Abstract
The synthesis of three Lex derivatives from one common protected trisaccharide is reported. These analogues will be used respectively for competitive binding experiments, conjugation to carrier proteins and immobilization on gold. An N-acetylglucosamine monosaccharide acceptor was first glycosylated at O-4 with a galactosyl imidate. This coupling was performed at 40 °C under excess of BF3·OEt2 activation and proceeded best if the acceptor carried a 6-chlorohexyl rather than a 6-azidohexyl aglycon. The 6-chlorohexyl disaccharide was then converted to an acceptor and submitted to fucosylation yielding the corresponding protected 6-chlorohexyl Lex trisaccharide. This protected trisaccharide was used as a precursor to the 6-azidohexyl, 6-acetylthiohexyl and 6-benzylthiohexyl trisaccharide analogues which were obtained in excellent yields (70–95%). In turn, we describe the deprotection of these intermediates in one single step using dissolving metal conditions. Under these conditions, the 6-chlorohexyl and 6-azidohexyl intermediates led respectively to the n-hexyl and 6-aminohexyl trisaccharide targets. Unexpectedly, the 6-acetylthiohexyl analogue underwent desulfurization and gave the n-hexyl glycoside product, whereas the 6-benzylthiohexyl analogue gave the desired disulfide trisaccharide dimer. This study constitutes a particularly efficient and convergent preparation of these three Lex analogues.

Graphical Abstract
Introduction
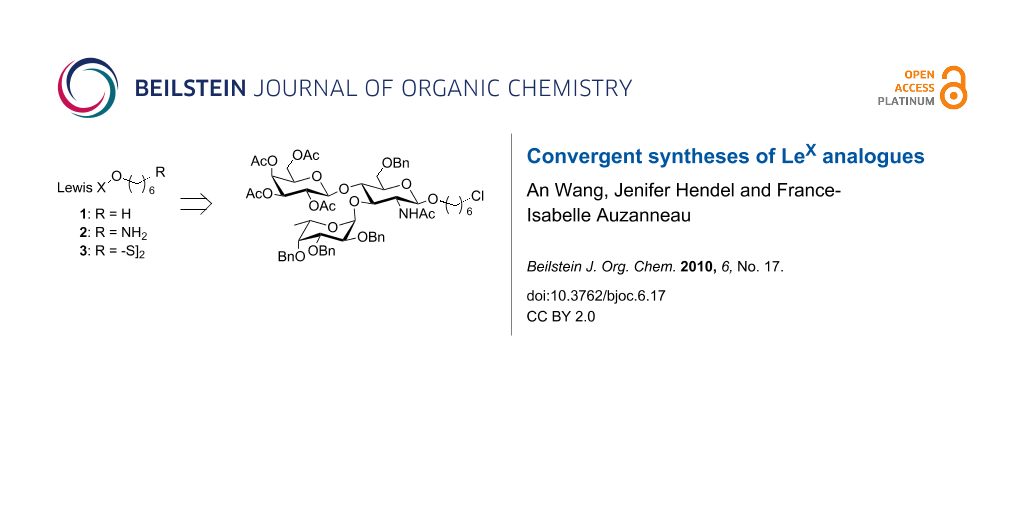
Our group is involved in the design of new anti-cancer vaccines based on the Tumor Associated Carbohydrate Antigen (TACA) dimeric Lex (dimLex) [1-6]. This tumor specific antigen consists of a hexasaccharide that displays the Lex trisaccharide antigen linked to O-3″ of the galactose residue of another Lex trisaccharide. Since it was first characterized [7,8], the Lex antigenic determinant, β-D-Galp(1,4)[α-LFucp(1,3)]-D-GlcNAcp, has been found on numerous cells and tissues such as kidney tubules, gastrointestinal epithelial cells, and cells of the spleen and brain [9-11]. Thus, there are numerous reports in the literature that deal with the chemical [12-36] or chemoenzymatic [37,38] preparation of Lex analogues as well as that of Lex intermediate building blocks to be further converted into the Sialyl Lex tetrasaccharide. The chemical syntheses usually follow one of three synthetic schemes: 1. a stepwise approach involving the successive galactosylation then fucosylation of a glucosamine acceptor [12-28]; 2. a stepwise approach in which the sequence of glycosylation of the glucosamine acceptor is reversed, i.e. the fucosylation is followed by the galactosylation [28-34]; 3. a block approach in which a lactosamine derivative prepared from lactose is subjected to fucosylation at O-3 [35,36]. Whereas these reports usually describe the preparation of one compound to be used in a specific experiment, we describe here the convergent synthesis of the three Lex derivatives 1–3 (Figure 1) from one common protected trisaccharide intermediate. These three Lex analogues (1–3) will be used respectively for competitive binding experiments (1), conjugation to carrier proteins (2) and immobilization to a gold plate (3).
![[1860-5397-6-17-1]](/bjoc/content/figures/1860-5397-6-17-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structure of Lex analogues 1–3.
Figure 1: Structure of Lex analogues 1–3.
Results and Discussion
Our synthetic approach to prepare these Lex derivatives began with the galactosylation at O-4 of glycosyl acceptor 4 with the known [39-41] galactosyl donor 7 followed by deprotection at O-3 of the glucosamine residue and fucosylation of the resulting disaccharide with the known [42] ethylthioglycoside 9. Since in addition to the Lex trisaccharide we are also interested in preparing fragments of the dimLex antigen, we examined the glycosylation at O-4 of glucosamine glycosyl acceptors with galactosyl donor 8, which is chloroacetylated rather than acetylated at O-3. Finally, we also investigated the reactivity towards glycosylation of the N-acetylated and phthalimido acceptors 5 and 6, respectively, that both carry a 6-azidohexyl aglycon (Figure 2).
![[1860-5397-6-17-2]](/bjoc/content/figures/1860-5397-6-17-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Monosaccharide glycosyl acceptors (4–6) and donors (7–9) used in this study.
Figure 2: Monosaccharide glycosyl acceptors (4–6) and donors (7–9) used in this study.
Synthesis of monosaccharide building blocks. The 6-chlorohexyl acceptor 4 was prepared in four steps from the known [43] chlorohexyl glucoside 10 (Scheme 1). Thus, peracetate 10 was deacetylated (NaOMe/MeOH) and converted to the benzylidene acetal 11 by reaction with benzaldehyde dimethyl acetal under camphorsulfonic acid (CSA) catalysis. Chloroacetylation of alcohol 11 gave the intermediate 12 which was converted to acceptor 4 via the reductive opening of the benzylidene acetal using NaCNBH3 and HCl·Et2O in anhydrous THF at 0 °C.
![[1860-5397-6-17-i1]](/bjoc/content/inline/1860-5397-6-17-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of monosaccharide glycosyl acceptors 4–6.
Scheme 1: Synthesis of monosaccharide glycosyl acceptors 4–6.
Both 6-azidohexyl acceptors 6 and 5 were prepared from the anomeric mixture of the known tetraacetate 13 [44]. Thus, tetraacetate 13 was reacted with 6-chlorohexanol (4 equiv) in the presence of BF3·OEt2 (5 equiv). To promote coupling, the reaction mixture was either stirred for 1 h at 50 °C in an oil bath (Supporting Information File 1, Method A) or submitted to microwave irradiation for 5 min at 50 °C (Supporting Information File 1, Method B). After acetylation of the excess chlorohexanol to ease its removal, pure glycoside 14 was isolated in excellent yield whether method A or B was followed. Thus, these syntheses of glycoside 14 constitute efficient alternatives to that reported by Nitz et al. in which the starting material was the corresponding anomeric bromide [45]. Nucleophilic displacement of the chlorine atom in glycoside 14 (NaN3, DMF, 80 °C) gave the known [46] 6-azidohexyl glycoside 15 quantitatively. Zemplén deacetylation of triacetate 15 followed by conversion of the triol to the 4,6-benzylidene acetal (16) and then chloroacetylation at O-3 gave intermediate 17 that was submitted to reductive opening of the benzylidene group (NaCNBH3, HCl·Et2O) to yield acceptor 6.
The triacetate 15 was also converted in seven steps to acceptor 5. The phthalimido group was first removed (ethylenediamine, EtOH) and the free amine acetylated. Zemplén deacetylation was followed by conversion of the triol to the 4,6-benzylidene acetal 18 which was chloroacetylated at O-3 to give the fully protected intermediate 19. Finally, the benzylidene acetal in compound 19 was reductively opened with Et3SiH and TfOH in CH2Cl2 at −30 °C to give acceptor 5.
The trichloroacetimidate glycosyl donor 8 was prepared from the p-thiotolyl glycoside 20 [47] (Scheme 2). Diol 20 was first acetylated to the diacetate 21 which was then treated with 90% AcOH at 70 °C to remove the isopropylidene group affording diol 22. The diol 22 was selectively acetylated at O-4 by converting it to the corresponding cyclic methylorthoacetate and opening the orthoacetate in situ by adding water to the reaction mixture. The resulting triacetate was chloroacetylated at O-3 and the resulting fully protected thioglycoside 23 was converted to the corresponding hemiacetal that was, in turn, treated with trichloroacetonitrile and DBU to give the α-trichloroacetimidate galactosyl donor 8.
![[1860-5397-6-17-i2]](/bjoc/content/inline/1860-5397-6-17-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of the galactosyl donor 8.
Scheme 2: Synthesis of the galactosyl donor 8.
Glycosylation at O-4 of glucosamine acceptors. It is well known that the hydroxyl group at C-4 of N-acetylglucosamine is a poor nucleophile and has reduced reactivity towards glycosylation when compared to other acceptors [48-50]. However, we have recently reported the successful O-4 glucosylation of an N-acetylglucosamine monosaccharide acceptor using a peracetylated glucopyranose α-trichloroacetimidate donor under activation with 2 equiv of BF3·OEt2 at room temperature [51]. We applied similar conditions: 2 equiv BF3·OEt2, 5 equiv of donor, 1 h at 40 °C for the coupling of donors 7 and 8 with the acceptors 4-6 (Table 1). As can be seen in Table 1 the 6-chlorohexyl glycoside acceptor 4 was easily glycosylated with either donors 7 or 8, affording the desired disaccharides 24 and 25 in about 70% yield for both reactions (entries 1 and 2).
