Abstract
Over the recent years, the nucleophilic manipulation of inactivated carbon–carbon double bonds has gained remarkable credit in the chemical community. As a matter of fact, despite lower reactivity with respect to alkynyl and allenyl counterparts, chemical functionalization of isolated alkenes, via carbon- as well as hetero atom-based nucleophiles, would provide direct access to theoretically unlimited added value of molecular motifs. In this context, homogenous [Au(I)] and [Au(III)] catalysis continues to inspire developments within organic synthesis, providing reliable responses to this interrogative, by combining crucial aspects such as chemical selectivity/efficiency with mild reaction parameters. This review intends to summarize the recent progresses in the field, with particular emphasis on mechanistic details.

Graphical Abstract
Review
1 Introduction
Homogeneous gold catalysis is emerged as one of the most powerful means for the activation of C–C multiple bonds toward a number of complexity-oriented transformations. In this segment, gold-catalyzed addition of carbon- and heteroatom-based nucleophiles to inactivated alkenes are widely recognized as “capricious” transformations due to alkyne and allene counterparts [1-5]. However, over the past few years, tremendous developments were made, and some of the major contributes will be summarized in the present review.
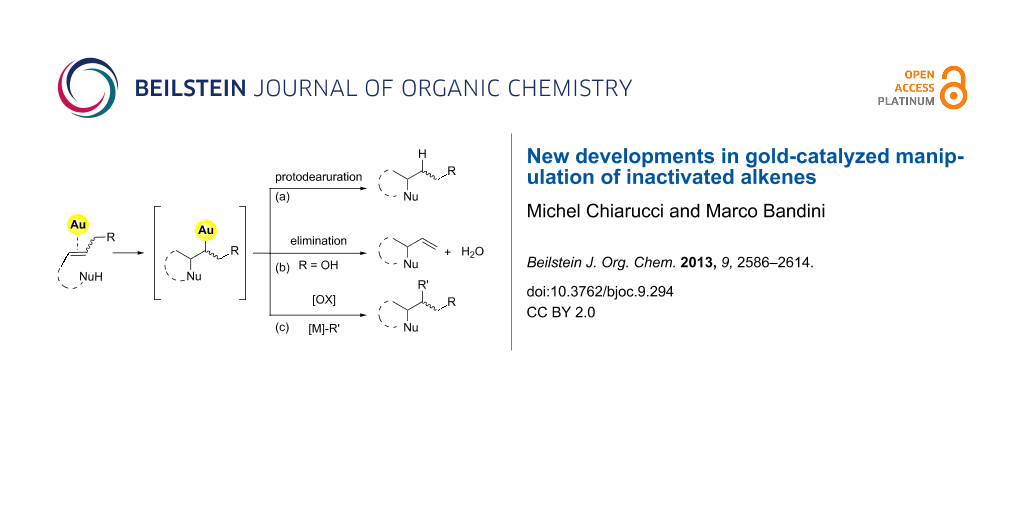
Mechanistically, it is generally accepted that the gold-catalyzed nucleophilic addition to alkenes proceeds through three elementary steps: i) activation of the C–C double bond by gold coordination. ii) anti-Nucleophilic attack with formation of an alkylgold intermediate (outer-sphere pathway). iii) Protodeauration with formation of the product and catalyst regeneration (Figure 1).
![[1860-5397-9-294-1]](/bjoc/content/figures/1860-5397-9-294-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Elementary steps in the gold-catalyzed nucleophilic addition to olefins.
Figure 1: Elementary steps in the gold-catalyzed nucleophilic addition to olefins.
The simplified mechanistic sketch depicted in Figure 1 accounts also for the lower reactivity of alkenes with respect to different π-systems. In particular, the intrinsic inertness of alkylgold intermediate 1 (i.e. Csp3–Au bond) towards protodeauration (path a, Figure 2) determines the scarce reactivity of alkenes in nucleophilic addition reactions [6].
The use of allylic alcohols as C=C surrogates opens up an eliminative pathway for the cleavage of the C–Au bond in the intermediate 1 (Figure 2, path b). This reaction channel is accessible under mild conditions and without external activation when proper gold catalysts are employed [7,8].
Furthermore, in recent years, advances in the gold-catalyzed functionalization of alkenes rely on the use of “oxidative strategies” exploiting the [Au(I)/Au(III)] or [Au(I)Au(I)/Au(II)Au(II)] catalytic couples that can be accessed through the use of a suitable exogenous oxidant (Figure 2, path c) [9].
![[1860-5397-9-294-2]](/bjoc/content/figures/1860-5397-9-294-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Different approaches for the gold-catalyzed manipulation of inactivated alkenes.
Figure 2: Different approaches for the gold-catalyzed manipulation of inactivated alkenes.
Last but not least, the potential role of Brønsted acid co-catalysis should always be considered when metal triflates are employed [10,11]. Indeed, it was demonstrated that catalytic amounts of TfOH could catalyze some specific additions of oxygen- and nitrogen-based nucleophiles to simple alkenes with comparable efficiency/selectivity as much as some metal triflates [12,13].
In this review some selected examples of gold-catalyzed nucleophilic additions to inactivated alkenes will be discussed with particular emphasis on the corresponding reaction machinery. In the context of this review, allylic alcohols will be treated as inactivated olefins, considering that normally activated equivalents, like halides, acetates, carbonates and phosphates are employed in metal-catalyzed Tsuji–Trost type alkylation [14,15].
2 Formation of C–O bonds
2.1 Mechanistic considerations
The addition of oxygen-based nucleophiles to C–C multiple bonds is an effective and atom-economical process for the formation of new C–O bonds. In this direction numerous examples of metal and Brønsted-acid catalyzed condensations of alcohols, phenols and carboxylic acids to inactivated olefins have been reported [16].
Interestingly, He and co-workers compared the catalytic attitude of TfOH and PPh3AuOTf in the addition of various nucleophiles to alkenes, demonstrating how the gold complex and triflic acid can exhibit complementary efficiency [17]. This finding suggests that although Brønsted acid co-catalysis is a possible competing process, under suitable conditions the gold-catalyzed pathway is still the dominating process. For example by adopting the addition of p-NO2- and p-MeO-phenol to aryl and alkyl olefins as a model process, it emerged that TfOH was an effective catalyst at room temperature and led to decomposition of the starting material at higher temperatures. On the contrary, cationic [Au(I)] species exhibited comparable activity to TfOH at 85 °C (Table 1).
![[Graphic 1]](/bjoc/content/inline/1860-5397-9-294-i41.svg?max-width=637&scale=1.0)
![[Graphic 2]](/bjoc/content/inline/1860-5397-9-294-i42.svg?max-width=637&scale=1.0)
![[Graphic 3]](/bjoc/content/inline/1860-5397-9-294-i43.svg?max-width=637&scale=1.0)
This complementary behavior can be ascribed to different activation modes of the double bond. Computational investigations on the [H+]-catalyzed reaction predicted a transition state with significant carbocation character [18,19], therefore more easily subjected to substrate isomerization or degradation. Notwithstanding, gold activation was shown to occur through the formation of a η2-complex [20-24].
An important contribution to this scenario, was also provided by Ujaque and co-workers, who performed a theoretical investigation on the Me3PAuOTf catalyzed addition of phenol to ethylene [25]. The proposed catalytic cycle initiated with the exchange of the TfO− ion with the alkene reveals that the poorly coordinating anion seems to form a “loose” ion pair lying far from the metal center (Figure 3). This step was energetically favorable and the activation of the alkene by the gold cation was confirmed by elongation of the C–C double bond [26]. After an exhaustive survey of plausible reaction channels, it turned out that the phenol is involved in assisting the concerted addition/protodeauration machinery. The presence of a second molecule of phenol, acting as a proton shuttle, considerably lowered the energetic barrier for the protodeauration (i.e. rate limiting step of the overall process), which takes place through a more favorable 6-membered cyclic transition state. Analogous calculations indicate that also water is a potential proton transfer agent in the protodeauration event of the catalytic cycle.
![[1860-5397-9-294-3]](/bjoc/content/figures/1860-5397-9-294-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Computed mechanistic cycle for the gold-catalyzed alkoxylation of ethylene with PhOH.
Figure 3: Computed mechanistic cycle for the gold-catalyzed alkoxylation of ethylene with PhOH.
2.2 Selected examples
Initial reports dealing with the gold-catalyzed addition of oxygen-based nucleophiles to isolated olefins required the use of relatively acidic nucleophiles.
In their seminal works, Yang and He reported on the Ph3PAuOTf assisted Markovnikov-like addition of phenols and carboxylic acids to C=C under mild reaction conditions (Scheme 1) [27]. Electron-rich and electron-poor phenols reacted smoothly in toluene (85 °C) with only 1 mol % of catalyst loading (Scheme 1a). Contrarily, carboxylic acids required a higher loading of catalyst (5 mol %), but an acceptable yield was obtained even when sterically demanding 2-methylpropionic acid was employed as the nucleophile (Scheme 1b).
![[1860-5397-9-294-i1]](/bjoc/content/inline/1860-5397-9-294-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: [Au(I)]-catalyzed addition of phenols and carboxylic acids to alkenes.
Scheme 1: [Au(I)]-catalyzed addition of phenols and carboxylic acids to alkenes.
Li and co-workers exploited the [Au(III)]-catalyzed addition of phenols and naphthols to conjugated dienes realizing an efficient synthesis of dihydrobenzofuran derivatives 3 (Scheme 2a) [28]. The protocol was assumed to proceed via a two-step mechanism: involving an initial hydroarylation of the double bond followed by an intramolecular phenol addition (Scheme 2b).
![[1860-5397-9-294-i2]](/bjoc/content/inline/1860-5397-9-294-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: [Au(III)] catalyzed annulations of phenols and naphthols with dienes.
Scheme 2: [Au(III)] catalyzed annulations of phenols and naphthols with dienes.
Shortly after, Zhang and Corma considerably expanded the scope of this transformation to aliphatic alcohols (Scheme 3) [29]. Under these conditions, the addition of primary and secondary alcohols to aryl and alkyl olefins 4 took place efficiently, with good yield and regioselectivity. However, poor diastereoselectivity was recorded in the presence of secondary alcohols.
![[1860-5397-9-294-i3]](/bjoc/content/inline/1860-5397-9-294-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: [Au(III)]-catalyzed addition of aliphatic alcohols to alkenes.
Scheme 3: [Au(III)]-catalyzed addition of aliphatic alcohols to alkenes.
The efficiency of the process relied on the stabilization of cationic [Au(III)] species by using a catalytic amount CuCl2 (16 mol %), which prevented gold deactivation via parasitic reductive side reactions [30,31].
