Abstract
Novel, functionalized piperazine derivatives were successfully synthesized and fully characterized by 1H/13C/19F NMR, MS, elemental analysis and lipophilicity. All piperazine compounds occur as conformers resulting from the partial amide double bond. Furthermore, a second conformational shape was observed for all nitro derivatives due to the limited change of the piperazine chair conformation. Therefore, two coalescence points were determined and their resulting activation energy barriers were calculated using 1H NMR. To support this result, single crystals of 1-(4-nitrobenzoyl)piperazine (3a, monoclinic, space group C2/c, a = 24.587(2), b = 7.0726(6), c = 14.171(1) Å, β = 119.257(8)°, V = 2149.9(4) Å3, Z = 4, Dobs = 1.454 g/cm3) and the alkyne derivative 4-(but-3-yn-1-yl)-1-(4-fluorobenzoyl)piperazine (4b, monoclinic, space group P21/n, a = 10.5982(2), b = 8.4705(1), c = 14.8929(3) Å, β = 97.430(1)°, V = 1325.74(4) Å3, Z = 4, Dobs = 1.304 g/cm3) were obtained from a saturated ethyl acetate solution. The rotational conformation of these compounds was also verified by XRD. As proof of concept for future labeling purposes, both nitropiperazines were reacted with [18F]F–. To test the applicability of these compounds as possible 18F-building blocks, two biomolecules were modified and chosen for conjugation either using the Huisgen-click reaction or the traceless Staudinger ligation.
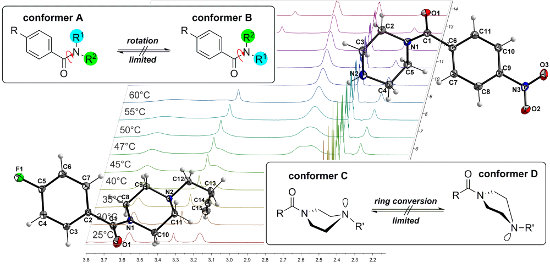
Graphical Abstract
Introduction
The development of new building blocks for specific and bioorthogonal labeling of biologically active compounds is of high importance. Depending on their size and composition, building blocks can influence (bio-)chemical parameters such as the lipophilicity (log P) affecting the solubility and the biological behavior of the resulting labeled biomolecule [1]. To diversify and regulate this behavior, compounds with a piperazine skeleton were chosen as excellent candidates for bispecific modification.
Simple alkylated and acylated secondary amines/amides are known from the literature [2] as well as their NMR properties [3]. The widely used solvent DMF is an intensely investigated example [4]. Functionalized piperazines [5], especially benzoylated derivatives, are our interest. In the past, mono or bisacylated piperazines with benzamide [6], nicotinamide [7] and isonicotinamide [8-11] residue were explored in terms of their NMR and complexation behavior. However, unsymmetrically substituted piperazines are rarely investigated [12,13].
N,N’-Unsymmetrically functionalized piperazines are the basis of the development of our new building blocks. In this case, one of the nitrogen atoms is used for the connection to the label (e.g., fluorescence dye, radionuclide) and the second is used for the introduction of a (bioorthogonal) functional group (e.g., azide, alkyne, phosphane, tetrazine) to later connect to the biomolecule via bioorthogonal ligation.
Our aim was the development of novel, N,N’-unsymmetrically functionalized piperazine derivatives using a high-yielding, simple synthesis with either a bioorthogonal alkyne or azide functionality for future labeling purposes. Furthermore, characterization of these piperazine derivatives was performed with emphasis on their particular NMR behavior. In order to demonstrate the Cu-catalyzed azide–alkyne click reaction (Huisgen 1,3-dipolar cycloaddition) and the traceless Staudinger ligation, a proof of concept study was performed for the site-selective labeling of a pharmacologically active peptide and a small organic compound. These compounds provide the respective reference compounds for later radiolabeling purposes. Finally, a procedure to synthesize the building blocks containing fluorine-18 was evaluated.
Results and Discussion
Synthesis of the piperazine compounds
Low-cost starting materials were applied for the preparation of all novel building blocks. First, piperazine (2) was reacted with functionalized benzoyl chlorides 1a,b to yield the mono-acylated amides 3a,b according to literature procedures [14]. Next, the necessary functional groups for the later click reactions to connect the resulting building blocks to biomolecules were introduced. Thus, 3a,b were alkylated with 4-tosylbutyne to give compounds 4a,b in high yields of 84% and 82%, respectively. These compounds are applicable in the classical Cu-catalyzed Huisgen-click reaction with azide-functionalized, biologically active molecules. Additionally, 3a,b were reacted with 3-azidopropyl tosylate to yield compounds 5a,b in high yields of 87% and 81%, respectively. These derivatives can be utilized for both variants of the Staudinger ligation in addition to the classical and strain-promoted variants of the Huisgen-click reaction. For future radiolabeling procedures, nitro derivatives 4a and 5a serve as starting material (precursor) whereas fluorine compounds 4b and 5b function as appendant reference compounds to analyze the prospective 18F-containing compounds. The reaction pathway for all piperazines is illustrated in Scheme 1.
![[1860-5397-12-242-i1]](/bjoc/content/inline/1860-5397-12-242-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Preparation of the nitro derivatives 4a and 5a and the fluorine-containing compounds 4b and 5b. Reagents and conditions: a) 1a, Et3N, CHCl3, 0 °C, 2 h, 0 °C then 2 h, rt; b) 1b, acetonitrile, 1 M HCl, rt, 4 h; c) 4-tosylbutyne, Et3N, THF, 60 °C, 3 d; d) 3-azidopropyl tosylate, Et3N, THF, 60 °C, 3 d.
Scheme 1: Preparation of the nitro derivatives 4a and 5a and the fluorine-containing compounds 4b and 5b. Rea...
Dynamic NMR studies
During the full characterization of our compounds, we observed a quite unusual behavior in the 1H and 13C NMR spectra. Four broad singlets (ratio: 1:1:1:1) were observed in the 1H NMR spectra of all nitro compounds 3a, 4a, and 5a (see Supporting Information File 1) and three broad singlets (ratio 1:1:2) were determined for all fluorine compounds 3b, 4b, and 5b measured in CDCl3 at 25 °C. Normally, under these conditions only two signals are expected for the NCH2-protons of unsymmetrically substituted piperazines [15-17].
To investigate this phenomenon, nitro compound 3a was chosen and 1H NMR spectra were measured in five different solvents to confirm this behavior. The results are illustrated in Figure 1. As an example, the spectrum of 3a shows four broad signals (δ = 2.81, 2.96, 3.33, 3.97 ppm in CDCl3 at 25 °C) for the piperazine NCH2 groups and evaluation of a H,H-COSY measured in CDCl3 showed an independent coupling of two NCH2 groups (Figure 2). Next, the HSQC spectra showed the independent coupling of the protons to the appropriate carbon signals (further detailed NMR spectra can be found in Supporting Information File 1). Additionally, four broad signals for the carbons of the NCH2 groups (e.g., 3a: δ = 43.7, 46.0, 46.3, 49.0 ppm) are found when analyzing the 13C NMR spectra.
![[1860-5397-12-242-1]](/bjoc/content/figures/1860-5397-12-242-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: 1H NMR spectra of compound 3a measured in five different solvents: (A) CDCl3, (B) DMSO-d6, (C) acetone-d6, (D) methanol-d4 and (E) acetonitrile-d3 (orange: region of interest).
Figure 1: 1H NMR spectra of compound 3a measured in five different solvents: (A) CDCl3, (B) DMSO-d6, (C) acet...
![[1860-5397-12-242-2]](/bjoc/content/figures/1860-5397-12-242-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: HSQC and H,H-COSY (small spectrum) of compound 3a measured in CDCl3 at 25 °C. The independent coupling of the NCH2 groups is marked.
Figure 2: HSQC and H,H-COSY (small spectrum) of compound 3a measured in CDCl3 at 25 °C. The independent coupl...
Two effects are responsible for this behavior. The first arises from the presence of two different conformers (rotamers); this is caused by the limited interconversion by rotation about the C–N amide bond resulting from the partial double bond character of N,N-dialkylated amides as shown in Figure 3. This observation is typical and can be found for symmetrically substituted amides. The best known and widest investigated example is DMF. Two distinct signals are observed in the 1H NMR spectrum of DMF as a result of two structurally different methyl groups (R1 ≠ R2) attached to the amide nitrogen [18,19]. In general, this behavior of symmetric N,N-dialkylamide spin systems is describable as first-order process on the NMR time scale [20].