Table 1: Glycosylation at O-4 of glucosamine acceptors 4–6a.
![[Graphic 1]](/bjoc/content/inline/1860-5397-6-17-i5.svg?max-width=637&scale=1.0)
|
|||
| Entry | Donor | Acceptor | Product (%) |
|---|---|---|---|
| 1 | 7 | 4 | 24 (69%) |
| 2 | 8 | 4 | 25 (72%) |
| 3 | 8 | 5 | 26 (27%)b |
| 4 | 8 | 6 | 27 (11%) |
aReagents and conditions: BF3·OEt2 (2 equiv), donor (5 equiv), CH2Cl2, 40 °C, 1 h.
bContaminated with degraded acceptor.
In contrast, the coupling of donor 8 with the 6-azidohexyl glycoside acceptor 5 did not proceed well (entry 3). Monitoring of the reaction by TLC showed degradation of the acceptor, and isolation of the desired disaccharide required both silica gel chromatography and RP-HPLC. Indeed, despite our efforts, and even though its structure was confirmed by NMR and HR-ESI mass spectrometry, disaccharide 26 could not be isolated free of degraded acceptor and/or disaccharide. To further test if the N-acetyl group was impacting negatively the glycosylation of acceptor 5, we attempted to couple trichloroacetimidate 8 with the phthalimido acceptor 6. However as can be seen in Table 1, entry 4, this glycosylation also gave disappointing results: TLC showed a considerable amount of degraded products and the isolation of the desired disaccharide from the reaction mixture required both silica gel chromatography and RP-HPLC. In this case, the disaccharide 27 could be obtained pure albeit in very low yield. These last two reactions suggest that the presence of the azido group on the hexyl aglycon carried by acceptors 5 and 6 is not compatible with the glycosylation conditions that we have established previously [51] for the glycosylation at O-4 of glucosamine acceptors. The disaccharide 24 was further used in the preparation of the Lex analogues 1–3.
Preparation of protected Lex analogues. The chloroacetate in disaccharide 24 was removed with thiourea (C5H5N/EtOH, 70 °C) to give the acceptor disaccharide 28 (61%), which was then fucosylated with the thioethyl glycoside 9 under copper (II) bromide–tetrabutylammonium bromide activation (Scheme 3). The desired Lex trisaccharide 29 was obtained in excellent yield and the α-configuration of the newly formed fucosidic bond was confirmed by 1H NMR (JH-1′,H-2′ = 3.7 Hz). The 6-chlorohexyl trisaccharide glycoside 29 was in turn used as a precursor to the 6-azidohexyl, 6-acetylthiohexyl and 6-thiobenzylhexyl trisaccharides 30–32 (Scheme 3).
![[1860-5397-6-17-i3]](/bjoc/content/inline/1860-5397-6-17-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Convergent synthesis of trisaccharides 29–32.
Scheme 3: Convergent synthesis of trisaccharides 29–32.
Thus, nucleophilic displacement of the chloride with sodium azide or potassium thioacetate was carried out in DMF at 80 °C and provided the 6-azidohexyl and 6-acetylthiohexyl trisaccharides 30 and 31, respectively. The introduction of the azido or thioacetyl groups into trisaccharides 30 and 31 was confirmed by HR-ESI mass spectrometry and by NMR. Indeed, the signals assigned to the methylene CH2Cl in trisaccharide 29 (1H NMR δ 3.50 ppm, 13C NMR δ 44.9 ppm) were no longer observed in trisaccharides 30 and 31. The methylene CH2N3 in trisaccharide 30 gave signals at 3.20 and 54.3 ppm in the 1H and 13C NMR spectra, respectively, whereas the methylene CH2SAc in trisaccharide 31 gave signals at 2.81 and around 28.5 ppm, in the 1H and 13C NMR spectra, respectively. In addition, signals corresponding to the thioacetyl group in trisaccharide 31 were identified at 2.29 ppm and 30.6 ppm in the 1H and 13C NMR spectra, respectively. Since, as will be described below, the deprotection of trisaccharide 31 under dissolving metal conditions did not provide the desired trisaccharide 3, the 6-benzylthiohexyl glycoside 32 was also prepared from the 6-chlorohexyl glycoside 29. Thus, the chloride 29 was allowed to react for 16 h with excess benzylthiol (15 equiv) and sodium hydride (15 equiv) in DMF at 80 °C. These reaction conditions led to the displacement of the chloride as well as to some deacetylation of the galactose residue. Thus, after acetylation of the crude product, the desired 6-benzylthiohexyl trisaccharide 32 was isolated in excellent yield (Scheme 3). It is important to point out that the 6-chlorohexyl glycoside 29 and the 6-benzylthiohexyl glycoside 32 co-eluted on silica gel and that only a very careful analysis of the NMR data recorded for the product could confirm the absence of unreacted starting material. Indeed, the large excess of benzylthiolate used to displace the chloride in trisaccharide 29 was essential for its complete conversion to the desired 6-benzylthiohexyl glycoside 32. The structure of trisaccharide 32 was confirmed by HR-ESI MS as well as by NMR. The methylene CH2SBn gave signals at 2.36 and 31.3 ppm, in the 1H and 13C NMR spectra, respectively whereas the S-benzyl group gave additional signals in the aromatic regions as well as signals corresponding to the SCH2Ph methylene around 3.70 and 36.3 ppm in the 1H and 13C NMR spectra, respectively.
Deprotection of trisaccharides 29–32 under dissolving metal conditions. As reported by Seeberger et al. [52], the removal of O- and S-benzyl groups as well as that of O-acetyl groups can be accomplished in one step and concurrently with the reduction of azido groups to the corresponding amines, using Birch reduction conditions. Thus we embarked on the one step deprotection of trisaccharides 29–32 with sodium in ammonia (Table 2).
Table 2: One step deprotection of trisaccharides 29–32a.
![[Graphic 2]](/bjoc/content/inline/1860-5397-6-17-i6.svg?max-width=637&scale=1.0)
|
|||
| Entry | Trisaccharide | Product | Yield (%) |
|---|---|---|---|
| 1 | 29 | 1 | 82 |
| 2 | 30 | 2 | 59 |
| 3 | 31 | 1 | 73 |
| 4 | 32 | 3 | 70 |
aReagents and conditions: Na/NH3(l), −78 °C, 50 min.
Treatment of trisaccharides 29 and 30 with sodium in liquid ammonia at −78 °C followed by neutralization of the reaction mixtures with AcOH gave the desired trisaccharides 1 and 2 (entries 1 and 2) that were isolated pure after chromatography on a Biogel P2 column eluted with water for compound 1, and 0.05 M ammonium acetate for the 6-aminohexyl compound 2. Whereas the structure of trisaccharide 1 was confirmed by HR-ESI mass spectrometry and NMR, the structure of the 6-aminohexyl glycoside 2 was confirmed by comparing its analytical data to that previously reported [31]. To our surprise, treatment of the 6-acetylthiohexyl trisaccharide 31 under Birch reduction conditions did not lead to the desired corresponding thiol or disulfide product but produced the hexyl glycoside 1. The mechanism proposed to explain this reductive desulfurization is shown in Scheme 4. It involves first a single electron transfer to the thioacetyl group that is followed by the cleavage of the carbon sulfur bond giving a thioacetate salt and an alkyl radical. The alkyl radical is then converted to the corresponding anion by a second electron transfer and the resulting anion is protonated by ammonia giving trisaccharide 1.
![[1860-5397-6-17-i4]](/bjoc/content/inline/1860-5397-6-17-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Proposed mechanism for the desulfurization of thioacetate 31 under dissolving metal conditions.
Scheme 4: Proposed mechanism for the desulfurization of thioacetate 31 under dissolving metal conditions.
In contrast to the thioacetate 31, treatment of the 6-benzylthiohexyl glycoside 32 under Birch reduction conditions did not lead to desulfurization and gave the disulfide trisaccharide dimer 3. Under these reductive conditions, and based on the work by Seeberger et al. [52], we did not expect the formation of the disulfide dimer as the major product but rather that of the corresponding thiol. However, the structure and homogeneity of disulfide dimer 3 was unequivocally confirmed by HR-ESI mass spectrometry and NMR. Interestingly this dimer gave a well resolved 1H NMR spectrum in D2O that did not support the formation of intramolecular Lex–Lex interactions such as those reported by de la Fuente and Penadés for a similar analogue [33]. Following published procedures, the disulfide dimer 3 will be reduced immediately prior to its conjugation to proteins [53] or immobilization on gold surface or gold nanoparticules [34].
In conclusion, we have reported above the efficient and convergent preparation of three Lex derivatives (1–3) from one common protected trisaccharide (29). Our results seem to indicate that glycosylation at O-4 of a glucosamine monosaccharide acceptor under excess BF3·OEt2 activation at 40 °C is compatible with a chlorinated aglycon but not with an aglycon carrying an azido group. We have also established that the fully protected precursors could be deprotected in one single step to give the final target compounds using dissolving metal conditions. However, we observed that a thioacetylated derivative will undergo an undesired reductive desulfurization. This study constitutes a particularly efficient convergent preparation of analogues that can each be used for a specific biochemical application.