Moreover, recent advances in the alkoxylation of olefins enabled the use of simple dimethyl acetals 6 in the carboalkoxylation of alkenes (Scheme 4) [32]. A 1:2 mixture of [picAuCl2] (7) and AgNTf2 efficiently catalyzed the double functionalization of aryl alkenes 4 in good yields and mild conditions. Dialkyl substituted olefins afforded the product only in moderate yields whereas monoalkyl olefins were completely unreactive under the optimized conditions. Although an activation of the acetals by the gold catalyst cannot be ruled out, a reaction pathway involving gold activation of the alkene, followed by addition of the alkyl gold intermediate 10 to the activated carbonyl compound 9 was also hypothesized (Scheme 4b).
![[1860-5397-9-294-i4]](/bjoc/content/inline/1860-5397-9-294-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: [Au(III)]-catalyzed carboalkoxylation of alkenes with dimethyl acetals 6.
Scheme 4: [Au(III)]-catalyzed carboalkoxylation of alkenes with dimethyl acetals 6.
3 Hydroamination of olefins
3.1 Mechanistic considerations
Due to the ubiquity of the C–N bond in organic compounds the development of efficient catalytic systems for the hydroamination of olefins is of particular significance from a practical point of view [33]. Although many metal and Brønsted acid assisted processes have been documented the high functional group tolerance of gold complexes combined with their high efficiency in the electrophilic activation of C–C multiple bonds have made gold catalysis an important tool for the hydroamination of alkenes. Detailed computational studies on the addition of benzyl carbamate to dienes revealed the protodeauration as the rate-determining step of the reaction [34]. Mechanistically, the initial stage was determined dealing with the energetically favourable substitution of the TfO− ligand from the gold coordination sphere with the diene (Figure 4). Preferred coordination geometry is the η2-type, with the gold cation coordinating to a single double bond. Either direct coordination of the nucleophile to the gold cation, or reaction of the PH3AuOTf with the carbamate to deliver TfOH and PH3AuNHCOOBn [35] were ruled out by experimental and computational observations. Differently, addition of the nucleophile to the gold activated double bond took place with Markovnikov regioselectivity affording the intermediate 11. At this point many possible reaction pathways were evaluated to shed light on the real mechanism of the protodeauration step. Interestingly the reaction profile with the lowest activation energy was distinguished in the TfO− promoted tautomerization of 11 to form 12 followed by direct proton transfer to afford the product 13 and regeneration of the catalyst.
![[1860-5397-9-294-4]](/bjoc/content/figures/1860-5397-9-294-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Postulated mechanism for the [Au(I)]-catalyzed hydroamination of olefins.
Figure 4: Postulated mechanism for the [Au(I)]-catalyzed hydroamination of olefins.
Intermediates of type 11 were isolated by Toste in the intramolecular hydroamination of alkenyl urea 14 with a stoichiometric amount of phosphine–gold complex [(PPh3Au)3O]BF4 (Scheme 5a) [2]. The reaction proceeded at room temperature and was found being favoured by electron-withdrawing ligands. Finally, the use of the deuterated compounds 14a,b permitted to confirm the anti-diastereoselective hydroamination of the double bond. (Scheme 5b). Interestingly, although 14 represents a likely intermediate of the reaction course, when it was treated with various Brønsted and Lewis acids, the expected product 16 was not observed and the elimination reaction to reform the starting material was the dominant process (Scheme 5c). Only reductive conditions afforded 16 in good yield (81%, Scheme 5d). This experimental evidence contributed substantially at the definition of the complex reaction machinery, with direct implication also in the correlated gold-catalyzed oxidative heteroarylation of unsaturated olefins (see section 6).
![[1860-5397-9-294-i5]](/bjoc/content/inline/1860-5397-9-294-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Isolation and reactivity of alkyl gold intermediates in the intramolecular hydroamination of alkenes.
Scheme 5: Isolation and reactivity of alkyl gold intermediates in the intramolecular hydroamination of alkene...
3.2 Selected examples
Initial reports on the gold-catalyzed hydroamination of olefins were presented by He [36] and Widenhoefer [37], independently. He and Brouwer disclosed the intermolecular addition of carbamates, sulfonamides and imidazolidinones to linear and cyclic dienes 17 in the presence of catalytic amounts of PPh3AuOTf (Scheme 6). The method featured excellent 1,2-regioselectivity and high chemoselectivity, providing protected allylamines 18, in good yields, using nearly equimolar amounts of the diene and nitrogen-based nucleophile.
![[1860-5397-9-294-i6]](/bjoc/content/inline/1860-5397-9-294-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: [Au(I)]-catalyzed intermolecular hydroamination of dienes.
Scheme 6: [Au(I)]-catalyzed intermolecular hydroamination of dienes.
Analogously, Widenhoefer and Han developed the intramolecular addition of carbamates to terminal alkenes affording N-protected pyrrolidines and piperidines 21 (Scheme 7). Best conditions involved the use of a 1:1 mixture of [Au(I)] complex 20a and AgOTf [38]. Although prolonged reaction times were required (up to 68 h in refluxing dioxane), remarkable tolerance in terms of carbamate protecting groups (Scheme 7a) and substituents at the carbon chain was recorded (Scheme 7b,c). Soon after the same team disclosed that the titled transformation could be extended to amide-based nucleophiles exploiting similar operational conditions [39].
![[1860-5397-9-294-i7]](/bjoc/content/inline/1860-5397-9-294-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Intramolecular [Au(I)]-catalyzed hydroamination of alkenes with carbamates.
Scheme 7: Intramolecular [Au(I)]-catalyzed hydroamination of alkenes with carbamates.
After these seminal works the scope of gold-catalyzed hydroamination of olefins was extend to other classes of nitrogen nucleophiles. Sulfonamides were successfully employed in the intra- and intermolecular hydroamination of alkenes catalyzed by Ph3PAuOTf (toluene, 85 °C, Scheme 8) [40]. In particular, primary and secondary sulfonamides reacted smoothly with mono and disubstituted alkenes delivering nitrogen compounds 22 and 23 with Markovnikov regioselectivity (Scheme 8a). Moreover, N-protected pyrrolidines 24 and 25 were accessed by intramolecular addition in excellent yields (Scheme 8b).
![[1860-5397-9-294-i8]](/bjoc/content/inline/1860-5397-9-294-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: [Au(I)]-catalyzed inter- as well as intramolecular addition of sulfonamides to isolated alkenes.
Scheme 8: [Au(I)]-catalyzed inter- as well as intramolecular addition of sulfonamides to isolated alkenes.
The combined use of more electrophilic phosphite-gold complexes [41,42] and enabling techniques such as microwave irradiation [43] led to consistent improvement in efficiency of the catalytic system. In particular when (Ph3O)PAuOTf was employed under microwave irradiation with a catalyst loading as low as 0.05 mol %, the condensation of TsNH2 to norbornene was realized in quantitative yield.
In addition, the introduction of carbene ligands [44] allowed performing the intramolecular hydroamination of N-alkenyl ureas, efficiently (Scheme 9) [45]. When IPrAuCl (27) and AgOTf were mixed together in 1:1 ratio, the intramolecular exo-addition of N’-alkyl and N’-arylurea to primary or secondary olefins took place smoothly at room temperature to afford the variously substituted pyrrolidines 28 and piperidines 30 (Scheme 9a,b). Even unsubstituted substrate 31 afforded the corresponding product 32 in excellent yield although more forcing conditions were necessary (Scheme 9c). Monosubstitution at carbon C1 or C2 of the chain led to the formation of the product 2,4-cis or 2,5-cis-32 with diastereomeric ratios up to 5.5:1.
![[1860-5397-9-294-i9]](/bjoc/content/inline/1860-5397-9-294-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Intramolecular hydroamination of N-alkenylureas catalyzed by gold(I) carbene complex.
Scheme 9: Intramolecular hydroamination of N-alkenylureas catalyzed by gold(I) carbene complex.
Enantioselective variant of this transformation was recently reported by Mikami and co-workers [46]. The protocol focuses on the use of a gold complex comprising the racemic 2,2'-bis(diphenylphosphino)-1,1'-biphenyl digold(I) complex 33 which, in combination with enantiopure silver phosphate 34, afforded the diastereopure complex 35. Interestingly, the strong [Au(I)]–[Au(I)] aurophilic interaction in 35 prevented racemization even if chiral anions were removed (Scheme 10a). (S)-33 found application in the intramolecular hydroamination of 36, delivering 37 in quantitative yield (98%) and moderate enantioselectivity (ee up to 48%, Scheme 10b). The efficiency of 33 was ascribed to double activation of the substrate by the binuclear gold complex 38.
![[1860-5397-9-294-i10]](/bjoc/content/inline/1860-5397-9-294-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Enantioselective hydroamination of alkenyl ureas with biphenyl tropos ligand and chiral silver phosphate.
Scheme 10: Enantioselective hydroamination of alkenyl ureas with biphenyl tropos ligand and chiral silver phos...
The synthetic versatility of alkenyl ureas was further demonstrated by Widenhoefer employing slightly different substrates, namely N-allyl-N’-arylureas. When 39 was treated with a 1:1 mixture of 20a and AgPF6, imidazolidin-2-ones 40 were obtained in high yields under mild reaction conditions (Scheme 11a) [47]. Analogously, substituted N-allylureas 41 provided 42 with high trans diastereoselectivity (dr = 50:1, Scheme 11b). Surprisingly, inversion of diastereoselection (i.e. cis stereoisomer as the major product) was recorded in the case of hydroxymethyl derivative 43.
![[1860-5397-9-294-i11]](/bjoc/content/inline/1860-5397-9-294-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 11: Intramolecular [Au(I)]-catalyzed hydroamination of N-allyl-N’-aryl ureas. (PNP = pNO2-C6H4, PMP = pMeO-C6H4).
Scheme 11: Intramolecular [Au(I)]-catalyzed hydroamination of N-allyl-N’-aryl ureas. (PNP = pNO2-C6H4, PMP = p...
The challenging task of direct hydroamination with simple amines was faced, by means of in situ protection of the basic functionality as an ammonium salt (Scheme 12) [48]. The best catalyst for the titled reaction turned out to be the complex 20b bearing an electron-rich phosphinic ligand. When 20b was mixed with AgOTf (1:1 ratio) the intramolecular hydroamination of alkenyl ammonium salts 45/47 took place at 80–100 °C affording pyrrolidines and piperidines 46/48 in moderate to good yields.
![[1860-5397-9-294-i12]](/bjoc/content/inline/1860-5397-9-294-i12.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 12: [Au(I)]-catalyzed hydroamination of alkenes with ammonium salts.
Scheme 12: [Au(I)]-catalyzed hydroamination of alkenes with ammonium salts.
In 2009 Widenhoefer reported the first example of enantioselective gold-catalyzed intermolecular hydroamination of simple olefins. Firstly, the authors demonstrated that functionalization of ethylene derivatives could be conveniently accessed by using cyclic ureas 49 as nucleophiles in combination with phosphine gold complexes. Starting from this consideration a protocol for the enantioselective addition of cyclic ureas to simple alkenes 4 was developed (Scheme 13) [49]. In spite of the forcing conditions (100 °C for 48 hours) the use of the chiral binuclear gold complex (S)-50(AuCl)2 ensured enantioselectivity up to 78% for the products 51.