![[1860-5397-12-242-3]](/bjoc/content/figures/1860-5397-12-242-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Illustration of the general partial double bond character of an amide bond and the limited isomerization of the amine nitrogen atom which results in possible conformers of piperazine compounds.
Figure 3: Illustration of the general partial double bond character of an amide bond and the limited isomeriz...
The same NMR behavior is observed for 4-nitrobenzoyl amides 3a, 4a and 5a. The expected single signal of the CH2 groups attached to the amide nitrogen is duplicated as seen for 3a in Figure 1. The coupling is pointed out by the analysis of a H,H-COSY of 3a (Figure 2, small spectrum).
The second effect is related to the reduced flipping of the piperazine ring at the amine nitrogen (Figure 3, bottom). In our case, interconversion of the amine is also reduced at room temperature. Normally, such formation of conformers is found for piperazines [21,22] and morpholines [21,23] only at lower temperatures (below −10 °C). Additionally, only the protons of 4-nitrobenzoylamides 3a, 4a and 5a exhibit this behavior at room temperature. Consequently, this phenomenon is strongly influenced by the substituent in the para-position of the benzoate (F vs NO2).
Both effects resulted in two different coalescence points dependent upon their different energy barriers. In order to further investigate the conformational behavior of the piperazines and to determine these energies, temperature-dependent NMR experiments [24] were performed for all nitro derivatives 3a, 4a, and 5a as well as for fluorine compound 3b. When monitoring compound 3a over a minimum range of 45 K, the four signals of the NCH2 groups gradually disappear and coalesce to the two expected signals at increased temperatures (>67 °C). At the coalescence temperature Tc, the exchange rate is given by the equation kexc = π·Δν/21/2 [25]. As a result, the activation energy (ΔG#exp) to the amide bond rotation can be calculated using the Eyring equation [14,16].
![[Graphic 1]](/bjoc/content/inline/1860-5397-12-242-i5.svg?max-width=637&scale=1.18182)
In general, the difference in chemical shifts Δδ (as well as Δν) strongly depends on the nature of the solvent [26]. Thus, the 1H NMR spectra seen in Figure 1 were recorded in CDCl3, DMSO-d6, acetone-d6, methanol-d4 and acetonitrile-d3 at 400 MHz and the results are summarized in Table 1. The NCH2 groups of 3a show the highest difference Δν1 and Δν2 when dissolved in CDCl3 and the lowest when dissolved in acetone-d6.
Table 1: NMR parameters Δν and kexc of nitro compound 3a measured in dependence of the solvent.
| solvent | Δν1 [Hz] | Δν2 [Hz] | kexc,1 [Hz] | kexc,2 [Hz] |
|---|---|---|---|---|
| CDCl3 | 59.5 | 176.2 | 132.2 | 391.4 |
| acetonitrile-d3 | 55.2 | 162.2 | 122.6 | 360.3 |
| DMSO-d6 | 49.5 | 157.6 | 110.0 | 350.1 |
| methanol-d4 | 53.3 | 155.8 | 118.4 | 346.1 |
| acetone-d6 | 30.4 | 133.2 | 67.5 | 295.9 |
Due to the low boiling point of most of these solvents, DMSO-d6 was chosen for the measurement of the coalescence temperature via temperature-dependent 1H NMR (Figure 4). For 3a, the Tc,1 was determined to be 50 °C (322 K) and Tc,2 was 67 °C (340 K). Using this data, the activation energies ΔG#exc were calculated to be 66.7 and 67.1 kJ/mol, respectively (Table 2).
![[1860-5397-12-242-4]](/bjoc/content/figures/1860-5397-12-242-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Temperature-dependent 1H NMR spectra of 3a measured in DMSO-d6 (aliphatic region of the piperazine protons from 2.00 to 4.00 ppm is shown).
Figure 4: Temperature-dependent 1H NMR spectra of 3a measured in DMSO-d6 (aliphatic region of the piperazine ...
Table 2: Evaluation of the Gibb’s energy ΔG#exc of compounds 3a, 3b, 4a and 5a from NMR data measured in DMSO-d6.
| Δν1 | Δν2 | kexc,1 | kexc,2 | Tc,1 [K] | Tc,2 [K] | ΔG#exc,1 [kJ/mol] | ΔG#exc,2 [kJ/mol] | |
|---|---|---|---|---|---|---|---|---|
| 3a | 49.5 | 157.6 | 110.0 | 350.1 | 323 | 340 | 66.7 | 67.1 |
| 3b | n.d. | 106.6 | n.d. | 236.8 | n.d. | 306 | n.d. | 61.1 |
| 4a | 48.5 | 154,0 | 107.7 | 342.1 | 320 | 338 | 66.1 | 66.7 |
| 5a | 48.6 | 154.9 | 108.0 | 344.1 | 315 | 338 | 65.0 | 66.7 |
For fluorine compound 3b, only the CH2 group attached to the amide nitrogen is split in two signals at room temperature and its Tc was found to be 33 °C (306 K) resulting in a ΔG#exc of 61.1 kJ/mol. This result nicely demonstrates the influence of the substituent in the ortho-position (F vs NO2) of the benzoate residue.
Next, the influence of alkyl groups on Tc and ΔG#exp were investigated using both nitrobenzoylpiperazines 4a and 5a. Both coalescence temperatures are found to be lower than those of the non-alkylated derivative 3a. This effect could arise due to the increased steric demand of the alkyl groups compared to the sole hydrogen connected to the amine nitrogen.
When comparing our results with the literature, Tc and ΔG#exp found for the amide site of the piperazines are in good agreement with the previously published (Tc = 330–340 K, ΔG#exp = 61–68 kJ/mol) [21,22,27]. In contrast, our values for Tc,1 and ΔG#exp,1 for the amine residues are much higher as found in the literature. For instance, N-alkylated morpholines showed a Tc of 248 K with a ΔG# of 11.1 kJ/mol [23] whereas piperidine shows a higher ΔG# of 42.3 and N-methylpiperidine a ΔG# = 49.8 kJ/mol [21,28].
X-ray structure analyses of 3a and 4b
Single crystals of 3a and 4b were obtained and their molecular structures determined using single crystal X-ray structure analysis. Crystals of 3a have monoclinic symmetry of the space group C2/c. Crystals of 4b have monoclinic symmetry of the space group P21/n. The C14–C15 distance of 1.188(1) Å and the C13–C14–C15 angle of 178.2(1)° clearly indicate this group to be an alkyne residue, thus enabling the use of the Huisgen-click reaction for binding to the target biomacromolecule. The molecular structures of these compounds and their atom numbering schemes are shown in Figure 5 and Figure 6.
![[1860-5397-12-242-5]](/bjoc/content/figures/1860-5397-12-242-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Molecular structure of compound 3a (ORTEP plot with 50% probability level).
Figure 5: Molecular structure of compound 3a (ORTEP plot with 50% probability level).
![[1860-5397-12-242-6]](/bjoc/content/figures/1860-5397-12-242-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Molecular structure of compound 4b (ORTEP plot with 50% probability level).
Figure 6: Molecular structure of compound 4b (ORTEP plot with 50% probability level).
The limited ability of the residues to rotate along the C1–N1 bond, as observed by NMR spectroscopy (see above), is supported by the results of both single-crystal structure determinations. The amine-type N2 atom in 4b has three bonds to the neighboring carbon atoms of about the same length (N2–C9: 1.466(1) Å, N2–C11: 1.466(1) Å, N2–C12: 1.464(1) Å), but the amide-type N1 atom has a significantly shorter distance to the C1 atom (1.350(1) Å), which carries the carbonyl O atom. The other two contacts of N1 to the C atoms of the piperazine ring are in the same distance range (N1–C8: 1.461(1) Å and N1–C10: 1.461(1) Å) as those of the N2 atom.