Experimental
General Methods: 1H (600.14, 400.13 or 300.13 MHz) and 13C NMR (150.9, 100.6 or 75.5 MHz) spectra were recorded for compounds solubilized in CDCl3 (internal standard, for 1H: residual CHCl3 δ 7.24; for 13C: CDCl3 δ 77.0) or D2O [external standard 3-(trimethylsilyl)-propionic acid-d4, sodium salt (TSP) for 1H δ 0.00, for 13C δ 0.00]. Chemical shifts and coupling constants were obtained from a first-order analysis of one-dimensional spectra. Assignments of proton and carbon resonances were based on COSY and 13C–1H heteronuclear correlated experiments. Mass spectra were obtained under electron spray ionization (ESI) on a high resolution mass spectrometer. TLC were performed on precoated aluminum plates with Silica Gel 60 F254 and detected with UV light and/or charred with a solution of 10% H2SO4 in EtOH. Compounds were purified by flash chromatography with Silica Gel 60 (230–400 mesh) unless otherwise stated. Solvents were distilled and dried according to standard procedures [54], and organic solutions were dried over Na2SO4 and concentrated under reduced pressure below 40 °C. HPLC purifications were run with HPLC grade solvents.
n-Hexyl 2-acetamido-2-deoxy-3-O-(α-L-fucopyranosyl)-4-O-(β-D-galactopyranosyl)-β-D-glucopyranoside (1). Trisaccharide 29 (20 mg, 0.017 mmol) or trisaccharide 31 (19 mg, 0.016 mmol) were dissolved in THF (5 mL) and liquid ammonia (20 mL) was condensed into the solution at −78 °C. Na (74 mg, 3.2 mmol) was added and the mixture was stirred for 50 min at −78 °C. The reaction was quenched with MeOH (5 mL) and the ammonia was allowed to evaporate at room temp. The remaining solution was neutralized with acetic acid (203 μL, 3.5 mmol), the solvent was evaporated and the residue was passed twice through a Biogel P2 column (100 × 1 cm) eluted with Milli-Q water to give the trisaccharide 1 (8.5 mg, 82% from 29; 7.0 mg, 73% from 31) as a white amorphous powder after lyophilization. [α]D = −47 (c 0.5, MeOH), 1H NMR (400 MHz, D2O): δ 5.12 (d, 1H, J = 4.5 Hz, H-1′); 4.83 (m, 1H, H-5′); 4.53 (d, 1H, J = 7.5 Hz, H-1); 4.46 (d, 1H, J = 7.5 Hz, H-1″); 4.00 (dd, 1H, J = 12.0, 1.0 Hz, H-6a); 3.83–3.95 (m, 7H, H-2, H-3, H-4, H-6b, H-3′, H-4″, OCHHCH2); 3.78 (d, 1H, J = 3.0 Hz, H-4′); 3.73 (m, 2H, H-6a″, H-6b″); 3.70 (m, 1H, H-2′); 3.66 (m, 1H, H-3″); 3.60 (m, 3H, H-5, H-5″, OCHHCH2,); 3.59 (m, 1H, H-2″); 2.03 (s, 3H, CH3CO); 1.55 (m, 2H, OCH2CH2); 1.24–1.37 (m, 6H, OCH2CH2CH2CH2CH2); 1.17 (d, 3H, J = 6.0 Hz, H-6′); 0.88 (t, 3H, J = 6.6 Hz, CH2CH3). 13C-NMR (100 MHz, D2O): 174.17 (C=O); 101.81 (C-1″); 100.91 (C-1); 98.61 (C-1′); 75.32 (C-5); 74.94, 74.88 (C-3, C-5″); 73.36 (C-4); 72.44 (C-3″); 71.89 (C-4′); 71.02 (C-2″); 70.66 (OCH2CH2); 69.19 (C-3′); 68.32 (C-4″); 67.68 (C-2′); 66.68 (C-5′); 61.47 (C-6″); 59.76 (C-6); 55.84 (C-2); 30.67, 28.53, 24.77, 22.00 (OCH2CH2CH2CH2CH2); 22.23 (CH3CO); 15.27 (C-6′); 13.30 (CH2CH3). HRESIMS Calcd for C26H48NO15 [M+H]+ 614.3024, found 614.3035.
6-Aminohexyl 2-acetamido-2-deoxy-3-O-(α-L-fucopyranosyl)-4-O-(β-D-galactopyranosyl)-β-D-glucopyranoside (2). The azidotrisaccharide 30 (19 mg, 0.16 mmol) was deprotected in the same conditions as described above for the deprotection of trisaccharide 29. After work up, the residue was passed twice through a Biogel P2 column (100 × 1 cm) eluted with 0.05 M ammonium acetate and after repeated lyophilization from Milli-Q water (3 × 10 mL) the known [31] trisaccharide 2 (6.5 mg, 59%) was obtained as the acetate salt in the form of a white amorphous powder. [α]D = −54 (c 0.9, H2O), lit. [31]: [α]D = −54.3 (c 1, H2O), 1H NMR (400 MHz, D2O): δ 5.12 (d, 1H, J = 4.5 Hz, H-1′); 4.83 (m, 1H, H-5′); 4.53 (d, 1H, J = 7.5 Hz, H-1); 4.46 (d, 1H, J = 7.5 Hz, H-1″); 4.00 (dd, 1H, J = 12.0, 1.0 Hz, H-6a); 3.83–3.95 (m, 7H, H-2, H-3, H-4, H-6b, H-3′, H-4″, OCHHCH2); 3.78 (d, 1H, J = 3.0 Hz, H-4′); 3.73 (m, 2H, H-6a″, H-6b″); 3.70 (m, 1H, H-2′); 3.66 (m, 1H, H-3″); 3.60 (m, 3H, H-5, H-5″, OCHHCH2); 3.59 (m, 1H, H-2″); 2.99 (t, 2H, J = 7.0 Hz, CH2NH2); 2.03, 2.01 (s, 6H, CH3CO); 1.57, 1.67 (m, 4H, OCH2CH2, CH2CH2NH2); 1.30–1.42 (m, 4H, OCH2CH2CH2CH2); 1.17 (d, 3H, J = 6.0 Hz, H-6′). 13C-NMR (100 MHz, D2O): 173.96 (C=O); 101.64 (C-1″); 100.81 (C-1); 98.45 (C-1′); 75.16 (C-5); 74.73 (C-3, C-5″); 73.17 (C-4); 72.28 (C-3″); 71.70 (C-4′); 70.85 (C-2″); 70.30 (OCH2CH2); 69.01 (C-3′); 68.15 (C-4″); 67.51 (C-2′); 66.53 (C-5′); 61.31 (C-6″); 59.57 (C-6); 55.65 (C-2); 39.21 (CH2NH2); 28.18, 26.46, 25.05, 24.45 [OCH2(CH2)4]; 22.05 (CH3CO); 15.10 (C-6′). HRESIMS calcd for C26H48N2O15 [M+H]+ 629.3133, found 629.3121.
6,6′-Dithio-bis(hexan-1,6-diyl)-bis[2-acetamido-2-deoxy-3-O-α-L-fucopyranosyl-4-O-(β-D-galactopyranosyl)-β-D-glucopyranoside] (3). The 6-benzylthiohexyl trisaccharide 32 (30 mg, 0.024 mmol) was deprotected in the same conditions as described above for the deprotection of trisaccharide 29. After work up, the residue was passed through a Biogel P2 column eluted with water to give the trisaccharide 3 (10.6 mg, 70%) as white amorphous powder after lyophilization. [α]D = −57 (c 0.7, MeOH), 1H NMR (600 MHz, D2O): δ 5.05 (d, 1H, J = 3.8 Hz, H-1′); 4.82–4.75 (m, 1H, H-5′); 4.47 (d, 1H, J = 7.7 Hz, H-1); 4.39 (d, 1H, J = 7.9 Hz, H-1″); 3.95 (d, 1H, J = 10.9 Hz, H-6a); 3.90–3.76 (m, 7H, H-2, H-3, H-4, H-6b, H-3′, H-4″, OCHHCH2); 3.75–3.71 (m, 1H, H-4′); 3.70–3.56 (m, 4H, H-2′H-3″, H-6a″, H-6b″); 3.55–3.49 (m, 3H, H-5, H-5″, OCHHCH2); 3.44 (t, 1H, J = 8.1 Hz, H-2″); 2.70 (t, 2H, J = 7.1 Hz, CH2S); 1.98 (s, 3H, CH3CO); 1.68–1.58 (m, 2H, SCH2CH2); 1.54–1.43 (m, 2H, OCH2CH2); 1.41–1.21 (m, 4H, OCH2CH2CH2CH2CH2CH2S); 1.12 (d, 3H, J = 6.6 Hz, H-6′. 13C-NMR (150 MHz, D2O): 174.09 (C=O); 101.83 (C-1″); 100.93 (C-1); 98.63 (C-1′); 75.35 (C-5); 74.96, 74.90 (C-3, C-5″); 73.41 (C-4); 72.48 (C-3″); 71.91 (C-4′); 71.05 (C-2″); 70.45 (OCH2CH2); 69.22 (C-3′); 68.34 (C-4″); 67.72 (C-2′); 66.71 (C-5′); 61.48 (C-6″); 59.81 (C-6); 55.86 (C-2); 38.16 (CH2S); 28.44, 28.30, 27.15, 24.67 (OCH2CH2CH2CH2CH2); 22.35 (CH3CO); 15.30 (C-6′). HRESIMS Calcd for C59H92N2O30S2Na [M+Na]+ 1311.5074, found 1311.5065.
6-Chlorohexyl 2-acetamido-4-O-(2,3,4,6-tetra-O-acetyl-β-D-galactopyranosyl)-6-O-benzyl-3-O-(chloroacetyl)-2-deoxy-β-D-glucopyranoside (24). BF3·Et2O (150 μL, 1.19 mmol, 2.0 equiv) was added to a solution of the acceptor 4 (300 mg, 0.59 mmol) and glycosyl donor 7 (1.46 g, 2.96 mmol, 5.0 equiv) [39-41] in anhyd CH2Cl2 (15 mL) at 40 °C. The reaction mixture was stirred for 1 h at 40 °C. The reaction was quenched with Et3N (170 μL, 1.22 mmol) and the solvent was evaporated. Flash chromatography of the residue (EtOAc–hexanes, 1:1 to 6:4) gave the disaccharide 24 (341 mg, 69%) as colorless oil. [α]D = −5 (c1.0, CHCl3), 1H NMR (400 MHz, CDCl3): δ 7.40–7.26 (m, 5H, Ar); 5.72 (d, 1H, J = 9.2 Hz, NH); 5.24 (bd, 1H, J = 3.4 Hz, H-4′); 5.11 (dd, 1H, J = 10.0, 8.9 Hz, H-3); 4.95 (dd, 1H, J = 10.4, 8.0 Hz, H-2′); 4.78 (dd, 1H, J = 10.4, 3.5 Hz, H-3′); 4.72 (d, 1H, J = 12.0 Hz, PhCHH); 4.50–4.41 (m, 3H, H-1, PhCHH); 4.39 (d, 1H, J = 8.0 Hz, H-1′); 4.15–4.01 (m, 4H, H-6a′, H-6b′, ClCH2CO); 4.01–3.88 (m, 2H, H-2, H-4); 3.86–3.78 (m, 1H, OCHH); 3.73–3.65 (m, 2H, H-6a, H-6b); 3.62 (t, 1H, J = 6.5 Hz, H-5′); 3.53–3.38 (m, 4H, H-5, OCHH, CH2Cl); 2.10, 2.04, 1.93, 1.92 (4 s, 15H, CH3CO); 1.77–1.67 (m, 2H, CH2CH2Cl); 1.61–1.49 (m, 2H, OCH2CH2); 1.44–1.27 (m, 4H, OCH2CH2CH2CH2). 13C NMR (100 MHz, CDCl3): δ 170.30, 170.16, 169.96, 168.96, 167.34 (C=O); 137.64, 128.57, 128.07, 127.97 (Ar); 100.87 (C-1); 100.12 (C-1′); 74.48, 74.40, 74.25 (C-3, C-4, C-5); 73.62 (PhCH2); 70.75, 70.60 (C-3′, C-5′); 69.27 (CH2O); 69.09 (C-2′); 67.35 (C-6); 66.81 (C-4′); 61.02 (C-6′); 53.45 (C-2); 44.97 (CH2Cl); 40.80 (ClCH2CO); 32.40 (CH2CH2Cl); 29.21 (OCH2CH2); 26.44, 25.14 (OCH2CH2CH2CH2); 23.25, 20.64, 20.58, 20.48, 20.48 (CH3CO). HRESIMS Calcd for C37H52Cl2NO16 [M+H]+ 836.2663, found 836.2634.
