5 |
| 8c | AuCl(PyC) | IBA | 0 | 9 |
| 9e | AuCl(PyC) | IBA | 3 | 36 |
![[Graphic 2]](/bjoc/content/inline/1860-5397-11-295-i4.svg?max-width=637&scale=1.18182)
aReaction conditions: 1a (0.20 mmol), 2a (0.20 mmol), Au catalyst (5 mol %), oxidant (0.20 mmol), (+)-10-camphorsulfonic acid (CSA, 0.20 mmol), CHCl3/MeOH (10:1, 1.1 mL), 65 °C. bDetermined by GC analysis with n-nonane as an internal standard. cWithout CSA. dTsOH·H2O was used instead of CSA. eCHCl3 (1.0 mL) was used as solvent.
Oxidative C–H arylation of heteroarenes with arylsilanes catalyzed by AuCl(PyC)
Having discovered the positive effect of using PyC as a ligand, we further examined the C–H arylation of various heteroarenes with arylsilanes (Table 2). It should be noted that all of the examined heteroarenes were not successfully applied in the previous gold-catalyzed C–H arylation. The reactions of 1a with halogenated aryltrimethylsilanes 2a and 2b afforded coupling products 3aa and 3ab in 14% and 15% isolated yields, respectively (Table 2, entries 1 and 2) [112]. 5-Methylisoxazole (1b) was arylated with bromo-, fluoro- and trifluoromethyl-substituted aryltrimethylsilanes, 2a, 2b and 2c, respectively, to give the corresponding 4-aryl-5-methylisoxazoles, 3ba, 3bb and 3bc, respectively, in higher efficiency as compared with 1a (Table 2, entries 3–5). This may be due to the higher tolerability of 1b than 1a toward undesired decomposition [113]. The introduction of the 3,5-dibromophenyl group onto methylisoxazole 1b resulted in lower yield of heterobiaryl 3bd (Table 2, entry 6). In the reaction of 5-phenylisoxazole (1c), the selective arylation at the C4 position occurred without any arylation at the phenyl group (Table 2, entry 7). 3,5-Dimethylisoxazole (1d) showed low reactivity, likely due to the steric hindrance, but the reaction gave sterically congested heterobiaryl 3da in 17% yield (Table 2, entry 8). In the case of the reaction of indole 1e, 3-arylindole 3ea was exclusively obtained in 44% yield (Table 2, entry 9). On the other hand, arylation of benzo[b]thiophene (1f) mainly afforded 2-arylbenzothiophene 3fa along with a small amount of 3-arylbenzothiophene 3fa' (Table 2, entry 10).
Table 2: AuCl(PyC)-catalyzed oxidative C–H arylation of heteroarenes with arylsilanes.a
![[Graphic 3]](/bjoc/content/inline/1860-5397-11-295-i5.svg?max-width=637&scale=1.0)
|
||||
| Entry | 1 | 2 | 3 | Yieldb |
|---|---|---|---|---|
| 1 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-11-295-i6.svg?max-width=637&scale=1.0)
1a |
![[Graphic 5]](/bjoc/content/inline/1860-5397-11-295-i7.svg?max-width=637&scale=1.0)
2a |
![[Graphic 6]](/bjoc/content/inline/1860-5397-11-295-i8.svg?max-width=637&scale=1.0)
3aa |
14% |
| 2 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-11-295-i9.svg?max-width=637&scale=1.0)
1a |
![[Graphic 8]](/bjoc/content/inline/1860-5397-11-295-i10.svg?max-width=637&scale=1.0)
2b |
![[Graphic 9]](/bjoc/content/inline/1860-5397-11-295-i11.svg?max-width=637&scale=1.0)
3ab |
15% |
| 3 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-11-295-i12.svg?max-width=637&scale=1.0)
1b |
![[Graphic 11]](/bjoc/content/inline/1860-5397-11-295-i13.svg?max-width=637&scale=1.0)
2a |
![[Graphic 12]](/bjoc/content/inline/1860-5397-11-295-i14.svg?max-width=637&scale=1.0)
3ba |
55% |
| 4 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-11-295-i15.svg?max-width=637&scale=1.0)
1b |
![[Graphic 14]](/bjoc/content/inline/1860-5397-11-295-i16.svg?max-width=637&scale=1.0)
2b |
![[Graphic 15]](/bjoc/content/inline/1860-5397-11-295-i17.svg?max-width=637&scale=1.0)
3bb |
54% |
| 5 |
![[Graphic 16]](/bjoc/content/inline/1860-5397-11-295-i18.svg?max-width=637&scale=1.0)
1b |
![[Graphic 17]](/bjoc/content/inline/1860-5397-11-295-i19.svg?max-width=637&scale=1.0)
2c |
![[Graphic 18]](/bjoc/content/inline/1860-5397-11-295-i20.svg?max-width=637&scale=1.0)
3bc |
33% |
| 6 |
![[Graphic 19]](/bjoc/content/inline/1860-5397-11-295-i21.svg?max-width=637&scale=1.0)
1b |
![[Graphic 20]](/bjoc/content/inline/1860-5397-11-295-i22.svg?max-width=637&scale=1.0)
2d |
![[Graphic 21]](/bjoc/content/inline/1860-5397-11-295-i23.svg?max-width=637&scale=1.0)
3bd |
13% |
| 7 |
![[Graphic 22]](/bjoc/content/inline/1860-5397-11-295-i24.svg?max-width=637&scale=1.0)
1c |
![[Graphic 23]](/bjoc/content/inline/1860-5397-11-295-i25.svg?max-width=637&scale=1.0)
2a |
![[Graphic 24]](/bjoc/content/inline/1860-5397-11-295-i26.svg?max-width=637&scale=1.0)
3ca |
28% |
| 8 |
![[Graphic 25]](/bjoc/content/inline/1860-5397-11-295-i27.svg?max-width=637&scale=1.0)
1d |
![[Graphic 26]](/bjoc/content/inline/1860-5397-11-295-i28.svg?max-width=637&scale=1.0)
2a |
![[Graphic 27]](/bjoc/content/inline/1860-5397-11-295-i29.svg?max-width=637&scale=1.0)
3da |
17% |
| 9 |
![[Graphic 28]](/bjoc/content/inline/1860-5397-11-295-i30.svg?max-width=637&scale=1.0)
1e |
![[Graphic 29]](/bjoc/content/inline/1860-5397-11-295-i31.svg?max-width=637&scale=1.0)
2a |
![[Graphic 30]](/bjoc/content/inline/1860-5397-11-295-i32.svg?max-width=637&scale=1.0)
3ea |
44% |
| 10 |
![[Graphic 31]](/bjoc/content/inline/1860-5397-11-295-i33.svg?max-width=637&scale=1.0)
1f |
![[Graphic 32]](/bjoc/content/inline/1860-5397-11-295-i34.svg?max-width=637&scale=1.0)
2a |
![[Graphic 33]](/bjoc/content/inline/1860-5397-11-295-i35.svg?max-width=637&scale=1.0)
3fa (C2):3fa’ (C3) = 83:17 |
22% |
aReaction conditions: 1 (0.20 mmol), 2 (0.20 mmol), AuCl(PyC) (5 mol %), IBA (0.20 mmol), CSA (0.20 mmol), CHCl3/MeOH (10:1, 1.1 mL), 65 °C, 18–48 h. bIsolated yield.
Reaction progress analysis
To further unveil the ligand effect of PyC, time-production profiles of coupling product 3ba were investigated for the reaction of 1b and 2a with AuCl(PyC), AuCl(PPh3) and AuCl(IPr). The yield of 3ba was determined by GC analysis, whereas the consumption of IBA (oxidant) was estimated by the production of methyl 2-iodobenzoate (5). The reaction plots with AuCl(PyC), AuCl(PPh3) and AuCl(IPr) are depicted in Figure 2. Noteworthy observations are as follows: (i) the reaction with AuCl(PyC) was fastest among those with three catalysts (Figure 2a), (ii) the induction periods with regard to the formation of 3ba were found in the reactions using AuCl(PyC) and AuCl(PPh3) (Figure 2a,b), and (iii) the oxidant consumption began at the reaction initiation for all catalysts (Figure 2c). In the reaction using AuCl(PyC), the coupling product 3ba was generated after a shorter induction period of about 3 h and reached 60% yield after 50 h (Figure 2a). On the other hand, the reaction using AuCl(PPh3) began after a longer induction period (ca. 5 h), and the yield of 3ba did not exceed the yield with AuCl(PyC) even after 100 h (see Supporting Information File 1 for details). No coupling product was produced with AuCl(IPr) although the consumption of about 10% of IBA was observed.
![[1860-5397-11-295-2]](/bjoc/content/figures/1860-5397-11-295-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Yield–time profiles of 4-(4-bromophenyl)-5-methylisoxazole (3ba) and methyl 2-iodobenzoate (5) with AuCl(PyC), AuCl(PPh3) and AuCl(IPr). (a) Yield of 3ba. (b) Magnified figure of (a). (c) Yield of 5. All yields were determined by GC analysis with n-nonane as an internal standard.
Figure 2: Yield–time profiles of 4-(4-bromophenyl)-5-methylisoxazole (3ba) and methyl 2-iodobenzoate (5) with...
Mechanistic considerations
Based on the above results and the literature [68-75], we propose the reaction mechanism of the gold-catalyzed C–H arylation of heteroarenes with arylsilanes as shown in Scheme 1. A gold(I) complex A is first oxidized to gold(III) species B by the iodine(III) reagent E derived from IBA by the exchange of a hydroxy group with an existing acid such as CSA, HCl and MeOH. We independently confirmed that the esterification of 2-iodobenzoic acid takes place to give 5 under the reaction conditions; 2-iodobenzoic acid was smoothly converted to 5 in chloroform/methanol solution at 65 °C. Transmetalation of gold(III) complex B with arylsilane 2 affords monoarylated gold(III) intermediate C. The electrophilic metalation of heteroarene 1 with C with concurrent generation of an acid (HX) produces diarylated gold(III) species D. Finally, the reductive elimination from D releases the coupling product 3 along with the regeneration of gold(I) species A. The side reaction leading to the homocoupling product of arylsilane 4 likely occurs via over-transmetalation of monoarylated gold(III) species C with arylsilane 2 or disproportionation of C [67-81].
![[1860-5397-11-295-i1]](/bjoc/content/inline/1860-5397-11-295-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Plausible reaction mechanism of gold-catalyzed oxidative C–H arylation of heteroarenes with arylsilanes.
Scheme 1: Plausible reaction mechanism of gold-catalyzed oxidative C–H arylation of heteroarenes with arylsil...