Furthermore, the environment of the N2 atom can best described as pyramidal with an average C–N2–C angle of 109.8°, whereas the environment of the N1 atom is almost planar, with an average C–N1–C angle of 119.9°. All these results indicate a partial double-bond character for the C1–N1 bond with a limited rotational ability. As discussed above, this results in two conformers, as shown for solutions of 4b by NMR spectroscopy. In the solid state, the molecules of the asymmetric unit are located on a side without any symmetry (besides identity). Through the symmetry element besides the molecule (i.e., inversion center) in the solid state, two conformers exist in a ratio of 1:1. Figure 7 shows a superimposed figure of the two conformers, where C1, the phenyl ring and F1 of both conformers are fitted on top of each other. The different structural arrangement of both conformers is clearly visible.
![[1860-5397-12-242-7]](/bjoc/content/figures/1860-5397-12-242-7.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: Superimposition fit of the two conformers, which exist in the ratio of 1:1 in the solid state structure of compound 4b (label A marks the atoms of the second conformer).
Figure 7: Superimposition fit of the two conformers, which exist in the ratio of 1:1 in the solid state struc...
Similar structural features are observed for 3a. The N1–C1 atom distance is again much shorter, 1.346(2) Å, than the other N–C bonds within the piperazine moiety, which range from 1.462(2) to 1.466(1) Å. Furthermore, the average bond angle of 119.8° of N1 indicates double-bond character and thereby limited rotational ability of the residues along the N1–C1 bond. As found for 4b, crystals of 3a contain two isomers in a 1:1 ratio as imposed by crystal symmetry.
Sample ligation using Huisgen-click and traceless Staudinger
As a proof of labeling concept, fluorine compound 4b was clicked to peptide 6 using the Cu-catalyzed Huisgen-click reaction. This SNEW peptide (SNEW: Ser-Asn-Glu-Trp) was chosen due to its biologically and pharmacologically activity and was modified with an azide moiety at the C-terminus to yield SNEWILPRLPQH-Azp 6 (the synthesis is reported elsewhere) [29]. The structure of 6 and the click reaction is shown in Scheme 2. For labeling purposes, building block 4b (10-fold excess) was added to peptide 6 which was dissolved in PBS buffer (pH 7.4), followed by addition of freshly prepared solutions of Na ascorbate and CuSO4. The mixture was stirred for 16 h at 40 °C and the desired product 7 was purified by semi-preparative HPLC.
![[1860-5397-12-242-i2]](/bjoc/content/inline/1860-5397-12-242-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Peptide labeling using the Huisgen-click reaction and building block 4b. Reagents and conditions: a) 4b, CuSO4, Na ascorbate, PBS buffer (pH 7.4), 40 °C, 16 h.
Scheme 2: Peptide labeling using the Huisgen-click reaction and building block 4b. Reagents and conditions: a...
The second proof of concept study was performed under Staudinger conditions using a small inhibitory molecule for the EphB4 receptor. For this purpose, the azide-containing building block 5b was reacted with 8 [30] to give 9. The resulting reference compound 9 was obtained after 3 h reaction time in a high yield of 76%. The reaction is shown in Scheme 3.
![[1860-5397-12-242-i3]](/bjoc/content/inline/1860-5397-12-242-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: The traceless Staudinger ligation to yield compound 9. Reagents and conditions: a) acetonitrile/water 10:1 (v/v), 60 °C, 3 h.
Scheme 3: The traceless Staudinger ligation to yield compound 9. Reagents and conditions: a) acetonitrile/wat...
Preparation of bioorthogonal 18F-containing building blocks
The development of new 18F-based radiotracers remains an ongoing goal in the field of radiopharmacy and provides tools for specific cancer diagnostics using positron emission tomography (PET) [31-33]. When radiotracers are based on tumor-specific peptides, proteins or antibodies [34,35], mild and fast methods for radiolabeling are mandatory because of the short half-live of [18F]fluoride (t1/2: 110 min). In most cases, indirect radiolabeling is used for these more sensitive biomacromolecules due to the harsh conditions (i.e., organic solvents, high temperatures and basic conditions) for the direct incorporation of [18F]fluoride, which can alter the biological/pharmacological behavior or, at least, destroy sensitive biomolecules [36-38]. For this purpose, bioorthogonal building blocks were developed using Huisgen-click or the Staudinger ligation.
Using these methods, a synthesis procedure for both bioorthogonal 18F-building blocks [18F]4b and [18F]5b was developed from their precursors 4a and 5a, respectively (Scheme 4). For this purpose, the nitro group of 4b and 5b was replaced by [18F]fluoride in a nucleophilic aromatic exchange reaction. To evaluate the radiolabeling process, the precursor 4a (approx. 3–4 mg) was first dissolved in anhydrous acetonitrile and added to dry [18F]fluoride (typically 0.5–1 GBq), but the radiochemical yield (RCY) did not exceed 5% after 60 min at 100 °C. Thus, anhydrous DMSO was chosen. After addition of [18F]fluoride the resulting mixture was stirred for 30 min at 150 °C and the 18F-building blocks were obtained in 12% RCY. An elongation of the reaction time to 60 min afforded [18F]4b in an elevated RCY of approx. 20% (80–155 MBq, decay corrected) after purification. Similar results were obtained for the labeling of azide-containing [18F]5b under the same reaction conditions. The radio-TLC of the reaction mixture of [18F]5b and the appropriate radio-TLC and (radio-)HPLC chromatograms are shown in Figure 8 and Figure 9. Compared to other alkyne and azide functionalized building blocks described in the literature [39], the radiochemical yields and the reaction times of [18F]4b and [18F]5b are similar.
![[1860-5397-12-242-i4]](/bjoc/content/inline/1860-5397-12-242-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Preparation of the radiolabeling building blocks [18F]4b and [18F]5b. Reagents and conditions: a) K[18F]F, Kryptofix K 2.2.2, DMSO, 150 °C, 60 min.
Scheme 4: Preparation of the radiolabeling building blocks [18F]4b and [18F]5b. Reagents and conditions: a) K[...
![[1860-5397-12-242-8]](/bjoc/content/figures/1860-5397-12-242-8.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: Radio-TLC (eluent: ethanol) of [18F]5b (Rf = 0.50; reaction mixture).
Figure 8: Radio-TLC (eluent: ethanol) of [18F]5b (Rf = 0.50; reaction mixture).
![[1860-5397-12-242-9]](/bjoc/content/figures/1860-5397-12-242-9.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 9: (Radio)HPLC of 5a (tR = 7.7 min, UV trace, red), 5b (tR = 6.7 min, UV trace: blue) and [18F]5b (tR = 7.1 min, γ-trace, green). The differences in the retention times of 5b and [18F]5b occurs due to the setup of UV- and γ-detector of the HPLC device.
Figure 9: (Radio)HPLC of 5a (tR = 7.7 min, UV trace, red), 5b (tR = 6.7 min, UV trace: blue) and [18F]5b (tR ...
Finally, the partition coefficients (log P) were determined to be 1.26 ± 0.01 (n = 3) for compound 4b and 1.45 ± 0.01 (n = 3) for 5b. Compared to other standard building blocks like [18F]SFB (log P = 1.8) [40], FBAM (log P = 2.7) [41], 4-fluorobenzaldehyde (log P = 1.9) [42] or the click labeling building block p-[18F]F-SA (log P = 1.7) [43], the lipophilicity of our 18F-building blocks is reduced. The hydrophilic character of the building blocks enables a radiolabeling in aqueous solutions and the influence of molecular size of the compounds to be labeled is negligible.
Conclusion
Novel piperazine derivatives were successfully synthesized with high yields using a convenient synthesis procedure. Evaluation of the NMR spectra showed doubled signals of the NCH2 groups of the piperazine moiety. One coalescence point was found in the spectra of the fluorine compounds whereas two different coalescence points were found for nitro compounds. A partial double bond in the amide residue and the limited ring conversion of the amine site led to rotational conformers. Activation energies ΔG#exc were calculated from coalescence temperatures Tc which were determined from dynamic 1H NMR measurements. The formation of conformers resulting from the partial double bond was additionally determined and verified using single crystal X-ray structure analysis. The second coalescence point is caused by the reduced flipping of the amine part in the nitro compounds. Furthermore, the coalescence temperature for the amine residue is higher than expected for amines. In contrast, the reduced flipping of the amine part was not found for the fluorine compounds. Thus, the coalescence is dependent on the substituent of the benzoyl moiety. Furthermore, a proof of concept was accomplished with a pharmacologically active peptide using the Huisgen-click reaction and compound 4b. Additionally, 5b was introduced in a small molecule using the traceless Staudinger ligation. The synthesis of the 18F-labeled building blocks [18F]4b and [18F]5b was accomplished using the SNAr concept. The appropriate precursors and reference compounds were prepared in two steps from simple commercially available starting materials in high yields, but at this stage the building blocks are not appropriate for further labeling purposes.