6-Chlorohexyl 2-acetamido-4-O-(2,3,4,6-tetra-O-acetyl-β-D-galactopyranosyl)-6-O-benzyl-2-deoxy-β-D-glucopyranoside (28). Thiourea (162 mg, 2.13 mmol, 6.0 equiv) was added to a solution of the disaccharide 24 (298 mg, 0.356 mmol) in a mixture of pyridine and EtOH (2:1, 15 mL). The solution was stirred for 10 h at 70 °C, the solvents removed by evaporation and the residue co-concentrated with toluene (2 × 10 mL). The crude residue was dissolved in CH2Cl2 (20 mL) and washed sequentially with 2 M HCl (10 mL), saturated aq NaHCO3 (10 mL) and brine (10 mL). The aq phases were re-extracted with CH2Cl2 and the combined organic layers were dried and concentrated. Flash chromatography of the residue (EtOAc-hexanes, 6:4) gave the pure disaccharide 28 (165 mg, 61%) as a white amorphous powder. [α]D = +1 (c 1.3, CHCl3), 1H NMR (400 MHz, CDCl3): δ 7.37–7.26 (m, 5H, Ar); 5.62 (d, 1H, J = 7.7 Hz, NH); 5.32 (bd, 1H, J = 3.4 Hz, H-4′); 5.13 (dd, 1H, J = 10.4, 8.0 Hz, H-2′); 4.90 (dd, 1H, J = 10.4, 3.4 Hz, H-3′); 4.74 (d, 1H, J = 8.2 Hz, H-1); 4.68 (d, 1H, J = 12.1 Hz, PhCHH); 4.47 (d, 1H, J = 12.1 Hz, PhCHH); 4.45 (d, 1H, J = 8.0 Hz, H-1′); 4.13–4.05 (m, 2H, H-6a′, H-6b′); 4.04–3.96 (m, 1H, H-3); 3.96–3.92 (bs, 1H, OH); 3.90–3.79 (m, 2H, H-5′, OCHH); 3.69–3.57 (m, 3H, H-4, H-6a, H-6b); 3.53–3.41 (m, 4H, H-5, OCHH, CH2Cl); 3.41–3.31 (m, 1H, H-2); 2.12, 2.03, 1.97, 1.95 (4 s, 15H, CH3CO); 1.78–1.69 (m, 2H, CH2CH2Cl); 1.62–1.50 (m, 2H, OCH2CH2); 1.46–1.29 (m, 4H, OCH2CH2CH2CH2). 13C NMR (100 MHz, CDCl3): δ 170.36, 170.07, 169.98, 169.91, 169.15 (C=O); 138.02, 128.48, 127.86, 127.78 (Ar); 101.13 (C-1′); 99.96 (C-1); 80.98 (C-4); 73.92 (C-5); 73.59 (PhCH2); 71.34 (C-3); 71.08 (C-5′); 70.67 (C-3′); 69.31 (CH2O); 68.73 (C-2′); 68.05 (C-6); 66.81 (C-4′); 61.31 (C-6′); 57.05 (C-2); 44.99 (CH2Cl); 32.44 (CH2CH2Cl); 29.28 (OCH2CH2); 26.49, 25.19 (OCH2CH2CH2CH2); 23.58, 20.65, 20.56, 20.53, 20.47 (CH3CO). HRESIMS Calcd for C35H51ClNO15 [M+H]+ 760.2947, found 760.2928.
6-Chlorohexyl 2-acetamido-4-O-(2,3,4,6-tetra-O-acetyl-β-D-galactopyranosyl)-6-O-benzyl-3-O-(2,3,4-tri-O-benzyl-α-L-fucopyranosyl)-2-deoxy-β-D-glucopyranoside (29). A solution of the disaccharide acceptor 28 (100 mg, 0.132 mmol) and fucosyl donor 9 (189 mg, 0.395 mmol, 3.0 equiv) [42] in a mixture of CH2Cl2 and DMF (1:1, 8 mL) containing activated powdered MS 4Å (400 mg) was stirred at room temp for 30 min. Cu(II)Br2 (88 mg, 0.394 mmol, 3.0 equiv) and Bu4NBr (131 mg, 0.409 mmol, 3.1 equiv) were added and the reaction mixture was stirred for 20 h at room temp. The reaction mixture was filtered over Celite® and the solids were washed with CH2Cl2 (5 mL). The filtrate was diluted with CH2Cl2 (60 mL) and washed sequentially with brine (50 mL) and saturated aq NaHCO3 (6 × 50 mL). The aq layers were re-extracted with CH2Cl2 (50 mL) and the combined organic layers were dried and concentrated. Flash chromatography of the residue (EtOAc–hexanes, 3:7) gave the trisaccharide 29 as colorless oil (126 mg, 81%). [α]D = −19 (c 0.5, CHCl3), 1H NMR (600 MHz, CDCl3): δ 7.45–7.24 (m, 20H, Ar); 6.02 (bs, 1H, NH); 5.29 (d, 1H, J = 3.2 Hz, H-4″); 5.09 (d, 1H, J = 3.7 Hz, H-1′); 5.04 (dd, 1H, J = 10.4, 8.2 Hz, H-2″); 4.98 (d, 1H, J = 11.9 Hz, PhCHH); 4.92–4.85 (m, 2H, H-1, PhCHH); 4.85–4.79 (m, 3H, H-3″, PhCH2); 4.75 (d, 1H, J = 11.9 Hz, PhCHH); 4.71 (d, 1H, J = 11.8 Hz, PhCHH); 4.69 (d, 1H, J = 12.0 Hz, PhCHH); 4.58 (d, 1H, J = 8.2 Hz, H-1″); 4.45 (d, 1H, J = 12.0 Hz, PhCHH); 4.43–4.38 (m, 1H, H-5′); 4.19 (t, 1H, J = 7.6 Hz, H-3); 4.16–4.11 (m, 2H, H-2′, H-6a″), 4.02 (dd, 1H, J = 10.9, 5.9 Hz, H-6b″); 3.97–3.90 (m, 2H, H-4, H-3′); 3.85–3.74 (m, 3H, H-6a, H-6b, OCHHCH2); 3.68 (s, 1H, H-4′); 3.58 (t, 1H, J = 7.0 Hz, H-5″); 3.55–3.45 (m, 4H, H-2, H-5, CH2Cl); 3.44–3.38 (m, 1H, OCHHCH2); 2.03, 2.02, 1.98, 1.84, 1.78 (5 s, 15H, CH3CO); 1.76–1.69 (m, 2H, CH2CH2Cl); 1.58–1.47 (m, 2H, OCH2CH2); 1.44–1.27 (m, 4H, OCH2CH2CH2CH2); 1.18 (d, 3H, J = 6.5 Hz, H-6′). 13C NMR (150 MHz, CDCl3): δ 170.45, 170.00, 169.88, 169.85, 169.21 (C=O); 138.72, 138.68, 138.47, 137.78, 128.46, 128.40, 128.30, 128.24, 128.11, 127.90, 127.73, 127.69, 127.61, 127.49, 127.28, 127.00 (Ar); 99.38, 99.34 (C-1, C-1″); 97.33 (C-1′); 79.82 (C-3′); 76.79 (C-4′); 76.31 (C-2′); 74.23 (PhCH2); 74.21 (C-5); 74.03 (C-4); 73.58, 73.35 (PhCH2); 73.32 (C-3); 72.38 (PhCH2); 70.52 (C-3″); 70.27 (C-5″); 69.23 (OCH2CH2); 68.73 (C-2″); 68.33 (C-6); 66.61 (C-4″); 66.39 (C-5′); 60.22 (C-6″); 56.20 (C-2); 44.92 (CH2Cl); 32.38 (CH2CH2Cl); 29.12 (OCH2CH2); 26.45, 25.08 (OCH2CH2CH2CH2); 22.95, 20.65, 20.52, 20.49, 20.44 (CH3CO). HRESIMS Calcd for C62H79ClNO19 [M+H]+ 1176.4935, found 1176.4933.