![[1860-5397-9-294-i13]](/bjoc/content/inline/1860-5397-9-294-i13.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 13: Enantioselective [Au(I)]-catalyzed intermolecular hydroamination of alkenes with cyclic ureas.
Scheme 13: Enantioselective [Au(I)]-catalyzed intermolecular hydroamination of alkenes with cyclic ureas.
A considerable enhancement of enantioselectivity was recently obtained by Toste’s group in the [Au(I)]-catalyzed intramolecular addition of sulfonamides to dienes (Table 2) [50]. The authors observed that, when the binuclear gold complex based on (R)-DTBM-segphos ligand 53 and AgBF4 was used in combination with alcoholic additives, the rate of the intramolecular hydroamination of 52 increased considerably, leading to the concomitant formation of 55 in combination with the expected product 54. A screening of various chiral alcohols and kinetic studies established that the use of 2.0 equivalents of (−)-menthol led to an increase in both the yield and ee of 55 leaving unaffected the formation of 54.
Table 2: Cooperative [Au(I)]/menthol catalysis for the enantioselective intramolecular hydroamination of dienes.
![[Graphic 4]](/bjoc/content/inline/1860-5397-9-294-i44.svg?max-width=637&scale=1.0)
|
||||
| Entry | 52 | Ra | Yield (%)b (54:55) | ee (%)c |
|---|---|---|---|---|
| 1 | a | Me | 95 (1:6.3) | 95 |
| 2 | b | H | 91 (1:6.1) | 85 |
| 3 | c | Ph | 93 (1:3.5) | 84 |
| 4 | d |
![[Graphic 5]](/bjoc/content/inline/1860-5397-9-294-i45.svg?max-width=637&scale=1.0)
|
99 (1:5.0) | 89 |
| 5 | e |
![[Graphic 6]](/bjoc/content/inline/1860-5397-9-294-i46.svg?max-width=637&scale=1.0)
|
80 (1:8.0) | 97 |
| 6 | f |
![[Graphic 7]](/bjoc/content/inline/1860-5397-9-294-i47.svg?max-width=637&scale=1.0)
|
77 (1:12.1) | 97 |
| 7 | g |
![[Graphic 8]](/bjoc/content/inline/1860-5397-9-294-i48.svg?max-width=637&scale=1.0)
|
42 (1:3.2) | 98 |
| 8 | h |
![[Graphic 9]](/bjoc/content/inline/1860-5397-9-294-i49.svg?max-width=637&scale=1.0)
|
99 (1:1.5) | 94 |
| 9 | i |
![[Graphic 10]](/bjoc/content/inline/1860-5397-9-294-i50.svg?max-width=637&scale=1.0)
|
67 (1:7.6) | 91 |
aMbs = p-MeO-benzenesulfonyl. bCombined yield of 54 and 55. cThe ee values are referred to (E)-55.
This observation found adequate rationale with the presence of two different competing mechanisms for the formation of the products. In particular, while 54 was formed via a standard hydroamination reaction (Scheme 14, path a), compound 55 was formed via Brønsted acid catalysis because of the enhancement of alcohol acidity due to gold coordination (Scheme 14, path b). Under these conditions various pyrrolidines and piperidines were obtained in excellent yields and high enantioselectivity (Table 2).
![[1860-5397-9-294-i14]](/bjoc/content/inline/1860-5397-9-294-i14.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 14: Mechanistic proposal for the cooperative [Au(I)]/menthol catalysis for the enantioselective intramolecular hydroamination of dienes.
Scheme 14: Mechanistic proposal for the cooperative [Au(I)]/menthol catalysis for the enantioselective intramo...
4 Formation of C–C bonds
Besides heteroatoms, the electrophilic activation of alkenes by gold catalysts can be exploited also to form new C–C bonds. Early achievements relied on the use of active methylene compounds or electron rich arenes as carbon nucleophiles.
4.1 Alkylation of active methylene compounds
Hydroalkylation of styrenes, indene and norbornene with 1,3-diketones was firstly reported in 2004 by using AuCl3 and AgOTf in 1:3 ratio as the catalyst (Scheme 15) [51]. The addition took place with Markovnikov selectivity in good yields, however optimal conditions suffered from severe limitations on 1,3-diketones as nucleophiles and unfunctionalized olefins. The same catalytic system was then employed with cyclic enols, dienes and trienes although lower yields were recorded [52].
![[1860-5397-9-294-i15]](/bjoc/content/inline/1860-5397-9-294-i15.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 15: [Au(III)]-catalyzed addition of 1,3-diketones to alkenes.
Scheme 15: [Au(III)]-catalyzed addition of 1,3-diketones to alkenes.
Remarkably, the intramolecular [Au(I)]-catalyzed addition of β-keto amides to inactivated alkenes was also exploited for the synthesis of highly substituted lactams 59, also in a preparative scale [53]. When alkenyl β-keto amides 60 were reacted with a 1:1 mixture of phosphine-gold complex 20a and AgOTf (toluene, 50–90 °C), exo-trig hydroalkylation of the C–C double bond took place, affording 5-, 6-membered lactams and spiro-lactams 61 in excellent yields and high trans diastereoselectivity (Scheme 16).
![[1860-5397-9-294-i16]](/bjoc/content/inline/1860-5397-9-294-i16.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 16: [Au(I)]-catalyzed intramolecular addition of β-keto amides to alkenes.
Scheme 16: [Au(I)]-catalyzed intramolecular addition of β-keto amides to alkenes.
4.2 The hydroarylation reaction
In addition to methylene active compounds, electron-rich benzenes and heteroaromatics can be added to gold activated alkenes under suitable conditions. In this context, in situ made [Ph3PAuOTf] was found to be an efficient catalyst for the addition of indoles 62 to styrenes and aryldienes in toluene at 85 °C (Scheme 17a,b). Less reactive aliphatic alkenes required harsher reaction conditions, however the corresponding hydroarylation products 66 were obtained from moderate to good yields under microwave irradiation (DCE, Scheme 17c) [54].
![[1860-5397-9-294-i17]](/bjoc/content/inline/1860-5397-9-294-i17.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 17: Intermolecular [Au(I)]-catalyzed addition of indoles to alkenes.
Scheme 17: Intermolecular [Au(I)]-catalyzed addition of indoles to alkenes.
Additionally, the stronger Lewis acid AuCl3/AgSbF6 was documented to catalyze the Markovnikov selective hydroarylation of aryl- and alkyl olefins with less-nucleophilic benzene derivatives and thiophene in good yields (Scheme 18, DCE, 50 °C) [55]. To be underlined that despite of efficiency, isomerization of the C=C was found competing with the desired Friedel–Crafts-type alkylation under the optimized reaction conditions.
![[1860-5397-9-294-i18]](/bjoc/content/inline/1860-5397-9-294-i18.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 18: Intermolecular [Au(III)]-catalyzed hydroarylation of alkenes with benzene derivatives and thiophene.
Scheme 18: Intermolecular [Au(III)]-catalyzed hydroarylation of alkenes with benzene derivatives and thiophene....
Analogously, [Au(III)] catalysis was employed in the intramolecular hydroarylation of olefins. It is woth mentioning, that a 1:1 mixture of AuCl3 and AgOTf promoted the ring-closing processes of arenes 68, delivering the corresponding dihydrobenzopyrans, tetralins and tetrahydroquinolines 69 in good yields (Scheme 19a). Experimental controls with deuterium labelled compounds suggested the step-wise mechanism described in Scheme 19b. In detail, initial gold activation of the olefin would trigger the outer-sphere attack of the aryl ring to the double bond followed by protodeauration of the alkyl–gold compound [56].
![[1860-5397-9-294-i19]](/bjoc/content/inline/1860-5397-9-294-i19.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 19: a) Intramolecular [Au(III)]-catalyzed hydroarylation of alkenes. b) A SEAr-type mechanism was hypothesized by the authors.
Scheme 19: a) Intramolecular [Au(III)]-catalyzed hydroarylation of alkenes. b) A SEAr-type mechanism was hypot...
4.3 Latest developments
Recent advances in the field of C–C bond forming processes through gold activated olefins allowed expanding the scope of the reaction to other carbon nucleophiles. For examples Che and co-workers reported on the hydroalkylation of alkenes with simple ketones [57]. Reaction of alkenyl ketones with IPrAuCl/AgClO4 (5 mol %) afforded functionalized cyclopentyl and cyclohexyl derivatives in excellent yields and good trans diastereoselectivity, via exo-trig cyclization (Scheme 20a–c).
![[1860-5397-9-294-i20]](/bjoc/content/inline/1860-5397-9-294-i20.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 20: Intramolecular [Au(I)]-catalyzed hydroalkylation of alkenes with simple ketones.
Scheme 20: Intramolecular [Au(I)]-catalyzed hydroalkylation of alkenes with simple ketones.
The proposed mechanism involved activation of the C–C double bond by the carbene-based cationic gold species to form the intermediate 76 which underwent gold-promoted tautomerization affording the enol 77. Nucleophilic attack of the enol on the gold activated alkenes led to a new C–C bond with subsequent releasing of the product 71 (Scheme 21).
![[1860-5397-9-294-i21]](/bjoc/content/inline/1860-5397-9-294-i21.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 21: Proposed reaction mechanism for the intramolecular [Au(I)]-catalyzed hydroalkylation of alkenes with ketones.
Scheme 21: Proposed reaction mechanism for the intramolecular [Au(I)]-catalyzed hydroalkylation of alkenes wit...
This protocol was also extended to an intermolecular variant (i.e. one-pot N-Michael addition/hydroalkylation). The use of [PPh3AuCl/AgClO4] (5/15 mol %) furnished corresponding functionalized pyrrolidines in good yields and moderate stereoselectivity (Scheme 22) [58].
![[1860-5397-9-294-i22]](/bjoc/content/inline/1860-5397-9-294-i22.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 22: Tandem Michael addition/hydroalkylation catalyzed by [Au(I)] and [Ag(I)] salts.
Scheme 22: Tandem Michael addition/hydroalkylation catalyzed by [Au(I)] and [Ag(I)] salts.
The preferred alkene activation versus allenes was recently observed in the cascade 1,3-migration/[2 + 2] cycloaddition of 1,7-enyne benzoates (Scheme 23) [59]. When 83 was heated in presence of the silver free gold complex 20c a variety of highly substituted azabicyclo[4.2.0]oct-5-enes 84 was obtained in good yields as a single regio- and diastereoisomer. The protocol displayed excellent functional group compatibility and efficient transfer of chirality was observed with enantiopure substrates. The proposed mechanism proceeded through 3,3-migration of the propargylic ester to form the allenoate 87. The expected activation of the allene was probably unfavoured for steric reasons, therefore gold activation of the alkene moiety triggered attack of the more nucleophilic double bound of the allene, forming the intermediate 88. Finally, condensation of the alkylgold 88 onto the carbonyl group led to the bicyclic product 84 (Scheme 23).