Experimental
General information
All chemicals were purchased from commercial suppliers and used without further purification unless otherwise specified. Anhydrous THF was purchased from Acros. NMR spectra of all compounds were recorded on an Agilent DD2-400 MHz NMR spectrometer. Chemical shifts of the 1H, 19F, and 13C spectra were reported in parts per million (ppm) using TMS as internal standard for 1H/13C and CFCl3 for 19F spectra. Mass spectrometric (MS) data were obtained on a Xevo TQ-S mass spectrometer (Waters) by electron spray ionization (ESI). The melting points were determined on a Galen III melting point apparatus (Cambridge Instruments & Leica) and are uncorrected. Microanalyses were carried out with an LECO CHNS 932 elemental analyzer. Chromatographic separations and TLC detections were performed using Merck Silica Gel 60 (63–200 μm) and Merck Silica Gel 60 F254 sheets, respectively. TLCs were developed by visualization under UV light (λ = 254 nm). Diffraction data were collected with a Bruker-Nonius Apex-II-diffractometer using graphite-monochromated Mo Kα radiation (λ = 0.71073 Å). The diffraction measurements were performed at −150 °C. The unit cell dimensions were recorded and refined using the angular settings of 7728 reflections for 4b and 6451 reflections for 3a. The structures were solved by direct methods and refined against F2 by full-matrix least-squares using the program suites from G. M. Sheldrick [44,45]. All non-hydrogen atoms were refined anisotropically; all hydrogen atoms except the one attached to the alkyne group (4b) were placed on geometrically calculated positions and refined using a riding model. The alkyne-H atom was refined isotropically. CCDC 1479835 and CCDC 1445857 contain the supplementary crystallographic data for compounds 3a and 4b. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif. Analytical (radio-)HPLC was performed on a VWR/Hitachi Elite LaChrome HPLC system, equipped with a reverse-phase column (Nucleosil 100-5C18 Nautilus, Machery Nagel), a UV diode array detector (250 nm), and a scintillation detector (Raytest, Gabi Star) at a flow rate of 1 mL/min. The radioactive compounds were identified with analytical radio-HPLC by comparison of the retention time of the reference compounds. Decay-corrected radiochemical yields (RCYs) were quantified by integration of radioactive peaks on a radio-TLC using a radio-TLC scanner (Fuji, BAS2000). [18F]Fluoride was produced using the PET cyclotron Cyclone 18/9 (IBA). [18O]H2O was irradiated with protons (18 MeV, 30 µA) exploiting the 18O(p,n)18F nuclear reaction.
CAUTION! Hazard warning for organic azides: risk of explosion by shock, friction, and fire upon heating. Store these compounds in a cool location.
Synthetical procedures
1-(4-Nitrobenzoyl)piperazine (3a): Piperazine (2, 1.0 g, 11.6 mmol) and Et3N (1.17g, 11.6 mmol) were dissolved in chloroform (30 mL) and the mixture was cooled to 0 °C. At this temperature, a solution of 4-nitrobenzoyl chloride (1a, 1.0 g, 5.4 mmol) dissolved in 30 mL of chloroform was added dropwise and the resulting mixture was allowed to stir at 0 °C for 2 h and at rt for 2 h. Afterwards, the precipitate was filtered and the chloroform solution was washed with saturated hydrogen carbonate solution (30 mL), with water (2 × 30 mL) and dried over Na2SO4. Finally, the solvent was removed to yield compound 3a (700 mg, 55%) as yellowish solid. mp 74 °C; 1H NMR (400 MHz, CDCl3) δ 1.72 (s, 1H, NH), 2.81 (br s, 2H, Pip-H), 2.96 (br s, 2H, Pip-H), 3.33 (br s, 2H, Pip-H), 3.77 (br s, 2H, Pip-H), 7.56 (d, 3J = 8.7 Hz, 2H, H-o), 8.27 (d, 3J = 8.7 Hz, 2H, H-m); 13C NMR (101 MHz, CDCl3) δ 43.5, 46.0, 46.6, 49.0 (4 × Pip-C), 124.0 (C-m), 128.2 (C-o), 142.2 (C-i), 148.5 (C-p), 163.1 (C=O); MS (ESI+) m/z (%): 258 (13) [M+ + Na], 236 (100) [M+ + H]; Anal. calcd for C11H13N3O3 (235.24): C, 56.16; H, 5.57; N, 17.86 %; found, C, 56.41; H, 5.35; N, 17.65.
1-(4-Fluorobenzoyl)piperazine (3b): Piperazine (2, 2.9 g, 33.67 mmol) was dissolved in HCl (1 M, 50 mL). A solution of 4-fluorobenzoyl chloride (1b, 1.1 g, 6.94 mmol) dissolved in acetonitrile (5 mL) was added dropwise and the resulting mixture was stirred for 4 h at rt. Afterwards, additional 9 mL of 1 M HCl were added and the aqueous layer was extracted with ethyl acetate (2 × 20 mL). Then, KOH was added to the aqueous phase until pH 8 was reached. The aqueous layer was again extracted with chloroform (2 × 25 mL), the combined organic layers were dried over Na2SO4 and the solvent was removed to yield 931 mg of compound 3b. Spectra are in agreement with those found in the literature [46].
4-(But-3-yn-1-yl)-1-(4-nitrobenzoyl)piperazine (4a): Compound 3a (110 mg, 0.47 mmol), but-3-yn-1-yl tosylate (150 mg, 0.67 mmol) and Et3N (100 mg, 0.98 mmol) were dissolved in anhydrous THF (10 mL) and the resulting mixture was stirred at 60 °C for 3 d. After reaction control by TLC, THF was changed by ethyl acetate (15 mL), water (15 mL) was added and the aqueous layer was extracted with ethyl acetate (3 × 15 mL). The combined organic layers were dried over Na2SO4, the solvent was removed and the crude product was purified via automated column chromatography (Biotage: 10 g KP-Sil, Gradient: petroleum ether → EtOAc) to yield 4a (112 mg, 84%) as yellow solid. Rf = 0.59 (ethanol); mp 76 °C; analytical HPLC: tR = 6.0 min (eluent: CH3CN/H2O, 15:85 + 0.1% TFA); 1H NMR (400 MHz, CDCl3) δ 1.97 (t, 4J = 2.5 Hz, 1H, ≡CH), 2.36 (dt, 3J = 7.3 Hz, 4J = 2.5 Hz, 2H, CH2C≡), 2.43 (br s, 2H, Pip-H), 2.51–2.65 (m, 4H, Pip-H, NCH2), 3.36 (br s, 2H, Pip-H), 3.79 (br s, 2H, Pip-H), 7.55 (d, 3J = Hz, 2H, H-o), 8.25 (d, 3J = Hz, 2H, H-m); 13C NMR (101 MHz, CDCl3) δ 17.0 (CH2C≡), 42.2, 47.6, 52.4, 53.1 (4 × Pip-C), 56.7 (NCH2), 69.5 (≡CH), 82.4 (CH2C≡), 124.0 (C-m), 128.2 (C-o), 142.0 (C-i), 148.4 (C-p), 167.9 (C=O); MS (ESI+) m/z (%): 288 (100) [M+ + H]; Anal. calcd for C15H17N3O3 (287.31): C, 62.71; H, 5.96; N, 14.63; found, C, 62.51; H, 5.95; N, 14.65.