6-Azidohexyl 2-acetamido-4-O-(2,3,4,6-tetra-O-acetyl-β-D-galactopyranosyl)-6-O-benzyl-3-O-(2,3,4-tri-O-benzyl-α-L-fucopyranosyl)-2-deoxy-β-D-glucopyranoside (30). NaN3 (17 mg, 0.26 mmol, 8.2 equiv) was added to a solution of the trisaccharide 29 (38 mg, 0.032 mmol) in anhyd DMF (2.5 mL) and the reaction mixture was heated at 80 °C for 36 h. The solvent was evaporated, the residue was dissolved in CH2Cl2 (50 mL) and washed with water (2 × 10 mL). The aq phases were re-extracted with CH2Cl2 and the combined organic layers were dried and concentrated. Flash chromatography of the residue (EtOAc-hexanes, 6:4) afforded the trisaccharide 30 as a clear glass (36 mg, 95%). [α]D = −47 (c 1.0, CH2Cl2), 1H NMR (400 MHz, CDCl3): δ 7.41–7.20 (m, 20H, Ar); 5.81 (d, 1H, J = 7.6 Hz, NH); 5.26 (d, 1H, J = 3.0 Hz, H-4″); 5.06 (d, 1H, J = 3.8 Hz, H-1′), 5.00 (dd, 1H, J = 10.4, 8.2 Hz, H-2″); 4.94 (d, 1H, J = 11.8 Hz, PhCHH); 4.91–4.83 (m, 2H, H-1, PhCHH); 4.82–4.74 (m, 3H, H-3″, PhCH2); 4.73–4.62 (m, 3H, PhCH2, PhCHH); 4.54 (d, 1H, J = 8.1 Hz, H-1″); 4.43–4.34 (m, 2H, H-5′, PhCHH); 4.20–4.05 (m, 3H, H-3, H-2′, H-6a″); 3.98 (dd, 1H, J = 10.8, 5.9 Hz, H-6b″); 3.95–3.87 (m, 2H, H-4, H-3′); 3.83–3.68 (m, 3H, H-6a, H-6b, OCHHCH2); 3.65 (d, 1H, J = 1.4 Hz, H-4′); 3.57–3.44 (m, 2H, H-5, H-5″); 3.43–3.31 (m, 2H, H-2, OCHHCH2); 3.20 (t, 2H, J = 6.9 Hz, CH2N3); 1.99, 1.98, 1.93, 1.89, 1.70 (5s, 15H, CH3CO); 1.58–1.43 (m, 4H, CH2CH2N3, OCH2CH2); 1.33–1.21 (m, 4H, OCH2CH2CH2CH2); 1.15 (d, 3H, J = 6.5 Hz, H-6′). 13C NMR (100 MHz, CDCl3): δ 170.09, 170.06, 169.96, 169.91, 169.20 (C=O); 138.88, 138.77, 138.57, 137.89, 128.53, 128.47, 128.38, 128.32, 128.19, 127.97, 127.77, 127.75, 127.66, 127.56, 127.36, 127.08 (Ar); 99.45 (C-1, C-1″); 97.44 (C-1′); 79.97 (C-3′); 76.90 (C-4′); 76.42 (C-2′); 74.19 (C-5, C-4); 73.70 (PhCH2); 73.43 (C-3, PhCH2); 72.46 (PhCH2); 70.62 (C-3″); 70.35 (C-5″); 69.29 (OCH2CH2); 68.80 (C-2″); 68.43 (C-6); 66.69 (C-4″); 66.42 (C-5′); 60.28 (C-6″); 56.60 (C-2); 51.32 (CH2N3); 29.23, 28.73, 26.41, 25.43 (OCH2CH2CH2CH2CH2CH2N3); 23.16, 20.70, 20.59, 20.56, 20.51 (CH3CO); 16.71 (C-6′). HRESIMS Calcd for C62H79N4O19 [M+H]+ 1183.5339, found 1183.5325.
6-Acetylthiohexyl 2-acetamido-4-O-(2,3,4,6-tetra-O-acetyl-β-D-galactopyranosyl)-6-O-benzyl-3-O-(2,3,4-tri-O-benzyl-α-L-fucopyranosyl)-2-deoxy-β-D-glucopyranoside (31). KSC(O)CH3 (26 mg, 0.22 mmol, 10 equiv) was added to a solution of the trisaccharide 29 (27 mg, 0.023 mmol) in anhyd DMF (1.5 mL) and the reaction mixture was heated at 80 °C for 16 h. Work up and chromatography (EtOAc–hexanes, 6:4), as described above for compound 30 gave the trisaccharide 31 as colorless glass (19 mg, 70%). [α]D = −43 (c 0.7, CH2Cl2), 1H NMR (400 MHz, CDCl3): δ 7.40–7.20 (m, 20H, Ar); 5.85 (d, 1H, J = 7.7 Hz, NH); 5.25 (d, 1H, J = 3.0 Hz, H-4″); 5.07 (d, 1H, J = 3.8 Hz, H-1′), 5.01 (dd, 1H, J = 10.4, 8.2 Hz, H-2″); 4.98 (d, 1H, J = 11.8 Hz, PhCHH); 4.90–4.83 (m, 2H, H-1, PhCHH); 4.82–4.74 (m, 3H, H-3″, PhCH2); 4.74–4.62 (m, 3H, PhCH2, PhCHH); 4.54 (d, 1H, J = 8.2 Hz, H-1″); 4.44–4.35 (m, 2H, H-5′, PhCHH); 4.18 (t, 1H, J = 7.7 Hz, H-3); 4.14–4.05 (m, 2H, H-2′, H-6a″); 3.97 (dd, 1H, J = 10.8, 5.9 Hz, H-6b″); 3.94–3.85 (m, 2H, H-4, H-3′); 3.83–3.68 (m, 3H, H-6a, H-6b, OCHHCH2); 3.65 (d, 1H, J = 2.7 Hz, H-4′); 3.53 (t, 1H, J = 6.8 Hz, H-5″); 3.50–3.44 (m, 1H, H-5); 3.43–3.30 (m, 2H, H-2, OCHHCH2); 2.81 (t, 2H, J = 7.2 Hz, CH2S); 2.29 (s, 3H, SCOCH3); 1.99, 1.97, 1.93, 1.89, 1.71 (5s, 15H, CH3CO); 1.59–1.41 (m, 4H, CH2CH2S, OCH2CH2); 1.33–1.20 (m, 4H, OCH2CH2CH2CH2); 1.15 (d, 3H, J = 6.5 Hz, H-6′). 13C NMR (100 MHz, CDCl3): δ 195.99, 170.16, 170.06, 169.97, 169.92, 169.19 (C=O); 138.86, 138.80, 138.59, 137.90, 128.53, 128.47, 128.37, 128.31, 128.19, 127.97, 127.80, 127.77, 127.67, 127.56, 127.34, 127.09 (Ar); 99.45, 99.42 (C-1, C-1″); 97.40 (C-1′); 79.94 (C-3′); 76.94 (C-4′); 76.40 (C-2′); 74.33 (C-5); 74.31 (PhCH2); 74.21 (C-4); 73.66, 73.44 (PhCH2); 73.36 (C-3); 72.49 (PhCH2); 70.64 (C-3″); 70.34 (C-5″); 69.39 (OCH2CH2); 68.81 (C-2″); 68.40 (C-6); 66.71 (C-4″); 66.40 (C-5′); 60.29 (C-6″); 56.60 (C-2); 30.61 (SCOCH3); 29.40, 29.40, 29.20, 28.96, 28.43, 25.36 (OCH2CH2CH2CH2CH2CH2S); 23.18, 20.70, 20.59, 20.56, 20.51 (CH3CO); 16.71 (C-6′). HRESIMS Calcd for C64H82NO20S [M+H]+ 1216.5151, found 1216.5151.
6-Benzylthiohexyl 2-acetamido-4-O-(2,3,4,6-tetra-O-acetyl-β-D-galactopyranosyl)-6-O-benzyl-3-O-(2,3,4-tri-O-benzyl-α-L-fucopyranosyl)-2-deoxy-β-D-glucopyranoside (32). PhCH2SH (60 µL, 0.44 mmol, 15 equiv) and NaH (21 mg, 0.44 mmol, 15 equiv) were added to a solution of the trisaccharide 29 (36 mg, 0.030 mmol) in anhyd DMF (3.0 mL) at room temp. After 10 min the reaction mixture was heated to 80 °C for 16 h, the solvent was evaporated and the residue was dissolved in Ac2O and pyridine (5 ml, 1:1). After 18 h the reaction mixture was co-concentrated with toluene (3 × 20 ml), the residue was dissolved in CH2Cl2 (30 mL) and the solution was washed with water (2 × 10 mL). The aq phases were re-extracted with CH2Cl2 and the combined organic layers were dried and concentrated. Flash chromatography of the residue (EtOAc–hexanes, 1:1) gave the trisaccharide 32 (35.6 mg, 94%) as a white solid. [α]D = −28 (c 1.0, CH2Cl2), 1H NMR (400 MHz, CDCl3): δ 7.44–7.13 (m, 25H, Ar); 5.79 (d, 1H, J = 7.6 Hz, NH); 5.25 (d, 1H, J = 3.0 Hz, H-4″); 5.05 (d, 1H, J = 3.8 Hz, H-1′), 5.00 (dd, 1H, J = 10.5, 8.2 Hz, H-2″); 4.94 (d, 1H, J = 11.8 Hz, PhCHH); 4.90–4.74 (m, 5H, H-1, H-3″, PhCH2, PhCHH); 4.73–4.64 (m, 3H, PhCHH, PhCH2); 4.54 (d, 1H, J = 8.2 Hz, H-1″); 4.43–4.36 (m, 2H, H-5′, PhCHH); 4.17 (t, 1H, J = 7.7 Hz, H-3); 4.14–4.06 (m, 2H, H-2′, H-6a″); 4.01–3.95 (m, 1H, H-6b″); 3.93–3.87 (m, 2H, H-4, H-3′); 3.79–3.64 (m, 6H, H-6a, H-6b, H-4′, SCH2Ph, OCHHCH2); 3.56–3.45 (m, 2H, H-5, H-5″); 3.41–3.31 (m, 2H, H-2, OCHHCH2); 2.36 (t, 2H, J = 7.3 Hz, CH2SBn); 1.99, 1.98, 1.94, 1.90, 1.70 (5s, 15H, CH3CO); 1.52–1.42 (m, 4H, CH2CH2S, OCH2CH2); 1.33–1.28 (m, 4H, OCH2CH2CH2CH2); 1.15 (d, 3H, J = 6.4 Hz, H-6′). 13C NMR (150 MHz, CDCl3): δ 170.14, 170.10, 170.02, 169.96, 169.20 (C=O); 140.06, 138.91, 138.81, 138.61, 137.93, 137.63, 130.14, 129.02, 128.97, 128.53, 128.49, 128.30, 128.00, 127.89, 127.73, 127.60, 127.39, 126.89 (Ar); 99.48, 99.41 (C-1, C-1″); 97.47 (C-1′); 80.05 (C-3′); 76.86 (C-4′); 76.42 (C-2′); 74.34 (C-5); 74.31 (PhCH2); 74.25 (C-4); 73.77, 73.46 (PhCH2); 73.35 (C-3); 72.47 (PhCH2); 70.67 (C-3″); 70.33 (C-5″); 69.49 (OCH2CH2); 68.81 (C-2″); 68.39 (C-6); 66.70 (C-4″); 66.41 (C-5′); 60.28 (C-6″); 52.07 (C-2); 36.30 (S-CH2Ph); 31.30 (CH2SBn); 29.29, 29.13, 29.06, 25.53 (OCH2CH2CH2CH2CH2CH2S); 23.23, 20.75, 20.64, 20.61, 20.57 (CH3CO); 16.76 (C-6′). HRESIMS Calcd for C69H86NO19S [M+H]+ 1264.5515, found 1264.5509.