Oxidation process of gold
In all reaction progress experiments with the three gold catalysts (Figure 2), the consumption of IBA (production of 5) was observed to some extent even in the induction period. Taking the possible reaction mechanism into consideration, the oxidation of gold(I) to gold(III) by the oxidant may occur during the induction period. While it is unclear what is oxidized in these reactions, we hypothesize that the highly electron-donating PyC ligand facilitates the oxidation of gold(I) to gold(III). As triphenylphosphine is known to be easily oxidized to triphenylphosphine oxide under the current oxidative conditions, the ligand-free gold(III) species is thought to be an active species in the arylation reaction with AuCl(PPh3) [69]. While the IPr-gold(I) complex is known to undergo oxidation to an IPr-gold(III) species [114], its inactiveness in the current reaction indicates that the electron-donating capability is not high enough to facilitate this process.
Direct observation and isolation of PyC-gold(III) complex
To verify our hypothesis that PyC accelerates the gold(I)-to-gold(III) oxidation, we attempted the direct observation and the isolation of the PyC-gold(III) complex. First of all, the gold(III) complex AuCl3(PyC) was newly synthesized by treating AuCl(PyC) with PhICl2 (see Experimental section and Supporting Information File 1 for details) [114]. The X-ray crystallographic analysis was successfully accomplished with a colorless single crystal of AuCl3(PyC), which was recrystallized from nitrobenzene and pentane (Figure 3) [115]. The X-ray crystal structure shows that the four gold bonds are in a planar surface, and the pyridylidene face and the added two chlorine atoms are in vertical positions. The ligand arrangement is quite similar to a series of reported NHC–AuCl3 complexes [114].
![[1860-5397-11-295-3]](/bjoc/content/figures/1860-5397-11-295-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: ORTEP drawing of AuCl3(PyC) with 50% probability. Hydrogen atoms and solvent are omitted for clarity.
Figure 3: ORTEP drawing of AuCl3(PyC) with 50% probability. Hydrogen atoms and solvent are omitted for clarit...
With the authentic AuCl3(PyC) in hand, we next carried out the direct observation of PyC-gold(III) species under the catalytic conditions. The treatment of AuCl(PyC) with 5-fluoroiodosobenzoic acid (5F-IBA) and CSA in CDCl3/CD3OD at 65 °C resulted in the full consumption of 5F-IBA within 30 min (monitored by 19F NMR). While the resulting mixture seemed to contain several PyC-gold(III) complexes, the formation of various gold(III) species bearing hydroxy, methoxy, sulfoxy and chloro groups made the analysis and isolation difficult. However, the subsequent addition of excess LiCl enabled us to detect the gold(III) species as AuCl3(PyC) by 1H and 13C NMR analyses. The 1H NMR analysis revealed that about 90% of AuCl(PyC) was consumed and AuCl3(PyC) was produced in 50% NMR yield. Fortunately, the isolation from the messy crude mixtures was accomplished to give AuCl3(PyC) in 24% isolated yield. We also conducted the same experiment with the AuCl(IPr) complex. From the 19F and 1H NMR analyses, approximately half of AuCl(IPr) and oxidant 5F-IBA remained unreacted after heating for 30 min, and AuCl3(IPr) was observed only in 34% 1H NMR yield [114] (Scheme 2).
![[1860-5397-11-295-i2]](/bjoc/content/inline/1860-5397-11-295-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Direct observation and isolation of carbene-gold(III) complex. Mes = 2,4,6-Me3C6H2, Xyl = 2,6-Me2C6H3, Dip = 2,6-iPr2C6H3.
Scheme 2: Direct observation and isolation of carbene-gold(III) complex. Mes = 2,4,6-Me3C6H2, Xyl = 2,6-Me2C6H...
These observations on gold(III) species support our hypothesis that the highly electron-donating PyC ligand strongly coordinates to a gold center and promotes the gold(I)-to-gold(III) oxidation by stabilizing a gold(III) species without dissociation. An IPr-gold(III) complex is known to be stable, but the lower electron-donation ability of IPr than that of PyC seems to result in the inefficient oxidation of AuCl(IPr). DFT calculations on the oxidation process of the AuCl(ligand) to AuCl3(ligand) also clarified the advantage of the PyC ligand over IPr by 3.6 kcal mol–1 (see Supporting Information File 1 for details). While it still remains unclear how the PyC ligand affects the transmetalation, C–H metalation and reductive elimination steps, we believe that the strongly electron-donating PyC not only facilitates gold(I)-to-gold(III) oxidation in catalysis but also prolongs the catalyst lifetime by preventing the ligand dissociation and formation of inactive gold nanoparticles.
Conclusion
In summary, we have developed the oxidative C–H arylation of heteroarenes with arylsilanes catalyzed by PyC-gold complex and revealed the advantageous features of using the PyC ligand. From the reaction progress, experiments and stoichiometric oxidation of gold(I) complexes, we conclude that the highly electron-donating PyC ligand promotes the gold(I)-to-gold(III) oxidation and stabilizes the gold(III) species, thereby facilitating the oxidative coupling reactions.
Experimental
Preparation of triarylpyridylidene-gold(I) chloride [AuCl(PyC)]: A 10 mL Schlenk tube containing a stir bar was dried under vacuum and filled with N2 after cooling to room temperature. Ag2O (232 mg, 1.0 mmol) and NBu4Cl·H2O (1.39 g, 5.0 mmol) were added to the solution of 3,5-bis(2,6-dimethylphenyl)-1-mesitylpyridin-1-ium triflate (730 mg, 1.0 mmol) in 1,2-dichloroethane (5.0 mL). The mixture was stirred at room temperature for 2 h, and AuCl(SMe2) (11.5 mg, 0.10 mmol) was then added to the reaction mixture. The reaction mixture was further stirred overnight, and the addition of CHCl3 (50 mL) to the mixture gave a white precipitate. The suspension was filtered off and the filtrate was concentrated under reduced pressure. The crude product was purified by column chromatography on silica gel (eluents: MeOH/CHCl3 1:20) and recrystallized from CHCl3/toluene at room temperature to give a pure AuCl(PyC)/toluene complex (286 mg, 40%) as a pale yellow crystal. The addition of CHCl3 and the concentration in vacuum yielded a pure AuCl(PyC) complex without toluene as white powder. The characterization data for AuCl(PyC) corresponded to the reported values [83].
General procedure for AuCl(PyC)-catalyzed oxidative C–H arylation of heteroarenes with arylsilanes: AuCl(PyC) (6.4 mg, 0.010 μmol, 5.0 mol %), heteroarene 1 (0.20 mmol), and aryltrimethylsilane 2 (0.20 mmol), 2-iodosobenzoic acid (IBA, 53 mg, 0.20 mmol), 10-camphorsulfonic acid (CSA) (47 mg, 0.20 mmol) and a stir bar were placed in a screw test tube, and dry CHCl3/MeOH (1.0 mL/0.10 mL) was added under N2 atmosphere. The tube was sealed with a cap equipped with a Teflon®-coated silicon rubber septum, and the mixture was stirred at 65 °C for 18–48 h. The reaction was quenched by addition of excess saturated aqueous NaHCO3, the aqueous layer was extracted with CH2Cl2, and the combined organic layers were dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel to afford the coupling product 3 (Table 2).
Oxidation of AuCl(PyC): The oxidation of AuCl(PyC) was performed according to the literature [16]. PhICl2 (54.8 mg, 0.20 mmol) was added into a solution of AuCl(PyC) (128 mg, 0.20 mmol) in CH2Cl2 (2.0 mL) under N2 atmosphere. After stirring at room temperature for 19 h, the reaction mixture was filtered through a pad of Celite®. The filtrate was poured into hexane and the resulting precipitate was collected by filtration to obtain pure AuCl3(PyC) as a white solid (140 mg, 99%). The colorless single crystal used for X-ray diffraction analysis was obtained by recrystallization from nitrobenzene and pentane. 1H NMR (CDCl3, 600 MHz) δ 8.17 (d, J = 2.1 Hz, 1H), 7.90 (d, J = 2.1 Hz, 1H), 7.29 (td, J = 7.6, 2.7 Hz, 2H), 7.19 (d, J = 7.6 Hz, 2H), 7.16 (d, J = 7.6 Hz, 2H), 7.07 (s, 2H), 2.36 (s, 3H), 2.28 (s, 12H), 2.15 (s, 6H); 13C NMR (CDCl3, 150 MHz) δ 162.5 (CH), 149.6 (4°), 146.6 (CH), 144.7 (CH), 141.9 (4°), 141.5 (4°), 138.5 (4°), 136.4 (4°), 135.6 (4°), 135.6 (4°), 133.2 (4°), 132.2 (4°), 130.4 (CH), 129.9 (CH), 129.7 (CH), 128.3 (4°), 128.3 (CH), 22.1 (CH3), 21.1 (CH3), 20.9 (CH3), 19.3 (CH3); HRMS (ESI+) m/z: [M − Cl + MeOH]+ calcd for C31H35AuCl2NO, 704.1756; found, 704.1722.
In situ observation and isolation of AuCl3(PyC): AuCl(PyC) (12.8 mg, 0.020 mmol), 5-fluoroiodosobenzoic acid (5F-IBA, 5.6 mg, 0.020 mmol) and CSA (4.6 mg, 0.020 mmol) were placed in an NMR tube, and CDCl3/CD3OD (10:1, 0.60 mL) was added under N2 atmosphere. The tube was sealed with a cap equipped with a Teflon®-coated silicon rubber septum and heated at 65 °C for 30 min. After cooling to room temperature, LiCl (8.4 mg, 0.20 mmol) was added. 1,1,2,2-Tetrachloroethane was added as an internal standard and an NMR yield of AuCl3(PyC) was estimated by 1H NMR spectroscopy. The solvent was removed in vacuum, and the residue was dissolved in EtOAc. The organic layer was washed with saturated aqueous NaHCO3 and brine, dried over Na2SO4, filtered, and concentrated in vacuum to afford the crude mixture. The crude mixture was further washed with Et2O to give pure AuCl3(PyC) as a white powder (3.4 mg, 24%, Scheme 2).
Supporting Information
| Supporting Information File 1: Experimental procedures, spectra of new compounds, CIF data, and details of the computational study. | ||
| Format: PDF | Size: 2.3 MB | Download |
Acknowledgements
This work was supported by the ERATO program from JST (K.I.) and the Funding Program for KAKENHI from MEXT (26810057 to H.I. and 23750038 to Y.S.). K.H. is a recipient of the JSPS research fellowship for young scientists. ITbM is supported by the World Premier International Research Center (WPI) Initiative (Japan).