![[1860-5397-9-294-i23]](/bjoc/content/inline/1860-5397-9-294-i23.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 23: Intramolecular [Au(I)]-catalyzed tandem migration/[2 + 2] cycloaddition of 1,7-enyne benzoates.
Scheme 23: Intramolecular [Au(I)]-catalyzed tandem migration/[2 + 2] cycloaddition of 1,7-enyne benzoates.
An interesting example of gold-catalyzed intramolecular cyclopropanation of olefins was recently documented by Maulide and coworkers [60]. In particular, a range of densely functionalized heterobicyclic and carbocycles 90 were readily accessible in high yields and high stereoselectivity starting from properly functionalized sulfonium ylides 89 (Scheme 24). Computational and experimental investigations suggested the initial 20d-based electrophilc activation of the C=C, with consequent nucleophilic attack by the ylidic carbon onto the internal carbon of the double bond. Finally, the intermediate lactone 92 underwent cyclopropanation, delivering SPh2 as a leaving group (Scheme 24b).
![[1860-5397-9-294-i24]](/bjoc/content/inline/1860-5397-9-294-i24.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 24: Intramolecular [Au(I)]-catalyzed cyclopropanation of alkenes.
Scheme 24: Intramolecular [Au(I)]-catalyzed cyclopropanation of alkenes.
5 Addition to allylic alcohols
The use of simple allylic alcohols as alkylating agents, in place of more activated analogues (i.e. halides, acetates, carbonates and phosphates) is highly desirable from a synthetic, environmental and economic point of view [61]. Late transition metal catalysts demonstrated efficiency in addressing the poor reactivity of these substrates [62]. Due to the ability of gold complexes to act as σ- and π-acids, gold catalysis gained a prominent role in the activation of allylic alcohols, delivering of water as the side-product. As the topic has been recently extensively reviewed by Aponick and Biannic [7], only the most recent examples of gold-catalyzed manipulation of inactivated allylic alcohols will be discussed here [63,64].
5.1 Mechanistic considerations
Very recently Ess and Aponick reported a detailed mechanistic study on the intramolecular hydroalkoxylation of hydroxy allylic alcohols 94, pointing out the key role of intramolecular hydrogen bond (H-bond) interactions for both reactivity and stereoselectivity [65]. Theoretical calculations and experimental evidences ruled out the SN1 reaction mechanism via allylic carbocation. Experiments with stereodefined hydroxy allylic alcohols revealed that the reaction was stereospecific: alcohols with the same configuration of the secondary carbinol atom, but different geometry of the double bond, afforded the cyclized product 95 with the opposite absolute configuration of the stereocenter, but the olefin had E configuration in both cases (Scheme 25).
![[1860-5397-9-294-i25]](/bjoc/content/inline/1860-5397-9-294-i25.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 25: Stereospecificity in [Au(I)]-catalyzed hydroalkoxylation of allylic alcohols.
Scheme 25: Stereospecificity in [Au(I)]-catalyzed hydroalkoxylation of allylic alcohols.
Whereas concerted SN2’ pathway was judged energetically unfavourable, two possible mechanistic channels were supposed to account for the observed stereoselectivity: anti addition followed by anti-elimination or syn addition followed by syn-elimination. Calculations showed that the former pathway was energetically favoured. Interestingly, a strong hydrogen bond was established during the catalytic cycle between the hydroxy groups. The H-bond turned out to be a key interaction for the high reactivity of 94 toward cyclization, leading to an intramolecular proton transfer resulting into a better leaving group. Furthermore, as the H-bond was conserved during the entire course of the reaction, it also determined the E geometry of the newly formed C–C double bond (Scheme 26).
![[1860-5397-9-294-i26]](/bjoc/content/inline/1860-5397-9-294-i26.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 26: Mechanistic investigation on the intramolecular [Au(I)]-catalyzed hydroalkoxylation of allylic alcohols.
Scheme 26: Mechanistic investigation on the intramolecular [Au(I)]-catalyzed hydroalkoxylation of allylic alco...
The importance of the H-bond network in gold-catalyzed enantioselective transformations of allylic alcohols has been recently highlighted also by Bandini and Miscione [66-68] that documented on the intramolecular enantioselective allylic alkylation of indoles. Combined computational and experimental studies revealed the presence of a complex H-bond network between the indole ring, the counterion (i.e. OTf−) and the leaving OH group along all the reaction profile (Scheme 27). Multiple functions were recognized in these interactions. Firstly, all the strong H-bonds conferred a constrained conformation to the substrate (U-fold) placing the two reacting moieties in close proximity (101). Secondly, after nucleophilic attack of the indole ring, the counterion facilitated the elimination of water (103) by shuttling one proton atom, from the indolyl ring to the leaving hydroxy group. The proposed catalytic cycle accounted also for the observed stereochemistry of the product 100 (R) when the chiral binuclear gold complex (R)-DTBM-MeO-biphep 50 was employed.
![[1860-5397-9-294-i27]](/bjoc/content/inline/1860-5397-9-294-i27.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 27: Mechanistic investigation on the intramolecular enantioselective [Au(I)]-catalyzed alkylation of indoles with allylic alcohols.
Scheme 27: Mechanistic investigation on the intramolecular enantioselective [Au(I)]-catalyzed alkylation of in...
5.2 Recent advances
Latest advances in gold-catalyzed transformations of allylic alcohols mainly concern the development of stereoselective processes. In particular, mild conditions allowed a more efficient development of enantioselective protocols and the scope of the process can be widen to more functionalized substrates, efficiently.
For examples Robertson and co-workers reported on the synthesis of (+)-isoaltholactone, by adopting an intramolecular stereoselective Ph3PAuOTf-catalyzed 5-exo alkoxylation of allylic alcohols, to form tetrahydrofuran rings (Scheme 28) [69]. Experiments with configurationally defined hydroxy allylic alcohols 105b,c demonstrated the stereospecificity of the process. Due to the complexity of the substrates, the stereochemical model proposed by Aponick [65] could only partially account for the stereochemical outcome of the reaction, since the importance of the configuration of the carbon bearing the nucleophilic OH group was not considered.
![[1860-5397-9-294-i28]](/bjoc/content/inline/1860-5397-9-294-i28.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 28: Synthesis of (+)-isoaltholactone via stereospecific intramolecular [Au(I)]-catalyzed alkoxylation of allylic alcohols.
Scheme 28: Synthesis of (+)-isoaltholactone via stereospecific intramolecular [Au(I)]-catalyzed alkoxylation o...
In this segment, Widenhoefer has recently documented on the enantioselective dehydrative amination of allylic alcohols with carbamates [70,71]. Pyrrolidines and piperidines 109 were obtained with enantiomeric excesses up to 94% by using a chiral gold complex based on (S)-DTBM-MeO-biphep (Scheme 29). Mechanistic investigation highlighted complete catalyst control on the configuration of the newly formed stereocenter. On the contrary, studies on stereochemically defined secondary allylic alcohols revealed that the configuration of the allylic carbon atom affected the configuration of the newly formed C–C double bond. The E configuration of the starting material turned out to be fundamental to achieve high enantioselectivity. Also in this case, a mechanism sketch consisting on anti-aminoauration followed by anti-elimination was invoked to account for the observed experimental outcome (Scheme 29d).
![[1860-5397-9-294-i29]](/bjoc/content/inline/1860-5397-9-294-i29.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 29: Intramolecular enantioselective dehydrative amination of allylic alcohols catalyzed by chiral [Au(I)]-complexes.
Scheme 29: Intramolecular enantioselective dehydrative amination of allylic alcohols catalyzed by chiral [Au(I...
A complementary approach for the enantioselective synthesis of 3,4-disubstituted pyrrolidines was recently reported by Bandini and co-workers [72]. Intramolecular hydroalkylation of allylic alcohols with aldehydes was performed under synergistic activation of the substrate by gold catalyst 20c and organocatalyst 116 (Scheme 30). The 5-membered hetero- and carbocycles 115 were obtained in moderate to good yield and interesting level of diastereo- and enantioselectivity, supporting the perfect compatibility between the amino-catalyst and the electrophilic gold complex. Experiments on chiral enriched secondary alcohols suggested the anti-attack of the in situ formed enamine 117 on gold activated allylic alcohol followed by anti-elimination of water.
![[1860-5397-9-294-i30]](/bjoc/content/inline/1860-5397-9-294-i30.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 30: Enantioselective intramolecular hydroalkylation of allylic alcohols with aldehydes catalyzed by 20c and chiral secondary amine.
Scheme 30: Enantioselective intramolecular hydroalkylation of allylic alcohols with aldehydes catalyzed by 20c...
6 Double functionalization of olefins by oxidative strategies
Differently from many transition metals, usually gold is not subjected to redox processes during the catalytic cycles. However, in recent years it has been demonstrated that both [Au(I)/Au(III)] (i.e. monometallic catalytic systems) and [Au(I)Au(I)/Au(II)Au(II)] (i.e. bimetallic catalytic systems) redox couples could be accessed using external stoichiometric oxidants (i.e. Selectfluor or hypervalent iodine compounds) [73]. As schematically shown in Figure 1 this approach allows a double functionalization of simple alkenes with subsequent formation of new C–X and C–C bonds in a single catalytic process.
In 2009 Muñiz and coworkers reported on the diamination of alkenes catalyzed by Ph3PAuOAc in presence of PhI(OAc)2 (Scheme 31) [74]. The process led to the formation of two new C–N bonds under mild conditions with excellent yields. A mechanistic rationale was also provided dealing with initial [Au(I)]-promoted anti-amination with formation of alkylgold intermediate 121, followed by oxidation to [Au(III)] species 122 by means of PhI(OAc)2. A final ring-closing step will provide product 120 (Scheme 31).
![[1860-5397-9-294-i31]](/bjoc/content/inline/1860-5397-9-294-i31.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 31: Gold-catalyzed intramolecular diamination of alkenes.
Scheme 31: Gold-catalyzed intramolecular diamination of alkenes.
Exploiting a similar strategy, Nevado developed a flexible protocol for the gold-assisted aminooxygenation of alkenes, intercepting the hydroamination intermediate with various nucleophiles (i.e. alcohols, ethers and esters, Scheme 32a) [75]. A remarkable variety of products could be accessed with minimal variation of the reaction conditions. For instance, by using nitriles as solvents and only 2 equivalents of water, a nucleophilic attack of the nitrile itself on the hydroamination intermediate took place, affording the corresponding aminoamidation product 127 in moderate to good yields (Scheme 32b). In absence of external nucleophiles, the phenyl rings of the substrate backbone could intercept the hydroamination intermediate affording tricyclic 3-benzazepines 129 (Scheme 32c).
![[1860-5397-9-294-i32]](/bjoc/content/inline/1860-5397-9-294-i32.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 32: Gold-catalyzed aminooxygenation and aminoarylation of alkenes.
Scheme 32: Gold-catalyzed aminooxygenation and aminoarylation of alkenes.