4-(But-3-yn-1-yl)-1-(4-fluorobenzoyl)piperazine (4b): Compound 3b (70 mg, 0.34 mmol), but-3-yn-1-yl tosylate (90 mg, 0.40 mmol) and Et3N (51 mg, 0.50 mmol) were dissolved in anhydrous THF (6 mL) and the resulting mixture was stirred at 60 °C for 3 d. After reaction control by TLC, THF was changed by ethyl acetate (15 mL), water (15 mL) was added and the aqueous layer was extracted with ethyl acetate (3 × 15 mL). The combined organic layers were dried over Na2SO4, the solvent was removed and the crude product was purified via automated column chromatography (Biotage: 10 g KP-Sil, Gradient: petroleum ether → EtOAc) to yield 4b (72 mg, 82%) as colorless solid. Rf = 0.60 (ethanol); analytical HPLC: tR = 5.5 min (eluent: CH3CN/H2O, 15:85 + 0.1% TFA); 1H NMR (400 MHz, CDCl3) δ 1.98 (t, 4J = 2.7 Hz, 1H, ≡CH), 2.38 (dt, 3J = 7.4 Hz, 4J = 2.7 Hz, 2H, CH2C≡), 2.50 (br s, 4H, Pip-H), 2.62 (t, 2H, 3J = 7.4 Hz, NCH2), 3.45 (br s, 2H, Pip-H), 3.75 (br s, 2H, Pip-H), 7.08 (“t”, 3JH,F = 3Jo,m = 8.7 Hz, H-m), 7.40 (dd, 2JH,F = 5.4 Hz, 3Jo,m = 8.7 Hz, H-o); 13C NMR (101 MHz, CDCl3) δ 17.0 (CH2C≡), 42.4, 47.8, 52.8 (4 x Pip-C), 56.9 (NCH2), 69.4 (≡CH), 82.5 (CH2C≡), 115.7 (d, 2JC,F = 21.8 Hz, C-m), 129.5 (d, 3JC,F = 8.5 Hz, C-o), 131.9 (d, 4JC,F = 3.3 Hz, C-i), 163.5 (d, 1JC,F = 249.9 Hz, C-p), 169.5 (C=O); 19F NMR (376 MHz, CDCl3) δ −110.3; MS (ESI+) m/z (%): 261 (100) [M+ + H]; Anal. calcd for C15H17FN2O (260.31): C, 69.21; H, 6.58; N, 10.76; found, C, 68.99; H, 6.61; N, 10.80.
4-(3-Azidopropyl)-1-(4-nitrobenzoyl)piperazine (5a): Compound 3a (150 mg, 0.64 mmol), 3-azidopropyl tosylate (195 mg, 0.77 mmol) and Et3N (129 mg, 1.28 mmol) were dissolved in anhydrous THF (10 mL) and the resulting mixture was stirred at 60 °C for 3 d at rt. After reaction control by TLC, THF was changed by ethyl acetate (15 mL), water (15 mL) was added and the aqueous layer was extracted with ethyl acetate (3 × 15 mL). The combined organic layers were dried over Na2SO4, the solvent was removed and the crude product was purified via automated column chromatography (Biotage: 10 g KP-Sil, Gradient: petroleum ether → EtOAc) to yield 5a (176 mg, 87%) as yellow solid. Rf = 0.49 (ethanol); analytical HPLC: tR = 7.7 min (eluent: CH3CN/H2O, 15:85 + 0.1% TFA); mp 64 °C; 1H NMR (400 MHz, CDCl3) δ 1.76 (qi, 3J = 6.8 Hz, 1H, CH2), 2.27–2.60 (m, 6H, Pip-H, NCH2), 3.25–3.47 (m, 4H, Pip-H, CH2N3), 3.80 (br s, 2H, Pip-H), 7.56 (d, 3J = Hz, 2H, H-o), 8.27 (d, 3J = Hz, 2H, H-m); 13C NMR (101 MHz, CDCl3) δ 26.2 (CH2), 42.3, 47.7, 52.7, 53.5 (4 × Pip-C), 49.5 (CH2N3), 55.1 (NCH2), 124.0 (C-m), 128.2 (C-o), 142.1 (C-i), 148.5 (C-p), 168.0 (C=O); MS (ESI+) m/z (%): 319 (100) [M+ + H]; Anal. calcd for C14H18FN6O3 (318.33): C, 52.82; H, 5.70; N, 26.40; found, C, 52.79; H, 5.66; N, 26.11.
4-(3-Azidopropyl)-1-(4-fluorobenzoyl)piperazine (5b): Compound 3b (83 mg, 0.40 mmol), 3-azidopropyl tosylate (122 mg, 0.48 mmol) and Et3N (61 mg, 0.60 mmol) were dissolved in anhydrous THF (6 mL) and the resulting mixture was stirred at 60 °C for 3 d. After reaction control by TLC, THF was changed by ethyl acetate (15 mL), water (15 mL) was added and the aqueous layer was extracted with ethyl acetate (3 × 15 mL). The combined organic layers were dried over Na2SO4, the solvent was removed and the crude product was purified via automated column chromatography (Biotage: 10 g KP-Sil, Gradient: petroleum ether → EtOAc) to yield 5b (94 mg, 81%) as colorless syrup. Rf = 0,48 (ethanol); analytical HPLC: tR = 6.7 min (eluent: CH3CN/H2O, 15:85 + 0.1% TFA); 1H NMR (400 MHz, CDCl3) δ 1.76 (qi, 2H, 3J = 6.7 Hz, CH2), 2.30–2.56 (m, 6H, NCH2+Pip-H), 3.35 (t, 2H, 3J = 6.7 Hz, CH2N3), 3.38–3.88 (br d, 4H, Pip-H), 7.08 (“t”, 3JH,F = 3Jo,m = 8.8 Hz, H-m), 7.40 (dd, 2JH,F = 5.4 Hz, 3Jo,m = 8.8 Hz, H-o); 13C NMR (101 MHz, CDCl3) δ 26.3 (CH2), 42.4, 48.0 (2 × Pip-C), 49.5 (CH2N3), 53.0, 53.5 (2 × Pip-C), 55.2 (CH2N), 115.7 (d, 2JC,F = 21.8 Hz, C-m), 129.5 (d, 3JC,F = 8.4 Hz, C-o), 131.9 (d, 4JC,F = 3.3 Hz, C-i), 163.5 (d, 1JC,F = 148.6 Hz, C-p), 163.5 (C=O); 19F NMR (376 MHz, CDCl3) δ −110.4; MS (ESI+) m/z (%): 292 (100) [M+ + H]; Anal. calcd for C14H18FN5O (291.32): C, 57.72; H, 6.23; N, 24.04; found, C, 57.90; H, 6.21; N, 24.24.
SNEW-peptide (7): Azide-functionalized peptide 6 (4.55 mg, 2.8 µmol) and 4b (1.00 mg, 3.84 µmol) were dissolved in PBS buffer (400 µL). Na ascorbate (50 µL, 0.6 M) and CuSO4 (50 µL, 0.4 M) were added and the reaction mixture was maintained at 40 °C for 16 h. Peptide 7 was obtained as colorless powder (4.01 mg, 76%) after purification using semi-preparative HPLC and lyophilization; MS (ESI+) m/z: calcd, 1886 [M]+; found, 944 [M + 2H]2+.
5-((3-((4-((5-Chlorobenzo[d][1,3]dioxol-4-yl)amino)pyrimidin-2-yl)amino)phenyl)sulfonyl)-N-(3-(4-(4-fluorobenzoyl)piperazin-1-yl)propyl)pentanamide (9): Compounds 5b (63 mg, 0.22 mmol) and 8 (165 mg, 22 mmol) were dissolved in a mixture of acetonitrile and water (5.5 mL, 10:1 v:v) and the resulting solution was maintained at 60 °C for 3 h. Afterwards, the solvent was removed and the crude product was purified via column chromatography to give 9 (112 mg, 69%) as a pale yellow syrup. Rf = 0.2 (EtOAc); 1H NMR (400 MHz, CDCl3) δ 1.41–1.52 (m, 2H, CH2), 1.57–1.67 (m, 2H, CH2), 1.72–1.81 (m, 2H, CH2), 1.93–2.00 (t, 3J = 7.2 Hz, 2H, CH2C=O), 2.33–2.54 (m, 6H, 3 × CH2N), 2.98 (t, 3J = 7.6 Hz, 2H, CH2S), 3.30–3.89 (m, 6H, 3 × CH2N), 5.93 (s, 2H, OCH2O), 6.05 (d, 3J = 5.9 Hz, 1H, Hdiox), 6.69 (d, 3J = 8.7 Hz, 1H, Hpyr), 6.92 (d, 3J = 8.7 Hz, 1H, Hpyr), 6.97 (s, 1H, NH), 7.07 (t, 3J = 8.5 Hz, 2H, H-o), 7.38–7.50 (m, 5H, H-m + HAr), 7.97 (d, 3J = 7.9 Hz, 1H, HAr), 8.05–8.10 (m, 2H, Hdiox + NH); 13C NMR (101 MHz, CDCl3) δ 22.0, 22.7, 26.1, 32.8 (4 × CH2), 42.3, 47.8 (br. s, 2 × CH2Pip), 49.3 (CH2N), 52.8, 53.3 (br. s, 2 × CH2Pip), 55.0, 55.6 (2 × CH2), 97.9 (CHAr), 102.2 (OCH2O), 106.7, 115.5 (d, 2JC,F = 21.5 Hz, C-m), 117.7, 118.6, 120.4, 122.0, 123.3, 123.7, 129.3 (d, 3JC,F = 8.6 Hz, C-o), 129.5, 131.9 (d, 4JC,F = 3.3 Hz, C-i), 139.2, 141.2, 143.5, 147.5, 156.9, 159.3, 161.1, 163.3 (d, 1JC,F = 251.0 Hz, C-p), 169.3 (C=O); 19F NMR (376 MHz, CDCl3) δ −110.3; MS (ESI+) m/z (%): 752 (80) [M + H]+; Anal. calcd for C36H39ClFN7O6S (752.25): C 57.48, H 5.23, N 13.03; found, C 57.66, H 5.21, N 13.31.