References
-
Arcadi, A. Chem. Rev. 2008, 108, 3266–3325. doi:10.1021/cr068435d
Return to citation in text: [1] -
Hashmi, A. S. K.; Rudolph, M. Chem. Soc. Rev. 2008, 37, 1766–1775. doi:10.1039/b615629k
Return to citation in text: [1] -
Li, Z.; Brouwer, C.; He, C. Chem. Rev. 2008, 108, 3239–3265. doi:10.1021/cr068434l
Return to citation in text: [1] -
Marion, N.; Nolan, S. P. Chem. Soc. Rev. 2008, 37, 1776–1782. doi:10.1039/b711132k
Return to citation in text: [1] -
Patil, N. T.; Yamamoto, Y. Chem. Rev. 2008, 108, 3395–3442. doi:10.1021/cr050041j
Return to citation in text: [1] -
Corma, A.; Leyva-Pérez, A.; Sabater, M. J. Chem. Rev. 2011, 111, 1657–1712. doi:10.1021/cr100414u
Return to citation in text: [1] -
Huang, H.; Zhou, Y.; Liu, H. Beilstein J. Org. Chem. 2011, 7, 897–936. doi:10.3762/bjoc.7.103
Return to citation in text: [1] -
Rudolph, M.; Hashmi, A. S. K. Chem. Soc. Rev. 2012, 41, 2448–2462. doi:10.1039/C1CS15279C
Return to citation in text: [1] -
Abbiati, G.; Marinelli, F.; Rossi, E.; Arcadi, A. Isr. J. Chem. 2013, 53, 856–868. doi:10.1002/ijch.201300040
Return to citation in text: [1] -
Chiarucci, M.; Bandini, M. Beilstein J. Org. Chem. 2013, 9, 2586–2614. doi:10.3762/bjoc.9.294
Return to citation in text: [1] -
López, F.; Mascareñas, J. L. Beilstein J. Org. Chem. 2013, 9, 2250–2264. doi:10.3762/bjoc.9.264
Return to citation in text: [1] -
Nunes dos Santos Comprido, L.; Hashmi, A. S. K. Isr. J. Chem. 2013, 53, 883–891. doi:10.1002/ijch.201300072
Return to citation in text: [1] -
Ohno, H. Isr. J. Chem. 2013, 53, 869–882. doi:10.1002/ijch.201300054
Return to citation in text: [1] -
Abbiati, G.; Rossi, E. Beilstein J. Org. Chem. 2014, 10, 481–513. doi:10.3762/bjoc.10.46
Return to citation in text: [1] -
Muratore, M. E.; Homs, A.; Obradors, C.; Echavarren, A. M. Chem. – Asian J. 2014, 9, 3066–3082. doi:10.1002/asia.201402395
Return to citation in text: [1] -
Xie, J.; Pan, C.; Abdukader, A.; Zhu, C. Chem. Soc. Rev. 2014, 43, 5245–5256. doi:10.1039/C4CS00004H
Return to citation in text: [1] [2] -
Yang, W.; Hashmi, A. S. K. Chem. Soc. Rev. 2014, 43, 2941–2955. doi:10.1039/c3cs60441a
Return to citation in text: [1] -
Dorel, R.; Echavarren, A. M. Chem. Rev. 2015, 115, 9028–9072. doi:10.1021/cr500691k
Return to citation in text: [1] -
Qian, D.; Zhang, J. Chem. Soc. Rev. 2015, 44, 677–698. doi:10.1039/C4CS00304G
Return to citation in text: [1] -
Soriano, E.; Marco-Contelles, J. Acc. Chem. Res. 2009, 42, 1026–1036. doi:10.1021/ar800200m
Return to citation in text: [1] -
Gaillard, S.; Cazin, C. S. J.; Nolan, S. P. Acc. Chem. Res. 2012, 45, 778–787. doi:10.1021/ar200188f
Return to citation in text: [1] -
Alcaide, B.; Almendros, P. Acc. Chem. Res. 2014, 47, 939–952. doi:10.1021/ar4002558
Return to citation in text: [1] -
Fensterbank, L.; Malacria, M. Acc. Chem. Res. 2014, 47, 953–965. doi:10.1021/ar4002334
Return to citation in text: [1] -
Fürstner, A. Acc. Chem. Res. 2014, 47, 925–938. doi:10.1021/ar4001789
Return to citation in text: [1] -
Hashmi, A. S. K. Acc. Chem. Res. 2014, 47, 864–876. doi:10.1021/ar500015k
Return to citation in text: [1] -
Obradors, C.; Echavarren, A. M. Acc. Chem. Res. 2014, 47, 902–912. doi:10.1021/ar400174p
Return to citation in text: [1] -
Wang, Y.-M.; Lackner, A. D.; Toste, F. D. Acc. Chem. Res. 2014, 47, 889–901. doi:10.1021/ar400188g
Return to citation in text: [1] -
Yeom, H.-S.; Shin, S. Acc. Chem. Res. 2014, 47, 966–977. doi:10.1021/ar4001839
Return to citation in text: [1] -
Zhang, D.-H.; Tang, X.-Y.; Shi, M. Acc. Chem. Res. 2014, 47, 913–924. doi:10.1021/ar400159r
Return to citation in text: [1] -
Zhang, L. Acc. Chem. Res. 2014, 47, 877–888. doi:10.1021/ar400181x
Return to citation in text: [1] -
For ligand effects in gold-catalyzed reactions, see references [32-36].
Return to citation in text: [1] -
Gorin, D. J.; Sherry, B. D.; Toste, F. D. Chem. Rev. 2008, 108, 3351–3378. doi:10.1021/cr068430g
Return to citation in text: [1] [2] -
Alcarazo, M.; Stork, T.; Anoop, A.; Thiel, W.; Fürstner, A. Angew. Chem., Int. Ed. 2010, 49, 2542–2546. doi:10.1002/anie.200907194
Return to citation in text: [1] [2] -
Nolan, S. P. Acc. Chem. Res. 2011, 44, 91–100. doi:10.1021/ar1000764
Return to citation in text: [1] [2] -
Cera, G.; Bandini, M. Isr. J. Chem. 2013, 53, 848–855. doi:10.1002/ijch.201300029
Return to citation in text: [1] [2] -
Gatineau, D.; Goddard, J.-P.; Mouriès-Mansuy, V.; Fensterbank, L. Isr. J. Chem. 2013, 53, 892–900. doi:10.1002/ijch.201300059
Return to citation in text: [1] [2] -
For selected reviews on gold-catalyzed oxidative C–C coupling reactions, see references [38-40].
Return to citation in text: [1] [2] -
Hopkinson, M. N.; Gee, A. D.; Gouverneur, V. Chem. – Eur. J. 2011, 17, 8248–8262. doi:10.1002/chem.201100736
Return to citation in text: [1] [2] [3] -
Wegner, H. A.; Auzias, M. Angew. Chem., Int. Ed. 2011, 50, 8236–8247. doi:10.1002/anie.201101603
Return to citation in text: [1] [2] [3] -
Brand, J. P.; Li, Y.; Waser, J. Isr. J. Chem. 2013, 53, 901–910. doi:10.1002/ijch.201300044
Return to citation in text: [1] [2] [3] -
For recent examples of oxidative heteroarylation of alkene through gold(I)/gold(III) catalytic cycle, see references [42-52].
Return to citation in text: [1] -
Zhang, G.; Peng, Y.; Cui, L.; Zhang, L. Angew. Chem., Int. Ed. 2009, 48, 3112–3115. doi:10.1002/anie.200900585
Return to citation in text: [1] [2] -
Ball, L. T.; Green, M.; Lloyd-Jones, G. C.; Russell, C. A. Org. Lett. 2010, 12, 4724–4727. doi:10.1021/ol1019162
Return to citation in text: [1] [2] -
Brenzovich, W. E., Jr.; Benitez, D.; Lackner, A. D.; Shunatona, H. P.; Tkatchouk, E.; Goddard, W. A., III; Toste, F. D. Angew. Chem., Int. Ed. 2010, 49, 5519–5522. doi:10.1002/anie.201002739
Return to citation in text: [1] [2] -
Brenzovich, W. E., Jr.; Brazeau, J.-F.; Toste, F. D. Org. Lett. 2010, 12, 4728–4731. doi:10.1021/ol102194c
Return to citation in text: [1] [2] -
Hopkinson, M. N.; Tessier, A.; Salisbury, A.; Giuffredi, G. T.; Combettes, L. E.; Gee, A. D.; Gouverneur, V. Chem. – Eur. J. 2010, 16, 4739–4743. doi:10.1002/chem.201000322
Return to citation in text: [1] [2] -
Melhado, A. D.; Brenzovich, W. E., Jr.; Lackner, A. D.; Toste, F. D. J. Am. Chem. Soc. 2010, 132, 8885–8887. doi:10.1021/ja1034123
Return to citation in text: [1] [2] -
Zhang, G.; Cui, L.; Wang, Y.; Zhang, L. J. Am. Chem. Soc. 2010, 132, 1474–1475. doi:10.1021/ja909555d
Return to citation in text: [1] [2] -
Zhang, G.; Luo, Y.; Wang, Y.; Zhang, L. Angew. Chem., Int. Ed. 2011, 50, 4450–4454. doi:10.1002/anie.201100293
Return to citation in text: [1] [2] -
Ball, L. T.; Lloyd-Jones, G. C.; Russell, C. A. Chem. – Eur. J. 2012, 18, 2931–2937. doi:10.1002/chem.201103061
Return to citation in text: [1] [2] -
Zhang, R.; Xu, Q.; Chen, K.; Gu, P.; Shi, M. Eur. J. Org. Chem. 2013, 7366–7371. doi:10.1002/ejoc.201300896
Return to citation in text: [1] [2] -
Zhu, S.; Ye, L.; Wu, W.; Jiang, H. Tetrahedron 2013, 69, 10375–10383. doi:10.1016/j.tet.2013.09.097
Return to citation in text: [1] [2] -
For recent examples of gold-catalyzed C–C bond forming oxidative coupling reaction, see references [54-66].
Return to citation in text: [1] -
Jones, C. J.; Taube, D.; Ziatdinov, V. R.; Periana, R. A.; Nielsen, R. J.; Oxgaard, J.; Goddard, W. A., III. Angew. Chem., Int. Ed. 2004, 43, 4626–4629. doi:10.1002/anie.200461055
Return to citation in text: [1] [2] -
Wegner, H. A.; Ahles, S.; Neuburger, M. Chem. – Eur. J. 2008, 14, 11310–11313. doi:10.1002/chem.200801848
Return to citation in text: [1] [2] -
Brand, J. P.; Charpentier, J.; Waser, J. Angew. Chem., Int. Ed. 2009, 48, 9346–9349. doi:10.1002/anie.200905419
Return to citation in text: [1] [2] -
Cui, L.; Zhang, G.; Zhang, L. Bioorg. Med. Chem. Lett. 2009, 19, 3884–3887. doi:10.1016/j.bmcl.2009.03.127
Return to citation in text: [1] [2] -
Peng, Y.; Cui, L.; Zhang, G.; Zhang, L. J. Am. Chem. Soc. 2009, 131, 5062–5063. doi:10.1021/ja901048w
Return to citation in text: [1] [2] -
Brand, J. P.; Waser, J. Angew. Chem., Int. Ed. 2010, 49, 7304–7307. doi:10.1002/anie.201003179
Return to citation in text: [1] [2] -
de Haro, T.; Nevado, C. J. Am. Chem. Soc. 2010, 132, 1512–1513. doi:10.1021/ja909726h
Return to citation in text: [1] [2] -
Hopkinson, M. N.; Ross, J. E.; Giuffredi, G. T.; Gee, A. D.; Gouverneur, V. Org. Lett. 2010, 12, 4904–4907. doi:10.1021/ol102061k
Return to citation in text: [1] [2] -
Qian, D.; Zhang, J. Beilstein J. Org. Chem. 2011, 7, 808–812. doi:10.3762/bjoc.7.92
Return to citation in text: [1] [2] -
Brand, J. P.; Chevalley, C.; Scopelliti, R.; Waser, J. Chem. – Eur. J. 2012, 18, 5655–5666. doi:10.1002/chem.201200200
Return to citation in text: [1] [2] -
Levin, M. D.; Toste, F. D. Angew. Chem., Int. Ed. 2014, 53, 6211–6215. doi:10.1002/anie.201402924
Return to citation in text: [1] [2] -
Ma, Y.; Zhang, S.; Yang, S.; Song, F.; You, J. Angew. Chem., Int. Ed. 2014, 53, 7870–7874. doi:10.1002/anie.201402475
Return to citation in text: [1] [2] -
Peng, H.; Xi, Y.; Ronaghi, N.; Dong, B.; Akhmedov, N. G.; Shi, X. J. Am. Chem. Soc. 2014, 136, 13174–13177. doi:10.1021/ja5078365
Return to citation in text: [1] [2] -
For gold-catalyzed oxidative C–H arylation of arenes, see references [68-72].