Supporting Information
| Supporting Information File 1: Experimental procedures and characteristics for compounds 4–6, 8, 11, 12, 14–19, 21–23, 25–27. | ||
| Format: PDF | Size: 134.4 KB | Download |
| Supporting Information File 2: 1H and 13C NMR spectra for compounds 1–6, 8, 11, 12, 16–19, 21–32, 1H NMR data for known compounds 14, 15. | ||
| Format: PDF | Size: 4.2 MB | Download |
References
-
Fukushi, Y.; Hakomori, S.-I.; Nudelman, E.; Cochran, N. J. Biol. Chem. 1984, 259, 4681–4685.
Return to citation in text: [1] -
Fukushi, Y.; Hakomori, S.-I.; Shepard, T. J. Exp. Med. 1984, 159, 506–520.
Return to citation in text: [1] -
Fukushi, Y.; Kannagi, R.; Hakomori, S.-I.; Shepard, T.; Kulander, B. G.; Singer, J. W. Cancer Res. 1985, 45, 3711–3717.
Return to citation in text: [1] -
Itzkowitz, S. H.; Yuan, M.; Fukushi, Y.; Palekar, A.; Phelps, P. C.; Shamsuddin, A. M.; Trump, B. T.; Hakomori, S.-I.; Kim, Y. S. Cancer Res. 1986, 46, 2627–2632.
Return to citation in text: [1] -
Nakasaki, H.; Mitomi, T.; Noto, T.; Ogoshi, K.; Hanaue, H.; Tanaka, Y.; Makuuchi, H.; Clausen, H.; Hakomori, S.-I. Cancer Res. 1989, 49, 3662–3669.
Return to citation in text: [1] -
Singhal, A. K.; Ørntoft, T. F.; Nudelman, E.; Nance, S.; Schibig, L.; Stroud, M. R.; Clausen, H.; Hakomori, S.-I. Cancer Res. 1990, 50, 1375–1380.
Return to citation in text: [1] -
Kobata, A.; Ginsburg, V. J. Biol. Chem. 1969, 244, 5496–5502.
Return to citation in text: [1] -
Yang, H.-J.; Hakomori, S.-I. J. Biol. Chem. 1971, 246, 1192–1200.
Return to citation in text: [1] -
Zhang, S.; Zhang, H. S.; Cordon-Cardo, C.; Reuter, V. E.; Singhal, A. K.; Lloyd, K. O.; Livingston, P. O. Int. J. Cancer 1997, 73, 50–56. doi:10.1002/(SICI)1097-0215(19970926)73:1<50::AID-IJC9>3.0.CO;2-0
Return to citation in text: [1] -
Satoh, J.; Kim, S. U. J. Neurosci. Res. 1994, 37, 466–474. doi:10.1002/jnr.490370406
Return to citation in text: [1] -
Croce, M. V.; Isla-Larrain, M.; Rabassa, M. E.; Demichelis, S.; Colussi, A. G.; Crespo, M.; Lacunza, E.; Segal-Eiras, A. Pathol. Oncol. Res. 2007, 13, 130–138. doi:10.1007/BF02893488
Return to citation in text: [1] -
Jacquinet, J.-C.; Sinaÿ, P. J. Chem. Soc. 1979, 314–318. doi:10.1039/P19790000314
Return to citation in text: [1] [2] -
Hindsgaul, O.; Norberg, T.; Pendu, J. L.; Lemieux, R. U. Carbohydr. Res. 1982, 109, 109–142. doi:10.1016/0008-6215(82)84034-2
Return to citation in text: [1] [2] -
Sato, S.; Ito, Y.; Nukada, T.; Nakahara, Y.; Ogawa, T. Carbohydr. Res. 1987, 167, 197–210. doi:10.1016/0008-6215(87)80279-3
Return to citation in text: [1] [2] -
Jain, R. K.; Matta, K. L. Carbohydr. Res. 1992, 226, 91–100. doi:10.1016/0008-6215(92)84057-Y
Return to citation in text: [1] [2] -
Numomura, S.; Iida, M.; Numata, M.; Sugimoto, M.; Ogawa, T. Carbohydr. Res. 1994, 263, C1–C6. doi:10.1016/0008-6215(94)00264-9
Return to citation in text: [1] [2] -
Jain, R. K.; Vig, R.; Rampal, R.; Chandrasekaran, E. V.; Matta, K. L. J. Am. Chem. Soc. 1994, 116, 12123–12124. doi:10.1021/ja00105a091
Return to citation in text: [1] [2] -
Yan, L.; Kahne, D. J. Am. Chem. Soc. 1996, 9239–9248. doi:10.1021/ja9608555
Return to citation in text: [1] [2] -
Lay, L.; Manzoni, L.; Schmidt, R. R. Carbohydr. Res. 1998, 310, 157–171. doi:10.1016/S0008-6215(98)00148-7
Return to citation in text: [1] [2] -
Figueroa-Pérez, S.; Verez-Bencomo, V. Tetrahedron Lett. 1998, 39, 9143–9146. doi:10.1016/S0040-4039(98)02104-2
Return to citation in text: [1] [2] -
Cao, S.; Gan, Z.; Roy, R. Carbohydr. Res. 1999, 318, 75–81. doi:10.1016/S0008-6215(99)00080-4
Return to citation in text: [1] [2] -
Gan, Z.; Cao, S.; Wu, Q.; Roy, R. J. Carbohydr. Chem. 1999, 18, 755–773. doi:10.1080/07328309908544034
Return to citation in text: [1] [2] -
Zhang, Y.-M.; Esnault, J.; Mallet, J.-M.; Sinaÿ, P. J. Carbohydr. Chem. 1999, 18, 419–427. doi:10.1080/07328309908544006
Return to citation in text: [1] [2] -
Zhu, T.; Boons, G.-J. J. Am. Chem. Soc. 2000, 122, 10222–10223. doi:10.1021/ja001930l
Return to citation in text: [1] [2] -
Zhu, T.; Boons, G.-J. Chem.–Eur. J. 2001, 7, 2382–2389. doi:10.1002/1521-3765(20010601)7:11<2382::AID-CHEM23820>3.0.CO;2-2
Return to citation in text: [1] [2] -
La Ferla, B.; Prosperi, D.; Lay, L.; Giovanni, R.; Panza, L. Carbohydr. Res. 2002, 337, 1333–1342. doi:10.1016/S0008-6215(02)00164-7
Return to citation in text: [1] [2] -
Xia, J.; Alderfer, J. L.; Locke, R. D.; Piskorz, C. F.; Matta, K. L. J. Org. Chem. 2003, 68, 2752–2759. doi:10.1021/jo020698u
Return to citation in text: [1] [2] -
Mukherjee, D.; Sarkar, S. K.; Chattopadhyay, P.; Chowdhury, U. S. J. Carbohydr. Chem. 2005, 24, 251–259. doi:10.1081/CAR-200058529
Return to citation in text: [1] [2] [3] -
Toepfer, A.; Schmidt, R. R. Tetrahedron Lett. 1992, 33, 5161–5164. doi:10.1016/S0040-4039(00)79122-2
Return to citation in text: [1] [2] -
Hummel, G.; Schmidt, R. R. Tetrahedron Lett. 1997, 38, 1173–1176. doi:10.1016/S0040-4039(97)00006-3
Return to citation in text: [1] [2] -
Kretzschmar, G.; Stahl, W. Tetrahedron 1998, 54, 6341–6358. doi:10.1016/S0040-4020(98)00294-4
Return to citation in text: [1] [2] [3] [4] [5] -
Manzoni, L.; Lay, L.; Schmidt, R. R. J. Carbohydr. Chem. 1998, 17, 769–758. doi:10.1080/07328309808002349
Return to citation in text: [1] [2] -
de la Fuente, J. M.; Penadés, S. Tetrahedron: Asymmetry 2002, 13, 1879–1888. doi:10.1016/S0957-4166(02)00480-9
Return to citation in text: [1] [2] [3] -
de Paz, J.-L.; Ojeda, R.; Barrientos, A. G.; Penadés, S.; Martín-Lomas, M. Tetrahedron: Asymmetry 2005, 16, 149–158. doi:10.1016/j.tetasy.2004.11.066
Return to citation in text: [1] [2] [3] -
Windmüller, R.; Schmidt, R. R. Tetrahedron Lett. 1994, 35, 7927–7930. doi:10.1016/0040-4039(94)80013-8
Return to citation in text: [1] [2] -
Sato, K.-I.; Seki, H.; Yoshimoto, A.; Nanaumi, H.; Takai, Y.; Ishido, Y. J. Carbohydr. Chem. 1998, 17, 703–727. doi:10.1080/07328309808002347
Return to citation in text: [1] [2] -
Ball, G. E.; O’Neill, R. A.; Schultz, J. E.; Lowe, J. B.; Weston, B. W.; Nagy, J. O.; Brown, E. G.; Hobbs, C. J.; Bednarski, M. D. J. Am. Chem. Soc. 1992, 114, 5449–5451. doi:10.1021/ja00039a080
Return to citation in text: [1] -
Nagahori, N.; Nishimura, S.-I. Chem.–Eur. J. 2006, 12, 6478–6485. doi:10.1002/chem.200501267
Return to citation in text: [1] -
Schmidt, R. R.; Stumpp, M. Liebigs Ann. Chem. 1983, 1249–1256. doi:10.1002/jlac.198319830717
Return to citation in text: [1] [2] -
Amvam-Zollo, P.-H.; Sinaÿ, P. Carbohydr. Res. 1986, 150, 199–212. doi:10.1016/0008-6215(86)80016-7
Return to citation in text: [1] [2] -
Toepfer, A.; Schmidt, R. R. J. Carbohydr. Chem. 1993, 12, 809–822. doi:10.1080/07328309308020096
Return to citation in text: [1] [2] -
Lönn, H. Carbohydr. Res. 1985, 139, 105–113. doi:10.1016/0008-6215(85)90011-4
Return to citation in text: [1] [2] -
Zemlyakov, A. E. Chem. Nat. Compd. 1998, 34, 80–85. doi:10.1007/BF02249693
Return to citation in text: [1] -
Lemieux, R. U.; Takeda, T.; Chung, B. Y. ACS Symp. Ser. 1976, 39, 90–115. doi:10.1021/bk-1977-0039.ch006
Return to citation in text: [1] -
Nitz, M.; Bundle, D. R. J. Org. Chem. 2000, 65, 3064–3073. doi:10.1021/jo991812k
Return to citation in text: [1] -
Joosten, J. A. F.; Kamerling, J. P.; Vliegenthart, J. F. G. Carbohydr. Res. 2003, 338, 2611–2627. doi:10.1016/S0008-6215(03)00313-6
Return to citation in text: [1] -
Choudhury, A. K.; Roy, N. J. Carbohydr. Chem. 1997, 16, 1363–1371. doi:10.1080/07328309708005755
Return to citation in text: [1] -
Paulsen, H. Angew. Chem., Int. Ed. Engl. 1982, 21, 155–173. doi:10.1002/anie.198201553
Return to citation in text: [1] -
Crich, D.; Dudkin, V. J. Am. Chem. Soc. 2001, 123, 6819–6825. doi:10.1021/ja010086b
Return to citation in text: [1] -
Liao, L.; Auzanneau, F.-I. Org. Lett. 2003, 5, 2607–2610. doi:10.1021/ol034669x
Return to citation in text: [1] -
Hendel, J. L.; Cheng, A.; Auzanneau, F.-I. Carbohydr. Res. 2008, 343, 2914–2923. doi:10.1016/j.carres.2008.08.025
Return to citation in text: [1] [2] -
Kwon, Y.; Soucy, R. L.; Snyder, D. A.; Seeberger, P. H. Chem.–Eur. J. 2005, 11, 2493–2504. doi:10.1002/chem.200400934
Return to citation in text: [1] [2] -
Ratner, D. M.; Adams, E. W.; Su, J.; O’Keefe, R. R.; Mrksich, M.; Seeberger, P. H. ChemBioChem 2004, 5, 379–383. doi:10.1002/cbic.200300804
Return to citation in text: [1] -
Armarego, W. L. F.; Chai, C. L. L. Purification of Laboratory Chemicals, 4th ed.; Elsevier, 2003.