4-(But-3-yn-1-yl)-1-(4-[18F]fluorobenzoyl)piperazine ([18F]4b): Similar as described in [47] an anion-exchange cartridge (Waters, Sep-Pak Light Accell Plus QMA) was activated by rinsing with 5 mL of a 1 M NaHCO3 solution and 10 mL of deionized H2O. It was charged with [18F]fluoride (0.5–1 GBq) and eluted with 1.5 mL of a solution of Kryptofix 2.2.2 (10 mg/mL) and K2CO3 (13 mM) in 7 mL CH3CN and 43 mL H2O. The solvents were evaporated azeotropically by subsequent addition of three portions of 1 mL each of anhydrous CH3CN under a stream of nitrogen at 110 °C. Precursor 4a (3–4 mg) was dissolved in 400 µL of anhydrous DMSO, and the mixture was added to the [18F]fluoride-containing sealed vial. The resulting solution was heated at 150 °C for 60 min. Afterwards, the mixture was treated with 25 mL deionized H2O and then passed through a C18 cartridge (LiChrolut RP-18/Merck, 500 mg) to yield 80–155 MBq (≈20% RCY, dc) of [18F]4b. Samples for analytical radio-TLC and radio-HPLC were taken. Analytical radio-TLC: Rf = 0.50 (ethanol). Analytical radio-HPLC: tR = 5.7 min (eluent: CH3CN/H2O, 15:85 + 0.1% TFA).
4-(3-Azidopropyl)-1-(4-[18F]fluorobenzoyl)piperazine ([18F]5b): Similar as described in [47] an anion-exchange cartridge (Waters, Sep-Pak Light Accell Plus QMA) was activated by rinsing with 5 mL of a 1 M NaHCO3 solution and 10 mL of deionized H2O. It was charged with [18F]fluoride (0.5–1 GBq) and eluted with 1.5 mL of a solution of Kryptofix 2.2.2 (10 mg/mL) and K2CO3 (13 mM) in 7 mL CH3CN and 43 mL H2O. The solvents were evaporated azeotropically by subsequent addition of three portions of 1 mL each of anhydrous CH3CN under a stream of nitrogen at 110 °C. Precursor 5a (3–4 mg) was dissolved in 400 µL of anhydrous DMSO, and the mixture was added to the [18F]fluoride-containing sealed vial. The resulting solution was heated at 150 °C for 60 min. Afterwards, the mixture was treated with 25 mL deionized H2O and then passed through a C18 cartridge (LiChrolut RP-18/Merck, 500 mg) to yield 69–134 MBq (≈17% RCY, dc) of [18F]5b. Samples for analytical radio-TLC and radio-HPLC were taken. Analytical radio-TLC: Rf = 0.49 (ethanol). Analytical radio-HPLC: tR = 7.1 min (eluent: CH3CN/H2O, 15:85 + 0.1% TFA).
Supporting Information
| Supporting Information File 1: Copies of NMR spectra of investigated piperazines, radioHPLC chromatograms, and separation methods for piperazines. | ||
| Format: PDF | Size: 2.5 MB | Download |
References
-
Hermanson, G. T., Ed. Bioconjugate Techniques, 2nd ed.; Elsevier – Academic Press: London, 2008.
Return to citation in text: [1] -
Lewis, F. D.; Burch, E. L. J. Phys. Chem. 1996, 100, 4055–4063. doi:10.1021/jp9526758
Return to citation in text: [1] -
Emir, N.; Bilge, M.; Tursun, M.; Keşan, G.; Parlak, C. Spectrochim. Acta, Part A 2014, 127, 388–395. doi:10.1016/j.saa.2014.02.085
Return to citation in text: [1] -
Gutowsky, H. S.; Holm, C. H. J. Chem. Phys. 1956, 25, 1228–1234. doi:10.1063/1.1743184
Return to citation in text: [1] -
Takasu, K.; Tanaka, K.; Fuji, K. Chem. Pharm. Bull. 2001, 49, 655–656. doi:10.1248/cpb.49.655
Return to citation in text: [1] -
Zhao, B.; Xiao, Y.; Yuan, D.; Lu, C.; Yao, Y. Dalton Trans. 2016, 45, 3880–3887. doi:10.1039/C5DT04217H
Return to citation in text: [1] -
Barnes, D. J.; Chapman, R. L.; Vagg, R. S.; Watton, E. C. J. Chem. Eng. Data 1978, 23, 349–350. doi:10.1021/je60079a006
Return to citation in text: [1] -
Wang, X.; Chang, Z.; Lin, H.; Tian, A.; Liu, G.; Zhang, J. Dalton Trans. 2014, 43, 12272–12278. doi:10.1039/C4DT01211A
Return to citation in text: [1] -
Mizzi, J. E.; LaDuca, R. L. Inorg. Chim. Acta 2014, 421, 183–190. doi:10.1016/j.ica.2014.05.038
Return to citation in text: [1] -
Sahoo, H. S.; Chand, D. K. Dalton Trans. 2010, 39, 7223–7225. doi:10.1039/c0dt00337a
Return to citation in text: [1] -
Tripathy, D.; Sahoo, H. S.; Ramkumar, V.; Chand, D. K. RSC Adv. 2014, 4, 18595–18599. doi:10.1039/c3ra47904h
Return to citation in text: [1] -
Iriepa, I.; Madrid, A. I.; Gálvez, E.; Bellanato, J. J. Mol. Struct. 2006, 787, 8–13. doi:10.1016/j.molstruc.2005.10.025
Return to citation in text: [1] -
Hirsch, J. A.; Augustine, R. L.; Koletar, G.; Wolf, H. G. J. Org. Chem. 1975, 40, 3547–3550. doi:10.1021/jo00912a017
Return to citation in text: [1] -
DeBernardis, J. F.; Kerkman, D. J.; Zinkowski, R. P. Certain substituted 1-aryl-3-piperazin-1’-yl propanones to treat Alzheimers Desease. U.S. Patent US5658909 A, Aug 19, 1997.