Return to citation in text: [1] [2] -
Ball, L. T.; Lloyd-Jones, G. C.; Russell, C. A. Science 2012, 337, 1644–1648. doi:10.1126/science.1225709
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Ball, L. T.; Lloyd-Jones, G. C.; Russell, C. A. J. Am. Chem. Soc. 2014, 136, 254–264. doi:10.1021/ja408712e
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11] -
Kar, A.; Mangu, N.; Kaiser, H. M.; Beller, M.; Tse, M. K. Chem. Commun. 2008, 386–388. doi:10.1039/B714928J
Return to citation in text: [1] [2] [3] [4] [5] -
Kar, A.; Mangu, N.; Kaiser, H. M.; Tse, M. K. J. Organomet. Chem. 2009, 694, 524–537. doi:10.1016/j.jorganchem.2008.11.016
Return to citation in text: [1] [2] [3] [4] [5] -
Wu, Q.; Du, C.; Huang, Y.; Liu, X.; Long, Z.; Song, F.; You, J. Chem. Sci. 2015, 6, 288–293. doi:10.1039/C4SC02070G
Return to citation in text: [1] [2] [3] [4] [5] -
For selected mechanistic investigations on gold(I)/gold(III) redox processes, see references [74-84].
Return to citation in text: [1] [2] [3] -
Mankad, N. P.; Toste, F. D. J. Am. Chem. Soc. 2010, 132, 12859–12861. doi:10.1021/ja106257n
Return to citation in text: [1] [2] [3] [4] -
Tkatchouk, E.; Mankad, N. P.; Benitez, D.; Goddard, W. A., III; Toste, F. D. J. Am. Chem. Soc. 2011, 133, 14293–14300. doi:10.1021/ja2012627
Return to citation in text: [1] [2] [3] [4] -
Hofer, M.; Nevado, C. Eur. J. Inorg. Chem. 2012, 1338–1341. doi:10.1002/ejic.201100956
Return to citation in text: [1] [2] [3] -
Cambeiro, X. C.; Boorman, T. C.; Lu, P.; Larrosa, I. Angew. Chem., Int. Ed. 2013, 52, 1781–1784. doi:10.1002/anie.201209007
Return to citation in text: [1] [2] [3] -
Hofer, M.; Nevado, C. Tetrahedron 2013, 69, 5751–5757. doi:10.1016/j.tet.2013.04.029
Return to citation in text: [1] [2] [3] -
Hofer, M.; Gomez-Bengoa, E.; Nevado, C. Organometallics 2014, 33, 1328–1332. doi:10.1021/om400884s
Return to citation in text: [1] [2] [3] -
Winston, M. S.; Wolf, W. J.; Toste, F. D. J. Am. Chem. Soc. 2014, 136, 7777–7782. doi:10.1021/ja503974x
Return to citation in text: [1] [2] [3] -
Wolf, W. J.; Winston, M. S.; Toste, F. D. Nat. Chem. 2014, 6, 159–164. doi:10.1038/nchem.1822
Return to citation in text: [1] [2] [3] -
For our contributions to the synthesis, complexation of pyridylidene ligands, see references [83] and [84].
Return to citation in text: [1] [2] -
Hata, K.; Segawa, Y.; Itami, K. Chem. Commun. 2012, 48, 6642–6644. doi:10.1039/c2cc33184e
Return to citation in text: [1] [2] [3] [4] [5] -
Yoshidomi, T.; Segawa, Y.; Itami, K. Chem. Commun. 2013, 49, 5648–5650. doi:10.1039/c3cc42655f
Return to citation in text: [1] [2] [3] -
For the synthesis and DFT calculation of pyridylidene, see references [86-92].
Return to citation in text: [1] -
Raubenheimer, H. G.; Toerien, J. G.; Kruger, G. J.; Otte, R.; van Zyl, W.; Olivier, P. J. Organomet. Chem. 1994, 466, 291–295. doi:10.1016/0022-328X(94)88058-1
Return to citation in text: [1] [2] -
Strasser, C. E.; Stander-Grobler, E.; Schuster, O.; Cronje, S.; Raubenheimer, H. G. Eur. J. Inorg. Chem. 2009, 1905–1912. doi:10.1002/ejic.200801180
Return to citation in text: [1] [2] -
Roselló-Merino, M.; Díez, J.; Conejero, S. Chem. Commun. 2010, 46, 9247–9249. doi:10.1039/c0cc03935g
Return to citation in text: [1] [2] -
Hollóczki, O.; Nyulászi, L. J. Org. Chem. 2008, 73, 4794–4799. doi:10.1021/jo8000035
Return to citation in text: [1] [2] -
Kassaee, M. Z.; Shakib, F. A.; Momeni, M. R.; Ghambarian, M.; Musavi, S. M. Tetrahedron 2009, 65, 10093–10098. doi:10.1016/j.tet.2009.09.043
Return to citation in text: [1] [2] -
Tukov, A. A.; Normand, A. T.; Nechaev, M. S. Dalton Trans. 2009, 7015–7028. doi:10.1039/b906969k
Return to citation in text: [1] [2] -
Wang, L.; Kong, L.; Li, Y.; Ganguly, R.; Kinjo, R. Chem. Commun. 2015, 51, 12419–12422. doi:10.1039/C5CC04091D
Return to citation in text: [1] [2] -
For the synthesis and characterization of nonclassical carbenes, see references [94-96].
Return to citation in text: [1] -
Canac, Y.; Soleilhavoup, M.; Conejero, S.; Bertrand, G. J. Organomet. Chem. 2004, 689, 3857–3865. doi:10.1016/j.jorganchem.2004.02.005
Return to citation in text: [1] [2] -
Melaimi, M.; Soleilhavoup, M.; Bertrand, G. Angew. Chem., Int. Ed. 2010, 49, 8810–8849. doi:10.1002/anie.201000165
Return to citation in text: [1] [2] -
Martin, D.; Melaimi, M.; Soleilhavoup, M.; Bertrand, G. Organometallics 2011, 30, 5304–5313. doi:10.1021/om200650x
Return to citation in text: [1] [2] -
For nonclassical carbene-gold complexes in catalytic reactions, see references [98-102]
Return to citation in text: [1] -
Zeng, X.; Frey, G. D.; Kinjo, R.; Donnadieu, B.; Bertrand, G. J. Am. Chem. Soc. 2009, 131, 8690–8696. doi:10.1021/ja902051m
Return to citation in text: [1] [2] -
Kinjo, R.; Donnadieu, B.; Bertrand, G. Angew. Chem., Int. Ed. 2011, 50, 5560–5563. doi:10.1002/anie.201100740
Return to citation in text: [1] [2] -
López-Gómez, M. J.; Martin, D.; Bertrand, G. Chem. Commun. 2013, 49, 4483–4485. doi:10.1039/c3cc41279b
Return to citation in text: [1] [2] -
Hu, X.; Martin, D.; Melaimi, M.; Bertrand, G. J. Am. Chem. Soc. 2014, 136, 13594–13597. doi:10.1021/ja507788r
Return to citation in text: [1] [2] -
Jin, L.; Weinberger, D. S.; Melaimi, M.; Moore, C. E.; Rheingold, A. L.; Bertrand, G. Angew. Chem., Int. Ed. 2014, 53, 9059–9063. doi:10.1002/anie.201404665
Return to citation in text: [1] [2] -
Electron-rich arenes show the reactivity toward dehydrogenative homo- and cross-coupling reaction under oxidative conditions without metal oxidants. For related examples, see references [104-107].
Return to citation in text: [1] -
Ito, H.; Ueda, K.; Itami, K. Cross-Dehydrogenative-Coupling Reactions without Metals. In From C–H to C–C Bonds: Cross-Dehydrogenative-Coupling; Li, C.-J., Ed.; The Royal Society of Chemistry, 2015; pp 153–196. doi:10.1039/9781782620082-00153
Return to citation in text: [1] [2] -
Jean, A.; Cantat, J.; Bérard, D.; Bouchu, D.; Canesi, S. Org. Lett. 2007, 9, 2553–2556. doi:10.1021/ol070941h
Return to citation in text: [1] [2] -
Kita, Y.; Morimoto, K.; Ito, M.; Ogawa, C.; Goto, A.; Dohi, T. J. Am. Chem. Soc. 2009, 131, 1668–1669. doi:10.1021/ja808940n
Return to citation in text: [1] [2] -
Morimoto, K.; Yamaoka, N.; Ogawa, C.; Nakae, T.; Fujioka, H.; Dohi, T.; Kita, Y. Org. Lett. 2010, 12, 3804–3807. doi:10.1021/ol101498r
Return to citation in text: [1] [2] -
For palladium-catalyzed C–H arylation of isoxazole, see references [109-111].
Return to citation in text: [1] -
Chiong, H. A.; Daugulis, O. Org. Lett. 2007, 9, 1449–1451. doi:10.1021/ol0702324
Return to citation in text: [1] [2] -
Fall, Y.; Reynaud, C.; Doucet, H.; Santelli, M. Eur. J. Org. Chem. 2009, 4041–4050. doi:10.1002/ejoc.200900309
Return to citation in text: [1] [2] -
Roger, J.; Požgan, F.; Doucet, H. Adv. Synth. Catal. 2010, 352, 696–710. doi:10.1002/adsc.200900793
Return to citation in text: [1] [2] -
Higher volatility of products resulted in the lower isolated yields.
Return to citation in text: [1] -
In the presence of 10 mol % of AuCl(PyC), the reaction of 1b and 2a gave lower yield of 3ba (33% GC yield) with significant amount of homo-coupling product of 2a (4,4’-dibromobiphenyl) being observed.