In the same field, Zhang reported the carboamination, carboalkoxylation and carbolactonization of terminal alkenes with arylboronic acids. Under best conditions, oxidative gold catalysis provided expedient access to various substituted N- or O-heterocycles in high yields (Scheme 33) [76]. Deuterium labeling experiments unambiguously demonstrated the anti-functionalization of the double bond and the use of neutral gold complexes suggested that [Au(I)] oxidation took place prior of C–X bond formation.
![[1860-5397-9-294-i33]](/bjoc/content/inline/1860-5397-9-294-i33.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 33: Gold-catalyzed carboamination, carboalkoxylation and carbolactonization of terminal alkenes with arylboronic acids.
Scheme 33: Gold-catalyzed carboamination, carboalkoxylation and carbolactonization of terminal alkenes with ar...
The same team also reported on the efficiency of gold catalysis in the functionalization of C–H bonds [77]. In particular, N-aryl-N’-allylureas 136 were selectively transformed into tricyclic indolines 137 in good yields. The reaction preceeded through the regioselective 5-exo anti-aminoauration of the C–C double bond (139) followed by oxidative coupling of the formed alkylgold moiety with the ortho C–H bond of the tethered phenyl group. Compound 137 was finally obtained via reductive elimination of intermediate 141 (Scheme 34). The formal [3 + 2] annulation between the aniline moiety and the C–C double bond constitutes the first example of C–H functionalization by alkylgold intermediates. The optimized conditions required the use of the electrophilic complex [(p-CF3Ph)3PAuNTf2] and 30 equivalents of water, in order to increase the solubility of Selectfluor in THF.
![[1860-5397-9-294-i34]](/bjoc/content/inline/1860-5397-9-294-i34.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 34: Synthesis of tricyclic indolines via gold-catalyzed formal [3 + 2] cycloaddition.
Scheme 34: Synthesis of tricyclic indolines via gold-catalyzed formal [3 + 2] cycloaddition.
Heteroarylation of alkenes with arylboronic acids under the assistance of redox gold catalysis was also elegantly investigated by Toste’s group. In particular, aminoarylation of terminal olefins was documented in the presence of catalytic amounts of [dppm(AuBr)2] (3 mol %) and Selectfluor as the stoichiometric oxidant [78]. Despite the undoubted synthetic interest relying on the synthetic approach (a wide range of densely functionalized nitrogen-based heterocyclic cores (143) were readily accessible) (Scheme 35), intriguing mechanistic insights derived from a detailed investigation based on experimental and computational observations [79].
![[1860-5397-9-294-i35]](/bjoc/content/inline/1860-5397-9-294-i35.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 35: Gold(I) catalyzed aminoarylation of terminal alkenes in presence of Selectfluor [dppm = bis(diphenylphosphino)methane].
Scheme 35: Gold(I) catalyzed aminoarylation of terminal alkenes in presence of Selectfluor [dppm = bis(dipheny...
In particular, the inability of [Au(I)] halide complexes in promoting the aminoauration of the double bond suggested the oxidation of the [Au(I)] complex as the first step of the catalytic cycle (Scheme 36 i). This hypothesis was further supported by the failure recorded when an isolated alkylgold compound was reacted with Selectfluor and PhB(OH)2 (ii). Moreover, Ph3PAuCl and PhB(OH)2 were mixed together, no formation of Ph3PAuPh was detected ruling out the transmetallation as initial stage of the catalytic cycle (iii). In addition, Ph3PAuPh proved to be a competent catalyst of the process but only in the presence of arylboronic acid (iv and v). The latter evidence prompted the authors to conclude that the aryl group was transferred from the boronic acid and not from the gold complex (i.e. the phenyl group on the in situ formed PPh3AuPh complex acts as a spectator in the process).
![[1860-5397-9-294-i36]](/bjoc/content/inline/1860-5397-9-294-i36.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 36: Mechanistic investigation on the aminoarylation of terminal alkenes by bimetallic gold(I) catalysis in presence of Selectfluor.
Scheme 36: Mechanistic investigation on the aminoarylation of terminal alkenes by bimetallic gold(I) catalysis...
Based on such observations, the mechanistic cycle depicted in the Scheme 37 was proposed. Oxidation of the bimetallic [dppm(Au2Br2)] complex by Selectfluor led to the preferred formation of the cationic [Au(II)Br2–Au(II)F]+ complex 144 also due to the instauration of a strong aurofilic interaction (σ-bond) between the two gold atoms. Then, intramolecular aminoauration of the C=C occurred via an anti-stereochemical reaction profile, leading to intermediate 145. Finally, arylation of 145 was supposed to occur via a concerted bimolecular elimination (146), in which the F–B bond assisted the formation of the new C–C bond (Scheme 37) [80].
![[1860-5397-9-294-i37]](/bjoc/content/inline/1860-5397-9-294-i37.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 37: Proposed mechanism for the aminoarylation of alkenes via [Au(I)-Au(I)]/[Au(II)-Au(II)] redox catalysis.
Scheme 37: Proposed mechanism for the aminoarylation of alkenes via [Au(I)-Au(I)]/[Au(II)-Au(II)] redox cataly...
It should be emphasized that the latter step of the proposed reaction machinery referred as “redox synergy” is in clear conflict with the commonly reported alternative invoking transmetalation/reductive elimination steps (see Schemes 33 and 34). The catalytic superiority of bimetallic systems with respect to PPh3AuOTf in the aminoarylation is clearly explainable by the latter mechanistic proposal, that found solid validations/analogies in the “Pd---Pd” cooperative catalysis [81].
The same catalytic system was also utilized in the three-component oxyarylation of olefins (Scheme 38) [82]. The reaction took place under mild conditions and exhibited a wide substrate scope, being highly tolerant towards a number of olefins, arylboronic acids and nucleophiles. In particular, primary, secondary, tertiary alcohols and even water could be employed as nucleophiles, affording the corresponding ethers and alcohols in moderate to good yields (up to 90%, Scheme 38a,c). Differently, although carboxylic acids were suitable nucleophiles as well, the corresponding products were isolated in lower extent (Scheme 38b).
![[1860-5397-9-294-i38]](/bjoc/content/inline/1860-5397-9-294-i38.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 38: Oxyarylation of terminal olefins via redox gold catalysis.
Scheme 38: Oxyarylation of terminal olefins via redox gold catalysis.
Furthermore, the present strategy was extended to arylsilanes, enabling the use of oxygen- and nitrogen-containing coupling partners [83,84]. Interestingly, due to the employment of Selectfluor as a stoichiometric exogenous oxidant, the addition of basic activators for silane reagents were not required. The ready availability of silane precursors, with respect to boronic acid counterparts, allowed an intermolecular variant to be successfully developed (Scheme 39a).
![[1860-5397-9-294-i39]](/bjoc/content/inline/1860-5397-9-294-i39.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 39: a) Intramolecular gold-catalyzed oxidative coupling reactions with aryltrimethylsilanes. b) Oxyarylation of alkenes catalyzed by gold in presence of iodine-(III) compound IBA as an external oxidant.
Scheme 39: a) Intramolecular gold-catalyzed oxidative coupling reactions with aryltrimethylsilanes. b) Oxyaryl...
A slight modification of the reaction conditions enabled to expand the oxidative coupling to a wider range of olefins. In particular the use of IBA (153) as the oxidant allowed substrates incompatible with Selectfluor, (i.e. styrene and gem-disubstituted olefins) to be efficiently employed (Scheme 39b) [85].
An innovative approach to the double functionalization of olefins was developed by Glorius and co-workers, very recently. The authors reported on the use of visible light-mediated photoredox catalysis to access the [Au(I)]/[Au(III)] redox couple during the intramolecular oxy- and aminoarylation of alkenes (Scheme 40) [86].
![[1860-5397-9-294-i40]](/bjoc/content/inline/1860-5397-9-294-i40.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 40: Oxy- and amino-arylation of alkenes by [Au(I)]/[Au(III)] photoredox catalysis.
Scheme 40: Oxy- and amino-arylation of alkenes by [Au(I)]/[Au(III)] photoredox catalysis.
Optimal conditions for the reaction involved the use of Ph3PAuNTf2 in presence of [Ru(bpy)3]2+ as redox photocatalyst and aryl diazonium salts 157. In the proposed tandem catalytic cycle, the initial anti oxy-auration of the double bond led to the alkylgold intermediate 159, that was oxidized to [Au(II)] 160 via a SET process by the aryl radical formed in the photoredox catalytic cycle. The highly reactive species 160 was promptly oxidized by [Ru(III)(bpy)3]3+ affording the [Au(III)] intermediate 161 and regenerating the [Ru(II)(bpy)3]2+ photocatalyst. Finally, arylated tetrahydrofuran 158 was obtained by reductive elimination with concomitant regeneration of the [Au(I)] catalyst.
Conclusion
Metal catalyzed electrophilic activation of isolated alkenes is by far considered among the most challenging metal-assisted nucleophilic manipulation of inactivated unsaturated hydrocarbons. Relative inertness of C=C with respect to alkynes or allenes accounts for this trend. In this scenario [Au(I)] and [Au(III)] catalysis is playing a major role leading to tremendous developments spanning from C-/hetero-nucleophilic manipulations, mono-/difunctionalizations, intra-/intermolecular transformations and regio-/stereoselective methodologies. In this chemistry, it should be mentioned that concomitant catalytic activity exerted by Brønsted acidity can not be ruled out a priori and it should not be underestimated. Therefore, practitioners should carefully determine the real role of gold complexes during the reaction course, with respect to [H+] sources, from time to time. Experimental controls would certainly contribute to unambiguously clarify the intrinsic mechanistic aspects of the process but they would also concur to a better optimization/rationalization of optical outcomes deriving from stereoselective transformations. Despite the apparent interchangeability between cationic gold(I) species and [H+] sources, a major breakthrough introduced by [Au(I)] complexes in the nucleophilic manipulation of inactivated alkenes relies on enantioselective processes, that still represent an unsolved issues in metal-free catalysis [87].
References
-
Hashmi, A. S. K.; Toste, F. D., Eds. Modern Gold Catalyzed Synthesis; Wiley-VCH: Weinheim, Germany, 2012.
Return to citation in text: [1] -
Toste, F. D.; Michelet, V., Eds. Gold Catalysis: An Homogeneous Approach; Imperial College Press: London, 2012.