Return to citation in text: [1] [2] -
Pretze, M.; Mamat, C. J. Fluorine Chem. 2013, 150, 25–35. doi:10.1016/j.jfluchem.2013.02.028
Return to citation in text: [1] -
Grosse-Gehling, P.; Wuest, F. R.; Peppel, T.; Köckerling, M.; Mamat, C. Radiochim. Acta 2011, 99, 365–373. doi:10.1524/ract.2011.1834
Return to citation in text: [1] [2] -
Mamat, C.; Flemming, A.; Köckerling, M. Crystals 2012, 2, 90–95. doi:10.3390/cryst2010090
Return to citation in text: [1] -
Wiberg, K. B.; Rablen, P. R.; Rush, D. J.; Keith, T. A. J. Am. Chem. Soc. 1995, 117, 4261–4270. doi:10.1021/ja00120a006
Return to citation in text: [1] -
LeMaster, C. B.; True, N. S. J. Phys. Chem. 1989, 93, 1307–1311. doi:10.1021/j100341a027
Return to citation in text: [1] -
Skorupska, E. A.; Nazarski, R. B.; Ciechańska, M.; Jóźwiak, A.; Kłys, A. Tetrahedron 2013, 69, 8147–8154. doi:10.1016/j.tet.2013.07.046
Return to citation in text: [1] -
Harris, R. K.; Spragg, R. A. Chem. Commun. 1966, 314–317. doi:10.1039/c19660000314
Return to citation in text: [1] [2] [3] [4] -
Hietapelto, V.; Laitinen, R. S.; Pursiainen, J.; Rahkamaa, E. Acta Chem. Scand. 1999, 53, 7–14. doi:10.3891/acta.chem.scand.53-0007
Return to citation in text: [1] [2] -
Baskar, R.; Baby, C.; Moni, M. S.; Subramanian, K. J. Mol. Struct. 2013, 1040, 90–97. doi:10.1016/j.molstruc.2013.02.029
Return to citation in text: [1] [2] -
Jackman, L. M.; Cotton, F. A. Dynamic Nuclear Magnetic Resonance Spectroscopy; Academic Press: New York, 1975.
Return to citation in text: [1] -
Friebolin, H. Ein- und zweidimensionale NMR-Spektroskopie – Eine Einführung; Wiley-VCH: Weinheim, 1999; 131 ff..
Return to citation in text: [1] -
Abraham, R. J.; Byrne, J. J.; Griffiths, L.; Perez, M. Magn. Reson. Chem. 2006, 44, 491–509. doi:10.1002/mrc.1747
Return to citation in text: [1] -
Miron, Y.; McGarvey, B. R.; Morawetz, H. Macromolecules 1969, 2, 154–161. doi:10.1021/ma60008a009
Return to citation in text: [1] -
Lambert, J. B.; Oliver, W. L., Jr.; Packard, B. S. J. Am. Chem. Soc. 1971, 93, 933–937. doi:10.1021/ja00733a025
Return to citation in text: [1] -
Pretze, M.; Kuchar, M.; Bergmann, R.; Steinbach, J.; Pietzsch, J.; Mamat, C. ChemMedChem 2013, 8, 935–945. doi:10.1002/cmdc.201300053
Return to citation in text: [1] -
Wodtke, R.; König, J.; Pigorsch, A.; Köckerling, M.; Mamat, C. Dyes Pigm. 2015, 113, 263–273. doi:10.1016/j.dyepig.2014.08.022
Return to citation in text: [1] -
Cole, E. L.; Stewart, M. N.; Littich, R.; Hoareau, R.; Scott, P. J. H. Curr. Top. Med. Chem. 2014, 14, 875–900. doi:10.2174/1568026614666140202205035
Return to citation in text: [1] -
Miller, P. W.; Long, N. J.; Vilar, R.; Gee, A. D. Angew. Chem., Int. Ed. 2008, 47, 8998–9033. doi:10.1002/anie.200800222
Return to citation in text: [1] -
Littich, R.; Scott, P. J. H. Angew. Chem., Int. Ed. 2012, 51, 1106–1109. doi:10.1002/anie.201106785
Return to citation in text: [1] -
Gu, Y.; Huang, D.; Liu, Z.; Huang, J.; Zeng, W. Med. Chem. 2011, 7, 334–344. doi:10.2174/157340611796799140
Return to citation in text: [1] -
Cai, L.; Lu, S.; Pike, V. W. Eur. J. Org. Chem. 2008, 2853–2873. doi:10.1002/ejoc.200800114
Return to citation in text: [1] -
Jacobson, O.; Kiesewetter, D. O.; Chen, X. Bioconjugate Chem. 2015, 21, 1–18. doi:10.1021/bc500475e
Return to citation in text: [1] -
Olberg, D. E.; Hjelstuen, O. K. Curr. Top. Med. Chem. 2010, 10, 1669–1679. doi:10.2174/156802610793176747
Return to citation in text: [1] -
Richter, S.; Wuest, F. Molecules 2014, 19, 20536–20556. doi:10.3390/molecules191220536
Return to citation in text: [1] -
Pretze, M.; Pietzsch, D.; Mamat, C. Molecules 2013, 18, 8618–8665. doi:10.3390/molecules18078618
Return to citation in text: [1] -
Wester, H.-J.; Hamacher, K.; Stoecklin, G. Nucl. Med. Biol. 1996, 23, 365–372. doi:10.1016/0969-8051(96)00017-0
Return to citation in text: [1] -
Berndt, M.; Pietzsch, J.; Wuest, F. Nucl. Med. Biol. 2007, 34, 5–15. doi:10.1016/j.nucmedbio.2006.09.009
Return to citation in text: [1] -
Poethko, T.; Schottelius, M.; Thumshirn, G.; Hersel, U.; Herz, M.; Henriksen, G.; Kessler, H.; Schwaiger, M.; Wester, H.-J. J. Nucl. Med. 2004, 45, 892–902.
Return to citation in text: [1] -
Ramenda, T.; Steinbach, J.; Wuest, F. Amino Acids 2013, 44, 1167–1180. doi:10.1007/s00726-012-1450-4
Return to citation in text: [1] -
Sheldrick, G. M. Acta Crystallogr., Sect. A: Found. Adv. 2008, 64, 112–122. doi:10.1107/S0108767307043930
Return to citation in text: [1] -
Sheldrick, G. M. SHELXL 2014/1; University of Göttingen: Germany, 2014.
Return to citation in text: [1] -
Verma, S. K.; Acharya, B. N.; Kaushik, M. P. Org. Lett. 2010, 12, 4232–4235. doi:10.1021/ol101604q
Return to citation in text: [1] -
Mamat, C.; Mosch, B.; Neuber, C.; Köckerling, M.; Bergmann, R.; Pietzsch, J. ChemMedChem 2012, 7, 1991–2003. doi:10.1002/cmdc.201200264
Return to citation in text: [1] [2]
| 39. | Pretze, M.; Pietzsch, D.; Mamat, C. Molecules 2013, 18, 8618–8665. doi:10.3390/molecules18078618 |
| 40. | Wester, H.-J.; Hamacher, K.; Stoecklin, G. Nucl. Med. Biol. 1996, 23, 365–372. doi:10.1016/0969-8051(96)00017-0 |
| 41. | Berndt, M.; Pietzsch, J.; Wuest, F. Nucl. Med. Biol. 2007, 34, 5–15. doi:10.1016/j.nucmedbio.2006.09.009 |
| 1. | Hermanson, G. T., Ed. Bioconjugate Techniques, 2nd ed.; Elsevier – Academic Press: London, 2008. |
| 5. | Takasu, K.; Tanaka, K.; Fuji, K. Chem. Pharm. Bull. 2001, 49, 655–656. doi:10.1248/cpb.49.655 |
| 21. | Harris, R. K.; Spragg, R. A. Chem. Commun. 1966, 314–317. doi:10.1039/c19660000314 |
| 23. | Baskar, R.; Baby, C.; Moni, M. S.; Subramanian, K. J. Mol. Struct. 2013, 1040, 90–97. doi:10.1016/j.molstruc.2013.02.029 |
| 4. | Gutowsky, H. S.; Holm, C. H. J. Chem. Phys. 1956, 25, 1228–1234. doi:10.1063/1.1743184 |
| 24. | Jackman, L. M.; Cotton, F. A. Dynamic Nuclear Magnetic Resonance Spectroscopy; Academic Press: New York, 1975. |