Return to citation in text: [1] -
Gaillard, S.; Bantreil, X.; Slawin, A. M. Z.; Nolan, S. P. Dalton Trans. 2009, 6967–6971. doi:10.1039/b907109a
See for the synthesis and characterization of AuCl3(IPr) complex.
Return to citation in text: [1] [2] [3] [4] -
Crystallographic data reported in this manuscript have been deposited with Cambridge Crystallographic Data Centre as supplementary publication No. CCDC-1045812 [AuCl3(PyC)]. Copies of the data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html.
Return to citation in text: [1]
| 32. | Gorin, D. J.; Sherry, B. D.; Toste, F. D. Chem. Rev. 2008, 108, 3351–3378. doi:10.1021/cr068430g |
| 33. | Alcarazo, M.; Stork, T.; Anoop, A.; Thiel, W.; Fürstner, A. Angew. Chem., Int. Ed. 2010, 49, 2542–2546. doi:10.1002/anie.200907194 |
| 34. | Nolan, S. P. Acc. Chem. Res. 2011, 44, 91–100. doi:10.1021/ar1000764 |
| 35. | Cera, G.; Bandini, M. Isr. J. Chem. 2013, 53, 848–855. doi:10.1002/ijch.201300029 |
| 36. | Gatineau, D.; Goddard, J.-P.; Mouriès-Mansuy, V.; Fensterbank, L. Isr. J. Chem. 2013, 53, 892–900. doi:10.1002/ijch.201300059 |
| 38. | Hopkinson, M. N.; Gee, A. D.; Gouverneur, V. Chem. – Eur. J. 2011, 17, 8248–8262. doi:10.1002/chem.201100736 |
| 39. | Wegner, H. A.; Auzias, M. Angew. Chem., Int. Ed. 2011, 50, 8236–8247. doi:10.1002/anie.201101603 |
| 40. | Brand, J. P.; Li, Y.; Waser, J. Isr. J. Chem. 2013, 53, 901–910. doi:10.1002/ijch.201300044 |
| 42. | Zhang, G.; Peng, Y.; Cui, L.; Zhang, L. Angew. Chem., Int. Ed. 2009, 48, 3112–3115. doi:10.1002/anie.200900585 |
| 43. | Ball, L. T.; Green, M.; Lloyd-Jones, G. C.; Russell, C. A. Org. Lett. 2010, 12, 4724–4727. doi:10.1021/ol1019162 |
| 44. | Brenzovich, W. E., Jr.; Benitez, D.; Lackner, A. D.; Shunatona, H. P.; Tkatchouk, E.; Goddard, W. A., III; Toste, F. D. Angew. Chem., Int. Ed. 2010, 49, 5519–5522. doi:10.1002/anie.201002739 |
| 45. | Brenzovich, W. E., Jr.; Brazeau, J.-F.; Toste, F. D. Org. Lett. 2010, 12, 4728–4731. doi:10.1021/ol102194c |
| 46. | Hopkinson, M. N.; Tessier, A.; Salisbury, A.; Giuffredi, G. T.; Combettes, L. E.; Gee, A. D.; Gouverneur, V. Chem. – Eur. J. 2010, 16, 4739–4743. doi:10.1002/chem.201000322 |
| 47. | Melhado, A. D.; Brenzovich, W. E., Jr.; Lackner, A. D.; Toste, F. D. J. Am. Chem. Soc. 2010, 132, 8885–8887. doi:10.1021/ja1034123 |
| 48. | Zhang, G.; Cui, L.; Wang, Y.; Zhang, L. J. Am. Chem. Soc. 2010, 132, 1474–1475. doi:10.1021/ja909555d |
| 49. | Zhang, G.; Luo, Y.; Wang, Y.; Zhang, L. Angew. Chem., Int. Ed. 2011, 50, 4450–4454. doi:10.1002/anie.201100293 |
| 50. | Ball, L. T.; Lloyd-Jones, G. C.; Russell, C. A. Chem. – Eur. J. 2012, 18, 2931–2937. doi:10.1002/chem.201103061 |
| 51. | Zhang, R.; Xu, Q.; Chen, K.; Gu, P.; Shi, M. Eur. J. Org. Chem. 2013, 7366–7371. doi:10.1002/ejoc.201300896 |
| 52. | Zhu, S.; Ye, L.; Wu, W.; Jiang, H. Tetrahedron 2013, 69, 10375–10383. doi:10.1016/j.tet.2013.09.097 |
| 1. | Arcadi, A. Chem. Rev. 2008, 108, 3266–3325. doi:10.1021/cr068435d |
| 2. | Hashmi, A. S. K.; Rudolph, M. Chem. Soc. Rev. 2008, 37, 1766–1775. doi:10.1039/b615629k |
| 3. | Li, Z.; Brouwer, C.; He, C. Chem. Rev. 2008, 108, 3239–3265. doi:10.1021/cr068434l |
| 4. | Marion, N.; Nolan, S. P. Chem. Soc. Rev. 2008, 37, 1776–1782. doi:10.1039/b711132k |
| 5. | Patil, N. T.; Yamamoto, Y. Chem. Rev. 2008, 108, 3395–3442. doi:10.1021/cr050041j |
| 6. | Corma, A.; Leyva-Pérez, A.; Sabater, M. J. Chem. Rev. 2011, 111, 1657–1712. doi:10.1021/cr100414u |
| 7. | Huang, H.; Zhou, Y.; Liu, H. Beilstein J. Org. Chem. 2011, 7, 897–936. doi:10.3762/bjoc.7.103 |
| 8. | Rudolph, M.; Hashmi, A. S. K. Chem. Soc. Rev. 2012, 41, 2448–2462. doi:10.1039/C1CS15279C |
| 9. | Abbiati, G.; Marinelli, F.; Rossi, E.; Arcadi, A. Isr. J. Chem. 2013, 53, 856–868. doi:10.1002/ijch.201300040 |
| 10. | Chiarucci, M.; Bandini, M. Beilstein J. Org. Chem. 2013, 9, 2586–2614. doi:10.3762/bjoc.9.294 |
| 11. | López, F.; Mascareñas, J. L. Beilstein J. Org. Chem. 2013, 9, 2250–2264. doi:10.3762/bjoc.9.264 |
| 12. | Nunes dos Santos Comprido, L.; Hashmi, A. S. K. Isr. J. Chem. 2013, 53, 883–891. doi:10.1002/ijch.201300072 |
| 13. | Ohno, H. Isr. J. Chem. 2013, 53, 869–882. doi:10.1002/ijch.201300054 |
| 14. | Abbiati, G.; Rossi, E. Beilstein J. Org. Chem. 2014, 10, 481–513. doi:10.3762/bjoc.10.46 |
| 15. | Muratore, M. E.; Homs, A.; Obradors, C.; Echavarren, A. M. Chem. – Asian J. 2014, 9, 3066–3082. doi:10.1002/asia.201402395 |
| 16. | Xie, J.; Pan, C.; Abdukader, A.; Zhu, C. Chem. Soc. Rev. 2014, 43, 5245–5256. doi:10.1039/C4CS00004H |
| 17. | Yang, W.; Hashmi, A. S. K. Chem. Soc. Rev. 2014, 43, 2941–2955. doi:10.1039/c3cs60441a |
| 18. | Dorel, R.; Echavarren, A. M. Chem. Rev. 2015, 115, 9028–9072. doi:10.1021/cr500691k |
| 19. | Qian, D.; Zhang, J. Chem. Soc. Rev. 2015, 44, 677–698. doi:10.1039/C4CS00304G |
| 20. | Soriano, E.; Marco-Contelles, J. Acc. Chem. Res. 2009, 42, 1026–1036. doi:10.1021/ar800200m |
| 21. | Gaillard, S.; Cazin, C. S. J.; Nolan, S. P. Acc. Chem. Res. 2012, 45, 778–787. doi:10.1021/ar200188f |
| 22. | Alcaide, B.; Almendros, P. Acc. Chem. Res. 2014, 47, 939–952. doi:10.1021/ar4002558 |
| 23. | Fensterbank, L.; Malacria, M. Acc. Chem. Res. 2014, 47, 953–965. doi:10.1021/ar4002334 |
| 24. | Fürstner, A. Acc. Chem. Res. 2014, 47, 925–938. doi:10.1021/ar4001789 |
| 25. | Hashmi, A. S. K. Acc. Chem. Res. 2014, 47, 864–876. doi:10.1021/ar500015k |
| 26. | Obradors, C.; Echavarren, A. M. Acc. Chem. Res. 2014, 47, 902–912. doi:10.1021/ar400174p |
| 27. | Wang, Y.-M.; Lackner, A. D.; Toste, F. D. Acc. Chem. Res. 2014, 47, 889–901. doi:10.1021/ar400188g |
| 28. | Yeom, H.-S.; Shin, S. Acc. Chem. Res. 2014, 47, 966–977. doi:10.1021/ar4001839 |
| 29. | Zhang, D.-H.; Tang, X.-Y.; Shi, M. Acc. Chem. Res. 2014, 47, 913–924. doi:10.1021/ar400159r |
| 30. | Zhang, L. Acc. Chem. Res. 2014, 47, 877–888. doi:10.1021/ar400181x |
| 69. | Ball, L. T.; Lloyd-Jones, G. C.; Russell, C. A. J. Am. Chem. Soc. 2014, 136, 254–264. doi:10.1021/ja408712e |
| 69. | Ball, L. T.; Lloyd-Jones, G. C.; Russell, C. A. J. Am. Chem. Soc. 2014, 136, 254–264. doi:10.1021/ja408712e |
| 94. | Canac, Y.; Soleilhavoup, M.; Conejero, S.; Bertrand, G. J. Organomet. Chem. 2004, 689, 3857–3865. doi:10.1016/j.jorganchem.2004.02.005 |
| 95. | Melaimi, M.; Soleilhavoup, M.; Bertrand, G. Angew. Chem., Int. Ed. 2010, 49, 8810–8849. doi:10.1002/anie.201000165 |
| 96. | Martin, D.; Melaimi, M.; Soleilhavoup, M.; Bertrand, G. Organometallics 2011, 30, 5304–5313. doi:10.1021/om200650x |
| 68. | Ball, L. T.; Lloyd-Jones, G. C.; Russell, C. A. Science 2012, 337, 1644–1648. doi:10.1126/science.1225709 |
| 69. | Ball, L. T.; Lloyd-Jones, G. C.; Russell, C. A. J. Am. Chem. Soc. 2014, 136, 254–264. doi:10.1021/ja408712e |
| 98. | Zeng, X.; Frey, G. D.; Kinjo, R.; Donnadieu, B.; Bertrand, G. J. Am. Chem. Soc. 2009, 131, 8690–8696. doi:10.1021/ja902051m |
| 99. | Kinjo, R.; Donnadieu, B.; Bertrand, G. Angew. Chem., Int. Ed. 2011, 50, 5560–5563. doi:10.1002/anie.201100740 |
| 100. | López-Gómez, M. J.; Martin, D.; Bertrand, G. Chem. Commun. 2013, 49, 4483–4485. doi:10.1039/c3cc41279b |
| 101. | Hu, X.; Martin, D.; Melaimi, M.; Bertrand, G. J. Am. Chem. Soc. 2014, 136, 13594–13597. doi:10.1021/ja507788r |