Return to citation in text: [1] [2] -
Rudolph, M.; Hashmi, A. S. K. Chem. Soc. Rev. 2012, 41, 2448–2462. doi:10.1039/c1cs15279c
Return to citation in text: [1] -
de Haro, T.; Nevado, C. Synthesis 2011, 2530–2539. doi:10.1055/s-0030-1260122
Return to citation in text: [1] -
Bandini, M. Chem. Soc. Rev. 2011, 40, 1358–1367. doi:10.1039/c0cs00041h
Return to citation in text: [1] -
LaLonde, R. L.; Brenzovich, W. E., Jr.; Benitez, D.; Tkatchouk, E.; Kelley, K.; Goddard, W. A., III; Toste, F. D. Chem. Sci. 2010, 1, 226–233. doi:10.1039/c0sc00255k
Return to citation in text: [1] -
Biannic, B.; Aponick, A. Eur. J. Org. Chem. 2011, 6605–6617. doi:10.1002/ejoc.201100858
Return to citation in text: [1] [2] -
Bandini, M.; Cera, G.; Chiarucci, M. Synthesis 2012, 504–512. doi:10.1055/s-0031-1289681
Return to citation in text: [1] -
Garcia, P.; Malacria, M.; Aubert, C.; Gandon, V.; Fensterbank, L. ChemCatChem 2010, 2, 493–497. doi:10.1002/cctc.200900319
Return to citation in text: [1] -
Hashmi, A. S. K. Catal. Today 2007, 122, 211–214. doi:10.1016/j.cattod.2006.10.006
Return to citation in text: [1] -
Taylor, J. G.; Adrio, L. A.; Hii, K. K. M. Dalton Trans. 2010, 39, 1171–1175. doi:10.1039/b918970j
Return to citation in text: [1] -
Schlummer, B.; Hartwig, J. F. Org. Lett. 2002, 4, 1471–1474. doi:10.1021/ol025659j
Return to citation in text: [1] -
Rosenfeld, D. C.; Shekhar, S.; Takemiya, A.; Utsunomiya, M.; Hartwig, J. F. Org. Lett. 2006, 8, 4179–4182. doi:10.1021/ol061174+
Return to citation in text: [1] -
Trost, B. M.; Van Vranken, D. L. Chem. Rev. 1996, 96, 395–422. doi:10.1021/cr9409804
Return to citation in text: [1] -
Trost, B. M.; Crawley, M. L. Chem. Rev. 2003, 103, 2921–2944. doi:10.1021/cr020027w
Return to citation in text: [1] -
Liu, C.; Bender, C. F.; Han, X.; Widenhoefer, R. A. Chem. Commun. 2007, 3607–3618. doi:10.1039/b615698c
Return to citation in text: [1] -
Li, Z.; Zhang, J.; Brouwer, C.; Yang, C.-G.; Reich, N. W.; He, C. Org. Lett. 2006, 8, 4175–4178. doi:10.1021/ol0610035
Return to citation in text: [1] -
Li, X.; Ye, S.; He, C.; Yu, Z.-X. Eur. J. Org. Chem. 2008, 4296–4303. doi:10.1002/ejoc.200800337
Return to citation in text: [1] -
Kovács, G.; Lledós, A.; Ujaque, G. Organometallics 2010, 29, 5919–5926. doi:10.1021/om1007192
Return to citation in text: [1] -
Brown, T. J.; Dickens, M. G.; Widenhoefer, R. A. Chem. Commun. 2009, 6451–6453. doi:10.1039/b914632f
Return to citation in text: [1] -
Hooper, T. N.; Green, M.; McGrady, J. E.; Patel, J. R.; Russell, C. A. Chem. Commun. 2009, 3877–3879. doi:10.1039/b908109g
Return to citation in text: [1] -
Brooner, R. E. M.; Widenhoefer, R. A. Organometallics 2011, 30, 3182–3193. doi:10.1021/om200291s
Return to citation in text: [1] -
Langseth, E.; Scheuermann, M. L.; Balcells, D.; Kaminsky, W.; Goldberg, K. I.; Eisenstein, O.; Heyn, R. H.; Tilset, M. Angew. Chem., Int. Ed. 2013, 52, 1660–1663. doi:10.1002/anie.201209140
Return to citation in text: [1] -
Jašíková, L.; Roithová, J. Organometallics 2012, 31, 1935–1942. doi:10.1021/om2012387
See for an experimental and theoretical study on bond energy in [Au(I)]-unsaturated hydrocarbons complexes.
Return to citation in text: [1] -
Kovács, G.; Lledós, A.; Ujaque, G. Organometallics 2010, 29, 3252–3260. doi:10.1021/om100063v
Return to citation in text: [1] -
Zuccaccia, D.; Belpassi, L.; Tarantelli, F.; Macchioni, A. J. Am. Chem. Soc. 2009, 131, 3170–3171. doi:10.1021/ja809998y
Return to citation in text: [1] -
Yang, C.; He, C.-G. J. Am. Chem. Soc. 2005, 127, 6966–6967. doi:10.1021/ja050392f
Return to citation in text: [1] -
Nguyen, R.-V.; Yao, X.; Li, C.-J. Org. Lett. 2006, 8, 2397–2399. doi:10.1021/ol0607692
Return to citation in text: [1] -
Zhang, X.; Corma, A. Chem. Commun. 2007, 3080–3082. doi:10.1039/b706961h
Return to citation in text: [1] -
Graf, T. A.; Anderson, T. K.; Bowden, N. B. Adv. Synth. Catal. 2011, 353, 1033–1038. doi:10.1002/adsc.201000859
Return to citation in text: [1] -
Zhang, X.; Corma, A. Dalton Trans. 2008, 397–403. doi:10.1039/b714617e
Return to citation in text: [1] -
Dong, W.; Zhang, M.; Xiao, F.; Wang, Y.; Liu, W.; Hu, X.; Yuan, Q.; Zhang, S. Synlett 2012, 23, 2799–2802. doi:10.1055/s-0032-1317492
Return to citation in text: [1] -
Müller, T. E.; Hultzsch, K. C.; Yus, M.; Foubelo, F.; Tada, M. Chem. Rev. 2008, 108, 3795–3892. doi:10.1021/cr0306788
Return to citation in text: [1] -
Kovács, G.; Ujaque, G.; Lledós, A. J. Am. Chem. Soc. 2008, 130, 853–864. doi:10.1021/ja073578i
Return to citation in text: [1] -
McBee, J. L.; Bell, A. T.; Tilley, T. D. J. Am. Chem. Soc. 2008, 130, 16562–16571. doi:10.1021/ja8030104
Return to citation in text: [1] -
Brouwer, C.; He, C. Angew. Chem., Int. Ed. 2006, 45, 1744–1747. doi:10.1002/anie.200504495
Return to citation in text: [1] -
Nieto-Oberhuber, C.; López, S.; Echavarren, A. M. J. Am. Chem. Soc. 2005, 127, 6178–6179. doi:10.1021/ja042257t
Return to citation in text: [1] -
Bender, C. F.; Widenhoefer, R. A. Chem. Commun. 2006, 4143–4144. doi:10.1039/b608638a
Return to citation in text: [1] -
Han, X.; Widenhoefer, R. A. Angew. Chem., Int. Ed. 2006, 45, 1747–1749. doi:10.1002/anie.200600052
Return to citation in text: [1] -
Zhang, J.; Yang, C.-G.; He, C. J. Am. Chem. Soc. 2006, 128, 1798–1799. doi:10.1021/ja053864z
Return to citation in text: [1] -
Giner, X.; Nàjera, C. Org. Lett. 2008, 10, 2919–2922. doi:10.1021/ol801104w
Return to citation in text: [1] -
Giner, X.; Nájera, C.; Kovács, G.; Lledós, A.; Ujaque, G. Adv. Synth. Catal. 2011, 353, 3451–3466. doi:10.1002/adsc.201100478
Return to citation in text: [1] -
Liu, X.-Y.; Li, C.-H.; Che, C.-M. Org. Lett. 2006, 8, 2707–2710. doi:10.1021/ol060719x
Return to citation in text: [1] -
Marion, N.; Nolan, S. P. Chem. Soc. Rev. 2008, 37, 1776–1782. doi:10.1039/b711132k
Return to citation in text: [1] -
Bender, C. F.; Widenhoefer, R. A. Org. Lett. 2006, 8, 5303–5305. doi:10.1021/ol062107i
Return to citation in text: [1] -
Kojima, M.; Mikami, K. Synlett 2012, 57–61. doi:10.1055/s-0031-1289875
Return to citation in text: [1] -
Li, H.; Song, F.; Widenhoefer, R. A. Adv. Synth. Catal. 2011, 353, 955–962. doi:10.1002/adsc.201000844
Return to citation in text: [1] -
Bender, C. F.; Widenhoefer, R. A. Chem. Commun. 2008, 2741–2743. doi:10.1039/b804081h
Return to citation in text: [1] -
Zhang, Z.; Lee, S. D.; Widenhoefer, R. A. J. Am. Chem. Soc. 2009, 131, 5372–5373. doi:10.1021/ja9001162
Return to citation in text: [1] -
Kanno, O.; Kuriyama, W.; Wang, Z. J.; Toste, F. D. Angew. Chem., Int. Ed. 2011, 50, 9919–9922. doi:10.1002/anie.201104076
For a non-stereoselective variant see: Baeza A; Nájera C. Synlett, 2011, 631–634. doi:10.1055/s-0030-1259680
Return to citation in text: [1] -
Yao, X.; Li, C.-J. J. Am. Chem. Soc. 2004, 126, 6884–6885. doi:10.1021/ja0482637
Return to citation in text: [1] -
Nguyen, R.-V.; Yao, X.-Q.; Bohle, D. S.; Li, C.-J. Org. Lett. 2005, 7, 673–675. doi:10.1021/ol047543o
Return to citation in text: [1] -
Zhou, C.-Y.; Che, C.-M. J. Am. Chem. Soc. 2007, 129, 5828–5829. doi:10.1021/ja070027j
Return to citation in text: [1] -
Wang, M.-Z.; Wong, M.-K.; Che, C.-M. Chem.–Eur. J. 2008, 14, 8353–8364. doi:10.1002/chem.200800040
Return to citation in text: [1] -
Xiao, Y.-P.; Liu, X.-Y.; Che, C.-M. J. Organomet. Chem. 2009, 694, 494–501. doi:10.1016/j.jorganchem.2008.07.035
Return to citation in text: [1] -
Jean, M.; van de Weghe, P. Tetrahedron Lett. 2011, 52, 3509–3513. doi:10.1016/j.tetlet.2011.04.122
Return to citation in text: [1] -