| 3. | Emir, N.; Bilge, M.; Tursun, M.; Keşan, G.; Parlak, C. Spectrochim. Acta, Part A 2014, 127, 388–395. doi:10.1016/j.saa.2014.02.085 |
| 20. | Skorupska, E. A.; Nazarski, R. B.; Ciechańska, M.; Jóźwiak, A.; Kłys, A. Tetrahedron 2013, 69, 8147–8154. doi:10.1016/j.tet.2013.07.046 |
| 47. | Mamat, C.; Mosch, B.; Neuber, C.; Köckerling, M.; Bergmann, R.; Pietzsch, J. ChemMedChem 2012, 7, 1991–2003. doi:10.1002/cmdc.201200264 |
| 2. | Lewis, F. D.; Burch, E. L. J. Phys. Chem. 1996, 100, 4055–4063. doi:10.1021/jp9526758 |
| 21. | Harris, R. K.; Spragg, R. A. Chem. Commun. 1966, 314–317. doi:10.1039/c19660000314 |
| 22. | Hietapelto, V.; Laitinen, R. S.; Pursiainen, J.; Rahkamaa, E. Acta Chem. Scand. 1999, 53, 7–14. doi:10.3891/acta.chem.scand.53-0007 |
| 47. | Mamat, C.; Mosch, B.; Neuber, C.; Köckerling, M.; Bergmann, R.; Pietzsch, J. ChemMedChem 2012, 7, 1991–2003. doi:10.1002/cmdc.201200264 |
| 12. | Iriepa, I.; Madrid, A. I.; Gálvez, E.; Bellanato, J. J. Mol. Struct. 2006, 787, 8–13. doi:10.1016/j.molstruc.2005.10.025 |
| 13. | Hirsch, J. A.; Augustine, R. L.; Koletar, G.; Wolf, H. G. J. Org. Chem. 1975, 40, 3547–3550. doi:10.1021/jo00912a017 |
| 15. | Pretze, M.; Mamat, C. J. Fluorine Chem. 2013, 150, 25–35. doi:10.1016/j.jfluchem.2013.02.028 |
| 16. | Grosse-Gehling, P.; Wuest, F. R.; Peppel, T.; Köckerling, M.; Mamat, C. Radiochim. Acta 2011, 99, 365–373. doi:10.1524/ract.2011.1834 |
| 17. | Mamat, C.; Flemming, A.; Köckerling, M. Crystals 2012, 2, 90–95. doi:10.3390/cryst2010090 |
| 44. | Sheldrick, G. M. Acta Crystallogr., Sect. A: Found. Adv. 2008, 64, 112–122. doi:10.1107/S0108767307043930 |
| 45. | Sheldrick, G. M. SHELXL 2014/1; University of Göttingen: Germany, 2014. |
| 8. | Wang, X.; Chang, Z.; Lin, H.; Tian, A.; Liu, G.; Zhang, J. Dalton Trans. 2014, 43, 12272–12278. doi:10.1039/C4DT01211A |
| 9. | Mizzi, J. E.; LaDuca, R. L. Inorg. Chim. Acta 2014, 421, 183–190. doi:10.1016/j.ica.2014.05.038 |
| 10. | Sahoo, H. S.; Chand, D. K. Dalton Trans. 2010, 39, 7223–7225. doi:10.1039/c0dt00337a |
| 11. | Tripathy, D.; Sahoo, H. S.; Ramkumar, V.; Chand, D. K. RSC Adv. 2014, 4, 18595–18599. doi:10.1039/c3ra47904h |
| 18. | Wiberg, K. B.; Rablen, P. R.; Rush, D. J.; Keith, T. A. J. Am. Chem. Soc. 1995, 117, 4261–4270. doi:10.1021/ja00120a006 |
| 19. | LeMaster, C. B.; True, N. S. J. Phys. Chem. 1989, 93, 1307–1311. doi:10.1021/j100341a027 |
| 46. | Verma, S. K.; Acharya, B. N.; Kaushik, M. P. Org. Lett. 2010, 12, 4232–4235. doi:10.1021/ol101604q |
| 7. | Barnes, D. J.; Chapman, R. L.; Vagg, R. S.; Watton, E. C. J. Chem. Eng. Data 1978, 23, 349–350. doi:10.1021/je60079a006 |
| 42. | Poethko, T.; Schottelius, M.; Thumshirn, G.; Hersel, U.; Herz, M.; Henriksen, G.; Kessler, H.; Schwaiger, M.; Wester, H.-J. J. Nucl. Med. 2004, 45, 892–902. |
| 6. | Zhao, B.; Xiao, Y.; Yuan, D.; Lu, C.; Yao, Y. Dalton Trans. 2016, 45, 3880–3887. doi:10.1039/C5DT04217H |
| 14. | DeBernardis, J. F.; Kerkman, D. J.; Zinkowski, R. P. Certain substituted 1-aryl-3-piperazin-1’-yl propanones to treat Alzheimers Desease. U.S. Patent US5658909 A, Aug 19, 1997. |
| 43. | Ramenda, T.; Steinbach, J.; Wuest, F. Amino Acids 2013, 44, 1167–1180. doi:10.1007/s00726-012-1450-4 |
| 26. | Abraham, R. J.; Byrne, J. J.; Griffiths, L.; Perez, M. Magn. Reson. Chem. 2006, 44, 491–509. doi:10.1002/mrc.1747 |
| 25. | Friebolin, H. Ein- und zweidimensionale NMR-Spektroskopie – Eine Einführung; Wiley-VCH: Weinheim, 1999; 131 ff.. |
| 14. | DeBernardis, J. F.; Kerkman, D. J.; Zinkowski, R. P. Certain substituted 1-aryl-3-piperazin-1’-yl propanones to treat Alzheimers Desease. U.S. Patent US5658909 A, Aug 19, 1997. |
| 16. | Grosse-Gehling, P.; Wuest, F. R.; Peppel, T.; Köckerling, M.; Mamat, C. Radiochim. Acta 2011, 99, 365–373. doi:10.1524/ract.2011.1834 |
| 34. | Gu, Y.; Huang, D.; Liu, Z.; Huang, J.; Zeng, W. Med. Chem. 2011, 7, 334–344. doi:10.2174/157340611796799140 |
| 35. | Cai, L.; Lu, S.; Pike, V. W. Eur. J. Org. Chem. 2008, 2853–2873. doi:10.1002/ejoc.200800114 |
| 36. | Jacobson, O.; Kiesewetter, D. O.; Chen, X. Bioconjugate Chem. 2015, 21, 1–18. doi:10.1021/bc500475e |
| 37. | Olberg, D. E.; Hjelstuen, O. K. Curr. Top. Med. Chem. 2010, 10, 1669–1679. doi:10.2174/156802610793176747 |
| 38. | Richter, S.; Wuest, F. Molecules 2014, 19, 20536–20556. doi:10.3390/molecules191220536 |
| 30. | Wodtke, R.; König, J.; Pigorsch, A.; Köckerling, M.; Mamat, C. Dyes Pigm. 2015, 113, 263–273. doi:10.1016/j.dyepig.2014.08.022 |
| 31. | Cole, E. L.; Stewart, M. N.; Littich, R.; Hoareau, R.; Scott, P. J. H. Curr. Top. Med. Chem. 2014, 14, 875–900. doi:10.2174/1568026614666140202205035 |
| 32. | Miller, P. W.; Long, N. J.; Vilar, R.; Gee, A. D. Angew. Chem., Int. Ed. 2008, 47, 8998–9033. doi:10.1002/anie.200800222 |
| 33. | Littich, R.; Scott, P. J. H. Angew. Chem., Int. Ed. 2012, 51, 1106–1109. doi:10.1002/anie.201106785 |
| 21. | Harris, R. K.; Spragg, R. A. Chem. Commun. 1966, 314–317. doi:10.1039/c19660000314 |
| 28. | Lambert, J. B.; Oliver, W. L., Jr.; Packard, B. S. J. Am. Chem. Soc. 1971, 93, 933–937. doi:10.1021/ja00733a025 |
| 29. | Pretze, M.; Kuchar, M.; Bergmann, R.; Steinbach, J.; Pietzsch, J.; Mamat, C. ChemMedChem 2013, 8, 935–945. doi:10.1002/cmdc.201300053 |
| 21. | Harris, R. K.; Spragg, R. A. Chem. Commun. 1966, 314–317. doi:10.1039/c19660000314 |
| 22. | Hietapelto, V.; Laitinen, R. S.; Pursiainen, J.; Rahkamaa, E. Acta Chem. Scand. 1999, 53, 7–14. doi:10.3891/acta.chem.scand.53-0007 |
| 27. | Miron, Y.; McGarvey, B. R.; Morawetz, H. Macromolecules 1969, 2, 154–161. doi:10.1021/ma60008a009 |
| 23. | Baskar, R.; Baby, C.; Moni, M. S.; Subramanian, K. J. Mol. Struct. 2013, 1040, 90–97. doi:10.1016/j.molstruc.2013.02.029 |
© 2016 Mamat et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)