| 102. | Jin, L.; Weinberger, D. S.; Melaimi, M.; Moore, C. E.; Rheingold, A. L.; Bertrand, G. Angew. Chem., Int. Ed. 2014, 53, 9059–9063. doi:10.1002/anie.201404665 |
| 37. | For selected reviews on gold-catalyzed oxidative C–C coupling reactions, see references [38-40]. |
| 38. | Hopkinson, M. N.; Gee, A. D.; Gouverneur, V. Chem. – Eur. J. 2011, 17, 8248–8262. doi:10.1002/chem.201100736 |
| 39. | Wegner, H. A.; Auzias, M. Angew. Chem., Int. Ed. 2011, 50, 8236–8247. doi:10.1002/anie.201101603 |
| 40. | Brand, J. P.; Li, Y.; Waser, J. Isr. J. Chem. 2013, 53, 901–910. doi:10.1002/ijch.201300044 |
| 41. | For recent examples of oxidative heteroarylation of alkene through gold(I)/gold(III) catalytic cycle, see references [42-52]. |
| 42. | Zhang, G.; Peng, Y.; Cui, L.; Zhang, L. Angew. Chem., Int. Ed. 2009, 48, 3112–3115. doi:10.1002/anie.200900585 |
| 43. | Ball, L. T.; Green, M.; Lloyd-Jones, G. C.; Russell, C. A. Org. Lett. 2010, 12, 4724–4727. doi:10.1021/ol1019162 |
| 44. | Brenzovich, W. E., Jr.; Benitez, D.; Lackner, A. D.; Shunatona, H. P.; Tkatchouk, E.; Goddard, W. A., III; Toste, F. D. Angew. Chem., Int. Ed. 2010, 49, 5519–5522. doi:10.1002/anie.201002739 |
| 45. | Brenzovich, W. E., Jr.; Brazeau, J.-F.; Toste, F. D. Org. Lett. 2010, 12, 4728–4731. doi:10.1021/ol102194c |
| 46. | Hopkinson, M. N.; Tessier, A.; Salisbury, A.; Giuffredi, G. T.; Combettes, L. E.; Gee, A. D.; Gouverneur, V. Chem. – Eur. J. 2010, 16, 4739–4743. doi:10.1002/chem.201000322 |
| 47. | Melhado, A. D.; Brenzovich, W. E., Jr.; Lackner, A. D.; Toste, F. D. J. Am. Chem. Soc. 2010, 132, 8885–8887. doi:10.1021/ja1034123 |
| 48. | Zhang, G.; Cui, L.; Wang, Y.; Zhang, L. J. Am. Chem. Soc. 2010, 132, 1474–1475. doi:10.1021/ja909555d |
| 49. | Zhang, G.; Luo, Y.; Wang, Y.; Zhang, L. Angew. Chem., Int. Ed. 2011, 50, 4450–4454. doi:10.1002/anie.201100293 |
| 50. | Ball, L. T.; Lloyd-Jones, G. C.; Russell, C. A. Chem. – Eur. J. 2012, 18, 2931–2937. doi:10.1002/chem.201103061 |
| 51. | Zhang, R.; Xu, Q.; Chen, K.; Gu, P.; Shi, M. Eur. J. Org. Chem. 2013, 7366–7371. doi:10.1002/ejoc.201300896 |
| 52. | Zhu, S.; Ye, L.; Wu, W.; Jiang, H. Tetrahedron 2013, 69, 10375–10383. doi:10.1016/j.tet.2013.09.097 |
| 53. | For recent examples of gold-catalyzed C–C bond forming oxidative coupling reaction, see references [54-66]. |
| 54. | Jones, C. J.; Taube, D.; Ziatdinov, V. R.; Periana, R. A.; Nielsen, R. J.; Oxgaard, J.; Goddard, W. A., III. Angew. Chem., Int. Ed. 2004, 43, 4626–4629. doi:10.1002/anie.200461055 |
| 55. | Wegner, H. A.; Ahles, S.; Neuburger, M. Chem. – Eur. J. 2008, 14, 11310–11313. doi:10.1002/chem.200801848 |
| 56. | Brand, J. P.; Charpentier, J.; Waser, J. Angew. Chem., Int. Ed. 2009, 48, 9346–9349. doi:10.1002/anie.200905419 |
| 57. | Cui, L.; Zhang, G.; Zhang, L. Bioorg. Med. Chem. Lett. 2009, 19, 3884–3887. doi:10.1016/j.bmcl.2009.03.127 |
| 58. | Peng, Y.; Cui, L.; Zhang, G.; Zhang, L. J. Am. Chem. Soc. 2009, 131, 5062–5063. doi:10.1021/ja901048w |
| 59. | Brand, J. P.; Waser, J. Angew. Chem., Int. Ed. 2010, 49, 7304–7307. doi:10.1002/anie.201003179 |
| 60. | de Haro, T.; Nevado, C. J. Am. Chem. Soc. 2010, 132, 1512–1513. doi:10.1021/ja909726h |
| 61. | Hopkinson, M. N.; Ross, J. E.; Giuffredi, G. T.; Gee, A. D.; Gouverneur, V. Org. Lett. 2010, 12, 4904–4907. doi:10.1021/ol102061k |
| 62. | Qian, D.; Zhang, J. Beilstein J. Org. Chem. 2011, 7, 808–812. doi:10.3762/bjoc.7.92 |
| 63. | Brand, J. P.; Chevalley, C.; Scopelliti, R.; Waser, J. Chem. – Eur. J. 2012, 18, 5655–5666. doi:10.1002/chem.201200200 |
| 64. | Levin, M. D.; Toste, F. D. Angew. Chem., Int. Ed. 2014, 53, 6211–6215. doi:10.1002/anie.201402924 |
| 65. | Ma, Y.; Zhang, S.; Yang, S.; Song, F.; You, J. Angew. Chem., Int. Ed. 2014, 53, 7870–7874. doi:10.1002/anie.201402475 |
| 66. | Peng, H.; Xi, Y.; Ronaghi, N.; Dong, B.; Akhmedov, N. G.; Shi, X. J. Am. Chem. Soc. 2014, 136, 13174–13177. doi:10.1021/ja5078365 |
| 67. | For gold-catalyzed oxidative C–H arylation of arenes, see references [68-72]. |
| 68. | Ball, L. T.; Lloyd-Jones, G. C.; Russell, C. A. Science 2012, 337, 1644–1648. doi:10.1126/science.1225709 |
| 69. | Ball, L. T.; Lloyd-Jones, G. C.; Russell, C. A. J. Am. Chem. Soc. 2014, 136, 254–264. doi:10.1021/ja408712e |
| 70. | Kar, A.; Mangu, N.; Kaiser, H. M.; Beller, M.; Tse, M. K. Chem. Commun. 2008, 386–388. doi:10.1039/B714928J |
| 71. | Kar, A.; Mangu, N.; Kaiser, H. M.; Tse, M. K. J. Organomet. Chem. 2009, 694, 524–537. doi:10.1016/j.jorganchem.2008.11.016 |
| 72. | Wu, Q.; Du, C.; Huang, Y.; Liu, X.; Long, Z.; Song, F.; You, J. Chem. Sci. 2015, 6, 288–293. doi:10.1039/C4SC02070G |
| 73. | For selected mechanistic investigations on gold(I)/gold(III) redox processes, see references [74-84]. |
| 74. | Mankad, N. P.; Toste, F. D. J. Am. Chem. Soc. 2010, 132, 12859–12861. doi:10.1021/ja106257n |
| 75. | Tkatchouk, E.; Mankad, N. P.; Benitez, D.; Goddard, W. A., III; Toste, F. D. J. Am. Chem. Soc. 2011, 133, 14293–14300. doi:10.1021/ja2012627 |
| 76. | Hofer, M.; Nevado, C. Eur. J. Inorg. Chem. 2012, 1338–1341. doi:10.1002/ejic.201100956 |
| 77. | Cambeiro, X. C.; Boorman, T. C.; Lu, P.; Larrosa, I. Angew. Chem., Int. Ed. 2013, 52, 1781–1784. doi:10.1002/anie.201209007 |
| 78. | Hofer, M.; Nevado, C. Tetrahedron 2013, 69, 5751–5757. doi:10.1016/j.tet.2013.04.029 |
| 79. | Hofer, M.; Gomez-Bengoa, E.; Nevado, C. Organometallics 2014, 33, 1328–1332. doi:10.1021/om400884s |
| 80. | Winston, M. S.; Wolf, W. J.; Toste, F. D. J. Am. Chem. Soc. 2014, 136, 7777–7782. doi:10.1021/ja503974x |
| 81. | Wolf, W. J.; Winston, M. S.; Toste, F. D. Nat. Chem. 2014, 6, 159–164. doi:10.1038/nchem.1822 |
| 103. | Electron-rich arenes show the reactivity toward dehydrogenative homo- and cross-coupling reaction under oxidative conditions without metal oxidants. For related examples, see references [104-107]. |
| 104. | Ito, H.; Ueda, K.; Itami, K. Cross-Dehydrogenative-Coupling Reactions without Metals. In From C–H to C–C Bonds: Cross-Dehydrogenative-Coupling; Li, C.-J., Ed.; The Royal Society of Chemistry, 2015; pp 153–196. doi:10.1039/9781782620082-00153 |
| 105. | Jean, A.; Cantat, J.; Bérard, D.; Bouchu, D.; Canesi, S. Org. Lett. 2007, 9, 2553–2556. doi:10.1021/ol070941h |
| 106. | Kita, Y.; Morimoto, K.; Ito, M.; Ogawa, C.; Goto, A.; Dohi, T. J. Am. Chem. Soc. 2009, 131, 1668–1669. doi:10.1021/ja808940n |
| 107. | Morimoto, K.; Yamaoka, N.; Ogawa, C.; Nakae, T.; Fujioka, H.; Dohi, T.; Kita, Y. Org. Lett. 2010, 12, 3804–3807. doi:10.1021/ol101498r |
| 84. | Yoshidomi, T.; Segawa, Y.; Itami, K. Chem. Commun. 2013, 49, 5648–5650. doi:10.1039/c3cc42655f |
| 31. | For ligand effects in gold-catalyzed reactions, see references [32-36]. |
| 32. | Gorin, D. J.; Sherry, B. D.; Toste, F. D. Chem. Rev. 2008, 108, 3351–3378. doi:10.1021/cr068430g |
| 33. | Alcarazo, M.; Stork, T.; Anoop, A.; Thiel, W.; Fürstner, A. Angew. Chem., Int. Ed. 2010, 49, 2542–2546. doi:10.1002/anie.200907194 |
| 34. | Nolan, S. P. Acc. Chem. Res. 2011, 44, 91–100. doi:10.1021/ar1000764 |
| 35. | Cera, G.; Bandini, M. Isr. J. Chem. 2013, 53, 848–855. doi:10.1002/ijch.201300029 |