Xiao, Y.-P.; Liu, X.-Y.; Che, C.-M. Angew. Chem., Int. Ed. 2011, 50, 4937–4941. doi:10.1002/anie.201100044
Return to citation in text: [1] -
Xiao, Y.-P.; Liu, X.-Y.; Che, C.-M. Beilstein J. Org. Chem. 2011, 7, 1100–1107. doi:10.3762/bjoc.7.126
Return to citation in text: [1] -
Rao, W.; Susanti, D.; Chan, P. W. H. J. Am. Chem. Soc. 2011, 133, 15248–15251. doi:10.1021/ja2052304
Return to citation in text: [1] -
Huang, X.; Klimczyk, S.; Veiros, L. F.; Maulide, N. Chem. Sci. 2013, 4, 1105–1110. doi:10.1039/c2sc21914j
Return to citation in text: [1] -
Lumbroso, A.; Cooke, M. L.; Brei, B. Angew. Chem., Int. Ed. 2013, 52, 1890–1932. doi:10.1002/anie.201204579
Return to citation in text: [1] -
Bandini, M. Angew. Chem., Int. Ed. 2011, 50, 994–995. doi:10.1002/anie.201006522
Return to citation in text: [1] -
Guo, S.; Song, F.; Liu, Y. Synlett 2007, 964–968. doi:10.1055/s-2007-973865
Return to citation in text: [1] -
Giner, X.; Trillo, P.; Nájera, C. J. Organomet. Chem. 2011, 696, 357–361. doi:10.1016/j.jorganchem.2010.09.072
Return to citation in text: [1] -
Ghebreghiorgis, T.; Biannic, B.; Kirk, B. H.; Ess, D. H.; Aponick, A. J. Am. Chem. Soc. 2012, 134, 16307–16318. doi:10.1021/ja306333a
Return to citation in text: [1] [2] -
Bandini, M.; Bottoni, A.; Chiarucci, M.; Cera, G.; Miscione, G. P. J. Am. Chem. Soc. 2012, 134, 20690–20700. doi:10.1021/ja3086774
Return to citation in text: [1] -
Bandini, M.; Eichholzer, A. Angew. Chem., Int. Ed. 2009, 48, 9533–9537. doi:10.1002/anie.200904388
Return to citation in text: [1] -
Bandini, M.; Gualandi, A.; Monari, M.; Romaniello, A.; Savoia, D.; Tragni, M. J. Organomet. Chem. 2011, 696, 338–347. doi:10.1016/j.jorganchem.2010.09.065
Return to citation in text: [1] -
Unsworth, W. P.; Stevens, K.; Lamont, S. G.; Robertson, J. Chem. Commun. 2011, 47, 7659–7661. doi:10.1039/c1cc11805f
Return to citation in text: [1] -
Mukherjee, P.; Widenhoefer, R. A. Org. Lett. 2010, 12, 1184–1187. doi:10.1021/ol902923e
Return to citation in text: [1] -
Mukherjee, P.; Widenhoefer, R. A. Angew. Chem., Int. Ed. 2012, 51, 1405–1407. doi:10.1002/anie.201107877
Return to citation in text: [1] -
Chiarucci, M.; di Lillo, M.; Romaniello, A.; Cozzi, P. G.; Cera, G.; Bandini, M. Chem. Sci. 2012, 3, 2859–2863. doi:10.1039/c2sc20478a
Return to citation in text: [1] -
Hopkinson, M. N.; Gee, A. D.; Gouverneur, V. Chem.–Eur. J. 2011, 17, 8248–8262. doi:10.1002/chem.201100736
Return to citation in text: [1] -
Iglesias, A.; Muñiz, K. Chem.–Eur. J. 2009, 15, 10563–10569. doi:10.1002/chem.200901199
Return to citation in text: [1] -
de Haro, T.; Nevado, C. Angew. Chem., Int. Ed. 2011, 50, 906–910. doi:10.1002/anie.201005763
Return to citation in text: [1] -
Zhang, G.; Cui, L.; Wang, Y.; Zhang, L. J. Am. Chem. Soc. 2010, 132, 1474–1475. doi:10.1021/ja909555d
Return to citation in text: [1] -
Zhang, G.; Luo, Y.; Wang, Y.; Zhang, L. Angew. Chem., Int. Ed. 2011, 50, 4450–4454. doi:10.1002/anie.201100293
Return to citation in text: [1] -
Brenzovich, W. E., Jr.; Benitez, D.; Lackner, A. D.; Shunatona, H. P.; Tkatchouk, E.; Goddard, W. A., III; Toste, F. D. Angew. Chem., Int. Ed. 2010, 49, 5519–5522. doi:10.1002/anie.201002739
Return to citation in text: [1] -
Tkatchouk, E.; Mankad, N. P.; Benitez, D.; Goddard, W. A., III; Toste, F. D. J. Am. Chem. Soc. 2011, 133, 14293–14300. doi:10.1021/ja2012627
Return to citation in text: [1] -
Mankad, N. P.; Toste, F. D. J. Am. Chem. Soc. 2010, 132, 12859–12861. doi:10.1021/ja106257n
See for a study on the reactivity of alkyl [Au(III)]-F complexes.
Return to citation in text: [1] -
Powers, D. C.; Ritter, T. Acc. Chem. Res. 2012, 45, 840–850. doi:10.1021/ar2001974
And reference therein.
Return to citation in text: [1] -
Melhado, A. D.; Brenzovich, W. E., Jr.; Lackner, A. D.; Toste, F. D. J. Am. Chem. Soc. 2010, 132, 8885–8887. doi:10.1021/ja1034123
Return to citation in text: [1] -
Ball, L. T.; Green, M.; Lloyd-Jones, G. C.; Russell, C. A. Org. Lett. 2010, 12, 4724–4727. doi:10.1021/ol1019162
Return to citation in text: [1] -
Brenzovich, W. E., Jr.; Brazeau, J. F.; Toste, F. D. Org. Lett. 2010, 12, 4728–4731. doi:10.1021/ol102194c
Return to citation in text: [1] -
Ball, L. T.; Lloyd-Jones, G. C.; Russell, C. A. Chem.–Eur. J. 2012, 18, 2931–2937. doi:10.1002/chem.201103061
Return to citation in text: [1] -
Sahoo, B.; Hopkinson, M. N.; Glorius, F. J. Am. Chem. Soc. 2013, 135, 5505–5508. doi:10.1021/ja400311h
Return to citation in text: [1] -
Shapiro, N. D.; Rauniyar, V.; Hamilton, G. L.; Wu, J.; Toste, F. D. Nature 2011, 470, 245–249. doi:10.1038/nature09723
Return to citation in text: [1]
| 41. | Giner, X.; Nàjera, C. Org. Lett. 2008, 10, 2919–2922. doi:10.1021/ol801104w |
| 42. | Giner, X.; Nájera, C.; Kovács, G.; Lledós, A.; Ujaque, G. Adv. Synth. Catal. 2011, 353, 3451–3466. doi:10.1002/adsc.201100478 |
| 43. | Liu, X.-Y.; Li, C.-H.; Che, C.-M. Org. Lett. 2006, 8, 2707–2710. doi:10.1021/ol060719x |
| 44. | Marion, N.; Nolan, S. P. Chem. Soc. Rev. 2008, 37, 1776–1782. doi:10.1039/b711132k |
| 87. | Shapiro, N. D.; Rauniyar, V.; Hamilton, G. L.; Wu, J.; Toste, F. D. Nature 2011, 470, 245–249. doi:10.1038/nature09723 |
| 51. | Yao, X.; Li, C.-J. J. Am. Chem. Soc. 2004, 126, 6884–6885. doi:10.1021/ja0482637 |
| 52. | Nguyen, R.-V.; Yao, X.-Q.; Bohle, D. S.; Li, C.-J. Org. Lett. 2005, 7, 673–675. doi:10.1021/ol047543o |
| 49. | Zhang, Z.; Lee, S. D.; Widenhoefer, R. A. J. Am. Chem. Soc. 2009, 131, 5372–5373. doi:10.1021/ja9001162 |
| 50. |
Kanno, O.; Kuriyama, W.; Wang, Z. J.; Toste, F. D. Angew. Chem., Int. Ed. 2011, 50, 9919–9922. doi:10.1002/anie.201104076
For a non-stereoselective variant see: Baeza A; Nájera C. Synlett, 2011, 631–634. doi:10.1055/s-0030-1259680 |
| 47. | Li, H.; Song, F.; Widenhoefer, R. A. Adv. Synth. Catal. 2011, 353, 955–962. doi:10.1002/adsc.201000844 |
| 48. | Bender, C. F.; Widenhoefer, R. A. Chem. Commun. 2008, 2741–2743. doi:10.1039/b804081h |
| 45. | Bender, C. F.; Widenhoefer, R. A. Org. Lett. 2006, 8, 5303–5305. doi:10.1021/ol062107i |
| 53. | Zhou, C.-Y.; Che, C.-M. J. Am. Chem. Soc. 2007, 129, 5828–5829. doi:10.1021/ja070027j |
| 54. | Wang, M.-Z.; Wong, M.-K.; Che, C.-M. Chem.–Eur. J. 2008, 14, 8353–8364. doi:10.1002/chem.200800040 |
| 55. | Xiao, Y.-P.; Liu, X.-Y.; Che, C.-M. J. Organomet. Chem. 2009, 694, 494–501. doi:10.1016/j.jorganchem.2008.07.035 |
| 62. | Bandini, M. Angew. Chem., Int. Ed. 2011, 50, 994–995. doi:10.1002/anie.201006522 |
| 7. | Biannic, B.; Aponick, A. Eur. J. Org. Chem. 2011, 6605–6617. doi:10.1002/ejoc.201100858 |
| 60. | Huang, X.; Klimczyk, S.; Veiros, L. F.; Maulide, N. Chem. Sci. 2013, 4, 1105–1110. doi:10.1039/c2sc21914j |
| 61. | Lumbroso, A.; Cooke, M. L.; Brei, B. Angew. Chem., Int. Ed. 2013, 52, 1890–1932. doi:10.1002/anie.201204579 |
| 58. | Xiao, Y.-P.; Liu, X.-Y.; Che, C.-M. Beilstein J. Org. Chem. 2011, 7, 1100–1107. doi:10.3762/bjoc.7.126 |
| 59. | Rao, W.; Susanti, D.; Chan, P. W. H. J. Am. Chem. Soc. 2011, 133, 15248–15251. doi:10.1021/ja2052304 |
| 56. | Jean, M.; van de Weghe, P. Tetrahedron Lett. 2011, 52, 3509–3513. doi:10.1016/j.tetlet.2011.04.122 |
| 57. | Xiao, Y.-P.; Liu, X.-Y.; Che, C.-M. Angew. Chem., Int. Ed. 2011, 50, 4937–4941. doi:10.1002/anie.201100044 |
| 65. | Ghebreghiorgis, T.; Biannic, B.; Kirk, B. H.; Ess, D. H.; Aponick, A. J. Am. Chem. Soc. 2012, 134, 16307–16318. doi:10.1021/ja306333a |
| 66. | Bandini, M.; Bottoni, A.; Chiarucci, M.; Cera, G.; Miscione, G. P. J. Am. Chem. Soc. 2012, 134, 20690–20700. doi:10.1021/ja3086774 |
| 67. | Bandini, M.; Eichholzer, A. Angew. Chem., Int. Ed. 2009, 48, 9533–9537. doi:10.1002/anie.200904388 |