Return to citation in text: [1]
| 39. | Schmidt, R. R.; Stumpp, M. Liebigs Ann. Chem. 1983, 1249–1256. doi:10.1002/jlac.198319830717 |
| 40. | Amvam-Zollo, P.-H.; Sinaÿ, P. Carbohydr. Res. 1986, 150, 199–212. doi:10.1016/0008-6215(86)80016-7 |
| 41. | Toepfer, A.; Schmidt, R. R. J. Carbohydr. Chem. 1993, 12, 809–822. doi:10.1080/07328309308020096 |
| 42. | Lönn, H. Carbohydr. Res. 1985, 139, 105–113. doi:10.1016/0008-6215(85)90011-4 |
| 1. | Fukushi, Y.; Hakomori, S.-I.; Nudelman, E.; Cochran, N. J. Biol. Chem. 1984, 259, 4681–4685. |
| 2. | Fukushi, Y.; Hakomori, S.-I.; Shepard, T. J. Exp. Med. 1984, 159, 506–520. |
| 3. | Fukushi, Y.; Kannagi, R.; Hakomori, S.-I.; Shepard, T.; Kulander, B. G.; Singer, J. W. Cancer Res. 1985, 45, 3711–3717. |
| 4. | Itzkowitz, S. H.; Yuan, M.; Fukushi, Y.; Palekar, A.; Phelps, P. C.; Shamsuddin, A. M.; Trump, B. T.; Hakomori, S.-I.; Kim, Y. S. Cancer Res. 1986, 46, 2627–2632. |
| 5. | Nakasaki, H.; Mitomi, T.; Noto, T.; Ogoshi, K.; Hanaue, H.; Tanaka, Y.; Makuuchi, H.; Clausen, H.; Hakomori, S.-I. Cancer Res. 1989, 49, 3662–3669. |
| 6. | Singhal, A. K.; Ørntoft, T. F.; Nudelman, E.; Nance, S.; Schibig, L.; Stroud, M. R.; Clausen, H.; Hakomori, S.-I. Cancer Res. 1990, 50, 1375–1380. |
| 37. | Ball, G. E.; O’Neill, R. A.; Schultz, J. E.; Lowe, J. B.; Weston, B. W.; Nagy, J. O.; Brown, E. G.; Hobbs, C. J.; Bednarski, M. D. J. Am. Chem. Soc. 1992, 114, 5449–5451. doi:10.1021/ja00039a080 |
| 38. | Nagahori, N.; Nishimura, S.-I. Chem.–Eur. J. 2006, 12, 6478–6485. doi:10.1002/chem.200501267 |
| 47. | Choudhury, A. K.; Roy, N. J. Carbohydr. Chem. 1997, 16, 1363–1371. doi:10.1080/07328309708005755 |
| 12. | Jacquinet, J.-C.; Sinaÿ, P. J. Chem. Soc. 1979, 314–318. doi:10.1039/P19790000314 |
| 13. | Hindsgaul, O.; Norberg, T.; Pendu, J. L.; Lemieux, R. U. Carbohydr. Res. 1982, 109, 109–142. doi:10.1016/0008-6215(82)84034-2 |
| 14. | Sato, S.; Ito, Y.; Nukada, T.; Nakahara, Y.; Ogawa, T. Carbohydr. Res. 1987, 167, 197–210. doi:10.1016/0008-6215(87)80279-3 |
| 15. | Jain, R. K.; Matta, K. L. Carbohydr. Res. 1992, 226, 91–100. doi:10.1016/0008-6215(92)84057-Y |
| 16. | Numomura, S.; Iida, M.; Numata, M.; Sugimoto, M.; Ogawa, T. Carbohydr. Res. 1994, 263, C1–C6. doi:10.1016/0008-6215(94)00264-9 |
| 17. | Jain, R. K.; Vig, R.; Rampal, R.; Chandrasekaran, E. V.; Matta, K. L. J. Am. Chem. Soc. 1994, 116, 12123–12124. doi:10.1021/ja00105a091 |
| 18. | Yan, L.; Kahne, D. J. Am. Chem. Soc. 1996, 9239–9248. doi:10.1021/ja9608555 |
| 19. | Lay, L.; Manzoni, L.; Schmidt, R. R. Carbohydr. Res. 1998, 310, 157–171. doi:10.1016/S0008-6215(98)00148-7 |
| 20. | Figueroa-Pérez, S.; Verez-Bencomo, V. Tetrahedron Lett. 1998, 39, 9143–9146. doi:10.1016/S0040-4039(98)02104-2 |
| 21. | Cao, S.; Gan, Z.; Roy, R. Carbohydr. Res. 1999, 318, 75–81. doi:10.1016/S0008-6215(99)00080-4 |
| 22. | Gan, Z.; Cao, S.; Wu, Q.; Roy, R. J. Carbohydr. Chem. 1999, 18, 755–773. doi:10.1080/07328309908544034 |
| 23. | Zhang, Y.-M.; Esnault, J.; Mallet, J.-M.; Sinaÿ, P. J. Carbohydr. Chem. 1999, 18, 419–427. doi:10.1080/07328309908544006 |
| 24. | Zhu, T.; Boons, G.-J. J. Am. Chem. Soc. 2000, 122, 10222–10223. doi:10.1021/ja001930l |
| 25. | Zhu, T.; Boons, G.-J. Chem.–Eur. J. 2001, 7, 2382–2389. doi:10.1002/1521-3765(20010601)7:11<2382::AID-CHEM23820>3.0.CO;2-2 |
| 26. | La Ferla, B.; Prosperi, D.; Lay, L.; Giovanni, R.; Panza, L. Carbohydr. Res. 2002, 337, 1333–1342. doi:10.1016/S0008-6215(02)00164-7 |
| 27. | Xia, J.; Alderfer, J. L.; Locke, R. D.; Piskorz, C. F.; Matta, K. L. J. Org. Chem. 2003, 68, 2752–2759. doi:10.1021/jo020698u |
| 28. | Mukherjee, D.; Sarkar, S. K.; Chattopadhyay, P.; Chowdhury, U. S. J. Carbohydr. Chem. 2005, 24, 251–259. doi:10.1081/CAR-200058529 |
| 29. | Toepfer, A.; Schmidt, R. R. Tetrahedron Lett. 1992, 33, 5161–5164. doi:10.1016/S0040-4039(00)79122-2 |
| 30. | Hummel, G.; Schmidt, R. R. Tetrahedron Lett. 1997, 38, 1173–1176. doi:10.1016/S0040-4039(97)00006-3 |
| 31. | Kretzschmar, G.; Stahl, W. Tetrahedron 1998, 54, 6341–6358. doi:10.1016/S0040-4020(98)00294-4 |
| 32. | Manzoni, L.; Lay, L.; Schmidt, R. R. J. Carbohydr. Chem. 1998, 17, 769–758. doi:10.1080/07328309808002349 |
| 33. | de la Fuente, J. M.; Penadés, S. Tetrahedron: Asymmetry 2002, 13, 1879–1888. doi:10.1016/S0957-4166(02)00480-9 |
| 34. | de Paz, J.-L.; Ojeda, R.; Barrientos, A. G.; Penadés, S.; Martín-Lomas, M. Tetrahedron: Asymmetry 2005, 16, 149–158. doi:10.1016/j.tetasy.2004.11.066 |
| 35. | Windmüller, R.; Schmidt, R. R. Tetrahedron Lett. 1994, 35, 7927–7930. doi:10.1016/0040-4039(94)80013-8 |
| 36. | Sato, K.-I.; Seki, H.; Yoshimoto, A.; Nanaumi, H.; Takai, Y.; Ishido, Y. J. Carbohydr. Chem. 1998, 17, 703–727. doi:10.1080/07328309808002347 |
| 48. | Paulsen, H. Angew. Chem., Int. Ed. Engl. 1982, 21, 155–173. doi:10.1002/anie.198201553 |
| 49. | Crich, D.; Dudkin, V. J. Am. Chem. Soc. 2001, 123, 6819–6825. doi:10.1021/ja010086b |
| 50. | Liao, L.; Auzanneau, F.-I. Org. Lett. 2003, 5, 2607–2610. doi:10.1021/ol034669x |
| 9. | Zhang, S.; Zhang, H. S.; Cordon-Cardo, C.; Reuter, V. E.; Singhal, A. K.; Lloyd, K. O.; Livingston, P. O. Int. J. Cancer 1997, 73, 50–56. doi:10.1002/(SICI)1097-0215(19970926)73:1<50::AID-IJC9>3.0.CO;2-0 |
| 10. | Satoh, J.; Kim, S. U. J. Neurosci. Res. 1994, 37, 466–474. doi:10.1002/jnr.490370406 |
| 11. | Croce, M. V.; Isla-Larrain, M.; Rabassa, M. E.; Demichelis, S.; Colussi, A. G.; Crespo, M.; Lacunza, E.; Segal-Eiras, A. Pathol. Oncol. Res. 2007, 13, 130–138. doi:10.1007/BF02893488 |