| 36. | Gatineau, D.; Goddard, J.-P.; Mouriès-Mansuy, V.; Fensterbank, L. Isr. J. Chem. 2013, 53, 892–900. doi:10.1002/ijch.201300059 |
| 108. | For palladium-catalyzed C–H arylation of isoxazole, see references [109-111]. |
| 109. | Chiong, H. A.; Daugulis, O. Org. Lett. 2007, 9, 1449–1451. doi:10.1021/ol0702324 |
| 110. | Fall, Y.; Reynaud, C.; Doucet, H.; Santelli, M. Eur. J. Org. Chem. 2009, 4041–4050. doi:10.1002/ejoc.200900309 |
| 111. | Roger, J.; Požgan, F.; Doucet, H. Adv. Synth. Catal. 2010, 352, 696–710. doi:10.1002/adsc.200900793 |
| 86. | Raubenheimer, H. G.; Toerien, J. G.; Kruger, G. J.; Otte, R.; van Zyl, W.; Olivier, P. J. Organomet. Chem. 1994, 466, 291–295. doi:10.1016/0022-328X(94)88058-1 |
| 87. | Strasser, C. E.; Stander-Grobler, E.; Schuster, O.; Cronje, S.; Raubenheimer, H. G. Eur. J. Inorg. Chem. 2009, 1905–1912. doi:10.1002/ejic.200801180 |
| 88. | Roselló-Merino, M.; Díez, J.; Conejero, S. Chem. Commun. 2010, 46, 9247–9249. doi:10.1039/c0cc03935g |
| 89. | Hollóczki, O.; Nyulászi, L. J. Org. Chem. 2008, 73, 4794–4799. doi:10.1021/jo8000035 |
| 90. | Kassaee, M. Z.; Shakib, F. A.; Momeni, M. R.; Ghambarian, M.; Musavi, S. M. Tetrahedron 2009, 65, 10093–10098. doi:10.1016/j.tet.2009.09.043 |
| 91. | Tukov, A. A.; Normand, A. T.; Nechaev, M. S. Dalton Trans. 2009, 7015–7028. doi:10.1039/b906969k |
| 92. | Wang, L.; Kong, L.; Li, Y.; Ganguly, R.; Kinjo, R. Chem. Commun. 2015, 51, 12419–12422. doi:10.1039/C5CC04091D |
| 85. | For the synthesis and DFT calculation of pyridylidene, see references [86-92]. |
| 86. | Raubenheimer, H. G.; Toerien, J. G.; Kruger, G. J.; Otte, R.; van Zyl, W.; Olivier, P. J. Organomet. Chem. 1994, 466, 291–295. doi:10.1016/0022-328X(94)88058-1 |
| 87. | Strasser, C. E.; Stander-Grobler, E.; Schuster, O.; Cronje, S.; Raubenheimer, H. G. Eur. J. Inorg. Chem. 2009, 1905–1912. doi:10.1002/ejic.200801180 |
| 88. | Roselló-Merino, M.; Díez, J.; Conejero, S. Chem. Commun. 2010, 46, 9247–9249. doi:10.1039/c0cc03935g |
| 89. | Hollóczki, O.; Nyulászi, L. J. Org. Chem. 2008, 73, 4794–4799. doi:10.1021/jo8000035 |
| 90. | Kassaee, M. Z.; Shakib, F. A.; Momeni, M. R.; Ghambarian, M.; Musavi, S. M. Tetrahedron 2009, 65, 10093–10098. doi:10.1016/j.tet.2009.09.043 |
| 91. | Tukov, A. A.; Normand, A. T.; Nechaev, M. S. Dalton Trans. 2009, 7015–7028. doi:10.1039/b906969k |
| 92. | Wang, L.; Kong, L.; Li, Y.; Ganguly, R.; Kinjo, R. Chem. Commun. 2015, 51, 12419–12422. doi:10.1039/C5CC04091D |
| 93. | For the synthesis and characterization of nonclassical carbenes, see references [94-96]. |
| 94. | Canac, Y.; Soleilhavoup, M.; Conejero, S.; Bertrand, G. J. Organomet. Chem. 2004, 689, 3857–3865. doi:10.1016/j.jorganchem.2004.02.005 |
| 95. | Melaimi, M.; Soleilhavoup, M.; Bertrand, G. Angew. Chem., Int. Ed. 2010, 49, 8810–8849. doi:10.1002/anie.201000165 |
| 96. | Martin, D.; Melaimi, M.; Soleilhavoup, M.; Bertrand, G. Organometallics 2011, 30, 5304–5313. doi:10.1021/om200650x |
| 97. | For nonclassical carbene-gold complexes in catalytic reactions, see references [98-102] |
| 98. | Zeng, X.; Frey, G. D.; Kinjo, R.; Donnadieu, B.; Bertrand, G. J. Am. Chem. Soc. 2009, 131, 8690–8696. doi:10.1021/ja902051m |
| 99. | Kinjo, R.; Donnadieu, B.; Bertrand, G. Angew. Chem., Int. Ed. 2011, 50, 5560–5563. doi:10.1002/anie.201100740 |
| 100. | López-Gómez, M. J.; Martin, D.; Bertrand, G. Chem. Commun. 2013, 49, 4483–4485. doi:10.1039/c3cc41279b |
| 101. | Hu, X.; Martin, D.; Melaimi, M.; Bertrand, G. J. Am. Chem. Soc. 2014, 136, 13594–13597. doi:10.1021/ja507788r |
| 102. | Jin, L.; Weinberger, D. S.; Melaimi, M.; Moore, C. E.; Rheingold, A. L.; Bertrand, G. Angew. Chem., Int. Ed. 2014, 53, 9059–9063. doi:10.1002/anie.201404665 |
| 68. | Ball, L. T.; Lloyd-Jones, G. C.; Russell, C. A. Science 2012, 337, 1644–1648. doi:10.1126/science.1225709 |
| 69. | Ball, L. T.; Lloyd-Jones, G. C.; Russell, C. A. J. Am. Chem. Soc. 2014, 136, 254–264. doi:10.1021/ja408712e |
| 70. | Kar, A.; Mangu, N.; Kaiser, H. M.; Beller, M.; Tse, M. K. Chem. Commun. 2008, 386–388. doi:10.1039/B714928J |
| 71. | Kar, A.; Mangu, N.; Kaiser, H. M.; Tse, M. K. J. Organomet. Chem. 2009, 694, 524–537. doi:10.1016/j.jorganchem.2008.11.016 |
| 72. | Wu, Q.; Du, C.; Huang, Y.; Liu, X.; Long, Z.; Song, F.; You, J. Chem. Sci. 2015, 6, 288–293. doi:10.1039/C4SC02070G |
| 74. | Mankad, N. P.; Toste, F. D. J. Am. Chem. Soc. 2010, 132, 12859–12861. doi:10.1021/ja106257n |
| 75. | Tkatchouk, E.; Mankad, N. P.; Benitez, D.; Goddard, W. A., III; Toste, F. D. J. Am. Chem. Soc. 2011, 133, 14293–14300. doi:10.1021/ja2012627 |
| 76. | Hofer, M.; Nevado, C. Eur. J. Inorg. Chem. 2012, 1338–1341. doi:10.1002/ejic.201100956 |
| 77. | Cambeiro, X. C.; Boorman, T. C.; Lu, P.; Larrosa, I. Angew. Chem., Int. Ed. 2013, 52, 1781–1784. doi:10.1002/anie.201209007 |
| 78. | Hofer, M.; Nevado, C. Tetrahedron 2013, 69, 5751–5757. doi:10.1016/j.tet.2013.04.029 |
| 79. | Hofer, M.; Gomez-Bengoa, E.; Nevado, C. Organometallics 2014, 33, 1328–1332. doi:10.1021/om400884s |
| 80. | Winston, M. S.; Wolf, W. J.; Toste, F. D. J. Am. Chem. Soc. 2014, 136, 7777–7782. doi:10.1021/ja503974x |
| 81. | Wolf, W. J.; Winston, M. S.; Toste, F. D. Nat. Chem. 2014, 6, 159–164. doi:10.1038/nchem.1822 |
| 82. | For our contributions to the synthesis, complexation of pyridylidene ligands, see references [83] and [84]. |
| 83. | Hata, K.; Segawa, Y.; Itami, K. Chem. Commun. 2012, 48, 6642–6644. doi:10.1039/c2cc33184e |
| 84. | Yoshidomi, T.; Segawa, Y.; Itami, K. Chem. Commun. 2013, 49, 5648–5650. doi:10.1039/c3cc42655f |
| 82. | For our contributions to the synthesis, complexation of pyridylidene ligands, see references [83] and [84]. |
| 83. | Hata, K.; Segawa, Y.; Itami, K. Chem. Commun. 2012, 48, 6642–6644. doi:10.1039/c2cc33184e |
| 84. | Yoshidomi, T.; Segawa, Y.; Itami, K. Chem. Commun. 2013, 49, 5648–5650. doi:10.1039/c3cc42655f |
| 68. | Ball, L. T.; Lloyd-Jones, G. C.; Russell, C. A. Science 2012, 337, 1644–1648. doi:10.1126/science.1225709 |
| 69. | Ball, L. T.; Lloyd-Jones, G. C.; Russell, C. A. J. Am. Chem. Soc. 2014, 136, 254–264. doi:10.1021/ja408712e |
| 83. | Hata, K.; Segawa, Y.; Itami, K. Chem. Commun. 2012, 48, 6642–6644. doi:10.1039/c2cc33184e |
| 37. | For selected reviews on gold-catalyzed oxidative C–C coupling reactions, see references [38-40]. |
| 38. | Hopkinson, M. N.; Gee, A. D.; Gouverneur, V. Chem. – Eur. J. 2011, 17, 8248–8262. doi:10.1002/chem.201100736 |
| 39. | Wegner, H. A.; Auzias, M. Angew. Chem., Int. Ed. 2011, 50, 8236–8247. doi:10.1002/anie.201101603 |
| 40. | Brand, J. P.; Li, Y.; Waser, J. Isr. J. Chem. 2013, 53, 901–910. doi:10.1002/ijch.201300044 |
| 54. | Jones, C. J.; Taube, D.; Ziatdinov, V. R.; Periana, R. A.; Nielsen, R. J.; Oxgaard, J.; Goddard, W. A., III. Angew. Chem., Int. Ed. 2004, 43, 4626–4629. doi:10.1002/anie.200461055 |
| 55. | Wegner, H. A.; Ahles, S.; Neuburger, M. Chem. – Eur. J. 2008, 14, 11310–11313. doi:10.1002/chem.200801848 |
| 56. | Brand, J. P.; Charpentier, J.; Waser, J. Angew. Chem., Int. Ed. 2009, 48, 9346–9349. doi:10.1002/anie.200905419 |
| 57. | Cui, L.; Zhang, G.; Zhang, L. Bioorg. Med. Chem. Lett. 2009, 19, 3884–3887. doi:10.1016/j.bmcl.2009.03.127 |