| 68. | Bandini, M.; Gualandi, A.; Monari, M.; Romaniello, A.; Savoia, D.; Tragni, M. J. Organomet. Chem. 2011, 696, 338–347. doi:10.1016/j.jorganchem.2010.09.065 |
| 63. | Guo, S.; Song, F.; Liu, Y. Synlett 2007, 964–968. doi:10.1055/s-2007-973865 |
| 64. | Giner, X.; Trillo, P.; Nájera, C. J. Organomet. Chem. 2011, 696, 357–361. doi:10.1016/j.jorganchem.2010.09.072 |
| 1. | Hashmi, A. S. K.; Toste, F. D., Eds. Modern Gold Catalyzed Synthesis; Wiley-VCH: Weinheim, Germany, 2012. |
| 2. | Toste, F. D.; Michelet, V., Eds. Gold Catalysis: An Homogeneous Approach; Imperial College Press: London, 2012. |
| 3. | Rudolph, M.; Hashmi, A. S. K. Chem. Soc. Rev. 2012, 41, 2448–2462. doi:10.1039/c1cs15279c |
| 4. | de Haro, T.; Nevado, C. Synthesis 2011, 2530–2539. doi:10.1055/s-0030-1260122 |
| 5. | Bandini, M. Chem. Soc. Rev. 2011, 40, 1358–1367. doi:10.1039/c0cs00041h |
| 10. | Hashmi, A. S. K. Catal. Today 2007, 122, 211–214. doi:10.1016/j.cattod.2006.10.006 |
| 11. | Taylor, J. G.; Adrio, L. A.; Hii, K. K. M. Dalton Trans. 2010, 39, 1171–1175. doi:10.1039/b918970j |
| 28. | Nguyen, R.-V.; Yao, X.; Li, C.-J. Org. Lett. 2006, 8, 2397–2399. doi:10.1021/ol0607692 |
| 75. | de Haro, T.; Nevado, C. Angew. Chem., Int. Ed. 2011, 50, 906–910. doi:10.1002/anie.201005763 |
| 9. | Garcia, P.; Malacria, M.; Aubert, C.; Gandon, V.; Fensterbank, L. ChemCatChem 2010, 2, 493–497. doi:10.1002/cctc.200900319 |
| 7. | Biannic, B.; Aponick, A. Eur. J. Org. Chem. 2011, 6605–6617. doi:10.1002/ejoc.201100858 |
| 8. | Bandini, M.; Cera, G.; Chiarucci, M. Synthesis 2012, 504–512. doi:10.1055/s-0031-1289681 |
| 26. | Zuccaccia, D.; Belpassi, L.; Tarantelli, F.; Macchioni, A. J. Am. Chem. Soc. 2009, 131, 3170–3171. doi:10.1021/ja809998y |
| 73. | Hopkinson, M. N.; Gee, A. D.; Gouverneur, V. Chem.–Eur. J. 2011, 17, 8248–8262. doi:10.1002/chem.201100736 |
| 6. | LaLonde, R. L.; Brenzovich, W. E., Jr.; Benitez, D.; Tkatchouk, E.; Kelley, K.; Goddard, W. A., III; Toste, F. D. Chem. Sci. 2010, 1, 226–233. doi:10.1039/c0sc00255k |
| 27. | Yang, C.; He, C.-G. J. Am. Chem. Soc. 2005, 127, 6966–6967. doi:10.1021/ja050392f |
| 74. | Iglesias, A.; Muñiz, K. Chem.–Eur. J. 2009, 15, 10563–10569. doi:10.1002/chem.200901199 |
| 17. | Li, Z.; Zhang, J.; Brouwer, C.; Yang, C.-G.; Reich, N. W.; He, C. Org. Lett. 2006, 8, 4175–4178. doi:10.1021/ol0610035 |
| 20. | Brown, T. J.; Dickens, M. G.; Widenhoefer, R. A. Chem. Commun. 2009, 6451–6453. doi:10.1039/b914632f |
| 21. | Hooper, T. N.; Green, M.; McGrady, J. E.; Patel, J. R.; Russell, C. A. Chem. Commun. 2009, 3877–3879. doi:10.1039/b908109g |
| 22. | Brooner, R. E. M.; Widenhoefer, R. A. Organometallics 2011, 30, 3182–3193. doi:10.1021/om200291s |
| 23. | Langseth, E.; Scheuermann, M. L.; Balcells, D.; Kaminsky, W.; Goldberg, K. I.; Eisenstein, O.; Heyn, R. H.; Tilset, M. Angew. Chem., Int. Ed. 2013, 52, 1660–1663. doi:10.1002/anie.201209140 |
| 24. |
Jašíková, L.; Roithová, J. Organometallics 2012, 31, 1935–1942. doi:10.1021/om2012387
See for an experimental and theoretical study on bond energy in [Au(I)]-unsaturated hydrocarbons complexes. |
| 70. | Mukherjee, P.; Widenhoefer, R. A. Org. Lett. 2010, 12, 1184–1187. doi:10.1021/ol902923e |
| 71. | Mukherjee, P.; Widenhoefer, R. A. Angew. Chem., Int. Ed. 2012, 51, 1405–1407. doi:10.1002/anie.201107877 |
| 16. | Liu, C.; Bender, C. F.; Han, X.; Widenhoefer, R. A. Chem. Commun. 2007, 3607–3618. doi:10.1039/b615698c |
| 25. | Kovács, G.; Lledós, A.; Ujaque, G. Organometallics 2010, 29, 3252–3260. doi:10.1021/om100063v |
| 72. | Chiarucci, M.; di Lillo, M.; Romaniello, A.; Cozzi, P. G.; Cera, G.; Bandini, M. Chem. Sci. 2012, 3, 2859–2863. doi:10.1039/c2sc20478a |
| 14. | Trost, B. M.; Van Vranken, D. L. Chem. Rev. 1996, 96, 395–422. doi:10.1021/cr9409804 |
| 15. | Trost, B. M.; Crawley, M. L. Chem. Rev. 2003, 103, 2921–2944. doi:10.1021/cr020027w |
| 69. | Unsworth, W. P.; Stevens, K.; Lamont, S. G.; Robertson, J. Chem. Commun. 2011, 47, 7659–7661. doi:10.1039/c1cc11805f |
| 12. | Schlummer, B.; Hartwig, J. F. Org. Lett. 2002, 4, 1471–1474. doi:10.1021/ol025659j |
| 13. | Rosenfeld, D. C.; Shekhar, S.; Takemiya, A.; Utsunomiya, M.; Hartwig, J. F. Org. Lett. 2006, 8, 4179–4182. doi:10.1021/ol061174+ |
| 18. | Li, X.; Ye, S.; He, C.; Yu, Z.-X. Eur. J. Org. Chem. 2008, 4296–4303. doi:10.1002/ejoc.200800337 |
| 19. | Kovács, G.; Lledós, A.; Ujaque, G. Organometallics 2010, 29, 5919–5926. doi:10.1021/om1007192 |
| 65. | Ghebreghiorgis, T.; Biannic, B.; Kirk, B. H.; Ess, D. H.; Aponick, A. J. Am. Chem. Soc. 2012, 134, 16307–16318. doi:10.1021/ja306333a |
| 33. | Müller, T. E.; Hultzsch, K. C.; Yus, M.; Foubelo, F.; Tada, M. Chem. Rev. 2008, 108, 3795–3892. doi:10.1021/cr0306788 |
| 30. | Graf, T. A.; Anderson, T. K.; Bowden, N. B. Adv. Synth. Catal. 2011, 353, 1033–1038. doi:10.1002/adsc.201000859 |
| 31. | Zhang, X.; Corma, A. Dalton Trans. 2008, 397–403. doi:10.1039/b714617e |
| 32. | Dong, W.; Zhang, M.; Xiao, F.; Wang, Y.; Liu, W.; Hu, X.; Yuan, Q.; Zhang, S. Synlett 2012, 23, 2799–2802. doi:10.1055/s-0032-1317492 |
| 78. | Brenzovich, W. E., Jr.; Benitez, D.; Lackner, A. D.; Shunatona, H. P.; Tkatchouk, E.; Goddard, W. A., III; Toste, F. D. Angew. Chem., Int. Ed. 2010, 49, 5519–5522. doi:10.1002/anie.201002739 |
| 79. | Tkatchouk, E.; Mankad, N. P.; Benitez, D.; Goddard, W. A., III; Toste, F. D. J. Am. Chem. Soc. 2011, 133, 14293–14300. doi:10.1021/ja2012627 |
| 76. | Zhang, G.; Cui, L.; Wang, Y.; Zhang, L. J. Am. Chem. Soc. 2010, 132, 1474–1475. doi:10.1021/ja909555d |
| 77. | Zhang, G.; Luo, Y.; Wang, Y.; Zhang, L. Angew. Chem., Int. Ed. 2011, 50, 4450–4454. doi:10.1002/anie.201100293 |
| 39. | Han, X.; Widenhoefer, R. A. Angew. Chem., Int. Ed. 2006, 45, 1747–1749. doi:10.1002/anie.200600052 |
| 40. | Zhang, J.; Yang, C.-G.; He, C. J. Am. Chem. Soc. 2006, 128, 1798–1799. doi:10.1021/ja053864z |
| 37. | Nieto-Oberhuber, C.; López, S.; Echavarren, A. M. J. Am. Chem. Soc. 2005, 127, 6178–6179. doi:10.1021/ja042257t |
| 85. | Ball, L. T.; Lloyd-Jones, G. C.; Russell, C. A. Chem.–Eur. J. 2012, 18, 2931–2937. doi:10.1002/chem.201103061 |
| 38. | Bender, C. F.; Widenhoefer, R. A. Chem. Commun. 2006, 4143–4144. doi:10.1039/b608638a |
| 86. | Sahoo, B.; Hopkinson, M. N.; Glorius, F. J. Am. Chem. Soc. 2013, 135, 5505–5508. doi:10.1021/ja400311h |
| 2. | Toste, F. D.; Michelet, V., Eds. Gold Catalysis: An Homogeneous Approach; Imperial College Press: London, 2012. |
| 82. | Melhado, A. D.; Brenzovich, W. E., Jr.; Lackner, A. D.; Toste, F. D. J. Am. Chem. Soc. 2010, 132, 8885–8887. doi:10.1021/ja1034123 |
| 36. | Brouwer, C.; He, C. Angew. Chem., Int. Ed. 2006, 45, 1744–1747. doi:10.1002/anie.200504495 |
| 83. | Ball, L. T.; Green, M.; Lloyd-Jones, G. C.; Russell, C. A. Org. Lett. 2010, 12, 4724–4727. doi:10.1021/ol1019162 |
| 84. | Brenzovich, W. E., Jr.; Brazeau, J. F.; Toste, F. D. Org. Lett. 2010, 12, 4728–4731. doi:10.1021/ol102194c |
| 34. | Kovács, G.; Ujaque, G.; Lledós, A. J. Am. Chem. Soc. 2008, 130, 853–864. doi:10.1021/ja073578i |
| 80. |
Mankad, N. P.; Toste, F. D. J. Am. Chem. Soc. 2010, 132, 12859–12861. doi:10.1021/ja106257n
See for a study on the reactivity of alkyl [Au(III)]-F complexes. |
| 35. | McBee, J. L.; Bell, A. T.; Tilley, T. D. J. Am. Chem. Soc. 2008, 130, 16562–16571. doi:10.1021/ja8030104 |
| 81. |
Powers, D. C.; Ritter, T. Acc. Chem. Res. 2012, 45, 840–850. doi:10.1021/ar2001974
And reference therein. |
© 2013 Chiarucci and Bandini; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)