| 45. | Nitz, M.; Bundle, D. R. J. Org. Chem. 2000, 65, 3064–3073. doi:10.1021/jo991812k |
| 7. | Kobata, A.; Ginsburg, V. J. Biol. Chem. 1969, 244, 5496–5502. |
| 8. | Yang, H.-J.; Hakomori, S.-I. J. Biol. Chem. 1971, 246, 1192–1200. |
| 46. | Joosten, J. A. F.; Kamerling, J. P.; Vliegenthart, J. F. G. Carbohydr. Res. 2003, 338, 2611–2627. doi:10.1016/S0008-6215(03)00313-6 |
| 39. | Schmidt, R. R.; Stumpp, M. Liebigs Ann. Chem. 1983, 1249–1256. doi:10.1002/jlac.198319830717 |
| 40. | Amvam-Zollo, P.-H.; Sinaÿ, P. Carbohydr. Res. 1986, 150, 199–212. doi:10.1016/0008-6215(86)80016-7 |
| 41. | Toepfer, A.; Schmidt, R. R. J. Carbohydr. Chem. 1993, 12, 809–822. doi:10.1080/07328309308020096 |
| 35. | Windmüller, R.; Schmidt, R. R. Tetrahedron Lett. 1994, 35, 7927–7930. doi:10.1016/0040-4039(94)80013-8 |
| 36. | Sato, K.-I.; Seki, H.; Yoshimoto, A.; Nanaumi, H.; Takai, Y.; Ishido, Y. J. Carbohydr. Chem. 1998, 17, 703–727. doi:10.1080/07328309808002347 |
| 44. | Lemieux, R. U.; Takeda, T.; Chung, B. Y. ACS Symp. Ser. 1976, 39, 90–115. doi:10.1021/bk-1977-0039.ch006 |
| 28. | Mukherjee, D.; Sarkar, S. K.; Chattopadhyay, P.; Chowdhury, U. S. J. Carbohydr. Chem. 2005, 24, 251–259. doi:10.1081/CAR-200058529 |
| 29. | Toepfer, A.; Schmidt, R. R. Tetrahedron Lett. 1992, 33, 5161–5164. doi:10.1016/S0040-4039(00)79122-2 |
| 30. | Hummel, G.; Schmidt, R. R. Tetrahedron Lett. 1997, 38, 1173–1176. doi:10.1016/S0040-4039(97)00006-3 |
| 31. | Kretzschmar, G.; Stahl, W. Tetrahedron 1998, 54, 6341–6358. doi:10.1016/S0040-4020(98)00294-4 |
| 32. | Manzoni, L.; Lay, L.; Schmidt, R. R. J. Carbohydr. Chem. 1998, 17, 769–758. doi:10.1080/07328309808002349 |
| 33. | de la Fuente, J. M.; Penadés, S. Tetrahedron: Asymmetry 2002, 13, 1879–1888. doi:10.1016/S0957-4166(02)00480-9 |
| 34. | de Paz, J.-L.; Ojeda, R.; Barrientos, A. G.; Penadés, S.; Martín-Lomas, M. Tetrahedron: Asymmetry 2005, 16, 149–158. doi:10.1016/j.tetasy.2004.11.066 |
| 12. | Jacquinet, J.-C.; Sinaÿ, P. J. Chem. Soc. 1979, 314–318. doi:10.1039/P19790000314 |
| 13. | Hindsgaul, O.; Norberg, T.; Pendu, J. L.; Lemieux, R. U. Carbohydr. Res. 1982, 109, 109–142. doi:10.1016/0008-6215(82)84034-2 |
| 14. | Sato, S.; Ito, Y.; Nukada, T.; Nakahara, Y.; Ogawa, T. Carbohydr. Res. 1987, 167, 197–210. doi:10.1016/0008-6215(87)80279-3 |
| 15. | Jain, R. K.; Matta, K. L. Carbohydr. Res. 1992, 226, 91–100. doi:10.1016/0008-6215(92)84057-Y |
| 16. | Numomura, S.; Iida, M.; Numata, M.; Sugimoto, M.; Ogawa, T. Carbohydr. Res. 1994, 263, C1–C6. doi:10.1016/0008-6215(94)00264-9 |
| 17. | Jain, R. K.; Vig, R.; Rampal, R.; Chandrasekaran, E. V.; Matta, K. L. J. Am. Chem. Soc. 1994, 116, 12123–12124. doi:10.1021/ja00105a091 |
| 18. | Yan, L.; Kahne, D. J. Am. Chem. Soc. 1996, 9239–9248. doi:10.1021/ja9608555 |
| 19. | Lay, L.; Manzoni, L.; Schmidt, R. R. Carbohydr. Res. 1998, 310, 157–171. doi:10.1016/S0008-6215(98)00148-7 |
| 20. | Figueroa-Pérez, S.; Verez-Bencomo, V. Tetrahedron Lett. 1998, 39, 9143–9146. doi:10.1016/S0040-4039(98)02104-2 |
| 21. | Cao, S.; Gan, Z.; Roy, R. Carbohydr. Res. 1999, 318, 75–81. doi:10.1016/S0008-6215(99)00080-4 |
| 22. | Gan, Z.; Cao, S.; Wu, Q.; Roy, R. J. Carbohydr. Chem. 1999, 18, 755–773. doi:10.1080/07328309908544034 |
| 23. | Zhang, Y.-M.; Esnault, J.; Mallet, J.-M.; Sinaÿ, P. J. Carbohydr. Chem. 1999, 18, 419–427. doi:10.1080/07328309908544006 |
| 24. | Zhu, T.; Boons, G.-J. J. Am. Chem. Soc. 2000, 122, 10222–10223. doi:10.1021/ja001930l |
| 25. | Zhu, T.; Boons, G.-J. Chem.–Eur. J. 2001, 7, 2382–2389. doi:10.1002/1521-3765(20010601)7:11<2382::AID-CHEM23820>3.0.CO;2-2 |
| 26. | La Ferla, B.; Prosperi, D.; Lay, L.; Giovanni, R.; Panza, L. Carbohydr. Res. 2002, 337, 1333–1342. doi:10.1016/S0008-6215(02)00164-7 |
| 27. | Xia, J.; Alderfer, J. L.; Locke, R. D.; Piskorz, C. F.; Matta, K. L. J. Org. Chem. 2003, 68, 2752–2759. doi:10.1021/jo020698u |
| 28. | Mukherjee, D.; Sarkar, S. K.; Chattopadhyay, P.; Chowdhury, U. S. J. Carbohydr. Chem. 2005, 24, 251–259. doi:10.1081/CAR-200058529 |
| 42. | Lönn, H. Carbohydr. Res. 1985, 139, 105–113. doi:10.1016/0008-6215(85)90011-4 |
| 52. | Kwon, Y.; Soucy, R. L.; Snyder, D. A.; Seeberger, P. H. Chem.–Eur. J. 2005, 11, 2493–2504. doi:10.1002/chem.200400934 |
| 51. | Hendel, J. L.; Cheng, A.; Auzanneau, F.-I. Carbohydr. Res. 2008, 343, 2914–2923. doi:10.1016/j.carres.2008.08.025 |
| 51. | Hendel, J. L.; Cheng, A.; Auzanneau, F.-I. Carbohydr. Res. 2008, 343, 2914–2923. doi:10.1016/j.carres.2008.08.025 |
| 31. | Kretzschmar, G.; Stahl, W. Tetrahedron 1998, 54, 6341–6358. doi:10.1016/S0040-4020(98)00294-4 |
| 31. | Kretzschmar, G.; Stahl, W. Tetrahedron 1998, 54, 6341–6358. doi:10.1016/S0040-4020(98)00294-4 |
| 34. | de Paz, J.-L.; Ojeda, R.; Barrientos, A. G.; Penadés, S.; Martín-Lomas, M. Tetrahedron: Asymmetry 2005, 16, 149–158. doi:10.1016/j.tetasy.2004.11.066 |
| 54. | Armarego, W. L. F.; Chai, C. L. L. Purification of Laboratory Chemicals, 4th ed.; Elsevier, 2003. |
| 33. | de la Fuente, J. M.; Penadés, S. Tetrahedron: Asymmetry 2002, 13, 1879–1888. doi:10.1016/S0957-4166(02)00480-9 |
| 53. | Ratner, D. M.; Adams, E. W.; Su, J.; O’Keefe, R. R.; Mrksich, M.; Seeberger, P. H. ChemBioChem 2004, 5, 379–383. doi:10.1002/cbic.200300804 |
| 31. | Kretzschmar, G.; Stahl, W. Tetrahedron 1998, 54, 6341–6358. doi:10.1016/S0040-4020(98)00294-4 |
| 52. | Kwon, Y.; Soucy, R. L.; Snyder, D. A.; Seeberger, P. H. Chem.–Eur. J. 2005, 11, 2493–2504. doi:10.1002/chem.200400934 |
© 2010 Wang et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)