| 58. | Peng, Y.; Cui, L.; Zhang, G.; Zhang, L. J. Am. Chem. Soc. 2009, 131, 5062–5063. doi:10.1021/ja901048w |
| 59. | Brand, J. P.; Waser, J. Angew. Chem., Int. Ed. 2010, 49, 7304–7307. doi:10.1002/anie.201003179 |
| 60. | de Haro, T.; Nevado, C. J. Am. Chem. Soc. 2010, 132, 1512–1513. doi:10.1021/ja909726h |
| 61. | Hopkinson, M. N.; Ross, J. E.; Giuffredi, G. T.; Gee, A. D.; Gouverneur, V. Org. Lett. 2010, 12, 4904–4907. doi:10.1021/ol102061k |
| 62. | Qian, D.; Zhang, J. Beilstein J. Org. Chem. 2011, 7, 808–812. doi:10.3762/bjoc.7.92 |
| 63. | Brand, J. P.; Chevalley, C.; Scopelliti, R.; Waser, J. Chem. – Eur. J. 2012, 18, 5655–5666. doi:10.1002/chem.201200200 |
| 64. | Levin, M. D.; Toste, F. D. Angew. Chem., Int. Ed. 2014, 53, 6211–6215. doi:10.1002/anie.201402924 |
| 65. | Ma, Y.; Zhang, S.; Yang, S.; Song, F.; You, J. Angew. Chem., Int. Ed. 2014, 53, 7870–7874. doi:10.1002/anie.201402475 |
| 66. | Peng, H.; Xi, Y.; Ronaghi, N.; Dong, B.; Akhmedov, N. G.; Shi, X. J. Am. Chem. Soc. 2014, 136, 13174–13177. doi:10.1021/ja5078365 |
| 69. | Ball, L. T.; Lloyd-Jones, G. C.; Russell, C. A. J. Am. Chem. Soc. 2014, 136, 254–264. doi:10.1021/ja408712e |
| 83. | Hata, K.; Segawa, Y.; Itami, K. Chem. Commun. 2012, 48, 6642–6644. doi:10.1039/c2cc33184e |
| 68. | Ball, L. T.; Lloyd-Jones, G. C.; Russell, C. A. Science 2012, 337, 1644–1648. doi:10.1126/science.1225709 |
| 69. | Ball, L. T.; Lloyd-Jones, G. C.; Russell, C. A. J. Am. Chem. Soc. 2014, 136, 254–264. doi:10.1021/ja408712e |
| 70. | Kar, A.; Mangu, N.; Kaiser, H. M.; Beller, M.; Tse, M. K. Chem. Commun. 2008, 386–388. doi:10.1039/B714928J |
| 71. | Kar, A.; Mangu, N.; Kaiser, H. M.; Tse, M. K. J. Organomet. Chem. 2009, 694, 524–537. doi:10.1016/j.jorganchem.2008.11.016 |
| 72. | Wu, Q.; Du, C.; Huang, Y.; Liu, X.; Long, Z.; Song, F.; You, J. Chem. Sci. 2015, 6, 288–293. doi:10.1039/C4SC02070G |
| 67. | For gold-catalyzed oxidative C–H arylation of arenes, see references [68-72]. |
| 68. | Ball, L. T.; Lloyd-Jones, G. C.; Russell, C. A. Science 2012, 337, 1644–1648. doi:10.1126/science.1225709 |
| 69. | Ball, L. T.; Lloyd-Jones, G. C.; Russell, C. A. J. Am. Chem. Soc. 2014, 136, 254–264. doi:10.1021/ja408712e |
| 70. | Kar, A.; Mangu, N.; Kaiser, H. M.; Beller, M.; Tse, M. K. Chem. Commun. 2008, 386–388. doi:10.1039/B714928J |
| 71. | Kar, A.; Mangu, N.; Kaiser, H. M.; Tse, M. K. J. Organomet. Chem. 2009, 694, 524–537. doi:10.1016/j.jorganchem.2008.11.016 |
| 72. | Wu, Q.; Du, C.; Huang, Y.; Liu, X.; Long, Z.; Song, F.; You, J. Chem. Sci. 2015, 6, 288–293. doi:10.1039/C4SC02070G |
| 73. | For selected mechanistic investigations on gold(I)/gold(III) redox processes, see references [74-84]. |
| 74. | Mankad, N. P.; Toste, F. D. J. Am. Chem. Soc. 2010, 132, 12859–12861. doi:10.1021/ja106257n |
| 75. | Tkatchouk, E.; Mankad, N. P.; Benitez, D.; Goddard, W. A., III; Toste, F. D. J. Am. Chem. Soc. 2011, 133, 14293–14300. doi:10.1021/ja2012627 |
| 76. | Hofer, M.; Nevado, C. Eur. J. Inorg. Chem. 2012, 1338–1341. doi:10.1002/ejic.201100956 |
| 77. | Cambeiro, X. C.; Boorman, T. C.; Lu, P.; Larrosa, I. Angew. Chem., Int. Ed. 2013, 52, 1781–1784. doi:10.1002/anie.201209007 |
| 78. | Hofer, M.; Nevado, C. Tetrahedron 2013, 69, 5751–5757. doi:10.1016/j.tet.2013.04.029 |
| 79. | Hofer, M.; Gomez-Bengoa, E.; Nevado, C. Organometallics 2014, 33, 1328–1332. doi:10.1021/om400884s |
| 80. | Winston, M. S.; Wolf, W. J.; Toste, F. D. J. Am. Chem. Soc. 2014, 136, 7777–7782. doi:10.1021/ja503974x |
| 81. | Wolf, W. J.; Winston, M. S.; Toste, F. D. Nat. Chem. 2014, 6, 159–164. doi:10.1038/nchem.1822 |
| 113. | In the presence of 10 mol % of AuCl(PyC), the reaction of 1b and 2a gave lower yield of 3ba (33% GC yield) with significant amount of homo-coupling product of 2a (4,4’-dibromobiphenyl) being observed. |
| 104. | Ito, H.; Ueda, K.; Itami, K. Cross-Dehydrogenative-Coupling Reactions without Metals. In From C–H to C–C Bonds: Cross-Dehydrogenative-Coupling; Li, C.-J., Ed.; The Royal Society of Chemistry, 2015; pp 153–196. doi:10.1039/9781782620082-00153 |
| 105. | Jean, A.; Cantat, J.; Bérard, D.; Bouchu, D.; Canesi, S. Org. Lett. 2007, 9, 2553–2556. doi:10.1021/ol070941h |
| 106. | Kita, Y.; Morimoto, K.; Ito, M.; Ogawa, C.; Goto, A.; Dohi, T. J. Am. Chem. Soc. 2009, 131, 1668–1669. doi:10.1021/ja808940n |
| 107. | Morimoto, K.; Yamaoka, N.; Ogawa, C.; Nakae, T.; Fujioka, H.; Dohi, T.; Kita, Y. Org. Lett. 2010, 12, 3804–3807. doi:10.1021/ol101498r |
| 68. | Ball, L. T.; Lloyd-Jones, G. C.; Russell, C. A. Science 2012, 337, 1644–1648. doi:10.1126/science.1225709 |
| 69. | Ball, L. T.; Lloyd-Jones, G. C.; Russell, C. A. J. Am. Chem. Soc. 2014, 136, 254–264. doi:10.1021/ja408712e |
| 70. | Kar, A.; Mangu, N.; Kaiser, H. M.; Beller, M.; Tse, M. K. Chem. Commun. 2008, 386–388. doi:10.1039/B714928J |
| 71. | Kar, A.; Mangu, N.; Kaiser, H. M.; Tse, M. K. J. Organomet. Chem. 2009, 694, 524–537. doi:10.1016/j.jorganchem.2008.11.016 |
| 72. | Wu, Q.; Du, C.; Huang, Y.; Liu, X.; Long, Z.; Song, F.; You, J. Chem. Sci. 2015, 6, 288–293. doi:10.1039/C4SC02070G |
| 73. | For selected mechanistic investigations on gold(I)/gold(III) redox processes, see references [74-84]. |
| 74. | Mankad, N. P.; Toste, F. D. J. Am. Chem. Soc. 2010, 132, 12859–12861. doi:10.1021/ja106257n |
| 75. | Tkatchouk, E.; Mankad, N. P.; Benitez, D.; Goddard, W. A., III; Toste, F. D. J. Am. Chem. Soc. 2011, 133, 14293–14300. doi:10.1021/ja2012627 |
| 109. | Chiong, H. A.; Daugulis, O. Org. Lett. 2007, 9, 1449–1451. doi:10.1021/ol0702324 |
| 110. | Fall, Y.; Reynaud, C.; Doucet, H.; Santelli, M. Eur. J. Org. Chem. 2009, 4041–4050. doi:10.1002/ejoc.200900309 |
| 111. | Roger, J.; Požgan, F.; Doucet, H. Adv. Synth. Catal. 2010, 352, 696–710. doi:10.1002/adsc.200900793 |
| 83. | Hata, K.; Segawa, Y.; Itami, K. Chem. Commun. 2012, 48, 6642–6644. doi:10.1039/c2cc33184e |
| 16. | Xie, J.; Pan, C.; Abdukader, A.; Zhu, C. Chem. Soc. Rev. 2014, 43, 5245–5256. doi:10.1039/C4CS00004H |
| 114. |
Gaillard, S.; Bantreil, X.; Slawin, A. M. Z.; Nolan, S. P. Dalton Trans. 2009, 6967–6971. doi:10.1039/b907109a
See for the synthesis and characterization of AuCl3(IPr) complex. |
| 114. |
Gaillard, S.; Bantreil, X.; Slawin, A. M. Z.; Nolan, S. P. Dalton Trans. 2009, 6967–6971. doi:10.1039/b907109a
See for the synthesis and characterization of AuCl3(IPr) complex. |
| 114. |
Gaillard, S.; Bantreil, X.; Slawin, A. M. Z.; Nolan, S. P. Dalton Trans. 2009, 6967–6971. doi:10.1039/b907109a
See for the synthesis and characterization of AuCl3(IPr) complex. |
| 115. | Crystallographic data reported in this manuscript have been deposited with Cambridge Crystallographic Data Centre as supplementary publication No. CCDC-1045812 [AuCl3(PyC)]. Copies of the data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html. |
| 69. | Ball, L. T.; Lloyd-Jones, G. C.; Russell, C. A. J. Am. Chem. Soc. 2014, 136, 254–264. doi:10.1021/ja408712e |
| 114. |
Gaillard, S.; Bantreil, X.; Slawin, A. M. Z.; Nolan, S. P. Dalton Trans. 2009, 6967–6971. doi:10.1039/b907109a
See for the synthesis and characterization of AuCl3(IPr) complex. |
© 2015 Hata et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)